

Abstract
Objective:
Genome sequencing (GS) is promising for unsolved leukodystrophies, but its efficacy has not been prospectively studied.
Methods:
A prospective time-delayed crossover design trial of GS to assess the efficacy of GS as a first-line diagnostic tool for genetic white matter disorders took place between December 1, 2015 and September 27, 2017. Patients were randomized to receive GS immediately with concurrent standard of care (SoC) testing, or to receive SoC testing for 4 months followed by GS.
Results:
Thirty-four individuals were assessed at interim review. The genetic origin of 2 patient’s leukoencephalopathy was resolved before randomization. Nine patients were stratified to the immediate intervention group and 23 patients to the delayed-GS arm. The efficacy of GS was significant relative to SoC in the immediate (5/9 [56%] vs 0/9 [0%]; Wild–Seber, p < 0.005) and delayed (control) arms (14/23 [61%] vs 5/23 [22%]; Wild–Seber, p < 0.005). The time to diagnosis was significantly shorter in the immediate-GS group (log-rank test, p = 0.04). The overall diagnostic efficacy of combined GS and SoC approaches was 26 of 34 (76.5%, 95% confidence interval = 58.8–89.3%) in <4 months, greater than historical norms of <50% over 5 years. Owing to loss of clinical equipoise, the trial design was altered to a single-arm observational study.
Interpretation:
In this study, first-line GS provided earlier and greater diagnostic efficacy in white matter disorders. We provide an evidence-based diagnostic testing algorithm to enable appropriate clinical GS utilization in this population.
Introduction
Pediatric onset white matter disorders, which include the leukodystrophies and genetic leukoencephalopathies,1 are caused by defects in any of the white matter structural components, including oligodendrocytes, astrocytes, microglia, axons, and blood vessels.2 Pediatric white matter disorders may be acquired (such as in the case of infection, multiple sclerosis, or trauma) or have a genetic etiology, and >100 unique genetic disorders affecting white matter growth, development, or maintenance have been described.3,4 The advent of high-resolution magnetic resonance imaging (MRI) and disease-specific pattern recognition has enabled an increase in overall diagnostic success, but these disorders remain challenging for clinicians.5–7 As recently as 2010, it was thought that only 50% of patients with white matter disorders had the genetic cause of their disorders successfully diagnosed,8,9 with time to diagnosis extending 8 years or more in the majority of cases.10 A rapid and accurate diagnosis in patients with white matter disorders can have a meaningful improvement in care, enabling appropriate therapeutic selection, avoidance of unnecessary interventions or diagnostic procedures, and prospective disease management.11
A growing number of studies have shown that next generation sequencing (NGS) can increase the overall diagnostic yield in patients with white matter abnormalities. For example, exome sequencing (ES) was able to resolve 42% of individuals in a cohort of individuals with white matter disorders for whom the underlying genetic cause had remained unresolved for long periods, and when retrospectively combined with standard of care (SoC) approaches yielded an overall 72% diagnostic success rate.7 A similar study of patients presenting to a pediatric neurology clinic with various neurodevelopmental disabilities found a 41% diagnostic success rate by ES alone, with half of the resolved individuals showing neuroimaging abnormalities.12 A clinical utility study of ES compared to conventional genetic testing in a cohort of individuals recruited from nongenetic subspecialty clinics found ES testing resulted in a higher diagnostic yield without an increase in costs.13 Combined with the increasing number of studies indicating that NGS investigations of unresolved individuals can yield novel insights and disease-associated genes,4,14 these findings strongly indicate a place for agnostic genomic testing approaches as a first-tier test for patients with neurological disorders.
Here we describe what is, to our knowledge, the first randomized control trial of genome sequencing (GS) in the pediatric neurology population, with a focus on individuals presenting with suspected genetic white matter abnormalities detected by MRI. GS is capable of detecting both small variants (single nucleotide variants [SNVs] and indels, typically detected by ES) and large copy number variants (CNVs; typically detected by chromosomal microarray) in both the nuclear and mitochondrial genome, and therefore holds potential as a unified genetic testing platform. Two recent studies have indicated that GS can improve diagnostic yield and clinical care when compared to SoC both in critically ill infants15 and in pediatric patients with a clinical phenotype indicative of an underlying genetic disorder.13 In this study, we compared GS to SoC testing with respect to both overall diagnostic yield and time to diagnosis, and find that GS should be considered as a first-line diagnostic tool for white matter disorder patients without a definitive diagnosis by clinical assessment and MRI.
Patients and Methods
Patient Recruitment and Study Design
Patients with a white matter disorder confirmed by an MRI performed no more than 2 months prior to enrollment were recruited to the LeukoSEQ Clinical Trial (NCT02699190). Additional enrollment criteria (Table 1) for this study established that the subject must show no evidence of an acquired cause for the white matter abnormalities, show no preexisting clinical or genetic diagnosis, be younger than 18 years, have both biologic parents available for trio GS, and have no prior or pending genomic testing (GS, ES, or large gene panels). The index MRI was reviewed by a minimum of 2 pediatric neurologists (A.V., M.S.v.d.K., or G.B.). This study was restricted to residents of the United States. No changes were made to the eligibility criteria after trial commencement for the data presented in this interim analysis. Written informed consent was given by all participants and obtained by the study coordinator or a certified genetic counselor. This study was approved by the institutional review board (IRB) at the Children’s Hospital of Philadelphia (IRB #16–013213).
TABLE 1.
Inclusion and Exclusion Criteria for the Study
| Inclusion Criteria (cases only) |
| Abnormalities of the white matter signal on neuroimaging (MRI) with T2 hyperintensity, which must be diffuse or involve specific anatomical tracts consistent with a genetic diagnosis |
| MRI abnormalities identified <2 months prior to enrollment |
| No evidence of an acquired cause for the white matter abnormalities (infection, trauma, birth related injury) |
| No preexisting diagnosis |
| Less than 18 years of age |
| Availability of both biologic parents for genomic testing |
| Exclusion Criteria (cases only) |
| Candidates with acquired disorders, including infection, ADEM, multiple sclerosis, vasculitis, or toxic leukoencephalopathies |
| Patients who have had previous genetic testing, including WES, WGS, or iterative panel testing of >20 cumulative genes; karyotype or microarray testing that did not yield a definitive diagnosis should not be considered as an excluding factor |
| Those with no third-party payer insurance, unable to receive standard of care diagnosis and therapeutic treatment |
| Candidates who have already received a definitive etiological diagnosis |
| Inclusion Criteria (parents) |
| Males or females of any age |
| Child with a suspected white matter disorder |
| No known/suspected disease or condition |
| Exclusion Criteria (parent) |
| Inability to provide consent |
ADEM = acute disseminated encephalomyelitis; MRI = magnetic resonance imaging; WES = whole exome sequencing; WGS = whole genome sequencing.
A randomized time-delayed crossover study design (Fig 1) was employed, and participants were assigned to either immediate-GS (“treatment arm”) or to the SoC diagnostic approaches with delayed-GS (“control arm” or delayed-GS arm) for 4 months followed by GS. Randomization was performed using a built-in feature on REDCap that relies on an allocation table provided by a statistician. The study used a centralized stratified permuted block randomization to minimize differences in diagnostic expertise between clinical sites, and to account for age and gender. The study staff performed assignment after recruitment and enrollment, which was blinded to coinvestigators, the referring physicians, and study participants and their families. SoC is defined as routine clinical testing employed by clinicians for testing disorders of expected genetic origin, including radiologic, enzymatic, biochemical analyte, chromosomal, targeted, or gene panel testing (including mitochondrial genome testing).14 Participants in the control arm who did not achieve a diagnosis in the 4-month control period were automatically assigned to receive GS.
FIGURE 1:
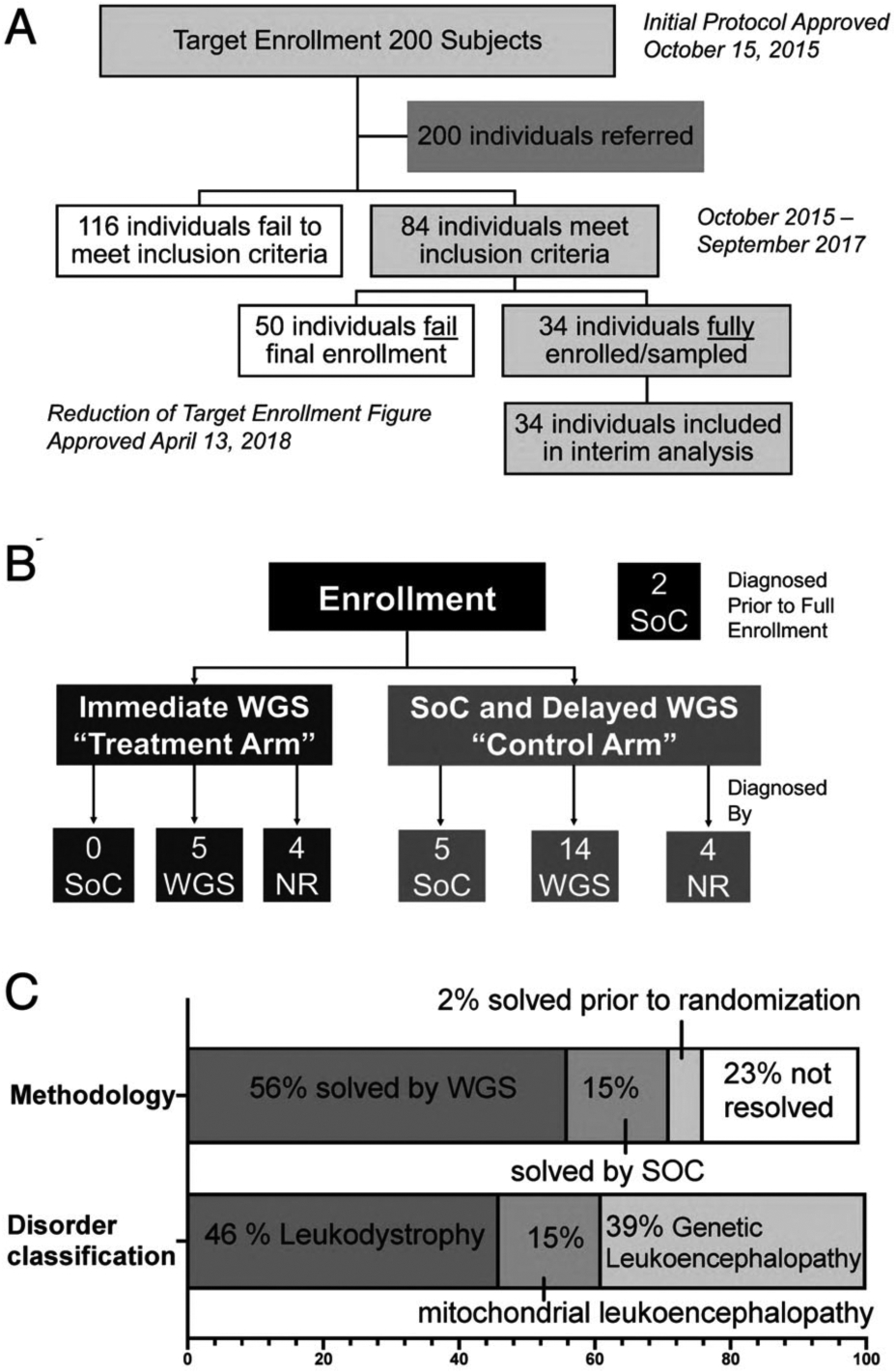
LeukoSeq trial design and principal results. (A) Overall recruitment and enrollment of the cohort examined in this study. (B) The trial employed a time-delayed crossover design with one-third of individuals assigned to the immediate genome sequencing (GS) arm and two-thirds of individuals receiving standard of care (SoC) for 4 months followed by GS. Of the 9 individuals assigned to immediate- GS, none received a diagnosis during the study period using SoC, 5 received a diagnosis using GS, and 4 did not achieve a diagnosis. Of the 23 individuals undergoing SoC with delayed-GS, only 5 individuals achieved a diagnosis with SoC approaches. Fourteen individuals achieved a diagnosis using GS, and 4 individuals did not achieve a diagnosis. Two patients noted in the black box at the top right received a diagnosis prior to randomization. (C) Distribution of cases solved by modality (SoC or GS) and broad class of white matter disorder. NR = not resolved; WGS = whole genome sequencing.
Clinical Genome Sequencing
Whole blood samples from enrolled participants and their biological parents were provided to the Illumina Clinical Services Laboratory (Illumina, San Diego, CA) for DNA extraction and clinical GS. Sequencing libraries were prepared using Illumina’s TruSeq DNA PCR-Free kit. Sequencing was performed on either a HiSeq2000 or a HiSeqX. Sequencing data were aligned to build 37.1 of the Human Reference Genome. All samples were sequenced to a minimum average coverage of ≥30-fold, with >99% of the genome covered at ≥10-fold coverage and ≥97% of the genome callable. Variant calling was performed using whole genome sequencing (WGS) workflow iSAAC 03.16.01.15, germline variant caller version 2.4.1 (Illumina), with SNVs and indels called using the Strelka germline caller16 and CNVs identified with Canvas.17,18 All variants were classified according to the American College of Medical Genetics standards and guidelines for interpretation of genetic sequence variants.19 In individuals where a variant of unknown significance was reported in a known gene, clinical confirmation (including orthogonal testing and detailed phenotypic evaluation) was performed prior to determining whether a case was classified as diagnosed. Disorders were classified as “canonical” leukodystrophies if they met inclusion criteria into the case description of leukodystrophies previously published based on consensus criteria.20
Statistical Analysis
Sample size and power analyses were calculated based on the assumption that the proportion of correct diagnosis using the current diagnostic approach is 50% and were determined using the nQuery Advisor Sample Size software version 7 (Statsols).21 Previous studies have shown high diagnostic efficacy using genome-wide testing approaches.7 Taking this into account, the LeukSeq study design anticipated that the rate of diagnosis using GS would exceed current SoC approaches, and our power analysis required randomization of two-thirds of all enrolled individuals to SoC and one-third to GS. To evaluate the diagnostic efficacy of delayed-GS in the control arm relative to SoC in each arm and in the entire cohort, we applied a test of proportions that allows for overlapping proportions,22 for example, those that are due to the presence of the same subjects in the SoC group and the delayed-GS group. Kaplan–Meier analysis with a log-rank test was used to compare the time to diagnosis between the immediate-GS in the treatment arm and delayed-GS in the control arm. A versatile log-rank test that is more sensitive to nonproportional hazards was also applied in a sensitivity analysis23; this confirmed the significant finding for the log-rank test. The median time to diagnosis in the control arm and treatment arms was computed for time following enrollment and then for time following initial MRI. The median time of diagnosis was estimated as the time at which the survival curve for the Kaplan–Meier curve crosses 0.50.
Secondary outcomes assessment of a comparison between expert MRI analysis and final molecular diagnosis for disorders defined as canonical leukodystrophies20 was achieved by computing the percentage of MRI analyses for leukodystrophies with 95% confidence intervals (CIs) based on the exact binomial distribution. Canonical leukodystrophies were defined by consensus opinion of leukodystrophy experts using a modified Delphi approach and considered those with primarily glial cell or myelin sheath involvement within the central nervous system.20 The overall diagnostic efficacy of the combination of GS and SoC approaches was evaluated by obtaining the percentage of diagnoses achieved in all subjects (with 95% CI) and employing a binomial test of proportions to assess whether the proportion of diagnoses achieved differed from the historical norm of 0.50.8
Two modifications have been made to the study design during the execution of this trial, and after the first subject was enrolled in November 2015. The first, planned in September of 2017 and IRB approved and updated in www.clinicaltrials.gov in April 2018, was a revision of the overall recruitment plan. In the original study protocol, enrollment was planned at 200 subjects, and a single interim analysis was planned to compare only the median time to diagnosis between those patients who received GS and those who received SoC. This was planned to occur after 100 patients had been enrolled in the study (of a total of 200). Slower than expected recruitment led to re-examination of the power analysis, and approval of a smaller cohort of 50 individuals with a planned interim analysis after at least half the cohort was collected. Interim analysis was therefore also conducted on a smaller number of individuals and included all subjects recruited until September 2017 (n = 34).
A second modification has been made subsequent to the interim analysis, and based on the loss of equipoise (evidence of benefit to individuals enrolled in the immediate cohort). The study was changed to a single arm study, in which all individuals will receive immediate-GS (approved in December 2018). Subjects enrolled after September 2017 are not included in the analysis described below and will be presented, along with clinical utility data, in a later publication at study closure.
Results
Of 200 candidates referred for study enrollment (see Fig 1A), we identified 84 patients who met the inclusion criteria of clinical history and MRI features consistent with a white matter disorder of possible genetic etiology (see Table 1). Fifty were ultimately excluded due to an MRI obtained >2 months prior to enrollment or due to preexisting broad-based genetic testing (exome sequencing or large panels). Thirty-four individuals were fully enrolled and had received results by the time of interim analysis (December 1, 2015 to September 27, 2017). The median age at enrollment was 1.4 years (interquartile range = 0.7–2.7 years). There were 20 females and 14 males in the study (Table 2). None of the affected children had a known family history of a defined genetic disease, although one individual had a sibling who had passed away in infancy without known cause and without genetic evaluation. Another individual was found, after diagnosis, to have a similarly although more mildly affected sibling and mother, and the parent of a third individual was found, after diagnosis, to have a possible family history of gait abnormalities. Clinical and neuroimaging of all solved cases is described in Supplementary Table 4, as well as Supplementary Tables 2 and 3.
TABLE 2.
LeukoSeq Study Demographics
| Demographic | Value in LeukoSeq Cohort |
|---|---|
| Referrals meeting inclusion criteria | 84 |
| Enrolled with results at time of analysis | 34 |
| Average age at enrollment | 3.34 years (range = 0.41–10.2 years) |
| Gender | 20 females, 14 males |
| Randomized to immediate-GS (“treatment arm”) | 3 females, 6 males (median age = 5.1 years) |
| Randomized to SoC with delayed-GS (“control arm”) | 16 females, 7 males (median age = 2.9 years) |
| Solved prior to randomization | 1 female, 1 male (median age = 1.5 years) |
GS = genome sequencing; SoC = standard of care.
The study used a time-delayed crossover design, with one-third of the patients randomized to receive immediate-GS in <30 days (treatment arm), and two-thirds to receive standard clinical care for 4 months followed by GS if they did not have a defined genetic diagnosis at crossover (control arm; see Fig 1C). Both arms of the study received SoC clinical diagnostic testing, including enzymatic and biochemical testing and targeted sequencing, throughout the course of the investigation. At the time of interim analysis, 9 patients were randomized to immediate-GS and 23 to delayed-GS, and 2 individuals had received a diagnosis by standard diagnostic approaches prior to randomization (see Fig 1B). In these 2 individuals, the diagnoses were based on urine organic acids (in an individual with Canavan disease [Mendelian Inheritance in Man (MIM) 271900]) or on leukocyte lysosomal enzyme analysis (in an individual with metachromatic leukodystrophy [MIM 250100]).
Of the 9 individuals in the immediate-GS arm, none received a diagnosis during the study period by SoC, and 5 received a diagnosis using GS (56% in a median of 5.4 weeks after enrollment and a median of 18.6 weeks after initial MRI). Four did not achieve a diagnosis (Figs 1B and 2A and Supplementary Table 4). Of the 23 individuals in the delayed-GS arm, only 5 individuals (22%) achieved a diagnosis with SoC approaches. Those who received a diagnosis by SoC in this arm were diagnosed by targeted gene or panel-based testing for leukodystrophy genes. Fourteen individuals achieved a diagnosis using GS (61% in a median of 22.1 weeks after enrollment and a median of 29.9 weeks after index MRI), and 4 individuals did not have genetic resolution of their conditions (see Figs 1B and 2A and Supplementary Table 4). Comparison of diagnostic efficacy of WGS versus SoC in the immediate group (5/9 vs 0/9; Wild–Seber test of overlapping proportions, p < 0.005), in the delayed group (14/23 vs 5/23; Wild–Seber test of overlapping proportions, p < 0.005) were significant. The diagnostic efficacy of GS in both the immediate (treatment; 5/9 [56%]) and delayed (control) arms (14/23 [61%]; total in both groups of 19/32) was significant relative to SoC (5/23 [22%]) overall (5/32 [16%] for SoC vs 19/32 [59%]; Wild–Seber test, p < 0.005). Likewise, the time to diagnosis was significantly shorter in the immediate-GS group than in the delayed-GS arm (9/9 participants who received immediate-GS were diagnosed within 5 weeks vs 5/22 participants who received SoC; p = 0.04, log-rank test; hazard ratio = 3.4) even when accounting for the rapid crossover to delayed-GS in unsolved individuals (4 months). Overall, 26 of 34 (76.5%; 95% CI = 58.8–89.3%) individuals were resolved, of whom 19 of 26 (73%) were resolved by GS. This was significantly different from the historical rate of diagnosis of <50% (p = 0.002, exact test of the hypothesis based on the binomial distribution).
FIGURE 2:
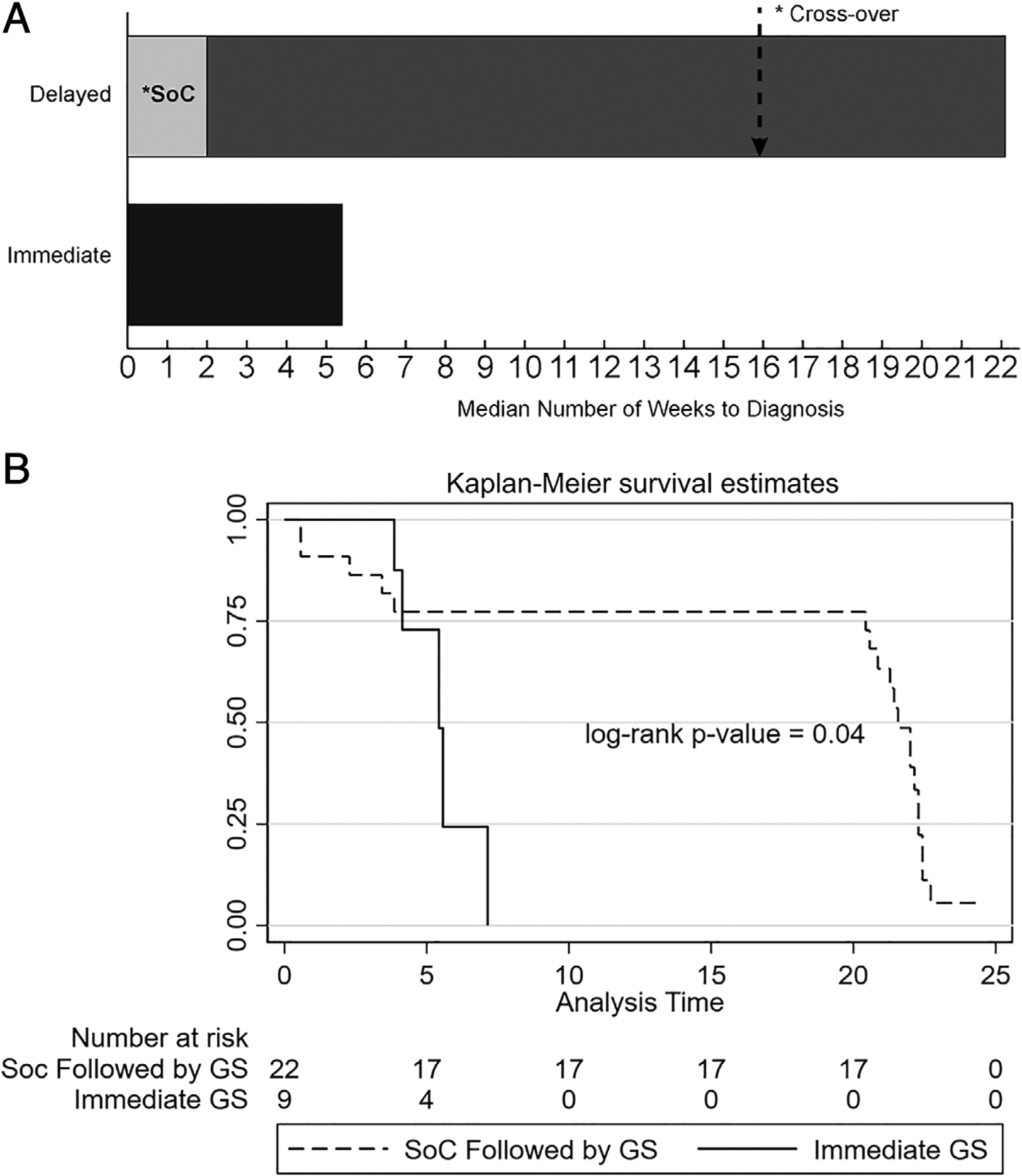
Time to diagnosis with genome sequencing (GS) or standard of care (SoC). (A) Median time to diagnosis (weeks) in either the immediate-GS or delayed-GS arms. Overall time to diagnosis correlates with time to access to GS for the majority of the cohort. Note that for patients solved by SoC diagnostic techniques in the SoC arm, all individuals who received a diagnosis received it within 2 weeks of enrollment, although for the remainder of the cohort SoC approaches continued until crossover. (B) Kaplan–Meier analysis of the proportion of individuals achieving a diagnosis for the immediate-GS versus delayed-GS arms, demonstrating time to diagnosis from enrollment in individuals achieving a diagnosis only. The time to diagnosis was significantly shorter in the immediate-GS group (p = 0.04, log-rank test). The likelihood of diagnosis using SoC approaches was greatest <5 weeks after the onset of testing; afterward, only GS resolved cases.
These findings demonstrate that time to diagnosis is closely correlated to the time at which GS is performed and are consistent with the observation that in the delayed-GS arm only 5 individuals were resolved by SoC (<5 weeks) and all further 14 diagnoses were made by GS at the time of crossover (see Supplementary Table 4). Thus, GS had a significantly larger overall diagnostic yield (19/34 [56%]) compared to SoC (7/34 [21%], including the 2 individuals resolved prior to randomization; p < 0.001 per Wild–Seber test of proportions).
The diagnoses facilitated by GS spanned a wide variety of white matter disorders and causal variant types (see Supplementary Table 4). Canonical leukodystrophies20 accounted for 12 of the 26 diagnoses (46%) made overall, but only one-quarter (26%) of the diagnoses achieved by GS. Mitochondrial pathway variants represented an additional small proportion (14/19 GS diagnoses [21%] and 4/26 total diagnoses [15%]), and the remainder (10/19 GS diagnoses [53%] and 10/26 total diagnoses [39%]) was composed of disorders associated with white matter abnormalities but not previously classified as a canonical leukodystrophy or a disorder of mitochondrial origin (see Table 2 and Fig 1C). The causal variants in 24 individuals were single nucleotide variants or indels in canonical protein-coding genes, but 2 individuals (10%) had variants in either small regulatory RNAs (ie, SNORD118) or CNVs (11q24.1q25 deletion) that typically are not detectable by ES. Of note, 2 additional individuals had CNVs of unknown significance, and these CNVs were not felt to contribute to a clinical diagnosis in these cases (see Supplementary Table 4). Finally, several individuals were found to have a missense variant of uncertain significance in a gene that was highly suggestive of their disorder. These disorders were considered resolved based on clinical expertise, where MRI, clinical presentation, or orthogonal study confirmed a diagnosis after discussion with a multi-disciplinary team performing the analysis of GS results.
The prospective diagnostic specificity of MRI pattern recognition in comparison to GS was also assessed in this study. MRIs were reviewed by at least 2 neuroimaging experts (A.V., M.S.v.d.K., and/or G.B.), and a provisional diagnosis was made based on disease-specific pattern analysis using a previously validated scoring system.9 If the ultimate molecular diagnosis was a canonical leukodystrophy,20 at least one of the reviewers correctly identified the MRI pattern and the associated genetic condition. In addition, reviewers were able to identify the category of disease if the final diagnosis was a mitochondrial leukoencephalopathy, although specific genotype could not be identified. However, if the final diagnosis was neither a mitochondrial disease nor a canonical leukodystrophy, MRI pattern recognition never identified the correct diagnosis (0/17 individuals; Fig 3). Comparison between expert MRI analysis and final molecular diagnosis indicated high concordance for disorders defined as leukodystrophies20 (80%, 95% CI = 44.39–97.48) but superior performance of GS in all other cases.
FIGURE 3:
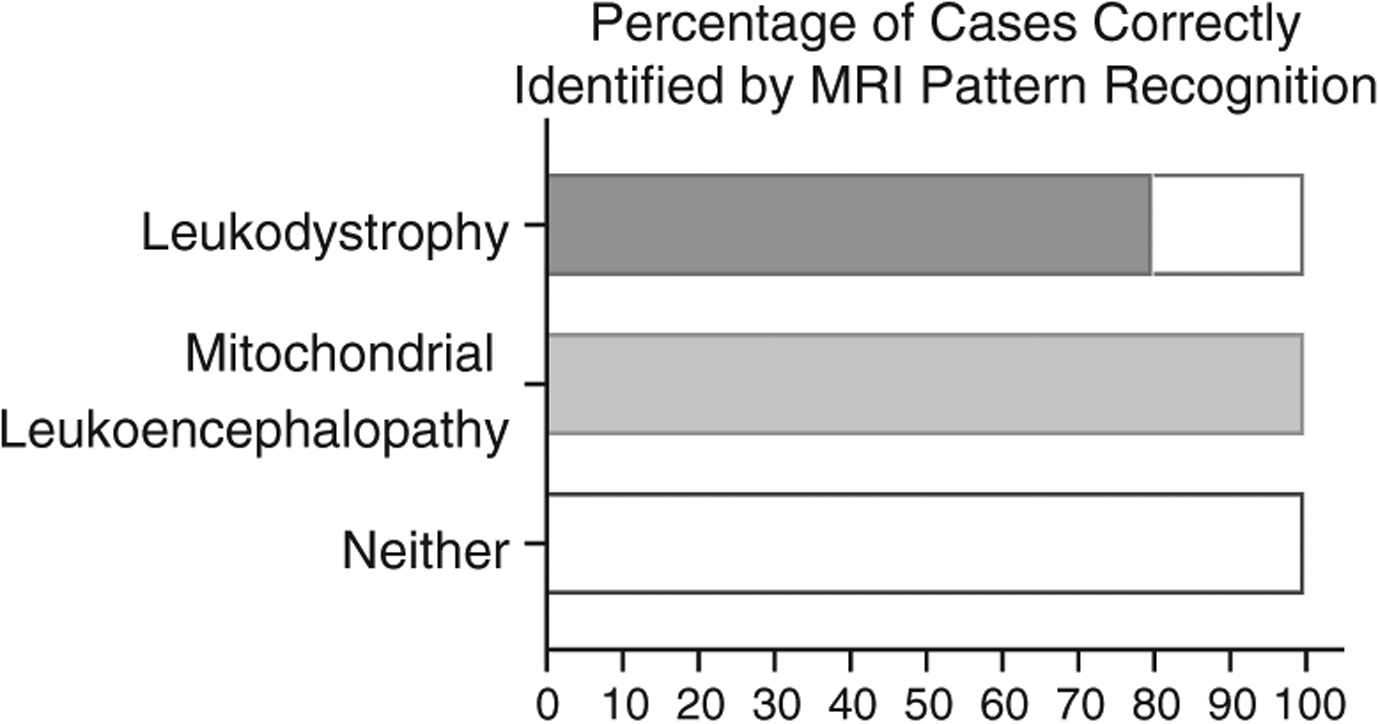
Accuracy of magnetic resonance imaging (MRI) pattern recognition prior to agnostic testing. Thirty-two MRIs were reviewed by clinicians experienced in the diagnosis of leukodystrophy patients. If the ultimate diagnosis was a canonical leukodystrophy, at least one of the reviewers correctly identified the MRI pattern and the diagnosis (8/10). High certainty of the category of disease if the final diagnosis was a mitochondrial leukoencephalopathy (4/4) was demonstrated. If the final diagnosis was neither a mitochondrial disease nor a leukodystrophy, MRI pattern recognition never identified the correct diagnosis (0/18 cases).
While a full assessment of the clinical utility of WGS in this setting is currently under study, it should be noted that in a proportion of cases, diagnosis in this cohort led to changes in clinical management related to their underlying disease. Specifically, for children affected by POLR3-related leukodystrophy, endocrinologic and ophthalmologic follow-up is warranted.24 Similarly, for individuals affected by vanishing white matter disease, careful attention to prevention of head injury and management of febrile infections provides important modification of the natural history of this disease. In children with mitochondrial disease, such as those identified as NDUFV1 and ISCA2, disease identification can allow for appropriate prognostication, management of extra neurologic organ involvement, and supportive care, whereas conversely, megalencephalic leukodystrophy with subcortical cysts associated with HEPACAM mutations has an often relatively benign phenotype and reassurance can be provided. Finally, in certain metabolic conditions, such as congenital disorders of glycosylation, disease management can provide screening for multisystem organ dysfunction commonly seen in these conditions. In addition, in our patient with SLC35A2 mutations, treatment with rare sugar galactose therapy was initiated.
Discussion
Here, we report on what is, to our knowledge, the first randomized controlled study of GS in pediatric patients with white matter disorders. Patients received either immediate first-line GS along with SoC diagnostics or SoC diagnostic tests for 4 months before receiving GS (if the individual had not achieved a definitive diagnosis by time of crossover). The findings indicate that implementation of GS as a first-line test in this population more than doubles the overall diagnostic yield compared to SoC in the same time period (21% vs 56%; p < 0.005, Wild–Seber test of overlapping proportions). Additionally, first-line GS significantly decreases the time to a diagnosis (p = 0.04, log-rank test) despite the rapid crossover to delayed-GS in unsolved individuals (4 months), and provides insights that may be missed by other molecular testing modalities including ES, including copy number and intergenic variants. In this cohort, only 12 of 26 resolved individuals were found to have variants in genes or enzymatic testing indicative of a canonical leukodystrophy.3,20 The remainder were unlikely to be identified on panel-based approaches commonly in use in this population. Importantly, GS also performs pattern recognition alone better than MRI in the context of patients who do not have a canonical leukodystrophy.20 Taken together, these data indicate that a combination of rapid biochemical testing and trio-GS can yield overall diagnostic success (26/34 individuals interrogated [76.5%]) in less than a month, which is a marked improvement compared to historical averages of 35–50% of white matter disorders resolved over 16 months to 8 years.3,10,25 This shift is particularly important given the growing number of rare diseases with disease-specific management, such that access to earlier diagnosis may ultimately change clinical care.
These findings prompt important changes to the diagnostic approach of suspected leukodystrophy patients as previously proposed by Parikh et al.14 First, unless MRI pattern allows one to be confident of diagnosis, lysosomal enzymes, cholestanol levels, urine organic acids (UOAs), or very long chain fatty acids (VLCFAs) should likely be performed. These tests permit diagnosis of several leukodystrophies where timely intervention may improve outcomes, including X-linked adrenoleukodystrophy (MIM 300100), cerebrotendinous xanthomatosis (MIM 213700), metachromatic leukodystrophy (MIM 250100), and Krabbe disease or globoid cell leukodystrophy (MIM 245200).26 Second, if the MRI pattern does not definitively reflect a known disorder, the patient should be tested by GS, in a trio setting to include both parents, which substantially increases the likelihood of achieving a diagnosis and reduces the overall time to diagnosis (see Fig 3).
Limitations in this study include that SoC approaches were determined by clinical care, and not by this research protocol. Thus, SoC was not standardized across sites and may have varied among individual physicians caring for affected individuals. To minimize potential ascertainment bias, individuals were recruited nationally across a range of care facilities. The individuals enrolled in the study came from a total of 21 care facilities (17 tertiary care centers, 4 regional clinics). This study is, however, representative of the variations in diagnostic testing observed in the clinical setting, and more likely to reflect the rate of diagnostic success in a real clinical setting. Additionally, this study did not directly compare GS and ES (or concurrent ES and chromosomal microarray plus mitochondrial genomic analysis). This study also did not assess the efficacy of proband only GS, which might be expected to be lower, particularly given the number of de novo variants observed in this cohort. Finally, although the findings are significant, the overall small sample size, as a result of the rarity of these conditions, may limit the ability to generalize these findings to other disorders. We anticipate, however, that for disorders with substantial phenotypic and genetic heterogeneity, as observed here, GS will outperform current serial SoC testing (biochemical, single gene, gene panels). Previous studies have shown that other genomic assays, including ES, result in improved diagnostic yield compared to SoC.7 In this cohort, if ES was used as a first-line test it may have detected the majority of pathogenic variants in this study, although GS provides better coverage in the exome itself27 and increases the chances of discovery of CNVs and pathogenic noncoding variants (see above). GS analyses were performed according to established analytical and American College of Medical Genetics and Genomics interpretation guidelines, and were returned to subjects by local clinical teams with relevant subspecialist (eg, pediatric neurology) or medical genetics training. We cannot rule out the possibility that the sites that participated in the LeukoSeq study did so because of their comfort with medical genetic findings.
The interim review findings described here led to the loss of clinical equipoise, and the trial design has been altered to a single arm prospective observation study, ensuring that after December 2018, all patients have access to GS testing in <30 days. The subsequent phase of this study includes a 12-month window during which health outcomes and resource utilization will be assessed. We will evaluate whether, consistent with other recent clinical utility studies,13,15,28 GS can refine health care utilization and associated cost of care.
Conclusions
Our results affirm the likely utility of first-line trio-GS provided in leukodystrophy patients, with measurable reductions in time to diagnosis and efficacy demonstrably higher than previous large cohort studies. A novel testing algorithm for the leukodystrophies is proposed based on this prospective evidence of testing outcomes (Fig 4). We continue to recommend first-line biochemical testing, including VLCFAs, lysosomal enzymes, and UOAs, and in the appropriate context cholestenol levels, to ensure that treatable entities are not missed.26 Targeted molecular testing may be considered if the clinical picture or MRI is characteristic of a single specific recognizable disorder. However, when the MRI pattern cannot be associated with a previously recognized leukodystrophy, rapid GS should be pursued to shorten the diagnostic odyssey and provide the greatest likelihood of definitive diagnosis.
FIGURE 4:
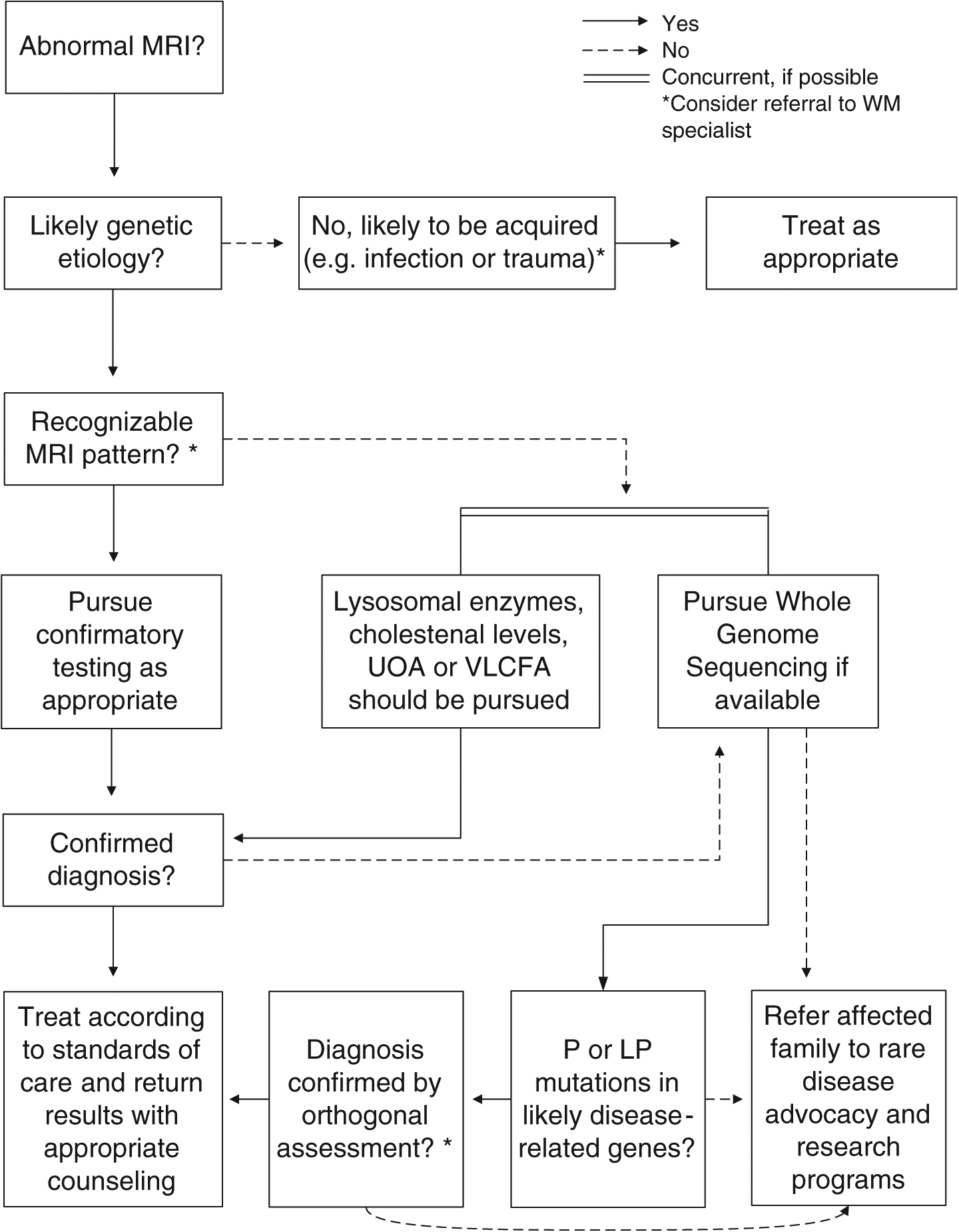
Diagnostic testing algorithm for patients with a suspected white matter (WM) disorder. A decision flow chart to determine appropriate diagnostic approaches given clinical assessment and magnetic resonance imaging (MRI) pattern analysis is shown. First- line biochemical testing, including very long chain fatty acids (VLCFAs), lysosomal enzymes, and urine organic acids (UOAs), and in the appropriate context cholestenol levels, are recommended to provide rapid diagnosis for treatable leukodystrophies. If the MRI pattern cannot be associated with a previously recognized leukodystrophy, rapid genome sequencing (GS; if available) should be pursued to shorten the diagnostic odyssey and provide the greatest likelihood of definitive diagnosis. Targeted molecular testing may be considered if the clinical picture or MRI is characteristic of a single specific recognizable disorder but does not demonstrate the same level of efficacy as broader exome sequencing or GS testing in the absence of salient clinical features. LP, likely pathogenic; P = pathogenic
Supplementary Material
Acknowledgments
Clinical genome sequencing, analysis, and interpretation were provided by Illumina and the Illumina Clinical Services Laboratory. The study is in part supported by the Pennsylvania Department of Health’s Commonwealth Universal Research Enhancement Program Tobacco Formula award SAP# 4100077047. Creation of a diagnostic algorithm was supported by the Leukodystrophy Care Network of the Hunter’s Hope Foundation. The Children’s Hospital of Philadelphia and the Jacob A. Kamens endowed chair supported all other study-related activities. No non-Illumina staff received any direct financial work or compensation for this effort. The participation of G.H. and C.S. is in part financed by the Australian National Health and Medical Research Council (1068278). G.B. has received the New Investigator Salary Award from the Canadian Institutes of Health Research (2017-2022).
We thank the patients and their families. The LeukoSEQ group dedicates this article to their coauthor and collaborator, James Weisfeld-Adams, MD (1979-2018).
Footnotes
Potential Conflicts of Interest
J.F. and R.J.T. are employees of Illumina. Illumina employees participated in trial design, review of genomic data, and revision of the manuscript. The other authors have no conflicts to report. Illumina provided in kind support by providing Clinical Laboratory Improvement Acts (CLIA)/College of American Pathologists (CAP) certified GS; however, all study activities, including randomization, patient review, trial coordination, and statistical analysis occurred at the academic institutions involved without direct input or support from Illumina.
Trial Registration: LeukoSeq Clinical Trial (NCT02699190), https://clinicaltrials.gov/ct2/show/NCT02699190
References
- 1.van der Knaap M, Valk J. Magnetic resonance of myelination and myelin disorders. 3rd ed. Berlin, Germany: Springer, 2005. [Google Scholar]
- 2.van der Knaap MS, Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol 2017;134:351–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richards J, Korgenski EK, Taft RJ, et al. Targeted leukodystrophy diagnosis based on charges and yields for testing. Am J Med Genet A 2015;167A:2541–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kevelam SH, Steenweg ME, Srivastava S, et al. Update on leukodystrophies: a historical perspective and adapted definition. Neuropediatrics 2016;47:349–354. [DOI] [PubMed] [Google Scholar]
- 5.Costello DJ, Eichler AF, Eichler FS. Leukodystrophies: classification, diagnosis, and treatment. Neurologist 2009;15:319–328. [DOI] [PubMed] [Google Scholar]
- 6.Boespflug-Tanguy O, Labauge P, Fogli A, Vaurs-Barriere C. Genes involved in leukodystrophies: a glance at glial functions. Curr Neurol Neurosci Rep 2008;8:217–229. [DOI] [PubMed] [Google Scholar]
- 7.Vanderver A, Simons C, Helman G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol 2016;79:1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonkowsky JL, Nelson C, Kingston JL, et al. The burden of inherited leukodystrophies in children. Neurology 2010;75:718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Knaap MS, Breiter SN, Naidu S, et al. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 1999;213:121–133. [DOI] [PubMed] [Google Scholar]
- 10.Vanderver A, Hussey H, Schmidt JL, et al. Relative incidence of inherited white matter disorders in childhood to acquired pediatric demyelinating disorders. Semin Pediatr Neurol 2012;19:219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein CJ, Foroud TM. Neurology individualized medicine: when to use next-generation sequencing panels. Mayo Clin Proc 2017;92: 292–305. [DOI] [PubMed] [Google Scholar]
- 12.Srivastava S, Cohen JS, Vernon H, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014;76:473–483. [DOI] [PubMed] [Google Scholar]
- 13.Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med 2018;20:435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parikh S, Bernard G, Leventer RJ, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab 2015;114:501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrikin JE, Cakici JA, Clark MM, et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom Med 2018;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–1817. [DOI] [PubMed] [Google Scholar]
- 17.Roller E, Ivakhno S, Lee S, et al. Canvas: versatile and scalable detection of copy number variants. Bioinformatics 2016;32:2375–2377. [DOI] [PubMed] [Google Scholar]
- 18.Ivakhno S, Roller E, Colombo C, et al. Canvas SPW: calling de novo copy number variants in pedigrees. Bioinformatics 2018;34:516–518. [DOI] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanderver A, Prust M, Tonduti D, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab 2015;114:494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elashoff JD. nQuery Advisor. Version 7.0 user’s guide. Chapel Hill, NC: UNC Information Technology Services, 2007. [Google Scholar]
- 22.Seber CJ, GAF W. Comparing two proportions from the same survey. Am Stat 1993;47:178–181. [Google Scholar]
- 23.Karrison T Versatile tests for comparing survival curves based on weighted log-rank statistics. Stata J 2016;16:678–690. [Google Scholar]
- 24.Adang LA, Sherbini O, Ball L, et al. Revised consensus statement on the preventive and symptomatic care of patients with leukodystrophies. Mol Genet Metab 2017;122:18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akwa Y, Hassett DE, Eloranta ML, et al. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol 1998;161:5016–5026. [PubMed] [Google Scholar]
- 26.Helman G, Van Haren K, Bonkowsky JL, et al. Disease specific therapies in leukodystrophies and leukoencephalopathies. Mol Genet Metab 2015;114:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belkadi A, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A 2015;112:5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan TY, Dillon OJ, Stark Z, et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr 2017;171:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.