

Abstract
Viruses are present in the intestinal microflora and are currently discussed as a potential causative mechanism for the development of inflammatory bowel disease. A number of viruses, such as Human Herpesvirus-8, express homologs to cellular FLIPs, which are major contributors for the regulation of epithelial cell death. In this study we analyzed the consequences of constitutive expression of HHV8-viral FLIP in intestinal epithelial cells (IECs) in mice. Surprisingly, expression of vFlip disrupts tissue homeostasis and induces severe intestinal inflammation. Moreover vFlipIEC-tg mice showed reduced Paneth cell numbers, associated with excessive necrotic cell death. On a molecular level vFlip expression altered classical and alternative NFκB activation. Blocking of alternative NFκB signaling by deletion of Ikka in vivo largely protected mice from inflammation and Paneth cell loss induced by vFLIP. Collectively, our data provide functional evidence that expression of a single viral protein in IECs can be sufficient to disrupt epithelial homeostasis and to initiate chronic intestinal inflammation.
INTRODUCTION
Intestinal immune homeostasis is maintained by a regulated balance of proliferation and cell death in the intestinal epithelium.1 Our previous studies have shown that a tight regulation of the cysteine protease Caspase-8 in intestinal epithelial cells (IECs) is indispensable for intestinal homeostasis: On the one hand, an uncontrolled activation of Caspase-8, such as by genetic deletion of cellular FLICE like inhibitory protein (cFLIP), leads to massive apoptotic death of IECs culminating in breakdown of the epithelial barrier and spontaneous mucosal inflammation.2 On the other hand, genetic deletion of Caspase-8 itself in IECs of mice (Casp8ΔIEC mice) promotes the development of spontaneous intestinal inflammation.3 Caspase-8 deficiency was associated with excessive caspase-independent Paneth cell death, which was identified as necroptosis.3,4 Necroptosis shares morphological features with necrosis, like organelle swelling and membrane breakdown, but in contrast to classical necrosis, it is regulated by an intracellular pathway and signaling molecules partially overlapping with apoptosis.5–8 Although necroptosis is not completely understood yet, it depends on the RIP kinases 1 and 3 which trigger activation and translocation of MLKL to the plasma membrane.9–11
Recent studies could further demonstrate that the NFκB signaling pathway plays an important role for cell death control in the epithelium: abrogation of NFκB signaling results in apoptosis and disruption of epithelial barrier integrity, associated with Paneth cell dysfunction,12,13 whereas constitutive activation of NFκB signaling in IECs also induces inflammation and Paneth cell dysfunction.14,15 Together these studies conclude that the NFκB pathway plays an important role for maintenance of epithelial homeostasis in the intestine.
Several studies have described that the intestinal microbiota and a microbial dysbiosis might contribute to the development of inflammatory bowel diseases (IBD).16 Beside bacteria, also viruses like Rotavirus, Norovirus, and Herpesviruses like Cytomegalovirus are known to cause intestinal pathology and symptoms, including inflammation, abdominal pain, and diarrhea.17–19 Interestingly, a number of viruses express molecules that can directly interfere with the host cell death machinery, suggesting that these viruses might have the potential to disrupt the balance between cell death and proliferation in the gut.20–22 One such virus, Human Herpesvirus-8 (HHV8), has been identified as the infectious cause of a number of diseases which are most often seen in patients with acquired immunodeficiency.23 HHV8 has the ability to express several proteins to overcome host defense reactions within the infected cell.24 Of note, one of these proteins shares structural homologies with the short isoform of the Caspase-8 regulator cFLIP.25,26 This viral homolog, termed vFLIP, has been suggested to negatively regulate Caspase-8 activity and to exert anti-apoptotic functions in vitro.26–28 Moreover vFLIP is suggested to block apoptosis in an NFκB-dependent manner, most likely due to direct interaction and activation of the NEMO protein complex.29–34 HHV8-vFLIP expression in B-cells triggered transdifferentiation and tumorigenesis, indicating that vFLIP might play an important role in the development of HHV8-induced lymphoproliferative disorders.35–37 Furthermore, mice with induced expression of HHV8-vFLIP in endothelial cells developed a profound inflammatory phenotype and showed remodeling of the myeloid lineage, demonstrating that HHV8-vFLIP expression caused changes in the host microenvironment during infection.38 Given the abundance of viruses in the intestinal microbiota, it can be reasoned that infection of IECs with viruses expressing cell death regulating proteins such as vFLIP might trigger barrier disruption and intestinal inflammation. In a proof-of-concept approach, we analyzed the consequences of HHV8-vFlip expression in epithelial cells of the gut in vivo (mice termed vFlipIEC-tg). We could demonstrate that vFlip expression spontaneously induced a severe and chronic intestinal inflammation. vFlipIEC-tg mice were characterized by reduced Paneth cell numbers and excessive non-apoptotic cell death, features which are also seen in patients suffering from IBD. Furthermore we could show in vivo, that HHV8-vFLIP expression strongly activated both canonical and alternative NFκB-signaling in IECs, leading to an upregulation of target gene expression. Strikingly, impairment of the alternative NFκB pathway by crossing vFlipIEC-tg mice to IkkaΔIEC mice reduced chronic inflammation and restored Paneth cell numbers. Taken together these data demonstrate that expression of a single viral protein can be sufficient to disrupt epithelial homeostasis and cause chronic intestinal inflammation.
RESULTS
Expression of vFlip in IECs triggers spontaneous development of inflammation
To generate mice expressing HHV8-vFlip in IECs, Rosa26.vFLIP mice were crossed to mice which express Cre Recombinase under the control of the IEC-specific Villin promoter (Fig. 1a). vFlipIEC-tg mice were born at the expected mendelian ratio. To confirm that the transgene is specifically expressed in IECs, GFP fluorescence was examined in the intestine and other organs of these animals. Accordingly, GFP was only detected in IECs from both small and large intestine of vFlipIEC-tg mice (Fig. 1b, c, Suppl. Fig. 1 A). vFlipIEC-tg mice showed reduced body size and weight as compared to control littermates (Suppl. Fig. 1 B, C), implicating gut pathology. To analyze the gut morphology in vivo, high resolution mini-endoscopy was performed, which revealed signs of intestinal inflammation, including erosions and crypt loss in the terminal ileum (Fig.1d). Endoscopy of the colon revealed thickening of the bowel wall, reduced translucency and loss of regular blood vessel structure in vFlipIEC-tg mice (Suppl. Fig. 1 D). Histological analysis of the intestinal architecture of vFlipIEC-tg mice compared to controls revealed signs of inflammation including increased cellularity and crypt loss in both small and large intestine (Fig. 1f, Suppl. Fig. 1 D, E). These findings were supported by gene expression analyses showing a significant upregulation of the proinflammatory markers Tnfa and S100a9 in vFlipIEC-tg mice as compared to controls (Fig. 1e, Suppl. Fig. 1 F). Furthermore, immunohistochemical stainings showed a high number of immune cells (macrophages (F4/80), CD4+ T-cells and CD11c+ dendritic cells) accumulating in the lamina propria of vFlipIEC-tg animals (Fig. 1g). Taken together, these data uncovered that expression of HHV8-vFlip in IECs is sufficient to cause spontaneous intestinal inflammation. Of note, we further observed that HHV8 is able to efficiently infect intestinal epithelial cells in vitro (Suppl. Fig. 2 A, B). To our knowledge, this is the first report demonstrating that a single viral molecule can drive gut inflammation with features similar to IBD in humans.
Fig. 1.
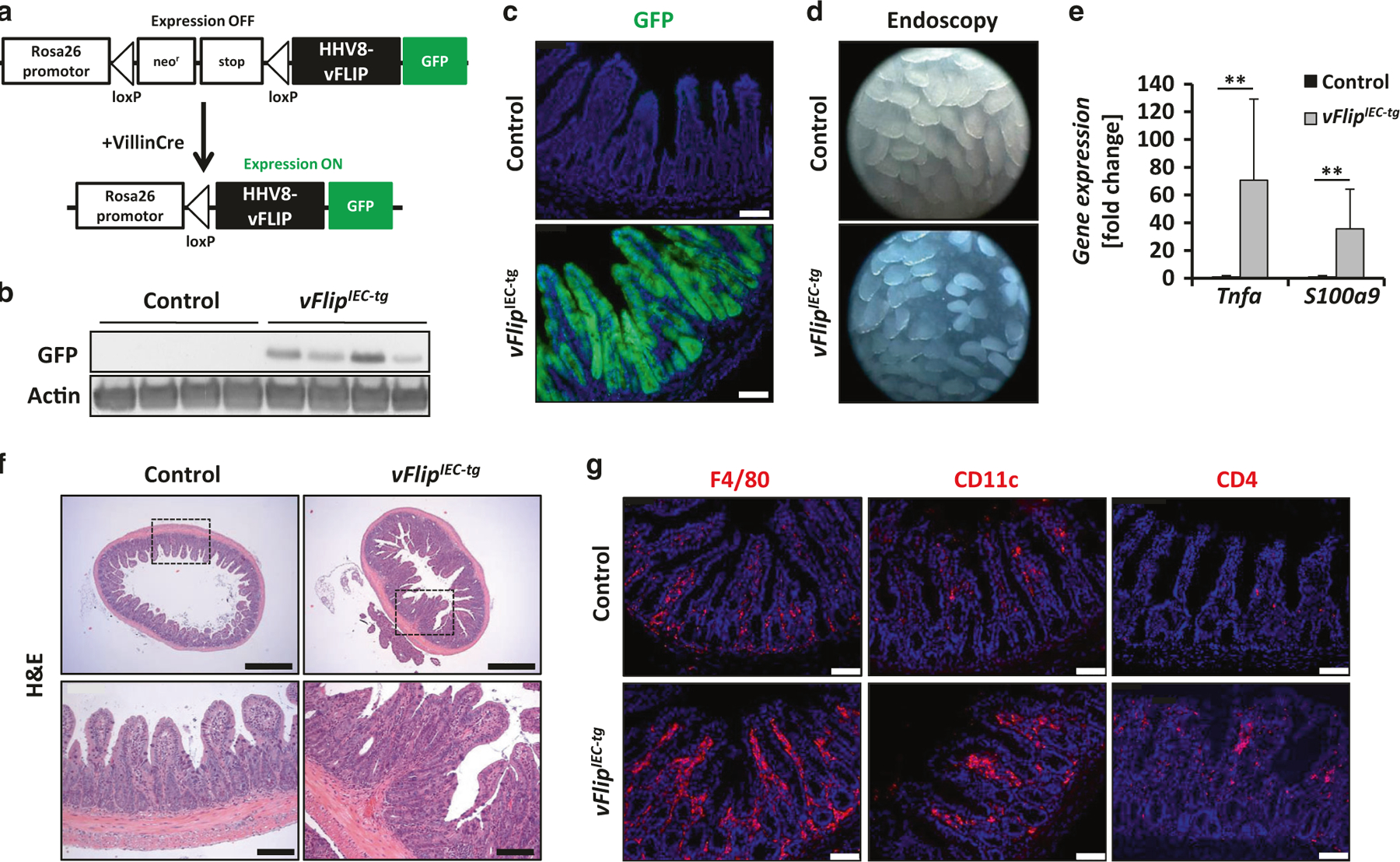
vFlipIEC-tg mice spontaneously develop severe intestinal inflammation. a Strategy for generating vFlipIEC-tg mice. Rosa26.vFLIP knockin mice were crossed to VillinCre mice to generate mice constitutively expressing vFlip in IECs. b Western Blot analysis of GFP using small intestinal IEC lysates from indicated mice. Actin was used as a loading control. c Representative pictures of GFP fluorescence staining of small intestinal cross sections from indicated mice. Scale bar 75 µm. d Representative endoscopic pictures from the small intestine of indicated mice. e Transcriptional analysis of Tnfa (n ≥ 10) and S100a9 (n ≥ 8) in the small intestine of control and vFlipIEC-tg mice. Values are shown + SD and were calculated relative to control mice. Hprt was used as internal standard. Data were pooled from 4 individual experiments, **p < 0.01. f Representative pictures of H&E stained small intestinal cross sections from indicated mice. Scale bar upper panel 500 µm, lower panel 100 µm. g Representative pictures of immunohistochemical stainings of small intestinal cross sections from indicated mice using antibodies against F4/80, CD11c and CD4. Scale bar 75 µm
Epithelial vFlip expression drives non-apoptotic Paneth cell loss
HHV8-vFLIP was described to inhibit Caspase-8 activity in vitro.26 Since we have previously demonstrated that mice with deficiency for caspase-8 in IECs (Casp8ΔIEC mice) showed decreased Paneth cell numbers due to necroptotic cell death,3 we hypothesized that vFlipIEC-tg mice might also show Paneth cell defects. Indeed we could observe a marked reduction of Paneth cells in the crypts of Lieberkühn of vFlipIEC-tg mice, as demonstrated by H&E staining and further confirmed by immunohistochemistry for Lysozyme, an antimicrobial peptide secreted by Paneth cells39 (Fig. 2a). In addition, gene expression analysis of the antimicrobial markers Ang4 and Lyz showed significantly reduced expression levels in vFlipIEC-tg animals as compared to controls (Fig. 2b).
Fig. 2.
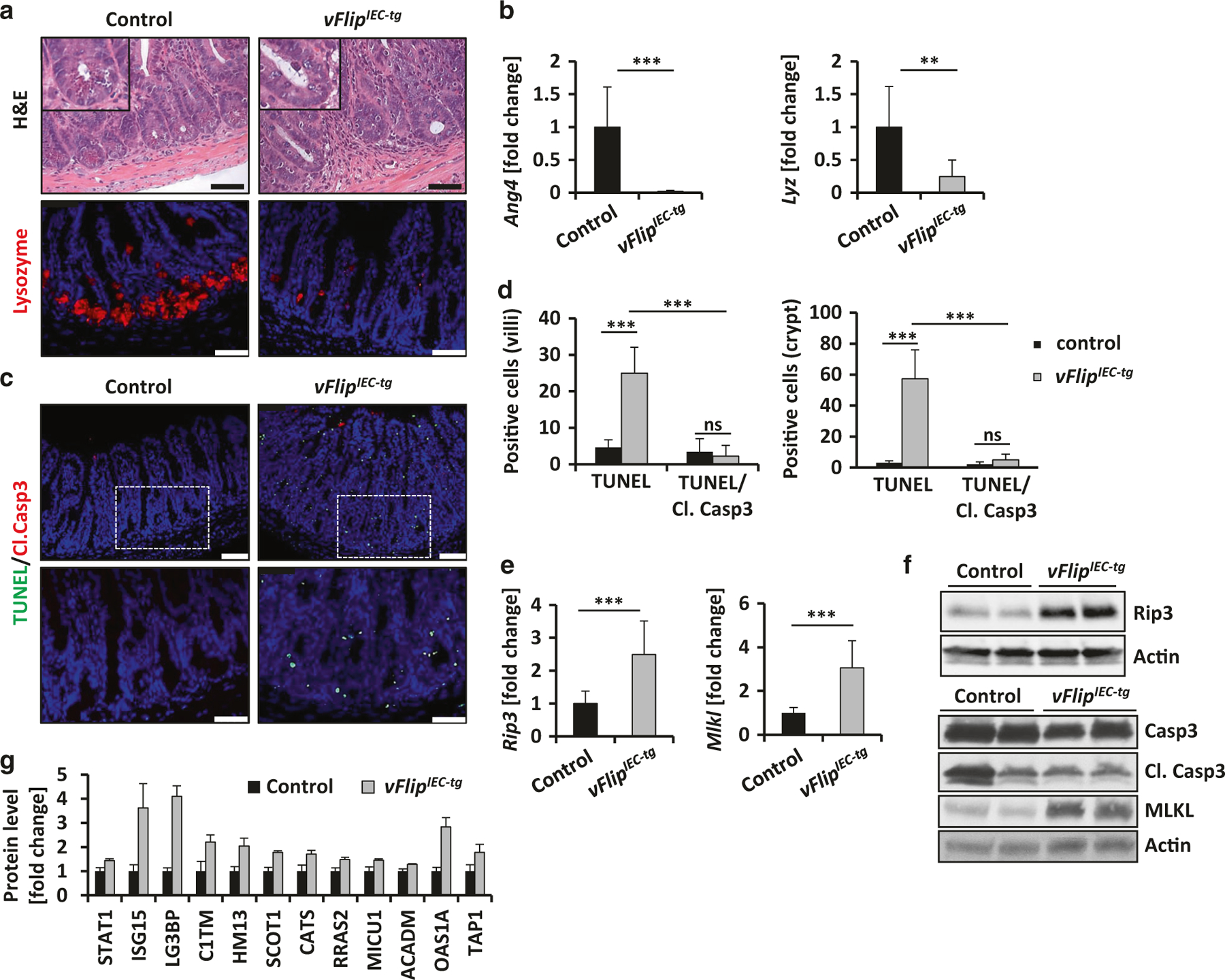
vFlipIEC-tg mice are characterized by reduced Paneth cell numbers and increased cell death. a Representative pictures of H&E and immunohistochemical Lysozyme staining of small intestinal cross sections of indicated mice. Scale bar 50 µm. b Transcriptional analysis of Ang4 (n = 11) and Lyz (n = 11) in the small intestine of indicated mice. Values are shown + SD and were calculated relative to control mice. Hprt was used as internal standard. Data were pooled from 4 individual experiments, **p < 0.01, ***p < 0.001. c Representative pictures of TUNEL/Cleaved Caspase-3 doublestaining of small intestinal cross sections from indicated mice. Scale bar upper panel: 75 µm, lower panel: 50 µm. d Quantification of cell death of IECs located at the villus or the crypt area. Villi: n(control) = 5, n(vFlipIEC-tg) = 10, crypts: n(control) = 8, n(vFlipIEC-tg) = 13, ***p < 0.001. e Transcriptional analysis of Rip3 (n ≥ 9) and Mlkl (n ≥ 9) in the small intestine of indicated mice. Values are shown + SD and were calculated relative to control mice. Hprt was used as internal standard. Data were pooled from 3 individual experiments, ***p < 0,001. f Western Blot analyses of small intestinal IEC lysates from indicated mice using antibodies against RIP3, Caspase-3, its cleaved forms and MLKL. Actin was used as a loading control. g Quantitative proteome analysis of known and putative STAT1 target genes in small intestinal epithelial lysates of control and vFlipIEC-tg mice (n = 3). Values are shown + SD and were calculated relative to control mice, p < 0.05 for all target genes between both groups
Due to the facts that HHV8-vFLIP was described to inhibit Caspase-8 activity26 and that Casp8ΔIEC and vFlipIEC-tg mice showed a strikingly similar phenotype regarding inflammation and Paneth cell reduction,3 we further investigated if lack of Paneth cells in vFlipIEC-tg mice resulted from augmented cell death. Indeed, vFlipIEC-tg animals showed increased epithelial cell death in the crypt area as compared to controls, which was indicated by TUNEL staining (Fig. 2c, d). Interestingly, transfection of human HT29 cells with two different vFlip expressing plasmids also induced augmented cell death after 48 h and 72 h as compared to cells transfected with control plasmids or untransfected cells (Suppl. Fig. 3). Moreover, vFlipIEC-tg animals showed increased cell death along the crypt villus axis. Of note, most of these dying cells were negative for cleaved Caspase-3 (Fig. 2c, d). Moreover western blot analysis of cleaved Caspase-3 showed no differences in IEC isolates of vFlipIEC-tg mice as compared to controls, suggesting a caspase-independent type of cell death, similar to cell death observed in Casp8ΔIEC mice (Fig. 2f). To investigate whether vFLIP-triggered cell death and inflammation might occur due to necroptosis, we first investigated the contribution of RIP3, an essential molecule for the canonical necroptosis pathway, to vFLIP-induced intestinal alterations. In fact, gene expression and protein levels of Rip3 were augmented in vFlipIEC-tg mice as compared to controls (Fig. 2e, f) RIP3 protein levels were also increased and in HHV8-infected HT29 cells as compared to non-infected cells (Suppl. Fig. 2 C, D). However, crossing of vFlipIEC-tg mice to Rip3−/− mice did not protect the animals from intestinal inflammation, which was demonstrated by colonoscopy and H&E staining of the small and large intestine of indicated mice (Suppl. Fig. 4). Moreover Rip3−/− x vFlipIEC-tg mice were also characterized by loss of Paneth cells in the crypts of the small intestine, which was shown by immunohistochemical staining of lysozyme (Suppl. Fig. 4).Beside RIP3, also MLKL is known to be an important molecule for canonical necroptosis. MLKL was recently described to be an important mediator of a non-canonical, RIP3-independent type of programmed necrotic cell death.40 Interestingly, in vFlipIEC-tg mice, gene expression and protein levels of MLKL were highly upregulated as compared to controls (Fig. 2e, f). In addition HHV8-infected HT29 cells were characterized by increased MLKL protein levels as compared to non-infected control cells (Suppl. Fig. 2 C, D). Moreover, by using a quantitative proteomics approach, we could identify that epithelial protein levels of STAT1, as well as protein levels of known and putative STAT1 target genes were significantly upregulated in vFlipIEC-tg mice as compared to controls (Fig. 2g). These data were in line with previous publications, showing increased vFLIP-induced STAT1 activation.41,42 The STAT1 pathway not only plays an important role during viral infection but it also represents an important mediator of RIP3-independent non-canonical programmed necrosis.40 Our data imply that vFLIP induces a RIP3-independent programmed cell death in the intestinal epithelium. Taken together, these data surprisingly show that excessive epithelial cell death in the intestine can be induced by expression of a viral FLIP protein.
Since vFlipIEC-tg mice are characterized by the dramatic loss of Paneth cells in the small intestine, we investigated if lack of Paneth cells and reduced expression of antimicrobial peptides induced changes in the microbial composition when compared to controls. Therefore we performed 16 S rRNA analyses of fecal samples of vFlipIEC-tg mice and controls. Interestingly, although the mice were co-housed, the microbiome of vFlipIEC-tg mice was characterized by slightly reduced species diversity as compared to controls based on the Shannon Index (Suppl. Fig. 5A). Moreover, the microbiota of vFlipIEC-tg mice clustered separately from control mice as shown by Bray Curtis dissimilarity (Suppl. Fig. 5 B) and heat map illustration of relative abundances of different bacterial genera (Suppl. Fig. 5 D), indicating differences in bacterial colonization in these mice. Interestingly, expression of vFlip in IECs promote an expansion of the phylum Proteobacteria, a condition previously linked to intestinal inflammation (Suppl. Fig. 5C).43 Taken together our data provide evidence that expression of a single viral protein in IECs can be sufficient to alter IEC homeostasis and drive intestinal dysbiosis that might trigger intestinal inflammation.
Gut inflammation and Paneth cell loss are triggered by vFLIP-induced activation of the NFκB pathway
HHV8-vFLIP has been shown to interact with NEMO and to activate the NFκB pathway.29,44 However, the relevance of these observations in vivo in the gut has not been demonstrated so far. Indeed, vFlip expression was associated with a strong activation of the classical NFκB signaling pathway in IECs, which was shown by immunohistochemical staining, demonstrating a translocation of P65 from the cytoplasm to the nucleus of IECs (Fig. 3a). Moreover western blot analysis revealed abundant phosphorylation of P65 and the NFκB inhibitor IκBα, as well as increased protein levels of the NFκB target gene iNOS in vFlipIEC-tg animals, but not in controls (Fig. 3b). Of note, we also observed a strong vFLIP-induced activation of the alternative NFκB pathway in IECs, as indicated by western blot analysis of P100/P52 (Fig. 3c). These observations are in accordance with previous studies, showing that under certain experimental conditions, vFLIP can directly bind to IKKα and induce cleavage of P100.45,46 Taken together, our analyses showed that HHV8-vFLIP expression is able to constitutively activate both classical and alternative NFκB signaling in vivo and that vFLIP via its interaction with the IKK complex might bridge both pathways to drive the inflammatory phenotype observed in vFlipIEC-tg animals.
Fig. 3.
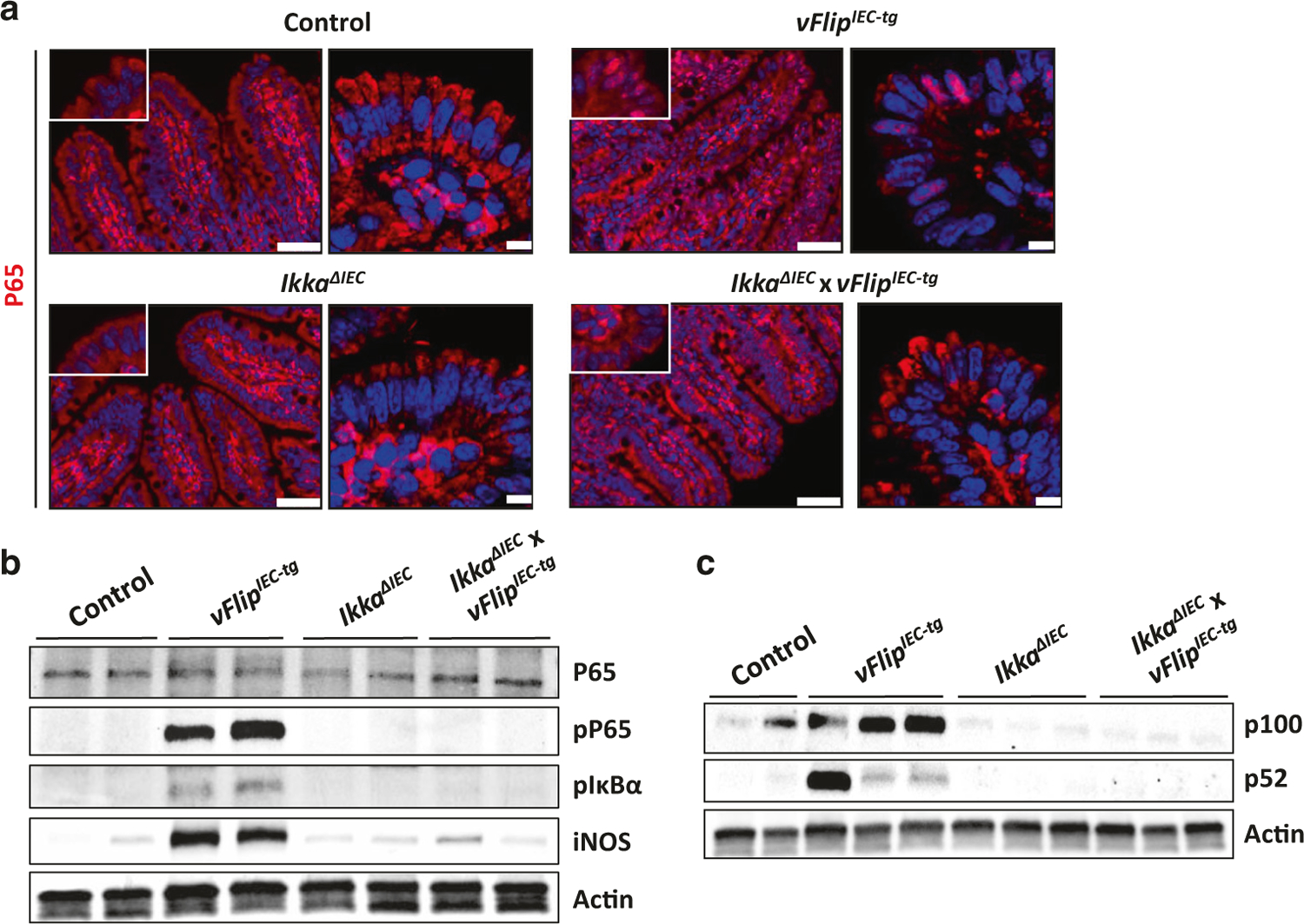
vFlip expression activates the NFκB pathway in IECs. a Representative pictures of an immunohistochemical P65 staining of small intestinal cross sections from indicated mice. Left panel: microscopic pictures, scale bar 50 µm. Right panel: confocal images of nuclear P65 translocation. Scale bar 7.5 µm. b Western Blot analyses of P65, phospho-P65, phospho-IκBα and iNOS using proteins isolated from small intestinal IECs of indicated mice. Actin was used as a loading control. c Western Blot analyses of P100 and P52 using proteins from small intestinal IECs of indicated mice. Actin was used as a loading control
Little is known about the role of IKKα in driving vFLIP-induced pathologies. As our data implicated a role for alternative NFκB signaling, we hypothesized that IKKα activation by vFLIP could be instrumental in mediating intestinal inflammation. In order to investigate if the inflammatory phenotype of vFlipIEC-tg animals functionally depends on alternative NFκB signaling, we impaired alternative NFκB signaling by additional deletion of Ikka (Suppl. Fig. 6A). As expected, these animals showed reduced alternative NFκB signaling as compared to vFlipIEC-tg animals (Fig. 3c). Interestingly, further analysis revealed reduced phosphorylation of P65 and IκBα in IkkaΔIEC x vFlipIEC-tg animals as compared to vFlipIEC-tg animals (Fig. 3b). Moreover protein levels of the NFκB target gene iNos were decreased in IkkaΔIEC x vFlipIEC-tg animals as compared to vFlipIEC-tg animals (Fig. 3b). In addition, immunohistochemical staining showed that Ikka deficiency in vFlipIEC-tg animals attenuated nuclear P65 translocation (Fig. 3a), collectively demonstrating reduced activation of classical NFκB signaling in IkkaΔIEC x vFlipIEC-tg mice.
Importantly, deletion of Ikka in vFlip-expressing animals diminished the inflammatory phenotype. In contrast to vFlipIEC-tg mice, IkkaΔIEC x vFlipIEC-tg mice showed similar body size at 5 weeks compared to controls (data not shown). High resolution endoscopy of the colon of IkkaΔIEC x vFlipIEC-tg mice showed reduced bowel wall thickening and increased translucency as compared to vFlipIEC-tg mice (Suppl. Fig. 6C). H&E staining revealed significantly reduced structural damage and decreased cellularity as compared to vFlipIEC-tg mice, which was further shown by a significantly decreased histology score of IkkaΔIEC x vFlipIEC-tg animals as compared to vFlipIEC-tg animals (Fig. 4a, b, Suppl. Fig. 6C). In addition, double mutant mice showed reduced amounts of infiltrating immune cells in the lamina propria, which was demonstrated by immunohistochemical stainings of the immune cell markers CD4 and F4/80 (Fig. 4a). Moreover, expression of the proinflammatory marker Tnfa was significantly diminished when compared to vFlipIEC-tg mice and were comparable to levels determined in wildtype controls (Fig. 4c). Interestingly, immunohistochemical staining of lysozyme further revealed increased Paneth cell numbers in IkkaΔIEC x vFlipIEC-tg mice compared to vFlipIEC-tg animals (Fig. 4d). Moreover the relative mRNA and protein levels of the antimicrobial peptide Lysozyme were upregulated in IkkaΔIEC x vFlipIEC-tg animals compared to vFlipIEC-tg mice (Fig. 4e, Suppl. Fig. 6 B). Of note, IkkaΔIEC animals in general showed slightly reduced lysozyme staining as compared to controls (Fig. 4d), confirming earlier reports that loss of Ikka in IECs drives Paneth cell dysfunction.47 In summary these data demonstrate that vFLIP-induced NFκB activation in IECs contributes to intestinal inflammation and Paneth cell reduction in vFlipIEC-tg mice and that IKKα might play a major role in vFLIP-induced pathologies.
Fig. 4.
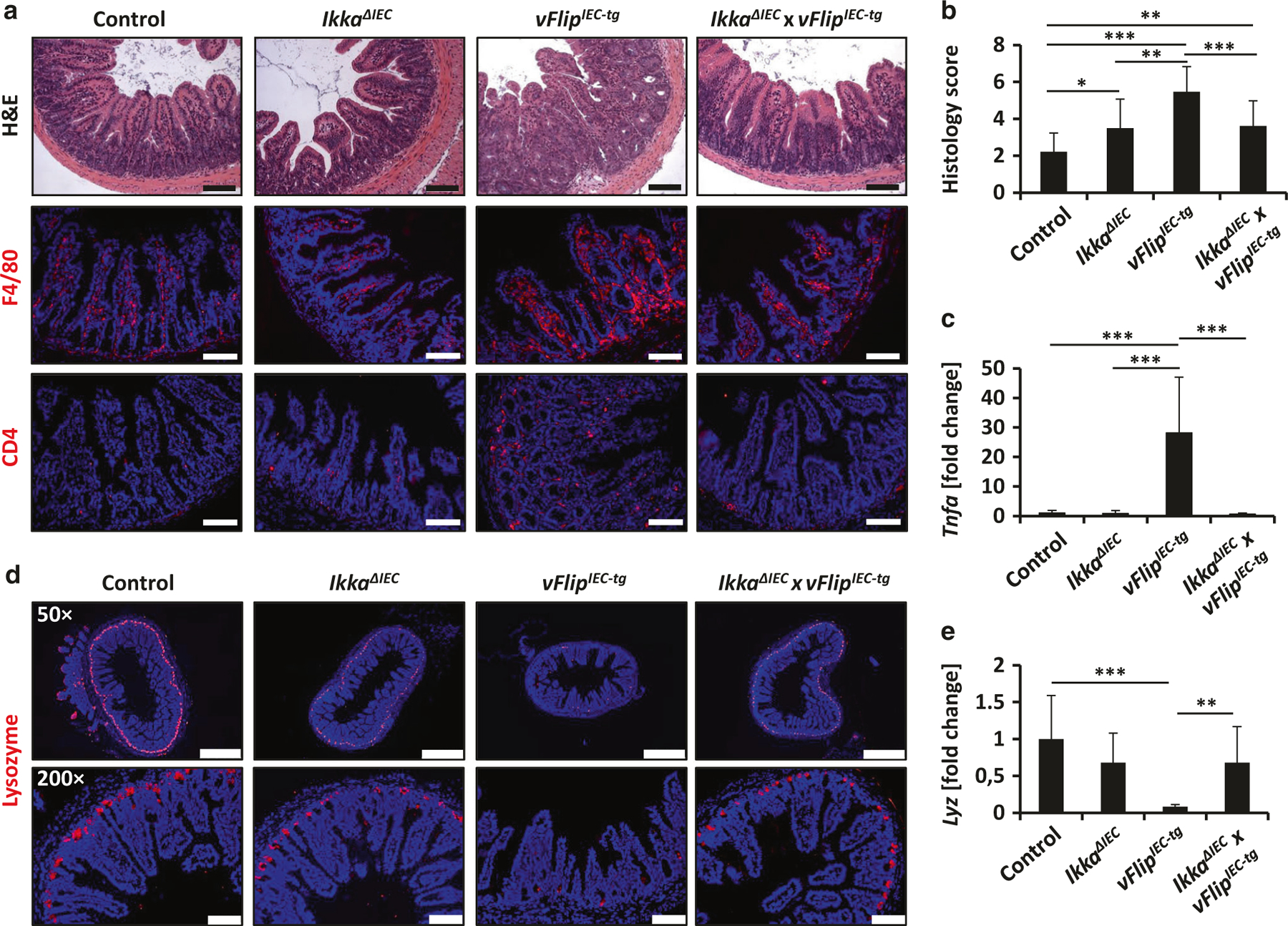
vFLIP-induced inflammation and Paneth cell loss depends on NFκB signaling. a Representative staining of H&E and immunohistochemical F4/80 and CD4 staining on small intestinal cross sections from indicated mice. Scale bar 100 µm. b Histology score of H&E stained small intestinal cross sections of control (n = 18), IkkaΔIEC (n = 10), vFlipIEC-tg (n = 17) and IkkaΔIEC x vFlipIEC-tg (n = 14) animals, *p < 0.05, **p < 0.01, ***p < 0.001. b Transcriptional analysis of Tnfa (n ≥ 9) in the small intestine of control and vFlipIEC-tg mice. Values are shown + SD and were calculated relative to control mice. Hprt was used as internal standard. Data were pooled from 3 individual experiments, ***p < 0.001. c Representative pictures of an immunohistochemical lysozyme staining of small intestinal cross sections from indicated mice. Scale bar upper panel: 500 µm, lower panel: 100 µm. d Transcriptional analysis of Lyz (n ≥ 9) in the small intestine of control and vFlipIEC-tg mice. Values are shown + SD and were calculated relative to control mice. Hprt was used as internal standard. Data were pooled from 3 individual experiments, **p < 0.01, ***p < 0.001
DISCUSSION
In this study, we investigated the effect of HHV8-vFlip expression in IECs on gut tissue homeostasis. One important regulator of cell death and homeostasis in the gut is Caspase-8. Our recent studies indicate that a tight regulation of Caspase-8 activity within certain limits is critical for gut immune homeostasis.2,3 The Caspase-8 gene so far has not been described as a risk locus for development of IBD, however caspase-8 activity is regulated by multiple cellular and pathogenic mechanisms. Along this line, it is highly interesting, that certain viruses, e.g., Herpesviridae, Poxviridae or Adenoviridea, which were found to be part of the enteric virome under non-pathogenic conditions48 encode molecules that can interfere with Caspase-8 activation and host cell death regulation.21,49 Whether or not such interactions might cause breakdown of gut immune homeostasis has not been shown. However it has already been described that Herpesviruses, like Cytomegalovirus (CMV) or Herpes Simplex virus (HSV), which express Caspase-8 inhibitors, may contribute to intestinal inflammation, like colitis or proctitis.18,50–52
On a molecular level, Caspase-8 activity can be regulated by cellular FLIP (cFLIP) proteins.25,53 Interestingly, several Herpesviruses like HHV8, Equine Herpesvirus-2, Bovine Herpesvirus-4, Herpesvirus Saimiri, as well as Molluscum Contagiosum Poxvirus express proteins which are characterized by two Death Effector Domains and therefore share structural homologies with the short isoform of cFLIP. These proteins are termed vFLIPs and have been suggested to regulate the host cell death machinery.28,54 Indeed our findings provide a proof-of-concept that expression of a single viral molecule can be sufficient to reprogram host cell death and survival pathways and to drive inflammation in susceptible tissues like the gut.
While there are several viruses expressing vFLIP and viruses interfering with caspase-8 activation by other mechanisms,49 there is little information in the literature linking HHV8 with gut pathology. However in vitro and in vivo studies have demonstrated that HHV8 has a broad cellular tropism and beside e.g. endothelial cells, fibroblasts or immune cells, also epithelial cells can be successfully infected by HHV8.55–61 Furthermore it was shown that proteins and molecules, which are important for HHV8 infection and entry into target cells, such as ephrin receptor tyrosine kinase A2 and heparan sulfate, are localized in the gastrointestinal tract.62–65 Being in line with these previous studies, our data provide evidence that HHV8 is also able to efficiently infect human intestinal epithelial cells. Moreover under pathogenic conditions there might be a direct connection between HHV8 infection and gut inflammation, since several case reports showed IBD patients suffering from HHV8-induced diseases.66–70 While our study was not intended to provide evidence for a role of HHV8 in IBD, we for the first time show, that expression of a single protein from a Human herpesvirus is able to disrupt intestinal homeostasis and to drive chronic inflammation in the gut.
The regulation of host cell death pathways by herpesviruses seems to be very complex and virus- and cell type-dependent.71 Beside regulation of Caspase-8 and apoptosis by expression of the herpesviral proteins vFLIP, vICA and UL39, herpesvirus infections can also trigger necrotic cell death: CMV and HSV1 infection can induce necrotic host cell death due to direct activation of the RIP3-MLKL axis by viral proteins or viral RNA.72–75 In accordance with these findings, we could identify an increased Rip3 and Mlkl mRNA expression in vFlip expressing animals, as well as increased RIP3 and MLKL protein levels. Surprisingly, genetic deletion of Rip3 in vFlipIEC-tg mice did not protect the animals from intestinal inflammation and Paneth cell loss. However, in line with previous studies,41,42 we could determine increased STAT1 signaling in vFlipIEC-tg mice as compared to controls. STAT1 not only plays an important role during viral infection, it was also recently shown to be a key mediator of non-canonical, RIP3-independent programmed cell death,40 suggesting that STAT1/MLKL-mediated non-apoptotic cell death might be a triggering factor for Paneth cell loss and inflammation in vFlipIEC-tg mice. Of note, vFLIP was previously described to inhibit apoptosis in vitro in certain cell lines.26–28,30,32,33 Belanger et al.26 showed that HHV8-vFLIP physically interacts with Caspase-8 in the cytoplasm in vitro and that vFLIP-binding to Caspase-8 blocked full activation of Caspase-8, a critical step for initiating the apoptosis cascade. Our data suggest that vFLIP expression in vivo might not only block apoptotic cell death, but also induce necrotic cell death, due to blocking of Caspase-8 activation, which is in line with excessive cell death observed in Casp8ΔIEC mice.3
Another important pathway critically involved in maintaining intestinal homeostasis and developing IBD is the NFκB signaling pathway.76 We and others have demonstrated that impaired NFκB signaling in IECs drives Paneth cell dysfunction and intestinal inflammation.12–15 Interestingly, HHV8-vFLIP also activates the NFκB pathway, as it was shown in vitro, that HHV8-vFLIP directly binds to the IKKγ-NEMO complex.29,31 We could show that expression of HHV8-vFLIP in IECs strongly induced both classical and alternative NFκB signaling. In accordance with our data, other studies also showed a vFLIP-induced activation of the alternative NFκB pathway and IKKα was suggested to play a major role for the activation of vFLIP-induced classical NFκB signaling.37,45,46 Interestingly, IEC-specific Ikka knockout in vFlipIEC-tg mice not only reduced the vFLIP-induced activation of both the classical and alternative NFκB pathway, it also led to decreased development of inflammation and largely restored Paneth cell numbers in the terminal ileum. Taken together these observations suggest that IKKα plays a fundamental role in vFLIP-induced pathologies and that both classical and alternative NFκB signaling contributed to the induction of inflammation and cell death in vFlipIEC-tg mice. However, we cannot exclude that impairment of the alternative NFκB pathway in vFlipIEC-tg mice influenced classical NFκB signaling in an indirect manner, due to reduced levels of inflammation and proinflammatory cytokine expression. Previous studies indeed demonstrated that persistent NFκB activation in IECs promotes the development of intestinal inflammation: whereas Guma et al.14 could not observe epithelial changes and ulcerations under steady state conditions, Vlantis et al.15 showed reduction of Paneth cells and spontaneous tumor development in aged mice with constitutive NFκB activation in IECs. However, in contrast to our data, increased epithelial cell death was not observed.
Collectively, our data demonstrate that expression of a single viral protein can be sufficient to disrupt epithelial homeostasis and cause chronic intestinal inflammation. Investigating the effect of viral protein expression in epithelial cells is important, since mucosal barriers are the main entry point of viruses in the human body and might be a target point for antiviral therapy.
MATERIAL AND METHODS
Mice
The generation of mice carrying a FLAG-tagged HHV8-vFLIP flanked by a loxP-flanked neoR-STOP cassette and a frt-flanked IRES-EGFP sequence in an ubiquitously expressed ROSA26 locus (Rosa26.vFLIP mice) was described earlier,36 as well as the generation of VillinCre, Rip3−/− and IkkaF/F mice.77–79 Transgenic mice expressing vFlip in intestinal epithelial cells were generated by breeding Rosa26.vFLIP mice to VillinCre mice (vFlipIEC-tg). To generate IkkaΔIEC mice, IkkaF/F mice were crossed to VillinCre mice.47 In all experiments, mice between 3–16 weeks were used. Mouse endoscopy was performed as previously described.80 Mice were routinely screened for pathogens according to FELASA guidelines. Animal protocols were approved by the Institutional Animal Care and Use Committee of the Regierung von Unterfranken.
Histology and Immunohistochemistry
Histopathological examinations were performed after Mayer’s H&E staining. Immunofluorescence staining was performed on formalin-fixed paraffin-embedded tissue or cryosections by using the Tyramide Signal Amplification (TSA) Cy3 system (Perkin&Elmer), according to the manufacturer’s protocol. The following primary antibodies were used: Anti-F4/80 (eBioscience), anti-CD11c (BD-Pharmingen), anti-CD4 (BD-Pharmingen), anti-Lysozyme (Dako), anti-P65 and anti-Cleaved Caspase-3 (both Cell Signaling). The following biotinylated secondary antibodies were used: anti-rabbit and anti-hamster (both Dianova), anti-rat (BD Biosciences). In experiments with murine samples, nuclei were counterstained with Hoechst 33342 (Invitrogen), in human cells, nuclei were stained with DAPI (Life technologies). For cell death analysis the In Situ Cell Death Detection Kit (Fluorescein) for TUNEL (TdT-mediated dUTP nick end labeling) from Roche was used. Bright-field and fluorescence pictures were taken by using the DMI4000 B microscope (Leica) together with the LEICA DFC360 FX or LEICA DFC420C camera. For confocal images the confocal fluorescence microscope Leica TCS SP5 II was used.
GFP fluorescence staining
For detection of GFP fluorescence ex vivo, fresh tissue was immediately fixed in 4 % Paraformaldehyde for 8 h (4 °C), transferred into 2% sucrose solution (8 h, 4 °C), embedded in Tissue-Tek OCT-compound (Sakura), cut at a Cryostat (Leica) and counterstained with Hoechst 33342 (Invitrogen).
Immunoblotting
Proteins were isolated from intestinal epithelial cells as previously described,81 separated by using a MiniProtean Precast gel (4–15 % polyacrylamide, Bio-Rad) and transferred to a nitrocellulose membrane (Millipore). For murine protein detection, the following primary antibodies were used: anti-P65 and anti-phospho-P65, anti-P100/P52, anti-IKKα, anti-IKKβ, anti-Caspase-3, anti-phospho-IκBα (all Cell Signaling), anti-GFP and anti-iNOS (both Abcam), anti-MLKL (Biorbyt) and a directly-labeled anti-Actin antibody (Abcam). For protein detection in human cells, an anti-MLKL and an anti-RIP3 antibody from Cell Signaling, as well as an anti-GAPDH antibody from Millipore were used. As a secondary antibody, an HRP-linked anti-rabbit antibody from Cell signaling was used.
Gene expression
Total RNA was isolated from the gut tissue by using the Peqgold Total RNA Kit (C-Line) from Peqlab. cDNA was synthesized by using the Script cDNA Synthesis Kit (Jena Bioscience) and analyzed by quantitative real time PCR with SYBRgreen reagent from Roche and QuantiTect primer assays from Qiagen. The results were normalized to the expression levels of the housekeeping gene hypoxanthine guanine phosphoribosyl transferase (Hprt).
Statistical analysis
Statistical analysis was performed using the two-tailed student t-test, *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
ACKNOWLEDGEMENTS
This work received funding from the DFG collaborative research centers SFB796 (subproject B9), SFB1181 (subproject C05), and TRR241 (subprojects A02, A03, A06). The work was furthermore supported by the DFG clinical research units KFO257 and the FOR2438 and by the Interdisciplinary Centre for Clinical Research (IZKF) of the University Erlangen-Nürnberg. The German Research Foundation DFG further supported this work within priority program SPP1656 (Intestinal Microbiota) and individual grant BE3686/2. We thank Kristina Urbanova, Melanie Zeitler, Stefan Wallmüller, Alexandra Wandersee, Christina Lindner and Gabriele Förtsch for excellent technical assistance. The present work was performed in (partial) fulfillment of the requirements for obtaining the degrees Dr. rer. nat. for Barbara Ruder and Vinay Murtadak.
Footnotes
ADDITIONAL INFORMATION
The online version of this article (https://doi.org/10.1038/s41385-018-0068-6) contains supplementary material, which is available to authorized users.
Competing interests: The authors declare no competing interests.
REFERENCES
- 1.Gunther C, Buchen B, Neurath MF & Becker C Regulation and pathophysiological role of epithelial turnover in the gut. Semin. Cell Dev. Biol 35, 40–50 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Wittkopf N et al. Cellular FLICE-like inhibitory protein secures intestinal epithelial cell survival and immune homeostasis by regulating caspase-8. Gastroenterology 145, 1369–1379 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Gunther C et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Degterev A et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol 1, 112–119 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Feoktistova M, Geserick P, Panayotova-Dimitrova D & Leverkus M Pick your poison: the Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 11, 460–467 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Vandenabeele P, Galluzzi L, Vanden Berghe T & Kroemer G Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol 11, 700–714 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Christofferson DE & Yuan J Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol 22, 263–268 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vercammen D et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med 188, 919–930 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Chen X et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24, 105–121 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Vlantis K et al. NEMO prevents RIP Kinase 1-mediated epithelial cell death and chronic intestinal inflammation by NF-kappaB-dependent and -independent functions. Immunity 44, 553–567 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nenci A et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Guma M et al. Constitutive intestinal NF-kappaB does not trigger destructive inflammation unless accompanied by MAPK activation. J. Exp. Med 208, 1889–1900 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vlantis K et al. Constitutive IKK2 activation in intestinal epithelial cells induces intestinal tumors in mice. J. Clin. Invest 121, 2781–2793 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker C, Neurath MF & Wirtz S The intestinal microbiota in inflammatory bowel disease. ILAR J 56, 192–204 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Karst SM, Zhu S & Goodfellow IG The molecular pathology of noroviruses. J. Pathol 235, 206–216 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Lawlor G & Moss AC Cytomegalovirus in inflammatory bowel disease: pathogen or innocent bystander? Inflamm. Bowel Dis 16, 1620–1627 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Lundgren O & Svensson L Pathogenesis of rotavirus diarrhea. Microbes Infect 3, 1145–1156 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahman MM & McFadden G Modulation of NF-kappaB signalling by microbial pathogens. Nat. Rev. Microbiol 9, 291–306 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galluzzi L et al. Viral strategies for the evasion of immunogenic cell death. J. Intern. Med Suppl. 267, 526–542 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Bohme L & Rudel T Host cell death machinery as a target for bacterial pathogens. Microbes Infect 11, 1063–1070 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Kaplan LD Human herpesvirus-8: Kaposi sarcoma, multicentric Castleman disease, and primary effusion lymphoma. Hematol. Am. Soc. Hematol. Educ. Program 2013, 103–108 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Lee HR, Lee S, Chaudhary PM, Gill P & Jung JU Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Future Microbiol 5, 1349–1365 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irmler M et al. Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Belanger C et al. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fasmediated apoptosis through binding and prevention of procaspase-8 maturation. J. Hum. Virol 4, 62–73 (2001). [PubMed] [Google Scholar]
- 27.Sarid R et al. vFLIP protects PC-12 cells from apoptosis induced by Sindbis virus: implications for the role of TNF-alpha. Cell Death Differ 8, 1224–1231 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Thome M et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386, 517–521 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Chaudhary PM, Jasmin A, Eby MT & Hood L Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18, 5738–5746 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Graham C et al. Kaposi’s sarcoma-associated herpesvirus oncoprotein K13 protects against B cell receptor-induced growth arrest and apoptosis through NF-kappaB activation. J. Virol 87, 2242–2252 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L et al. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem 277, 13745–13751 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Sun Q, Matta H & Chaudhary PM The human herpes virus 8-encoded viral FLICE inhibitory protein protects against growth factor withdrawal-induced apoptosis via NF-kappa B activation. Blood 101, 1956–1961 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Thurau M et al. Viral inhibitor of apoptosis vFLIP/K13 protects endothelial cells against superoxide-induced cell death. J. Virol 83, 598–611 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tolani B, Matta H, Gopalakrishnan R, Punj V & Chaudhary PM NEMO is essential for Kaposi’s sarcoma-associated herpesvirus-encoded vFLIP K13-induced gene expression and protection against death receptor-induced cell death, and its N-terminal 251 residues are sufficient for this process. J. Virol 88, 6345–6354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sin SH & Dittmer DP Viral latency locus augments B-cell response in vivo to induce chronic marginal zone enlargement, plasma cell hyperplasia, and lymphoma. Blood 121, 2952–2963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballon G, Chen K, Perez R, Tam W & Cesarman E Kaposi sarcoma herpesvirus (KSHV) vFLIP oncoprotein induces B cell transdifferentiation and tumorigenesis in mice. J. Clin. Invest 121, 1141–1153 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chugh P et al. Constitutive NF-kappaB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc. Natl Acad. Sci. USA 102, 12885–12890 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ballon G, Akar G & Cesarman E Systemic expression of Kaposi sarcoma herpesvirus (KSHV) Vflip in endothelial cells leads to a profound proinflammatory phenotype and myeloid lineage remodeling in vivo. PLoS. Pathog 11, e1004581 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peeters T & Vantrappen G The Paneth cell: a source of intestinal lysozyme. Gut 16, 553–558 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gunther C et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Invest 126, 4346–4360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alkharsah KR et al. Deletion of Kaposi’s sarcoma-associated herpesvirus FLICE inhibitory protein, vFLIP, from the viral genome compromises the activation of STAT1-responsive cellular genes and spindle cell formation in endothelial cells. J. Virol 85, 10375–10388 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakakibara S, Pise-Masison CA, Brady JN & Tosato G Gene regulation and functional alterations induced by Kaposi’s sarcoma-associated herpesvirus-encoded ORFK13/vFLIP in endothelial cells. J. Virol 83, 2140–2153 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin NR, Whon TW & Bae JW Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 33, 496–503 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Field N et al. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci 116, 3721–3728 (2003). Pt 18. [DOI] [PubMed] [Google Scholar]
- 45.Matta H & Chaudhary PM Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc. Natl. Acad. Sci. U. S. A 101, 9399–9404 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matta H, Sun Q, Moses G & Chaudhary PM Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein-induced NF-kappaB activation. J. Biol. Chem 278, 52406–52411 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Diamanti MA et al. IKKalpha controls ATG16L1 degradation to prevent ER stress during inflammation. J. Exp. Med 214, 423–437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popgeorgiev N, Temmam S, Raoult D & Desnues C Describing the silent human virome with an emphasis on giant viruses. Intervirology 56, 395–412 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Mocarski ES, Upton JW & Kaiser WJ Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol 12, 79–88 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dimitroulia E, Spanakis N, Konstantinidou AE, Legakis NJ & Tsakris A Frequent detection of cytomegalovirus in the intestine of patients with inflammatory bowel disease. Inflamm. Bowel Dis 12, 879–884 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Lavery EA & Coyle WJ Herpes simplex virus and the alimentary tract. Curr. Gastroenterol. Rep 10, 417–423 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Sigle GW & Kim R Sexually transmitted proctitis. Clin. Colon Rectal Surg 28, 70–78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsuchiya Y, Nakabayashi O & Nakano H FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int. J. Mol. Sci 16, 30321–30341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krueger A, Baumann S, Krammer PH & Kirchhoff S FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol. Cell Biol 21, 8247–8254 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ambroziak JA et al. Herpes-like sequences in HIV-infected and uninfected Kaposi’s sarcoma patients. Science 268, 582–583 (1995). [DOI] [PubMed] [Google Scholar]
- 56.Bechtel JT, Liang Y, Hvidding J & Ganem D Host range of Kaposi’s sarcoma-associated herpesvirus in cultured cells. J. Virol 77, 6474–6481 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duus KM, Lentchitsky V, Wagenaar T, Grose C & Webster-Cyriaque J Wildtype Kaposi’s sarcoma-associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J. Virol 78, 4074–4084 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Foreman KE et al. Propagation of a human herpesvirus from AIDS-associated Kaposi’s sarcoma. N. Engl. J. Med 336, 163–171 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Kaleeba JA & Berger EA Broad target cell selectivity of Kaposi’s sarcoma-associated herpesvirus glycoprotein-mediated cell fusion and virion entry. Virology 354, 7–14 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Pauk J et al. Mucosal shedding of human herpesvirus 8 in men. N. Engl. J. Med 343, 1369–1377 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Webster-Cyriaque J Development of Kaposi’s sarcoma in a surgical wound. N. Engl. J. Med 346, 1207–1210 (2002). [DOI] [PubMed] [Google Scholar]
- 62.Hahn AS et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat. Med 18, 961–966 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hafner C et al. Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin. Chem 50, 490–499 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Oshiro M et al. Immunohistochemical localization of heparan sulfate proteoglycan in human gastrointestinal tract. Histochem. Cell Biol 115, 373–380 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Akula SM, Wang FZ, Vieira J & Chandran B Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282, 245–255 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Hamzaoui L et al. Iatrogenic colorectal Kaposi sarcoma complicating a refractory ulcerative colitis in a human immunodeficiency negative-virus patient. Pan Afr. Med. J 15, 154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rodriguez-Pelaez M et al. Kaposi’s sarcoma: an opportunistic infection by human herpesvirus-8 in ulcerative colitis. J. Crohn’s Colitis 4, 586–590 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Girelli CM, Serio G, Rocca E & Rocca F Refractory ulcerative colitis and iatroge nic colorectal Kaposi’s sarcoma. Dig. Liver Dis 41, 170–174 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Tedesco M et al. Colorectal Kaposi’s sarcoma in an HIV-negative male in association with ulcerative rectocolitis: a case report. Anticancer Res 19, 3045–3048 (1999). 4B. [PubMed] [Google Scholar]
- 70.Weber JN et al. Kaposi’s sarcoma of the bowel–presenting as apparent ulcerative colitis. Gut 26, 295–300 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo H, Kaiser WJ & Mocarski ES Manipulation of apoptosis and necroptosis signaling by herpesviruses. Med. Microbiol. Immunol 204, 439–448 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang Z et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host. Microbe 17, 229–242 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Maelfait J et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J 36, 2529–2543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Upton JW, Kaiser WJ & Mocarski ES DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl Acad. Sci. USA 111, 15438–15443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Atreya I, Atreya R & Neurath MF NF-kappaB in inflammatory bowel disease. J. Intern. Med Suppl. 263, 591–596 (2008). [DOI] [PubMed] [Google Scholar]
- 77.el Marjou F et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Liu B et al. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell 14, 212–225 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Newton K, Sun X & Dixit VM Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell Biol 24, 1464–1469 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Becker C et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21, 491–501 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Gunther C et al. Caspase-8 controls the gut response to microbial challenges by Tnf-alpha-dependent and independent pathways. Gut 64, 601–610 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.