

Abstract
Recently, short synthetic peptides have gained interest as targeting agents in the design of site-specific nanomedicines. In this context, our work aimed at developing new tools for the diagnosis and/or therapy of hepatocellular carcinoma (HCC) by grafting the hepatotropic George Baker (GB) virus A (GBVA10-9) and Plasmodium circumsporozoite protein (CPB)-derived peptides to the biocompatible poly(benzyl malate), PMLABe. We successfully synthesized PMLABe derivatives end-functionalized with peptides GBVA10-9, CPB, and their corresponding scrambled peptides through a thiol/maleimide reaction. The corresponding nanoparticles (NPs), varying by the nature of the peptide (GBVA10-9, CPB, and their scrambled peptides) and the absence or presence of poly(ethylene glycol) were also successfully formulated using nanoprecipitation technique. NPs were further characterized by dynamic light scattering (DLS), electrophoretic light scattering (ELS) and transmission electron microscopy (TEM), highlighting a diameter lower than 150 nm, a negative surface charge, and a more or less spherical shape. Moreover, a fluorescent probe (DiD Oil) has been encapsulated during the nanoprecipitation process. Finally, preliminary in vitro internalisation assays using HepaRG hepatoma cells demonstrated that CPB peptide-functionalized PMLABe NPs were efficiently internalized by endocytosis, and that such nanoobjects may be promising drug delivery systems for the theranostics of HCC.
Keywords: poly(malic acid) derivatives, GB virus A, circumsporozoite protein of Plasmodium berghei, (Co)polymers end-functionalized with peptide, multifunctional nanoparticles, human hepatoma cells
1. Introduction
At the beginning of the 20th century, Paul Ehrlich coined the term “Magic Bullet”, postulating that the targeting of drug towards specific diseased cells/tissues could be achieved without affecting healthy ones [1]. Ever since, tremendous research work has been done for designing efficient site-specific drugs using various ligand molecules, such as monoclonal antibodies (mAbs) [2,3,4], vitamins [2,5,6,7], carbohydrates [2,8], and aptamers [9,10,11]. More recently, short synthetic peptides have gained interest as targeting agents in the design of site-specific nanomedicines for therapy and/or diagnosis of several pathologies, especially cancers [2,12,13,14,15,16].
The discovery of peptides dates back to the turn of the 20th century, when Emil Fisher first synthesized in 1901 what he called a dipeptide, the glycyl-glycine, followed a few years later (1907) by the synthesis of an octadecapeptide, the Leu-Gly3-Leu-Gly3-Leu-Gly9 [17]. Almost in parallel, in 1902 Theodor Curtius synthesized benzoyl-glycyl-glycine using the azide reaction he developed [17]. Following these pioneer works, researchers worked to increase the number of amino acids in the peptide’s chain, which was possible thanks to the use of the removable benzyloxycarbonyl protecting group developed by Max Bergmann and Leonidas Zervas [17]. The uses of peptides as therapeutic molecules emerged in the 1950s when some pure peptides (oxytocin, vasopressin, for example) were synthesized by Vincent du Vigneaud [18,19,20]. However, their synthesis was long and tedious until the development by Merrifield of a functionalized resin, bearing his name, composed by styrene/divinylbenzene block copolymers, thus allowing faster and easier solid-phase peptides’ synthesis [18,19]. Since then, peptides (and proteins) are used for several indications under various therapeutic classes as a result of their high activity and specificity, and their low systemic toxicity resulting from their short in vivo half-life [18,21,22,23]. However, their fast degradation by peptidases and poor biodistribution at the targeted tissues/organs limit their uses. To overcome such drawbacks, strategies have been developed to increase peptides’ stability in vivo and to improve their biodistribution, including introduction of non-natural amino acids (D-amino acids, for example), cyclization, N- and C-termini and side-chain chemical modifications, etc. [21,22,23,24].
Beside their uses as therapeutic molecules [18,22,23,25], short peptides have been quite recently developed as targeting agents, since it was shown that peptide receptors are overexpressed, or even exclusively expressed, in tumors [12]. Such increasing interest has been catalyzed by the discovery, at the end of the 1980s, of specific receptors to somatostatin (SSTR1-5) expressed by human neuroendocrine tumors (NETs) and their metastases [12,26]. Octreotide, a somatostatin analog, grafted to diethylenetriamine pentaacetic acid (DTPA) chelating indium 111 (111In) was approved by the US Food and Drug Administration (FDA) in 1994 under the name of OctreoScan® for the diagnosis and localization of NETs overexpressing the somatostatin receptor SSTR2 [12,27]. Twenty years later (2018), Lutathera®, which consists of octreotate, another somatostatin analog, grafted to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) chelating the lutetium 177 (177Lu), was the first FDA-approved radiopharmaceutical for the therapy of SSTR2-positive NETs [12,28]. Since the emergence of somatostatin and its analogs, numerous peptides have been identified by different technologies, such as phage-display, for their high degree of specificity towards cancer cells [12,13,14,15,16]. Such peptides, which are usually easily synthesized on large scales at a low cost, can be chemically modified to be grafted on various materials, such as lipids or polymers, to engineer nanocarriers characterized by their good biocompatibility in addition to their excellent targeting properties [12,13,14,15,16,17].
The advanced stages of hepatocellular carcinoma (HCC), the major primary liver cancer, still have a very poor prognosis despite the recent progress in the therapeutic management of this cancer [29]. In addition, only a few patients are eligible for curative treatments in the early stages of the disease, or can be treated downstream to decrease the stage of their tumor so that they may be eligible for curative treatments [30,31]. Consequently, there is an urgent need for tools allowing effective therapies and/or early diagnosis of HCC.
In this context, the use of peptides able to specifically target hepatoma cells to detect as soon as possible even small nodules and/or treat early and advanced stages of HCC might be of great interest. In order to identify peptides exhibiting a strong tropism towards human hepatoma cells, in a previous work, we elected 12 peptides reported in the literature to bind hepatoma cells, including GB virus A (GBVA10-9) and Plasmodium circumsporozoite protein (CPB), and launched a comparative study of the in vitro internalization by human hepatocytes in primary culture and hepatoma cells of the corresponding biotinylated peptides binding fluorescent streptavidin to form a model bioconjugate [32]. The GBVA10-9- and CPB-streptavidin complexes showed the strongest binding to hepatoma cells [32]. We next demonstrated that the same biotinylated peptides, when bridged via the streptavidin to biotinylated nanoparticles (NPs) constituted by the amphiphilic block copolymer biotin-poly(ethylene glycol)-block-poly(benzyl malate) (BiotPEG-b-PMLABe), significantly enhanced the cell uptake by human hepatoma cells but not by normal human hepatocytes [32]. These data suggested that GBVA10-9 and CPB peptides were good candidates to functionalize polymeric nanoparticles to efficiently target human hepatic cancer cells.
Our main objective of the present study was to establish the synthesis of peptides end-functionalized poly(benzyl malate) and poly(ethylene glycol)-block-poly(benzyl malate) through a thiol/maleimide addition reaction in order to eliminate the streptavidin used in our previous study [32]. The corresponding nanoparticles varying by the nature of the peptide (GBVA10-9, CPB or their scrambled control counterparts, Figure 1) present at their surfaces and by the absence or presence of poly(ethylene glycol) were also prepared and characterized by dynamic light scattering (DLS) (Malvern PANalytical Ltd., Malvern UK), electrophoretic light scattering (ELS) (Malvern PANalytical Ltd., Malvern UK) and transmission electron microscopy (TEM) (JEOL, Tokyo, Japan). Moreover, a fluorescent probe (DiD Oil) was encapsulated and preliminary in vitro internalisation assays using HepaRG hepatoma cell lines were performed.
Figure 1.

Structure of: (A) GBVA10-9 and its scrambled control (GBVA10-9scr), and (B) CPB and its scrambled control (CPBscr).
2. Materials and Methods
2.1. Materials and Apparatus
All chemicals were used as received. α-maleimide, ω-carboxylic acid PEG62 (Mw = 3000 g/mol, n = 62) and α-methoxy, ω-carboxylic acid PEG42 (Mw = 2015 g/mol, n = 42) were purchased from Iris Biotech GmbH (Marktredwitz, Germany). Tetraethylammonium hydroxide and 6-maleimidohexanoic acid were purchased from Sigma-Aldrich (Saint-Louis, MO, USA). Peptides were provided by Eurogentec (Liege, Belgium). 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo dicarbocyanine perchlorate (DiD Oil) was purchased from Invitrogen (Thermo Fisher Scientific, Illkrich-Graffenstaden, France). Dry THF was purchased from Acros Organics (Thermo Fischer Scientific, Illkrich-Graffenstaden, France). Dimethylformamide (DMF) was purchased from Sigma-Aldrich (Saint-Quentin-Fallavier, France). Spectra/Por 4 Dialysis Membrane was acquired from Repligen (Breda, The Netherlands). The monomer, benzyl malolactonate (MLABe), was synthesized as described previously [33].
Nuclear Magnetic Resonance spectroscopy: The standard temperature was adjusted to 298 K. NMR spectra were recorded on a Bruker Avance III 400 spectrometer (Bruker, Wissembourg, France) operating at 400.13 MHz for 1H, equipped with a BBFO probe with a Z-gradient coil and a GREAT 1/10 gradient unit. The zg30 Bruker pulse program was used for 1D 1H NMR, with a TD of 64 k, a relaxation delay d1 = 2 s and 8 scans. The spectrum width was set to 18 ppm. Fourier transform of the acquired FID was performed without any apodization in most cases.
Infrared spectroscopy: FT-IR spectra were recorded on an Avatar 320FT-IR Thermo Nicolet spectrometer (Nicolet, Montigny le Bretonneux, France) between 500 and 4000 cm−1 by direct measurement.
Size Exclusion Chromatography: Weight average molar mass (Mw) and dispersity (Đ = Mw/Mn) values were measured by size exclusion chromatography (SEC) in THF at 40 °C (flow rate = 1.0 mL/min) on a GPC2502 Viscotek apparatus (Malvern Instruments Ltd., Malvern, UK), country) equipped with a refractive index detector Viscotek VE 3580 RI (Malvern Instruments Ltd., Malvern, UK), a guard column Viscotek TGuard (Malvern Instruments Ltd., Malvern, UK), Org 10 × 4.6 mm, a LT5000L gel column (for samples soluble in organic medium) 300 × 7.8 mm and a GPC/SEC OmniSEC Software (OmniSEC™ 4.6.1, Malvern Instruments Ltd., Malvern, UK). The polymer samples were dissolved in THF (2 mg/mL). All elution curves were calibrated with polystyrene standards.
Dynamic Light Scattering and Electrophoretic Light Scattering: Dynamic light scattering (DLS) and zeta potential (electrophoretic light scattering, ELS) measurements are performed on a Nano-sizer ZS90 (Malvern Instruments Ltd., Malvern, UK) at 25 °C, with a He-Ne laser (Malvern Instruments Ltd., Malvern, UK) at 633 nm and a detection angle of 90 °C.
Transmission electron microscopy: Transmission electron microscopy (TEM) images were recorded using a Jeol 2100 microscope (JEOL, Tokyo, Japan) equipped with a Glatan Orius 200D camera using a 80 KeV accelerating voltage on the THEMIS platform (ISCR—Rennes) (Univ Rennes, Rennes, France). Each sample was deposited on a Formvar-carbon film coated on a 300-mesh copper grid. After 6 min, the excess of the sample was removed and staining was realized with phosphotungstic acid (1 v%).
Flow cytometry: Cells were analyzed by flow cytometry (Becton Dickinson LSRFortessa™ X-20), using the cytometry core facility of the Biology and Health Federative research structure Biosit, Rennes, France, to quantify the fluorescence emitted by the DiD Oil-loaded NPs within cells. Cytometry data were analyzed using FACSDivaTM software (Software version 8.0.1, Beckton Dickinson, East Rutherford, NJ, USA).
Confocal microscopy: Images of NPs in human HepaRG hepatoma cells were acquired with Leica TCS SP8 confocal microscope piloted by the Las AF software (Software version 3.3, Leica Microsystems, Wetzlar, Germany) (microscopy Rennes imaging center facility of the Biology and Health Federative research structure Biosit, Rennes, France) and processed with the ImageJ software (Open Source Software version 1.51n, Laboratory for Optical and Computational Instrumentation, University of Wisconsin, Madison, WI, USA).
2.2. Synthesis of PMLABe Derivatives
Synthesis of tetraethylammonium 6-maleimidohexanoate (MalHexCOO− +NEt4): The methanol of 947 μL (1.42 × 10−3 mol) of tetraethylammonium hydroxide solution (25 wt% in methanol) was evaporated under vacuum, and 4 mL of anhydrous chloroform was added. In parallel, 300 mg (1.42 × 10−3 mol) of 6-maleimidohexanoic acid were solubilized in anhydrous chloroform (4 mL) under N2 stream, and the flask was put in an ice bath. The chloroform solution of tetraethylammonium hydroxide was then added to this solution, the mixture was stirred for 1 h, and the chloroform was evaporated under vacuum leading to the expected tetraethylammonium maleimidohexanoate as a violet oil (Yield: 93%) that was characterized by 1H NMR. 1H NMR (400 MHz, Acetone-d6), δ (ppm): 1.26–1.34 (m, 2H); 1.35–1.36 (m, 12H); 1.53–1.61 (m, 4H); 2.25–2.29 (t, 2H); 3.44–3.48 (t, 2H); 3.51–3.56 (d, 8H); 6.84 (s, 2H).
Synthesis of tetraethylammonium α-methoxy-ω-carboxy poly(ethylene glycol) (MeOPEG42-COO− +NEt4): In this process, 200 mg (9.93 × 10−5 mol) of α-methoxy-ω-carboxylic acid PEG42 (M = 2015 g/mol) was dissolved in methanol (2 mL). Then, 58 μL (9.93 × 10−5 mol) of tetraethylammonium hydroxide solution in methanol (25 wt%) was added, and the mixture was stirred for 15 min at room temperature before the evaporation of methanol under vacuum. The crude product was solubilized in acetone (2 mL) and precipitated into diethyl ether (200 mL). The obtained white precipitate was dried under vacuum (Yield: 97%) and characterized by 1H NMR. 1H NMR (400 MHz, Acetone-d6), δ (ppm): 1.36–1.41 (m, 2H); 3.50–3.55 (d, 8H); 3.59 (s, 4x42H).
Synthesis of tetraethylammonium α-maleimido-ω-carboxy poly(ethylene glycol) (MalPEG62-COO− +NEt4): In this process, 300 mg (0.1 × 10−3 mol) of α-maleimido-ω-carboxylic acid poly(ethylene glycol) was dissolved in methanol (2.2 mL). Then, 67 μL (0.1 × 10−3 mol) of tetraethylammonium hydroxide solution in methanol (25 wt%) was added, and the mixture was stirred for 15 min at room temperature before the evaporation of methanol under vacuum. The crude product was solubilized in acetone (2 mL) and precipitated into diethyl ether (200 mL). The obtained orange precipitate was dried under vacuum (Yield: 83%) and characterized by 1H NMR. 1H NMR (400 MHz, Acetone-d6), δ (ppm): 1.36–1.41 (m, 12H); 3.48–3.55 (d, 8H); 3.59 (s, 4x62H). MNMR = 2860 g/mol.
Synthesis of poly(benzyl malate) (PMLABe73): PMLABe73 was synthesized as previously described, with slight modifications, with a monomer/initiator ratio chosen to obtain a theoretical molar mass of 15,000 g/mol (n = 73) [33]. Briefly, 16.7 mg (6.64 × 10−5 mol) of tetraethylammonium benzoate were solubilized into 700 μL of anhydrous ethanol freshly distilled over natrium. The initiator solution was placed in the polymerization flask, ethanol was eliminated under vacuum at room temperature and the tetraethylammonium benzoate was dried under vacuum at room temperature overnight. MLABe (1 g, 4.85 × 10−3 mol) of kept under N2 stream was transferred into the polymerization flask containing the initiator. The polymerization was conducted at 37 °C for one day (disappearance of the lactone peak at 1844 cm−l from the FT-IR spectrum). The polymer was dissolved in acetone (2 mL), one drop of concentrated HCI was added and the polymer was precipitated into a large excess of ethanol (200 mL). The white precipitate was dried under vacuum (Yield: 89%) and characterized by 1H NMR and SEC. 1H NMR (400 MHz, Acetone-d6), δ (ppm): 2.97 (m, 2nH); 5.14 (m, 1nH); 5.53 (m, 2nH); 7.33 (m, 5nH); 8.04–8.06 (d, 2H). SEC (THF, 40 °C, polystyrene standards): Mw = 7,300 g/mol, Ð = 1.46.
Synthesis of maleimide poly(benzyl malate) (MalPMLABe73): In this process, 11 mg (3.33 × 10−5 mol) of MalHexCOO− +NEt4 was put under N2 stream. MLABe (0.5 g, 2.43 × 10−3 mol) was dissolved into 0.15 mL of anhydrous THF under N2 stream, and this solution was then transferred into the polymerization flask containing the initiator. The monomer/initiator ratio was chosen to obtain a theoretical molar mass of 15,000 g/mol (n = 73). The polymerization was conducted for 18 h at 37 °C (disappearance of the lactone peak at 1844 cm−l from the FT-IR spectrum). The polymer was dissolved in acetone (1 mL), one drop of concentrated HCI was added and the polymer was precipitated into a large excess of ethanol (200 mL). The white precipitate was dried under vacuum (Yield: 82%) and characterized by 1H NMR and SEC. 1H NMR (400 MHz, Acetone-d6), δ (ppm): 2.97 (m, 2nH); 5.14 (m, 1nH); 5.53 (m, 2nH); 7.33 (m, 5nH). SEC (THF, 40 °C, polystyrene standards): Mw = 5730 g/mol, Ð = 1.48.
Synthesis of poly(ethylene glycol)-block-poly(benzyl malate) (PEG42-b-PMLABe73): In this process, 134 mg (6.65 × 10−5 mol) of PEG42COO− +NEt4 (M = 2015 g/mol) was put under N2 stream. MLABe (1 g, 4.85 × 10−3 mol) was dissolved into 0.15 mL of anhydrous THF under N2 stream, and this solution was then transferred into the polymerization flask containing the initiator. The monomer/initiator ratio was chosen to obtain a theoretical molar mass of 15,000 g/mol (n = 73). The polymerization was conducted for 22 h at 37 °C (disappearance of the lactone peak at 1844 cm−l from the FT-IR spectrum). The polymer was dissolved in acetone (1 mL), one drop of concentrated HCI was added and the polymer was precipitated into a large excess of ethanol (200 mL). The white precipitate was dried under vacuum (Yield: 76%) and characterized by 1H NMR and SEC. 1H NMR (400 MHz, Acetone-d6), δ (ppm): 2.97 (m, 2nH); 3.58 (m, 4x42H); 5.14 (m, 1nH); 5.54 (m, 2nH); 7.33 (m, 5nH). MNMR = 12,980 g/mol. SEC (THF, 40 °C, polystyrene standards): Mw = 6460 g/mol, Ð = 1.45.
Synthesis of maleimide poly(ethylene glycol)-block-poly(β-benzyl malate) (MalPEG62-b-PMLABe73): In this process, 104 mg (3.33 × 10−5 mol) MalPEG62-COO− +NEt4 was put under N2 stream, and solubilized in 0.5 mL of anhydrous THF. MLABe (0.5 g, 2.43 × 10−3 mol) was dissolved into 0.4 mL of anhydrous THF under N2 stream, and this solution was then transferred into the polymerization flask containing the initiator. The monomer/initiator ratio was chosen to obtain a theoretical molar mass of 15,000 g/mol (n = 73). The polymerization was conducted for 22 h at 27 °C (disappearance of the lactone peak at 1844 cm−l from the FT-IR spectrum). The polymer was dissolved in acetone (2 mL), one drop of concentrated HCI was added and the polymer was precipitated into a large excess of ethanol (200 mL). The orange precipitate was dried under vacuum (Yield: 82%) and characterized by 1H NMR and SEC. 1H NMR (400 MHz, Acetone-d6), δ (ppm): 2.94 (m, 2nH); 3.59 (m, 4x62H); 5.14 (m, 1nH); 5.54 (m, 2nH); 7.33 (m, 5nH). MNMR = 13,800 g/mol. SEC (THF, 40 °C, polystyrene standards): Mw = 8470 g/mol, Ð = 1.59.
Grafting of peptides onto synthesized polymers MalPMLABe73 and MalPEG67-b-PMLABe73: The four selected peptides (GBVA10-9, CPB, GBVA10-9scr and CPBscr) were grafted onto the maleimide end-functionalized MalPMLABe73 and MalPEG62-b-PMLABe73. Typically, (co)polymer (1 equivalent (eq.)) solubilized in 1.4 mL of DMF was added to peptide (1 eq.) solubilized in 1.4 mL of DMF. A phosphate buffer solution (PBS, pH = 7.2) was then added dropwise (54 µL of PBS at 0.01 M in 1 mL of DMF). The reaction was allowed to proceed overnight at room temperature, and excess reactant was removed by extensive dialysis against DMF for 24 h (molecular weight cut off (MWCO) of 12,000–14,000 g/mol). After DMF elimination, the purified peptide-modified (co)polymers were characterized by 1H NMR. The following eight peptide-modified (co)polymers were obtained: GBVA10-9PMLABe73, GBVA10-9PEG62-b-PMLABe73, CPBPMLABe73, CPBPEG62-b-PMLABe73, GBVA10-9scrPMLABe73, GBVA10-9scrPEG62-b-PMLABe73, CPBScrPMLABe73 and CPBScrPEG62-b-PMLABe73.
2.3. Formulation of Nanoparticles
Peptide-decorated nanoparticles (Pept-NPs) and native nanoparticles (NPs) encapsulating the fluorescence probe DiD Oil were formulated using the nanoprecipitation method initially described by Fessi et al. with slight modifications [34]. Twelve batches of Pept-NPs were thus formulated starting from 10 wt% of peptide-modified (co)polymers and 90 wt% of PMLABe73 or PEG42-b-PMLABe73 and named as indicated in Table 1.
Table 1.
Composition of peptide-decorated nanoparticles (Pept-NPs).
| Peptide | Code | Composition |
|---|---|---|
| GBVA10-9 | NPs 1 | 10 wt% GBVA10-9PMLABe73 + 90 wt% PMLABe73 |
| NPs 2 | 10 wt% GBVA10-9PEG62-b-PMLABe73 + 90 wt% PMLABe73 | |
| NPs 3 | 10 wt% GBVA10-9PEG62-b-PMLABe73 + 90 wt% PEG42-b-PMLABe73 | |
| CPB | NPs 4 | 10 wt% CPBPMLABe73 + 90 wt% PMLABe73 |
| NPs 5 | 10 wt% CPBPEG62-b-PMLABe73 + 90 wt% PMLABe73 | |
| NPs 6 | 10 wt% CPBPEG62-b-PMLABe73 + 90 wt% PEG42-b-PMLABe73 | |
| GBVA10-9scr | NPs 7 | 10 wt% GBVA10-9scrPMLABe73 + 90 wt% PMLABe73 |
| NPs 8 | 10 wt% GBVA10-9scrPEG62-b-PMLABe73 + 90 wt% PMLABe73 | |
| NPs 9 | 10 wt% GBVA10-9scrPEG62-b-PMLABe73 + 90 wt% PEG42-b-PMLABe73 | |
| CPBscr | NPs 10 | 10 wt% CPBscrPMLABe73 + 90 wt% PMLABe73 |
| NPs 11 | 10 wt% CPBscrPEG62-b-PMLABe73 + 90 wt% PMLABe73 | |
| NPs 12 | 10 wt% CPBscrPEG62-b-PMLABe73 + 90 wt% PEG42-b-PMLABe73 |
The different batches of NPs 1, 2, 3, 7, 8 and 9 were prepared as follows: 5 µL of a solution of peptide-modified (co)polymer at a concentration of 20 mg/mL in DMF was mixed with 135 µL of a solution of (co)polymers at a concentration of 6.67 mg/mL in DMF. Then, 10 µL of a DiD Oil solution in DMF at a concentration of 0.1 mg/mL was added. This blue solution was rapidly added to 1 mL of distilled water under vigorous stirring and the resulting suspension was stirred at room temperature for 1 h. The resulting bluish suspensions were put on the top of a Sephadex G25 column. After penetration of the sample, 2.5 mL of distilled water was added. After penetration, 3.5 mL of distilled water was added and the blue NP suspensions were collected in vials and analyzed by DLS, ELS and TEM.
The NPs 4, 5, 6, 10, 11 and 12 were prepared as follows: 5 µL of a solution of peptide-modified (co)polymer at a concentration of 20 mg/mL in DMF was mixed with 135 µL of a solution of (co)polymers at a concentration of 6.67 mg/mL in DMF. Then, 10 µL of a DiD Oil solution in DMF at a concentration of 0.1 mg/mL was added. This blue solution was rapidly added to 2 mL of distilled water under vigorous stirring and the resulting suspension was stirred at room temperature for 15 min. The resulting bluish suspensions were put on the top of a Sephadex G25 column. After penetration of the sample, 0.5 mL of distilled water was added. After penetration, 3.5 mL of distilled water was added and the blue NP suspensions were collected in vials and analyzed by DLS, ELS and TEM.
In parallel, three batches of NPs were formulated starting from PMLABe73, PEG42-b-PMLABe73 and a mixture of PMLABe73/PEG42-b-PMLABe73 (Table 2).
Table 2.
Composition of NPs without peptide.
| Code | Composition |
|---|---|
| NPs 13 | PMLABe73 |
| NPs 14 | 10 wt% PEG42-b-PMLABe73 + 90 wt% PMLABe73 |
| NPs 15 | PEG42-b-PMLABe73 |
NPs 13 were prepared as follows: 5 µL of a solution of PMLABe73 at a concentration of 20 mg/mL in DMF was mixed with 135 µL of a solution of PMLABe73 at a concentration of 6.67 mg/mL in DMF. Then, 10 µL of a DiD Oil solution in DMF at a concentration of 0.1 mg/mL was added. This blue solution was added rapidly into 1 mL of distilled water under vigorous stirring. The bluish suspension was stirred at room temperature for 1 h. NPs 14 were prepared as follows: 5 µL of a solution of PEG42-b-PMLABe73 at a concentration of 20 mg/mL in DMF was mixed with 135 µL of a solution of PMLABe73 at a concentration of 6.67 mg/mL in DMF. Then, 10 µl of a DiD Oil solution in DMF at a concentration of 0.1 mg/mL was added. This blue solution was rapidly added to 1 mL of distilled water under vigorous stirring and the resulting suspension was stirred at room temperature for 1 h. NPs 15 were prepared as follows: 5 µL of a solution of PEG42-b-PMLABe73 at a concentration of 20 mg/mL in DMF was mixed with 135 µL of a solution of PEG42-b-PMLABe73 at a concentration of 6.67 mg/mL in DMF. Then, 10 µL of a DiD Oil solution in DMF at a concentration of 0.1 mg/mL was added. This blue solution was rapidly added to 1 mL of distilled water under vigorous stirring and the resulting suspension was stirred at room temperature for 1 h. Suspensions of NPs 13, 14 and 15 thus obtained were then put on the top of a Sephadex G25 column. After penetration of the sample, 2.5 mL of distilled water was added. After penetration, 3.5 mL of distilled water was added and the blue NP suspensions were collected in vial and analyzed by DLS, ELS and TEM.
The concentration of loaded DiD oil was evaluated by UV at 660 nm using a calibration curve of DiD oil solution in a mixture of DMF/H2O (80/20). Then, 400 µL of the DiD oil loaded nanoparticles were mixed with 1600 µL of DMF and the resulting solution was analyzed by UV at 660 nm, giving access to the encapsulation efficiency (E.E.). The encapsulation efficiency was calculated using the following equation:
| E.E% = [DiD oil]encapsulated/[DiD oil]initial × 100 | (1) |
2.4. Cell Culture and In Vitro Uptake Assay of NPs
The human HepaRG hepatoma cells were grown in William’s E medium supplemented with 10% fetal calf serum (FCS), 2% L-glutamine, 5 mg/L insulin, hydrocortisone hemisuccinate at 5 × 10−5 M, 100 units/mL penicillin and 100 µg/mL streptomycin. The fetal calf serum (FCS) was a mixture of two different FCS batches: 75% from Biosera and 25% of FetalClone® II (HyClone). The HepaRG cells were cultured at 37 °C in a humidified atmosphere of 5% CO2, and the medium was renewed every 2 days.
For the NPs’ uptake assay, 105 HepaRG cells were seeded in 24-well plates the day before the incubations with the different batches of NPs. The culture media of the cells were renewed with media containing NPs at a final concentration of (co)polymers of 25 µg/mL. The cells were incubated with NPs for 24 h, then were detached with trypsin-EDTA and resuspended in complete medium for flow cytometry analysis and evaluation of the NPs’ uptake by measuring the fluorescence emitted by the DiD Oil-loaded NPs. For the experiments of cell uptake inhibition, cells were incubated with CPBPMLABe73/PMLABe73 (NPs 4) for 4 h in two distinct conditions: (1) at 4 and 37 °C, (2) in the absence or presence of genistein (200 μM). For the detection of fluorescent NPs within the cells by confocal microscopy, the HepaRG cells (104 cells) were seeded in 8-well Lab-Tek chamber slides (cover glass slide) the day before incubation with NPs. The cells were incubated with NPs for 24 h, then, the nuclear DNA was stained with Hoechst 33342 (Sigma-Aldrich, 1 mg/mL, diluted at 1:5000) for 30 min at RT in the dark. Fluorescent NPs and nuclei were visualized by confocal microscopy.
3. Results and Discussion
3.1. Synthesis and Characterization of Peptides End-Functionalized PMLABe Derivatives
The design of nanocarriers for therapy and/or diagnosis of cancers has attracted and continues to attract huge amount of research works as highlighted by the very recent reviews published on this subject [35,36,37,38]. Such nanocarriers can take various forms (polymeric nanoparticles and micelles, inorganic nanoparticles, liposomes, etc.), be constituted by very different organic and inorganic materials, be sensitive to external (magnetic field, for example) or internal (pH variations) stimuli, and can encapsulate anti-cancer drugs and/or fluorescent probes and/or radionucleides [35,36,37,38]. However, whatever the materials used to formulate such nanocarriers, they have to respect very strict specifications: they have to be biocompatible, (bio)degradable and/or fully eliminated from the organism, to encapsulate a large amount of drugs, to specifically release their cargo at the site of action, etc. [35,36,37,38]. To fulfil such specifications, biodegradable polymeric materials must be selected to construct the nanocarriers, such as poly(amino acids) or polyesters [38]. Among the polyesters, one attracted our attention several years ago: poly(malic acid) (Figure 2) [39].
Figure 2.
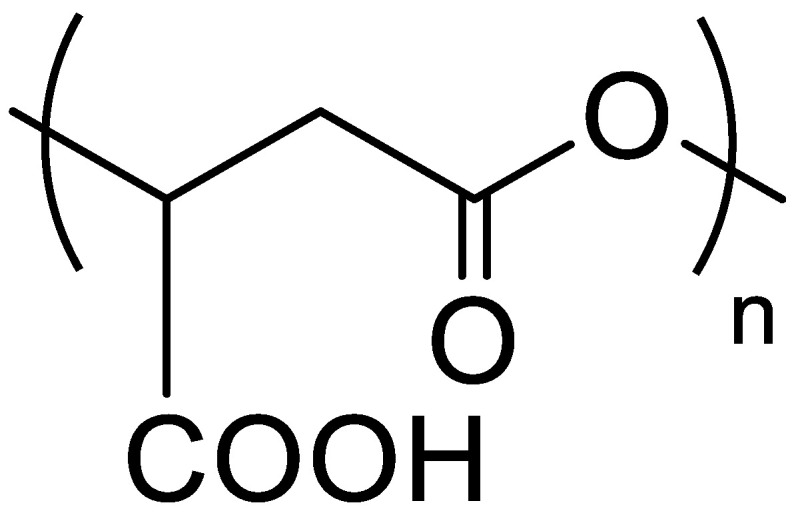
Structure of poly(malic acid) (PMLA).
Indeed, a large family varying by the nature of lateral chains, the molar masses and/or the type of polymers (i.e., homopolymers, block/random/grafted copolymers) can be obtained by anionic ring-opening polymerization (aROP) of β-substituted β-lactones or by chemical modifications of naturally available PMLA. A wide variety of nanocarriers can therefore be formulated with properties adjusted to the targeted applications [39]. We have chosen to synthesize the selected PMLA derivatives by aROP of benzyl malolactonate (MLABe) in the presence of different carboxylate salts of tetraethylammonium as initiators (Scheme 1), because this method allows us to control both the polymers’ molar masses (fixed by the ratio monomer/initiator) and the nature of the end chains [33,39,40,41,42].
Scheme 1.

Synthesis of the selected PMLA derivatives.
3.1.1. Synthesis of PMLABe Derivatives
In the present work, four poly(benzyl malate) (PMLABe) derivatives (Scheme 1), varying by the nature of one of their end-chains (benzoate, methoxy or maleimide) and by their structure (hydrophobic homopolymers or amphiphilic block copolymers), were synthesized and characterized. While the tetraethylammonium benzoate is commercially available, the other three initiators (Figure 3) were synthesized by simply mixing the selected carboxylic acid with tetraethylammonium hydroxide.
Figure 3.

Structure of synthesized initiators.
1H NMR spectra of the resulting purified products allowed us to confirm that the expected initiators have been successfully synthesized and can be further used as initiators for the aROP of benzyl malolactonate (MLABe) as shown by Scheme 1. Molar mass of the PMLABe block was controlled by the ratio monomer/initiator that was adjusted to obtain PMLABe block with a theoretical molar mass of 15,000 g/mol (n = 73) [33]. After purification by precipitation in an excess of ethanol, a solvent in which both initiators and free PEG (if any) are soluble, the resulting homopolymers and block copolymers were characterized by 1H NMR (structure and molar mass of the PMLABe block) and size exclusion chromatography (SEC, weight average molar mass (Mw) and dispersity (Ð)). For PMLABe73 and MalPMLABe73, it was impossible to calculate its molar mass from 1H NMR spectra. Indeed, even if the end-chain protons were visible at 8.03–8.05 ppm (Figure S1) on the 1H NMR spectrum, their integration was not accurate, since these two small peaks represented only two protons over the ones of the polymer main chain. For PMLABe end-terminated by the maleimide group, the two protons of the maleimide end-groups were not, as expected, visible on the 1H NMR spectrum (Figure 4A).
Figure 4.

1H NMR spectra in acetone-d6 of: (A) MalPMLABe73, and (B) MalPEG62-b-PMLABe73.
For amphiphilic block copolymers, the molar mass of the PMLABe block was calculated using the integration of the methylene protons of the PEG block (δ = 3.59 ppm, Figure 4B and Figure S1), whose molar mass was given by the supplier, and the integration of the methylene protons of the PMLABe block (δ = 5.14 ppm, Figure 4B and Figure S1). As shown by results gathered in Table 3, calculated molar masses of PMLABe block were in good agreement with the theoretical ones fixed by the monomer/initiator ratio, thus highlighting the quite good control of the polymerization reaction [30].
Table 3.
Molar masses of PMLABe blocks calculated from 1H NMR spectra and of synthesized (co)polymers measured by SEC in THF.
| Mtheo PMLABe a (g/mol) | MNMR PMLABe b (g/mol) | Mw c (g/mol) | Ð c | |
|---|---|---|---|---|
| PMLABe73 | 15,000 | nd d | 7280 | 1.46 |
| MalPMLABe73 | 15,000 | nd d | 5730 | 1.48 |
| PEG42-b-PMLABe73 | 15,000 | 12,980 | 6460 | 1.45 |
| MalPEG62-b-PMLABe73 | 15,000 | 13,800 | 8470 | 1.59 |
(a) Theoretical molar mass fixed by the ratio monomer/initiator; (b) molar mass calculated from the 1H NMR spectrum; (c) measured by SEC in THF (1 mL/min, Polystyrene standards, 40 °C), (d) nd: not determined.
Nevertheless, SEC measurements gave weight average molar masses (Mw) of the synthesized homopolymers and amphiphilic block copolymers, that are lower than the theoretical ones. We have already observed such a phenomenon for amphiphilic block copolymers [33], a phenomenon that can be attributed to the conditions used for SEC measurements (solvent and standards’ nature) together with a possible absorption of the (co)polymers onto the SEC column, leading to an underestimation of molar masses. However, SEC analyses allowed us to demonstrate the good control of the polymerization procedures because the dispersity values are lower than 1.5 (Table 3), and only one peak has been observed on all the chromatograms.
We can conclude that aROP of MLABe using the selected initiators leads to the expected (co)polymers with adequate structures, molar masses, and dispersity.
3.1.2. Peptide Grafting
As widely recognized, the thiol-maleimide reaction, also known as Michael addition, allows us to easily conjugate peptides with a thiol function at either their N- or C-terminal position to polymeric materials, lipids, nanoparticles, liposomes or biomolecules, presenting an accessible maleimide group [16,43,44,45,46,47,48,49]. Such Michael addition has been described to take place under mild conditions of pH and temperature, and to be conducted in aqueous and/or organic solutions [16,43,44,45,46,47,48,49]. Moreover, maleimide functions are also well-known to have a very strong affinity and specificity towards thiol groups. Therefore, Michael addition can be realized with (macro)molecules and/or nanoobjects without reacting with other functional groups such as amines and carboxylic acids [16,43,44,45,46,47,48,49].
In this context, maleimide end-terminated PMLABe derivatives previously synthesized were further used to graft mercaptopropanoic acid C-terminus modified GBVA10-9, CPB, GBVA10-9scr (scrambled control of GBVA10-9) and CPBscr (scrambled control of CPB) peptides through Michael addition, as shown by Scheme 2.
Scheme 2.

Synthetic route to peptide modified PMLABe derivatives (Pept: GBVA10-9, GBVA10-9scr, CPB and CPBscr).
As described in the Materials and Methods section, the Michael addition between the selected peptides C-terminated by a thiol function and the synthesized (co)polymers end-terminated by a maleimide group was conducted in DMF in the presence of phosphate buffer solution (PBS) at pH 7.2 at room temperature. The unreacted peptides were eliminated by dialysis against DMF. After elimination of DMF, peptide-grafted (co)polymers were analyzed by 1H NMR (Figure 5 and Figure S2).
Figure 5.

1H NMR spectrum of: (A). CPBPMLABe73, and (B). CPBPEG62-b-PMLABe73.
The 1H NMR spectra of the purified peptide-grafted (co)polymers showed the presence of peaks corresponding to the considered peptide together with the ones corresponding to the (co)polymers. Based on the relative integration of methyl protons belonging to the peptides (0.81 ppm for GBVA10-9, 0.92 ppm for GBVA10-9scr, 0.90 ppm for CPB and 0.86 ppm for CPBscr) and of methylene groups of PMLABe at 5.14 ppm, we assumed that 80 to 95% of either PMLABe73 or PEG62-b-PMLABe73 were modified with the selected peptides (Table 4).
Table 4.
Conjugation percentage of each peptide on each maleimide (co)polymer.
| (Co)Polymer | Conjugation Percentage (%) |
|---|---|
| GBVA10-9PMLABe73 | 90 |
| GBVA10-9PEG62-b-PMLABe73 | 81 |
| CPBPMLABe73 | 95 |
| CPBPEG62-b-PMLABe73 | 95 |
| GBVA10-9scrPMLABe73 | 95 |
| GBVA10-9scrPEG62-b-PMLABe73 | 77 |
| CPBscrPMLABe73 | 83 |
| CPBscrPEG62-b-PMLABe73 | 77 |
To make sure that free peptides were efficiently eliminated using dialysis, mixtures of PMLABe73 and thiol-modified GBVA10-9 and PMLABe73 and thiol-modified CPB, solubilized in DMF at the same concentrations as the ones used for Michael addition, were dialyzed against DMF. The solutions contained in the dialysis bags were then analyzed by 1H NMR after elimination of the DMF. The 1H NMR spectra thus obtained showed the absence of peptides’ peaks at the precision of proton NMR, while peaks corresponding to the PMLABe73 were observed (Figure S3).
All together, these results demonstrate the success of the peptide grafting onto maleimide end-functionalized PMLABe73 and PEG62-b-PMLABe73.
3.2. Preparation and Characterization of Peptides Functionalized Nanoparticles
Several methods have been described to prepare well-defined NPs starting from either hydrophobic homopolymers or amphiphilic block copolymers [50,51]. Among all the available techniques, nanoprecipitation of hydrophobic homopolymers or amphiphilic block copolymers solubilized into organic solvent miscible to water into an aqueous solution is a simple but robust method leading to well-defined NP suspensions [34,50,51]. Several years ago, we applied this technique, firstly described by Fessi et al. [34], to the formulation of hydrophobic and amphiphilic PMLABe derivatives, allowing us to reproducibly obtain well-defined NPs [33]. We therefore applied this technique to prepare peptide-decorated NPs starting from the successfully synthesized peptide-modified PMLABe derivatives. We, however, had to adjust the nanoprecipitation technique, i.e., the nature of the organic solvent and (co)polymer concentration in both the organic solvent and aqueous solution, to such PMLABe derivatives, also taking into account the fact that the fluorescence probe DiD Oil also had to be encapsulated into all NP batches for further in vitro cellular uptake studies.
As all the peptide-modified (co)polymers and DiD Oil are well-soluble in DMF (solvent miscible with water), we therefore used this organic solvent for DiD Oil-loaded peptide-decorated NP formulation. Twelve batches of peptide-decorated NP suspensions in water were thus prepared (Table 1), together with three batches of native NP suspensions (NPs without peptide, Table 2). After nanoprecipitation, the aqueous suspensions were passed through a Sephadex column to eliminate non-encapsulated DiD Oil. Indeed, Sephadex columns, usually used for proteins desalting, allow products’ separation based on steric exclusion. In this context, low-molar mass molecules remain on the top of the column, while high-molar mass compounds are eluted as a function of their molar mass: the higher the molar mass, the faster the molecule is eluted. Free low-molar mass DiD Oil is therefore retained at the top of the Sephadex column while DiD oil-loaded NPs are eluted at the exclusion volume of the column. Peptide-decorated NP suspensions were then characterized (Table 5) by dynamic light scattering (DLS) and electrophoretic light scattering (ELS), giving access to the hydrodynamic diameter (Dh), sample dispersity (PDI), and surface charge (zeta potential). Finally, DiD Oil encapsulation efficiency (E.E.) was determined by UV analysis at 660 nm (Table 5).
Table 5.
Characteristics of peptide-decorated and native NPs.
| Code | Composition (wt%) | Dh a (nm) | PDI a | ζ b (mV) | E.E. c (%) |
|---|---|---|---|---|---|
| NPs 1 | GBVA10-9PMLABe73/PMLABe73 (10/90) | 108 ± 60 | 0.18 | −48 ± 7 | 76 |
| NPs 2 | GBVA10-9PEG62-b-PMLABe73/PMLABe73 (10/90) | 66 ± 30 | 0.21 | −46 ± 8 | 68 |
| NPs 3 | GBVA10-9PEG62-b-PMLABe73/PEG42-b-PMLABe73 (10/90) | 71 ± 50 | 0.28 | −37 ± 5 | 80 |
| NPs 4 | CPBPMLABe73/PMLABe73 (10/90) | 139 ± 60 | 0.12 | −54 ± 10 | 55 |
| NPs 5 | CPBPEG62-b-PMLABe73/PMLABe73 (10/90) | 128 ± 60 | 0.13 | −50 ± 6 | 93 |
| NPs 6 | CPBPEG62-b-PMLABe73/PEG42-b-PMLABe73 (10/90) | 102 ± 60 | 0.30 | −43 ± 6 | 81 |
| NPs 7 | GBVA10-9scrPMLABe73/PMLABe73 (10/90) | 115 ± 50 | 0.16 | −58 ± 8 | 66 |
| NPs 8 | GBVA10-9scrPEG62-b-PMLABe73/PMLABe73 (10/90) | 106 ± 60 | 0.17 | −56 ± 7 | 95 |
| NPs 9 | GBVA10-9scrPEG62-b-PMLABe73/PEG42-b-PMLABe73 (10/90) | 70 ± 50 | 0.28 | −51 ± 8 | 71 |
| NPs 10 | CPBscrPMLABe73/PMLABe73 (10/90) | 163 ± 60 | 0.10 | −53 ± 8 | 76 |
| NPs 11 | CPBscrPEG62-b-PMLABe73/PMLABe73 (10/90) | 146 ± 70 | 0.13 | −52 ± 6 | 95 |
| NPs 12 | CPBscrPEG62-b-PMLABe73/PEG42-b-PMLABe73 (10/90) | 74 ± 50 | 0.23 | −47 ± 10 | 77 |
| NPs 13 | PMLABe73 | 71 ± 40 | 0.21 | −62 ± 8 | 74 |
| NPs 14 | PEG42-b-PMLABe73/PMLABe73 (10/90) | 62 ± 40 | 0.22 | −59 ± 9 | 49 |
| NPs 15 | PEG42-b-PMLABe73 | 64 ± 50 | 0.27 | −55 ± 7 | 68 |
a. Measured by DLS; b. Zeta pontential measured by DLS; c DiD Oil Encapsulation Efficiency measured by UV at 660 nm.
As shown by the results gathered in Table 5, peptide-decorated NPs have hydrodynamic diameter varying from 66 to 163 nm, while native NPs have Dh between 62 and 71 nm. Pearce et al. recently reviewed the evolution of NPs designed as cancer drug delivery systems, and more importantly they gave the adequate size range to gain satisfactory blood circulation time and adapted in vivo biodistribution. Pearce et al. thus reported that NPs should have a size between 5 and 200 nm [52]. In terms of diameter, all peptide-decorated and native NP batches therefore are in the range of nanovector’s sizes that might be used for biomedical applications [52,53]. If the majority of the NP suspensions have a moderate dispersity (PDI lower or equal to 0.25), some NP suspensions seem to be quite dispersed with PDI above 0.25 (but lower or equal to 0.30). However, the quality of these samples remains acceptable to perform in vitro assays [51]. On the other hand, all the prepared NPs showed a negative surface charge, as illustrated by negative zeta potential values varying from −37 to −59 mV (Table 5). Both the high absolute values and negative charges highlighted the high stability of the prepared peptide-decorated and native NP suspensions. Indeed, it is commonly accepted that: (i) nanovectors with an absolute value of zeta potential equal or greater than 30 mV are electrostatically stabilized, and (ii) negatively charged nanoobjects might reduce non-specific liver/spleen uptake [51,52,53]. The results gathered in Table 5 show that zeta potential values measured for NPs without peptides were slightly higher than those measured for NPs decorated with peptides. Such differences might be due to the charges present in the peptide molecules through the positively charged amino acids that are relatively abundant in all the considered peptides. These positive charges might partially neutralize the negative charges of PMLABe derivatives coming from their carboxylate end-chain. Moreover, the configuration of peptidic chains at the surface of the corresponding NPs probably plays a role on the values of zeta potentials. Indeed, peptides might be either sticking out of NPs or stuck to the surface of NPs, thus masking the negative charges coming from the (co)polymers. The hydrophobic fluorescence probe DiD Oil was efficiently encapsulated in all the prepared NP suspensions, as highlighted by the values of calculated encapsulation efficiency (E.E.) that varied from 49 to 95% (Table 5). In addition, no DiD Oil residual traces were observed on the Sephadex column for all NP suspensions, meaning that no free DiD Oil was present in the prepared suspensions.
To check the robustness of the above-described process, several batches of all peptide-decorated and native NPs were prepared and analyzed by DLS. As shown by the results gathered in Figure 6, quite reproducible results in terms of both diameters (Figure 6A) and dispersity (Figure 6B) were obtained, as shown by the reasonable error bar values.
Figure 6.

Variations in: (A) diameter, and (B) dispersity of peptide-decorated and native NPs.
The data given by Figure 6A also clearly show the influence of the peptide’s nature on the diameter values for NPs composed of PeptPMLABe73/PMLABe73. Indeed, a significant increase in NP diameters can be observed on one side between NPs decorated with GBVA10-9 and those decorated with its scrambled control (GBVA10-9scr), and on the other side between NPs decorated with CPB and those decorated with its scrambled control (CPBscr). Such behavior might be explained by the different physico-chemical properties of the peptides as a result of their different amino acids sequences and natures. However, more experiments need to be performed to confirm this conclusion, in particular the evaluation of the hydrophobic or hydrophilic character of each peptide. Fully PEGylated NPs decorated or not with peptides showed similar diameters with values around 80 nm, probably because of the presence of PEG chains that forced the hydrophobic inner core to be more condensed, thus lowering the influence of the peptide nature on the corresponding NPs size and/or forced the entrapment of the peptides into or close to the hydrophobic inner-core. On the other hand, quite surprising results were observed for NPs composed of the mixture Pept-PEG62-b-PMLABe73/PMLABe73 (10/90). Indeed, as shown by Figure 6, GBVA10-9scr, CPB and CPBscr-decorated NPs prepared from the above mentioned (co)polymers mixture have similar diameters above 120 nm, while GBVA10-9-decorated NPs based on the mixture Pept-PEG62-b-PMLABe73/PMLABe73 (10/90) have diameters that are close to those measured for NPs based on the mixture PEG42-b-PMLABe73/PMLABe73 (10/90), with values around 80 nm, similar to those observed for fully PEGylated NPs. It seems that the presence of the flexible PEG62 block leads to a partial entrapment of GBVA10-9 into the inner-core of the NPs because of the higher hydrophobic character of the GBVA10-9 peptide.
The morphology of both peptide-decorated and native NPs was assessed by transmission electron microscopy (TEM) (JEOL, Tokyo, Japan) using phosphotungstic acid as a staining agent (Figure 7).
Figure 7.

TEM images of peptide-decorated and native NPs.
TEM allows us to visualize nanoobjects’ morphology and size together with sample dispersity. As shown by TEM images, all prepared peptide-decorated and native NPs have more or less spherical shapes with dispersity similar to those measured by DLS. Diameters evaluated from TEM images were globally lower than those measured by DLS as a result of TEM samples’ preparation. Indeed, while DLS measurements considered NPs in suspensions, TEM measurements gave images of NPs under a dried state, thus leading to an under-estimation of their diameters.
3.3. In Vitro Internalization Assays
Peptide-functionalized NPs were next used in preliminary in vitro human hepatoma cell uptake assays. The HepaRG cells were incubated with peptide-modified NPs and native NPs at the same concentration (25 µg/mL) for 24 h, and analyzed by flow cytometry to evaluate the influence of both peptide nature and NPs composition on the cell uptake. As shown by Figure 8A, all the cells were positive regardless of the NPs’ composition, further demonstrating our previous data indicating that PMLABe-based NPs were efficiently internalized in human cells [34,40]. Nevertheless, fluorescence intensity, representing accumulation levels of NPs in HepaRG cells, significantly varied with NPs composition, the absence or presence of peptides on their surfaces, and peptides’ nature (Figure 8B). Fully PEGylated NPs (NPs 3, NPs 6; NPs 9, NPs 12, and NPs 15) were poorly internalized by HepaRG cells, even for NPs prepared with GBVA10-9PEG62-b-PMLABe73/PEG42-b-PMLABe73 and CPBPEG62-b-PMLABe73/PEG42-b-PMLABe73 copolymers. It seems that the peptides are buried into the PEG corona, as illustrated by their low average diameters measured by DLS (Table 4 and Figure 6). On the contrary, NPs constituted by CPBPMLABe73/PMLABe73 mixture (NPs 4) are highly internalized by HepaRG cells, and, as expected, the NPs with the CPB scrambled control (CPBscr, NPs 10) are significantly less internalized by HepaRG cells. Surprisingly, NPs decorated with GBVA10-9 (NPs 1) and those with GBVA10-9scr (NPs 7) on their surfaces were internalized at the same level as NPs based on PMLABe73 (NPs 13). GBVA10-9 and GBVA10-9scr are composed by several hydrophobic amino acids, thus probably leading to the entrapment of these peptides within the hydrophobic NPs’ inner core constituted by poly(benzyl malate).
Figure 8.

Flow cytometry analysis of NP uptake by HepaRG cells: (A) percentage of positive cells, and (B) fluorescence intensity, and impact of cold (4 °C) and genistein on NPs 4 uptake.
Finally, the results obtained with NPs formulated from the mixture PeptPEG62-b-PMLABe73/PMLABe73 (10/90) are quite surprising. Indeed, both the GBVA10-9scr- and CPBscr-decorated NPs (NPs 8 and NPs 11, respectively) seem to be more efficiently internalized into HepaRG cells than similar NPs decorated with GBVA10-9 and CPB (NPs 2 and NPs 5, respectively). As already described in the literature, the nature of the linker between the peptide and the carrier (whatever its nature) has a great influence on the specificity of the peptide [54,55,56]. In this study, the linker between selected peptides and PMLABe73 is either the hexyl group (NPs 1, NPs 4, NPs 7 and NPs 10) or PEG62 (NPs 2, NPs 5, NPs 8 and NPs 11), thus probably leading to great differences in the presentation and/or presence of all the peptide amino acids sequence at the surface of concerned NPs, and therefore on the recognition ability of the considered peptide.
The cellular uptake of nanoparticles is usually an energy-dependent mechanism of endocytosis [57,58,59,60]. In order to investigate whether the uptake of peptide-functionalized NPs in HepaRG cells could also involve endocytosis (Figure 8B-insert), the energy-dependent uptake and the endocytosis were studied by comparing the uptake of CPBPMLABe73/PMLABe73-based NPs (NPs 4) at 4 and 37 °C, and in the absence or presence of the endocytosis inhibitor, genistein, in HepaRG cells. The intensity of fluorescence was strongly decreased in cells maintained in the cold at 4 °C, suggesting that the cellular entry of NPs required an energy-dependent process. In addition, the treatment with genistein also strongly reduced (~70% decrease in fluorescence) the cell accumulation of NPs 4, further demonstrating the internalization of NPs by endocytosis. Genistein is known to mainly inhibit calveolae-mediated endocytosis. Our data thus suggest that internalization of NPs 4 involves this pathway, as we previously reported for other PMLA-based NPs [61].
In addition to in vitro uptake assays, confocal microscopy studies were realized on HepaRG cells after their incubation with Pept-NPs constituted by the mixtures PeptPEG62-b-PMLABe73/PMLABe73 encapsulating the fluorescent probe DiD Oil (red fluorescence) in order to visualize the localization of Pept-NPs, the cell nucleus being stained in blue with Hoechst 33342 reagent (Figure 9). These images confirmed cell internalization of NPs with a bright red fluorescence observed as cytoplasmic dots for all NPs, with a more intense staining for NPs 11 around the cells’ nuclei.
Figure 9.

Images obtained by confocal microscopy.
4. Conclusions
Thanks to the versatility of the PMLABe synthesis method, several peptide-functionalized derivatives were successfully obtained and characterized. Two hepatotropic peptides (GBVA10-9 and CPB) and their scrambled controls (GBVA10-9scr and CPBscr, respectively) have been grafted at the end chain of two PMLABe derivatives, MalPMLABe73 and MalPEG62-b-PMLABe73, through Michael addition. Peptide-modified (co)polymers were then formulated with unmodified (co)polymers in order to introduce around 10 wt% of peptides at NPs’ surfaces. Prepared NPs showed hydrodynamic diameters, dispersity, zeta potential and morphology adapted to their uses as drug carriers in the frame of cancer therapy. Preliminary in vitro assays (flow cytometry and confocal microscopy) demonstrated the great influence of the nature of the linker between the peptide and the (co)polymer, the NPs compositions, and the nature of amino acid sequences on their uptake by HepaRG hepatoma cells. The mechanism of internalization, at least for NPs 4, involves a calveolae-dependent endocytosis. Further studies are underway to highlight the presence or absence of all the amino acid sequences at the surface of NPs: 1H NMR spectra in deuterated water (D2O) of all the formulated NPs are currently in progress. The first results obtained with NPs without peptides showed that: (i) for NPs based on PMLABe73, no peaks of polymers can be observed on the 1H NMR spectrum highlighting that the polymer is well-condensed as a hydrophobic core; (ii) for NPs based on PEG42-b-PMLABe73, only the peak corresponding to the PEG chains is visible on the 1H NMR spectrum showing the core-shell structure of such NPs; and (iii) for the NPs constituted by the mixture PMLABe73/PEG42-b-PMLABe73 (90/10), a small peak corresponding to the PEG chain is visible on the 1H NMR spectra, thus demonstrating the presence of PEG chains at the surface of these NPs, but in a smaller amount than the fully PEGylated NPs.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/nano11040958/s1, Figure S1: 1H NMR spectra (CD3COCD3) of A. PMLABe73, B. PEG42-b-PMLABe73. Figure S2: 1H NMR spectra of A. GBVA10-9PMLABe73 (DMSO-d6), B. GBVA10-9PEG62-b-PMLABe73 (DMSO-d6), C. GBVA10-9scrPMLABe73 (DMF-d7), D. GBVA10-9scrPEG62-b-PMLABe73 (DMF-d7), E. CPBscr-PMLABe73 (DMF-7), E. CPBscr-PEG62-b-PMLABe73 (DMF-d7). Figure S3: 1H NMR spectra of products obtained after dialysis of: A. a mixture of PMLABe73 and GBVA10-9-SH, and B. a mixture of PMLABe73 and CPB-SH.
Author Contributions
Conceptualization, S.C.-M., N.L. and P.L.; methodology, C.B., V.D., E.V., C.R. and M.V.; software, C.B., V.D. and M.V.; validation, S.C.-M., P.L. and N.L.; formal analysis, S.C.-M., P.L. and N.L.; investigation, C.B., V.D., E.V., C.R. and M.V.; resources, C.R. and E.V.; data curation, C.B., V.D., M.V., S.C.-M., N.L. and P.L.; writing—original draft preparation, S.C.-M., P.L. and N.L.; writing—review and editing, S.C.-M., P.L., N.L. and N.N.; visualization, S.C.-M., P.L. and N.L.; supervision, S.C.-M., P.L., N.L. and N.N.; project administration, S.C.-M., P.L. and N.L.; funding acquisition, S.C.-M., P.L. and N.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Fondation pour la Recherche Médicale (FRM N°DCM20181039540), Cancéropôle Grand Ouest (CGO n° 17/028), Labex IRON (n° ANR-11-LABX-0018), Institut National de la Santé et de la Recherche Médicale (Inserm), l’Ecole Nationale Supérieure de Chimie de Rennes (ENSCR, France), the Centre National de la Recherche Scientifique (CNRS). Clarisse Brossard was recipient of a PhD fellowship from the Fondation pour la Recherche Médicale (FRM N°DCM20181039540).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Winau F., Westphal O., Winau R. Paul Ehrlich—In search of the magic bullet. Microbes Infect. 2004;6:786–789. doi: 10.1016/j.micinf.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Toporkiewicz M., Meissner J., Matusewicz L., Czogalla A., Sikorski A.F. Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: Principles, hopes, and challenges. Int. J. Nanomed. 2015;10:1399–1414. doi: 10.2147/IJN.S74514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panowski S., Bhakta S., Raab H., Polakis P., Junutula J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs. 2014;6:34–45. doi: 10.4161/mabs.27022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maso K., Grigolettob A., Vicenta M.J., Pasut G. Molecular platforms for targeted drug delivery. Int. Rev. Cell. Mol. Biol. 2019;346:1–50. doi: 10.1016/bs.ircmb.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Russell-Jones G., McTavish K., McEwan J., Rice J., Nowotnik D. Vitamin-mediated as a potential mechanism to increase drug uptake by tumor. J. Inorg. Biochem. 2004;98:1625–1633. doi: 10.1016/j.jinorgbio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Tripodo G., Mandracchia D., Collina S., Rui M., Rossi D. New Perspectives in Cancer Therapy: The Biotin-Antitumor Molecule Conjugates. Med. Chem. 2014;S1:1–8. [Google Scholar]
- 7.Saxera A., Kori M.L. Preparation and characterization of paclitaxel-folic acid conjugates to target cancer cells. J. Adv. Sci. Res. 2019;10:362–369. [Google Scholar]
- 8.Shukla R.K., Tiwari A. Carbohydrate Molecules: An Expanding Horizon in Drug Delivery and Biomedicine. Crit. Rev. Therap. Drug Carr. Syst. 2011;28:255–292. doi: 10.1615/critrevtherdrugcarriersyst.v28.i3.20. [DOI] [PubMed] [Google Scholar]
- 9.Cadinoiu A.N., Rata D.M., Atanase L.I., Daraba O.M., Gherghel D., Vochita G., Popa M. Aptamer-Functionalized Liposomes as a Potential Treatment for Basal Cell Carcinoma. Polymers. 2019;11:1515. doi: 10.3390/polym11091515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cesarini V., Scopa C., Silvestris D.A., Scafidi A., Petrera V., Del Baldo G., Gallo A. Aptamer-Based In Vivo Therapeutic Targeting of Glioblastoma. Molecules. 2020;25:4267. doi: 10.3390/molecules25184267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rata D.M., Cadinoiu A.N., Atanase L.I., Popa M., Mihai C.T., Solcan C., Ochiuz L., Vochita G. Topical formulations containing aptamer-functionalized nanocapsules loaded with 5-fluorouracil—An innovative concept for the skin cancer therapy. Mater. Sci. Eng. C Mater. Biol. Appl. 2021;119:111591. doi: 10.1016/j.msec.2020.111591. [DOI] [PubMed] [Google Scholar]
- 12.Reubi J.C. Peptide Receptors as Molecular Targets for Cancer Diagnosis and Therapy. Endocr. Rev. 2003;24:389–427. doi: 10.1210/er.2002-0007. [DOI] [PubMed] [Google Scholar]
- 13.Roveri M., Bernasconi M., Leroux J.M., Luciani P. Peptides for tumor-specific drug targeting: State of the art and beyond. J. Mat. Chem. B. 2017;5:4348–4364. doi: 10.1039/C7TB00318H. [DOI] [PubMed] [Google Scholar]
- 14.Sun H., Dong Y., Jan F., Zhong Z. Peptide-decorated polymeric nanomedicines for precision cancer therapy. J. Control. Release Off. J. Control. Release Soc. 2018;290:11–27. doi: 10.1016/j.jconrel.2018.09.029. [DOI] [PubMed] [Google Scholar]
- 15.Kim J.H., Bae C., Kim M.J., Song I.H., Ryu J.H., Choi J.H., Lee C.J., Nam J.S., Kim J.I. A novel nucleolin-binding peptide for Cancer Theranostics. Theranostics. 2020;10:9153–9171. doi: 10.7150/thno.43502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang S.Y., Wei H., Yu C.U. Peptide-functionalized delivery vehicles for enhanced cancer therapy. Int. J. Pharm. 2021;593:120141. doi: 10.1016/j.ijpharm.2020.120141. [DOI] [PubMed] [Google Scholar]
- 17.Goodman M., Cai W., Smith N.D. The bold legacy of Emil Fisher. J. Pept. Sci. 2003;9:594–603. doi: 10.1002/psc.476. [DOI] [PubMed] [Google Scholar]
- 18.Loffet A. Peptides as drugs: Is there a market? J. Pept. Sci. 2002;8:1–7. doi: 10.1002/psc.366. [DOI] [PubMed] [Google Scholar]
- 19.du Vigneaud V. Hormones of the Posterior Pituitary Gland: Oxytocin and Vasopressin, The Harvey Lectures 1954–1955. Academic Press; New York, NY, USA: 1956. pp. 1–26. [PubMed] [Google Scholar]
- 20.du Vigneaud V., Ressler C., Swan C.J.M., Roberts C.W., Katsoyannis P.G., Gordon S. The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J. Am. Chem. Soc. 1953;75:4879–4880. doi: 10.1021/ja01115a553. [DOI] [Google Scholar]
- 21.Adessi C., Soto C. Converting a peptide into a drug: Strategies to improve stability and bioavailability. Curr. Med. Chem. 2002;9:963–978. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- 22.McGregor D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008;8:616–619. doi: 10.1016/j.coph.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 23.Lau J.L., Dunn M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018;26:2700–2707. doi: 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 24.Vans B.J., King A.T., Katsifis A., Matesic L., Jamie J.F. Methods to enhance the metabolic stability of peptide-based PET radiopharmaceuticals. Molecules. 2020;25:2314. doi: 10.3390/molecules25102314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stone T.A., Deber C. Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochem. Biophys. Acta. 2017;1859:577–585. doi: 10.1016/j.bbamem.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Eychenne R., Bouvry C., Bourgeois M., Loyer P., Benoist E., Lepareur N. Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy. Molecules. 2020;25:4012. doi: 10.3390/molecules25174012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsen J.O., Pozderac R.V., Hinkle G., Hill T., O’Dosisio T.M., Schirmer W.J., Ellison E.C., O’Dorisio M.S. Somatostatin receptor imaging of neuroendocrine tumors with Indium-111 pentetreotide (OctreoScan) Semin. Nucl. Med. 1995;XXV:251–261. doi: 10.1016/S0001-2998(95)80014-X. [DOI] [PubMed] [Google Scholar]
- 28.Hennirch U., Kopka K. Lutathera®: The first FDA- and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy. Pharmaceuticals. 2019;12:114. doi: 10.3390/ph12030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faivre S., Rimassa L., Finn R.S. Molecular therapies for HCC: Looking outside the box. J. Hepatol. 2020;72:342–352. doi: 10.1016/j.jhep.2019.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Ghavimi S., Apfel T., Azimi H., Persaud A., Pyrsopoulos N.T. Management and Treatment of Hepatocellular Carcinoma with Immunotherapy: A Review of Current and Future Options. J. Clin. Transl. Hepatol. 2020;8:168–176. doi: 10.14218/JCTH.2020.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akateh C., Black S.M., Conteh L., Miller E.D., Noonan A., Elliott E., Pawlik T.M., Tsung A., Cloyd J.M. Neoadjuvant and adjuvant treatment strategies for hepatocellular carcinoma. World J. Gastroenterol. 2019;25:3704–3721. doi: 10.3748/wjg.v25.i28.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vène E., Jarnouen K., Ribault C., Vlach M., Verres Y., Bourgeois M., Lepareur N., Cammas-Marion S., Loyer P. Hepatotropic circumsporozoite protein of Plasmodium berghei- and GB Virus A-derived peptides strongly enhance cell uptake of bioconjugates and functionalized polymeric nanoparticles by human hepatoma cells: Influence of the amino acid sequence and opsonization. Biomaterials. 2021 under review. [Google Scholar]
- 33.Fessi H., Puisieux F., Devissaguet J.P., Ammoury N., Benita S. Nanocapsule formation by interfacial deposition following solvent displacement. Int. J. Pharm. 1989;55:R1–R4. doi: 10.1016/0378-5173(89)90281-0. [DOI] [Google Scholar]
- 34.Huang Z.W., Laurent V., Chetouani G., Ljubimova J.Y., Holler E., Benvegnu T., Loyer P., Cammas-Marion S. New functional degradable and bio-compatible nanoparticles based on poly(malic acid) derivatives for site-specific anti-cancer drug delivery. Int. J. Pharm. 2012;423:84–92. doi: 10.1016/j.ijpharm.2011.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su S., Kang P.M. Recent advances in nanocarrier-assisted therapeutics delivery systems. Pharmaceutics. 2020;12:837. doi: 10.3390/pharmaceutics12090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siddique S., Chow J.C.L. Application of nanomaterials in biomedical imaging and cancer therapy. Nanomaterials. 2020;10:1700. doi: 10.3390/nano10091700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Indoria S., Singh V., Hsieh M.F. Recent advances in theranostic polymeric nanoparticles for cancer treatment: A review. Int. J. Pharm. 2020;582:119314. doi: 10.1016/j.ijpharm.2020.119314. [DOI] [PubMed] [Google Scholar]
- 38.Hwang D., Ramsey J.D., Kabanov A.V. Polymeric micelles for the delivery of poorly soluble drugs: From nanoformulation to clinical approval. Adv. Drug Del. Rev. 2020;156:80–118. doi: 10.1016/j.addr.2020.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cammas-Marion S., Loyer P. Natural and synthetic poly(malic acid)-based derivates: A family of versatile biopolymers for the design of drug nanocarriers. J. Drug Targ. 2014;22:556–575. doi: 10.3109/1061186X.2014.936871. [DOI] [PubMed] [Google Scholar]
- 40.Loyer P., Bedhouche W., Huang Z.W., Cammas-Marion S. Degradable and Biocompatible Nanoparticles Decorated with Cyclic RGD Peptide for Efficient Drug Delivery to Hepatoma Cells In Vitro. Int. J. Pharm. 2013;454:727–737. doi: 10.1016/j.ijpharm.2013.05.060. [DOI] [PubMed] [Google Scholar]
- 41.Vene E., Jarnouen K., Huang Z.W., Bedhouche W., Montier T., Cammas-Marion S., Loyer P. In vitro Toxicity Evaluation and in vivo Biodistribution of Polymeric Micelles Derived from Poly(ethylene glycol)-b-poly(benzyl malate) Copolymers. Pharm. Nanotech. 2016;4:24–37. doi: 10.2174/221173850401160413171933. [DOI] [Google Scholar]
- 42.Venkatraj N., Nanjan M.J., Loyer P., Chandrashekar M.J.N., Cammas-Marion S. Poly(malic acid) bearing Doxorubicin and N-Acetyl Galactosamine as a site-specific prodrugs for targeting HepatoCellular Carcinoma. J. Biomat. Sci. Polym. Ed. 2017;28:1140–1157. doi: 10.1080/09205063.2017.1311294. [DOI] [PubMed] [Google Scholar]
- 43.Pounder R.J., Stanford M.J., Books P., Richards S.P., Dove A.P. Metal free thiol-maleimide ‘click’ reaction as a mild functionalization strategy for degradable polymers. Chem. Commun. 2008:5158–5160. doi: 10.1039/b809167f. [DOI] [PubMed] [Google Scholar]
- 44.Liu Z., Dong C., Wang X., Wang H., Li W., Tan J., Chang J. Self-assembled biodegradable protein-polymer vesicle as a tumor-targeted nanocarrier. ACS Appl. Mater. Interfaces. 2014;6:2393–2400. doi: 10.1021/am404734c. [DOI] [PubMed] [Google Scholar]
- 45.Petrelli A., Borsali R., Fort S., Halila S. Oligosaccharide-based block copolymers: Metal-free thiol-maleimide click conjugation and self-assembly into nanoparticles. Carbohydr. Polym. 2015;124:109–116. doi: 10.1016/j.carbpol.2015.01.079. [DOI] [PubMed] [Google Scholar]
- 46.Gunnoo S.B., Madder A. Chemical protein modification through cysteine. ChemBioChem. 2016;17:529–553. doi: 10.1002/cbic.201500667. [DOI] [PubMed] [Google Scholar]
- 47.Nawroth J.F., McDaniel J.R., Chilkoti A., Jordan R., Luxenhofer R. Maleimide-functionalized poly(2-oxazoline)s and their conjugation to elastin-like polypeptides. Macromol. Biosci. 2016;16:322–333. doi: 10.1002/mabi.201500376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patil N.A., Karas J.A., Turner B.J., Shabanpoor F. Thiol-cyanobenzothiazole ligation for the efficient preparation of peptide-PNA conjugates. Bioconjugate Chem. 2019;30:793–799. doi: 10.1021/acs.bioconjchem.8b00908. [DOI] [PubMed] [Google Scholar]
- 49.Lee J.C., Donahue N.D., Mao A.S., Karim A., Komarneni M., Thomas E.E., Francek E.R., Yang W., Wilhem S. Exploring maleimide-based nanoparticle surface engineering to control cellular interactions. ACS Appl. Nano. Mater. 2020;3:2421–2429. doi: 10.1021/acsanm.9b02541. [DOI] [Google Scholar]
- 50.Vauthier C., Bouchemal K. Methods for the Preparation and Manufacture of Polymeric Nanoparticles. Pharm. Res. 2009;26:1025–1058. doi: 10.1007/s11095-008-9800-3. [DOI] [PubMed] [Google Scholar]
- 51.Patel V.R., Agrawal Y.K. Nanosuspension: An approach to enhance solubility of drugs. J. Adv. Pharm. Technol. Res. 2011;2:81–87. doi: 10.4103/2231-4040.82950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearce A.K., O’Reilly R.K. Insight into active targeting of nanoparticles in drug delivery: Advances in clinical studies and design considerations for cancer nanomedicine. Bioconjugate Chem. 2019;30:2300–2311. doi: 10.1021/acs.bioconjchem.9b00456. [DOI] [PubMed] [Google Scholar]
- 53.Elsabahy M., Wooley K.L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012;41:2545–2561. doi: 10.1039/c2cs15327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tornesello A.L., Buonaguro L., Tornesello M.L., Buonaguro F.M. New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules. 2017;22:1282. doi: 10.3390/molecules22081282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benesova M., Schafer M., Bauder-Wust U., Afshar-Oromieh A., Kratochwil C., Mier W., Haberkorn U., Kopka K., Eder M. Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. J. Nucl. Med. 2015;56:914–920. doi: 10.2967/jnumed.114.147413. [DOI] [PubMed] [Google Scholar]
- 56.Liu S. Bifunctional coupling agents for radiolabeling of biomolecules and target-specific delivery of metallic radionuclides. Adv. Drug Deliv. Rev. 2008;60:1347–1370. doi: 10.1016/j.addr.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sahay G., Alakhova D.Y., Kabanov A.V. Endocytosis of nanomedicines. J. Control. Release. 2010;145:182–195. doi: 10.1016/j.jconrel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iversen T.G., Skotland T., Sandvig K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today. 2011;6:176–185. doi: 10.1016/j.nantod.2011.02.003. [DOI] [Google Scholar]
- 59.Ho L.W.C., Yung W.Y., Sy K.H.S., Li H.Y., Choi C.K.K., Leung K.C.F., Lee T.W.Y., Choi C.H.J. Effect of alkylation on the cellular uptake of polyethylene glycol-coated gold nanoparticles. ACS Nano. 2017;11:6058–6101. doi: 10.1021/acsnano.7b02044. [DOI] [PubMed] [Google Scholar]
- 60.Jiang Z., He H., Liu H., Thayumanavan S. Cellular uptake evaluation of amphiphilic polymer assemblies: Importance of interplay between pharmacological and genetic approaches. Biomacromolecules. 2019;20:4407–4418. doi: 10.1021/acs.biomac.9b01073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vene E., Barouti G., Jarnouen K., Gicquel T., Rauch C., Ribault C., Guillaume S.M., Cammas-Marion S., Loyer P. Opsonisation of nanoparticles prepared from poly(β-hydroxybutyrate) and poly(trimethylene carbonate)-b-poly(malic acid) amphiphilic diblock copolymers: Impact on the in vitro cell uptake by primary human macrophages and HepaRG hepatoma cells. Int. J. Pharm. 2016;513:438–452. doi: 10.1016/j.ijpharm.2016.09.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.