

Abstract
Glycosylation, the enzymatic attachment of carbohydrates to proteins and lipids, regulates nearly all cellular processes and is critical in the development and function of the nervous system. Axon pathfinding, neurite outgrowth, synaptogenesis, neurotransmission, and many other neuronal processes are regulated by glycans. Over the past 25 years, studies analyzing post-mortem brain samples have found evidence of aberrant glycosylation in individuals with schizophrenia. Proteins involved in both excitatory and inhibitory neurotransmission display altered glycans in the disease state, including AMPA and kainate receptor subunits, glutamate transporters EAAT1 and EAAT2, and the GABAA receptor. Polysialylated NCAM (PSA-NCAM) and perineuronal nets (PNNs), highly glycosylated molecules critical for axonal migration and synaptic stabilization, are both downregulated in multiple brain regions of individuals with schizophrenia. Additionally, enzymes spanning several pathways of glycan synthesis show differential expression in brains of individuals with schizophrenia. These changes may be due to genetic predisposition, environmental perturbations, medication use, or a combination of these factors. However, the recent association of several enzymes of glycosylation with schizophrenia by genome-wide association studies underscores the importance of glycosylation in this disease. Understanding how glycosylation is dysregulated in the brain will further our understanding of how this pathway contributes to the development and pathophysiology of schizophrenia.
Introduction
Glycosylation, the enzymatic attachment of sugars to proteins and lipids, is a major regulator of health and disease [1]. Mutations in glycosylation enzymes and associated genes cause over 130 known unique Mendelian conditions termed congenital disorders of glycosylation or CDGs, resulting in a broad set of symptoms affecting numerous organs [2, 3]. Despite their clinical heterogeneity, nearly 80% of patients with CDGs have neurologic symptoms including seizures and intellectual disability, highlighting the importance of glycosylation in the nervous system [4]. Neuronal glycosylation has been studied extensively, and plays important roles in cell-cell recognition, adhesion, migration, neurite outgrowth, axon pathfinding, synaptogenesis, and the regulation of neurotransmission [5]. For example, mutating N-glycosylation sites in neurotransmitter receptors and transporters can impair subunit assembly, intracellular trafficking, cell surface expression, protein stability, and ligand binding [6–11]. Glycan structures are differentially expressed during development of the brain, and changes in glycan abundance have been observed in several neuropsychiatric conditions [12].
Genome-wide association studies (GWAS) have identified over one hundred genes associated with schizophrenia, enabling researchers to study the molecular underpinnings that contribute to this complex polygenic disorder [13]. In addition to genes involved in synaptic transmission and the immune system, several glycosylation enzymes are directly implicated in the pathogenesis of the disorder (reviewed in linked publication, Mealer et al, 2020, submitted for publication). Though it has received less attention than other possible pathogenic mechanisms, a substantial literature exists documenting dysregulated glycosylation in schizophrenia, primarily in post-mortem studies. These studies suggest that changes in glycosylation may, at least in part, contribute to abnormal neuronal signaling and connectivity observed in schizophrenia. However, such changes may also result from exposures associated with the disease. Given the known importance of glycosylation in brain development and the neurodevelopmental origins of schizophrenia, targeting the glycosylation pathway represents a novel area of research for schizophrenia therapeutics. Understanding both the proximal genetic vulnerabilities and subsequent functional changes in individuals with schizophrenia will be necessary to inform such studies. Here, following a brief discussion on the synthesis of glycans as well as the tools used to study them, we review prior studies of glycosylation changes in individuals with schizophrenia.
Overview of glycan assembly
Glycosylation requires the concerted effort of more than 300 enzymes to generate the rich diversity of glycans necessary for proper protein folding, protein trafficking, cell-cell recognition, cell migration, and countless other cellular processes [14, 15]. Glycosylation enzymes often function in a single pathway with only one or occasionally a few enzymes capable of completing each step. For glycoproteins, the majority of glycans are attached to asparagine (N-linked) or serine/threonine (O-linked) residues [16]. N-linked glycosylation of proteins occurs post-translationally in the endoplasmic reticulum (ER) with the “en bloc” transfer of a high-mannose “precursor” to target proteins [17, 18]. N-glycans are then further processed in the ER and Golgi apparatus by glycosidases that trim sugars from the structure and glycosyltransferases that add additional monosaccharides, resulting in a diverse array of hybrid- and complex-type N-glycans. Initiation of O-glycosylation is more variable and can occur in the ER or Golgi depending on the primary monosaccharide being attached, which can then be similarly extended by various glycosyltransferases [19–21]. Synthesis of glycolipids, including galactosylceramide, the most common in the brain, and glucosylceramide, a common precursor to more complex glycolipid structures, also occurs in a stepwise fashion through the ER and Golgi [22, 23]. Galactosylceramide can be sulfated to generate a 3-O-sulfated derivative termed sulfatide, which is a major component of myelin [24]. Additional glycosylated structures commonly found in the brain are proteoglycans, which consist of a protein core modified by linear polysaccharide chains termed glycosaminoglycans (GAGs) [25]. GAGs can attach to asparagine, serine or threonine resides on the core protein and vary in length, monosaccharide composition, and sulfate modifications, giving rise to potentially hundreds of polysaccharide structures [26]. Because the final product in each pathway is a culmination of sequential enzymatic reactions akin to an assembly line, improper function or expression of a single glycosylation enzyme at any point can result in altered glycans and a range of downstream effects such as a CDG or subtle changes in the risk of common phenotypes [27, 28].
Biological tools for assessing glycosylation
Before reviewing the evidence of altered glycobiology in schizophrenia, it will be useful to understand how such alterations are assayed. Glycobiologists employ a variety of techniques to analyze glycoproteins and glycoenzymes from post-mortem brain tissue and other human samples [29]. Deglycosylation assays typically utilize two glycosidases that release either all N-linked glycans (PNGase F or N-glycanase), or preferentially high-mannose “precursor” and hybrid N-glycans (Endo H or Endoglycosidase H) from glycoproteins of interest, resulting in a molecular mass shift when the digested proteins are run on a gel, thus indicating the presence of N-glycans. Indirect assays use carbohydrate-binding proteins (lectins) with specific glycan affinities to quantify the abundance of such motifs within a sample based on the amount of bound lectin. Antibodies specific to glycan epitopes or the glycosylated form of a protein are also used in both immunohistochemistry and western blot analysis. Further, expression of glycosylation enzymes is measured at both the protein level by western blot and RNA level using microarrays. Finally, glycomic analysis using mass spectrometry (MS) and/or high-performance liquid chromatography (HPLC) has also been employed in patient samples to detect glycans within blood and cerebrospinal fluid (CSF) cleaved by PNGase F. Together, these techniques have identified altered N-glycans on neurotransmitter receptors and transporters [30–33], reduced expression of polysialylated NCAM [34, 35] and perineuronal nets [36–41], and differential expression of several glycosylation enzymes in the brains of individuals with schizophrenia [42–44], as well as changes in the relative abundance of glycan classes in the CSF [45] (Table 1 and Figure 1).
Table 1:
Glycoproteins altered in post-mortem studies of schizophrenia
| Substrate | Glycosylation Change in Schizophrenia | Methodology | Reference |
|---|---|---|---|
| AMPA Receptors | Decreased high mannose N-glycans on GluA2 subunit | Deglycosylation Lectin-affinity | [30] |
| NMDA Receptors | Increased high mannose N-glycans on kainate GluK2 subunit | Deglycosylation Lectin-affinity | [31] |
| EAATs | Decreased complex N-glycans on EAAT1 and EAAT2 | Deglycosylation | [32] |
| GABAA Receptor | Decreased high mannose structures on α1 subunit; Increased high mannose N-glycans on β1 subunit; Altered total N-glycans on β2 subunit | Deglycosylation | [33] |
| PSA-NCAM | Reduction of PSA-NCAM in hippocampus and DLPFC | Immunostaining | [34, 35] |
| PNN | Reduction of glycosaminoglycans (GAGs) detected in the amygdala, olfactory epithelium, entorhinal cortex, and prefrontal cortex | Immuno- and lectin staining | [36–41] |
Figure 1. Glycoproteins and glycoenzymes altered in schizophrenia through gene expression and post-mortem studies.
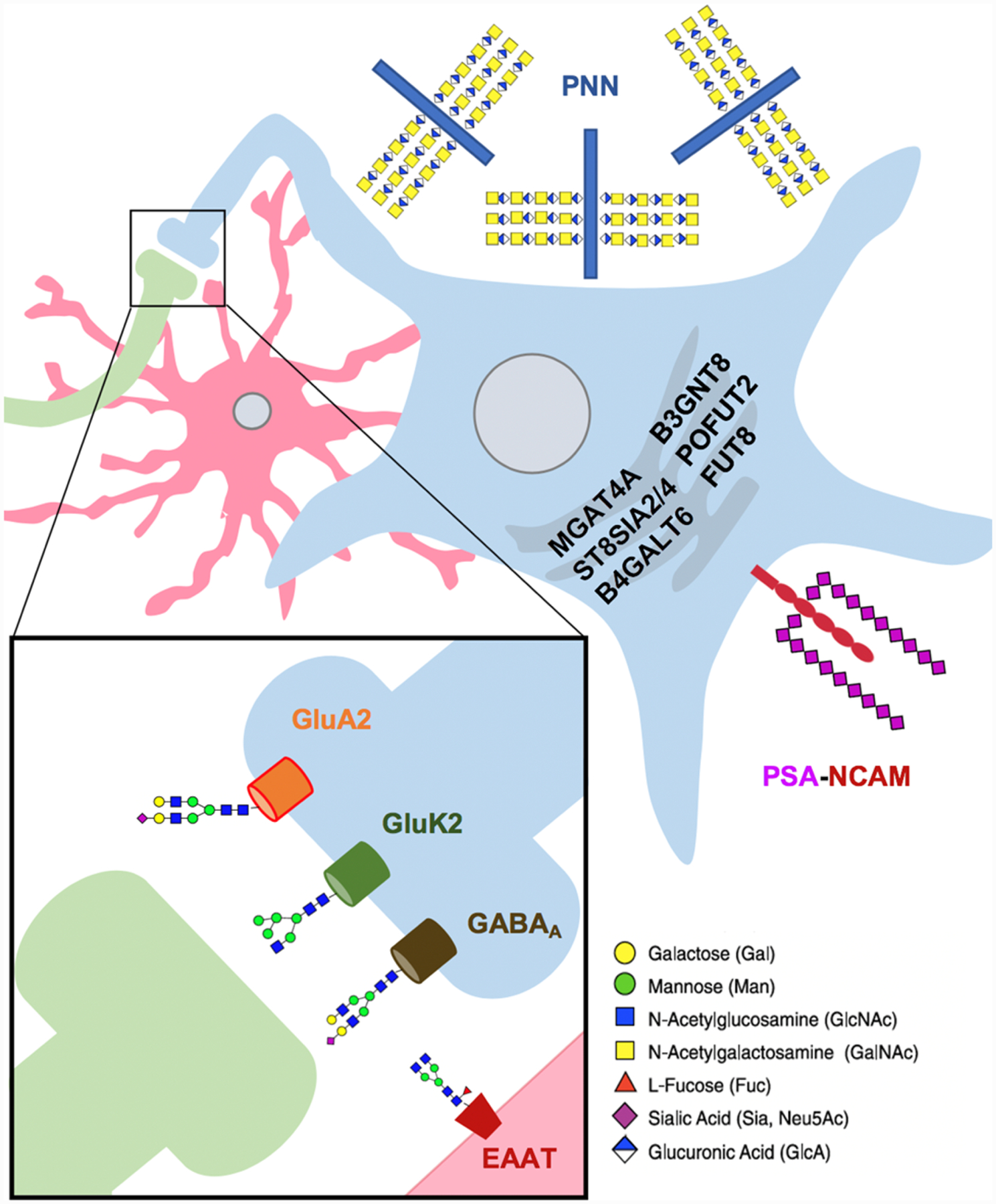
Numerous studies primarily in post-mortem brain samples have identified altered glycosylation of multiple neuronal proteins, as well as changes in the expression of glycosyltransferase enzymes. Presynaptic neuron is shown in green, postsynaptic neuron in blue, astrocyte in red. Glycans included for illustrative purposes, with monosaccharides coded according to SNFG guidelines (https://www.ncbi.nlm.nih.gov/glycans/snfg.html). Perineuronal Nets - PNN; Polysialylated Neuronal Cell Adhesion Molecule - PSA-NCAM.
Differential N-glycosylation of glutamate- and GABA-associated proteins
The balance between excitatory and inhibitory signaling has long been the focus of extensive research in schizophrenia, with dysfunction thought to underlie changes in network activity across the brain [46–50]. Analysis of post-mortem brains from individuals with schizophrenia has revealed differences in the N-glycosylation of proteins involved in excitatory glutamate and inhibitory gamma aminobutyric acid (GABA) transmission. However, the reported changes vary across pathways, proteins, and subunits, limiting a clear explanation for how such changes contribute to schizophrenia.
Glycosylation studies of the glutamate system have analyzed three different types of receptors – AMPA, NMDA, and kainate [30, 31] – as well as the excitatory amino acid transporters EAAT1, 2, and 3 [32] for differences in N-glycosylation. Of the four AMPA receptor subunits (GluA1–4) isolated from dorsolateral prefrontal cortex, GluA2 and GluA4 were sensitive to Endo H and PNGase F digestions, indicating the presence of both high-mannose/hybrid and complex-type N-glycans on these subunits [30]. It is important to note that all four subunits have putative N-glycosylation sites based on sequence analysis [51], suggesting that all four subunits are glycosylated though likely at levels lower than the detection limit of this assay. Compared to controls, GluA2 subunits isolated from individuals with schizophrenia showed significantly lower levels of high-mannose/hybrid glycans revealed by EndoH treatment as well as decreased binding to ConA, a lectin that binds high mannose and complex N-glycans [52]. The functional implications aberrant GluA2 glycosylation were not tested, but the authors hypothesized that the change could impair protein trafficking of the subunit and disrupt the formation of AMPA receptor complexes, therefore impacting overall glutamate signaling [30].
NMDA and kainate receptors have also been examined, with five out of six subunits shown to be N-glycosylated; GluN1, GluN2A, GluN2b, GluK2 and GluK5 are all sensitive to both PNGase F and EndoH [31]. Of the glycosylated proteins, the GluK2 kainate receptor subunit from individuals with schizophrenia displayed increased sensitivity to EndoH, suggesting greater abundance of high-mannose/hybrid-type N-glycans in the disease state. While this result is in contrast to the analysis of AMPA receptors, taken together the data suggests an overall dysregulation of N-glycosylation on glutamate receptors in schizophrenia.
The glutamate transporters EAAT1, EAAT2, and EAAT3 remove this excitatory amino acid from the synaptic cleft, halting signaling and recycling the neurotransmitter. Analysis of these transporters isolated from the dorsolateral prefrontal and cingulate cortices revealed no change after treatment with EndoH, suggesting a lack of high-mannose/hybrid N-glycans on all three transporters [32]. However, EAAT1 and EAAT2, which are primarily expressed in astrocytes, were sensitive to PNGase F, consistent with the presence of complex N-glycans. These transporters from individuals with schizophrenia were less sensitive to PNGase F treatment, indicating a decrease in complex N-glycans on EAAT1 and EAAT2 compared to controls.
Differential glycosylation of GABA-associated proteins has also been observed in post-mortem samples from individuals with schizophrenia. Enzymatic deglycosylation and lectin-affinity analysis of GABAA receptor subunits isolated from the superior temporal gyrus revealed the presence of N-glycans on the α1, α4, β1, β2, and β3 subunits [33]. Interestingly, each subunit displayed a different glycosylation change in individuals with schizophrenia compared to controls, with the α1 subunit showing decreased high-mannose N-glycans, the β1 subunit showing increased high-mannose N-glycans, and the β2 subunit showing an increase in total N-glycosylation. A follow up study investigating the trafficking and assembly of these subunits demonstrated abnormal localization of the β1 and β2 subunits isoforms in schizophrenia [53]. These authors attributed abnormalities in GABAA subunit trafficking to altered glycosylation and proposed that this phenomenon could be contributing to inhibitory signaling deficits observed in schizophrenia.
Decreased polysialylation of NCAM
Neural cell adhesion molecule (NCAM) is a glycoprotein critical for many processes in brain development including cell-cell adhesion, axonal migration, cellular differentiation, and synapse formation [54]. This protein is expressed in multiple isoforms including the “embryonic” polysialylated isoform (PSA-NCAM) and three “adult” isoforms which lack polysialic acid (NCAM-180, NCAM-140, and NCAM-120), though small amounts of PSA-NCAM is found in the adult brain as well [38, 55, 56]. PSA-NCAM is used as a marker for developing and migrating neurons and can be modified with linear chains of up to 400 negatively charged sialic acid carbohydrate monomers, which are thought to regulate adhesion properties of the protein [57, 58].
NCAM isoforms have been studied in the hippocampi of post-mortem brains from individuals with schizophrenia and controls using immunohistochemistry with antibodies recognizing either all isoforms of NCAM or PSA-NCAM specifically [34]. The overall expression of NCAM was not different between the two groups, but hippocampi from individuals with schizophrenia displayed lower levels of PSA-NCAM in the dentate gyrus and hilar regions. A separate analysis of NCAM in the dorsolateral prefrontal cortex of post-mortem samples also showed significantly reduced levels of PSA-NCAM in individuals with schizophrenia, and this change was not observed in cases of bipolar disorder or major depressive disorder [35].
The two enzymes responsible for polysialylation of NCAM – ST8SIA2 and ST8SIA4 – have been investigated in candidate gene analyses of schizophrenia, though they have not shown statistically significant association in large-scale GWAS results. A small candidate gene study in a Japanese sample reported nominal association between schizophrenia and two SNPs in the ST8SIA2 promoter region [59]. A subsequent candidate gene study in a Han Chinese sample also reported association between schizophrenia and ST8SIA2 coding variants, as well as haplotype comprising SNPs reported in the Japanese study [60]. In a follow-up study, the two missense mutations in ST8SIA2 were generated and assayed for polysialylation activity and binding using a heterologous cell model [61]. Both mutations resulted in less polysialic acid on NCAM, which in turn affected the protein function and binding to BDNF and dopamine. Mouse models with combinations of variants in St8sia2, St8sia4 and Ncam1 demonstrated disrupted brain connectivity measured by morphometric histology in the anterior commissure, corpus callosum, and internal capsule, which was shown to depend on the extent of NCAM polysialylation rather than the expression level of the protein [62]. However, as noted, variants in these genes have not been convincingly associated with schizophrenia. While candidate gene studies have well-known limitations, these results are at least consistent with the hypothesis that variation in polysialylation may affect neuronal function.
Alterations in perineuronal nets and extracellular matrix components
Perineuronal nets (PNNs) are heterogeneous glycoprotein aggregates in the neural extracellular matrix (ECM) that regulate synaptic plasticity and have been the subject of extensive investigation in relation to schizophrenia [63, 64]. Multiple studies have demonstrated decreased levels of PNNs in the amygdala, entorhinal cortex, olfactory epithelium, and layers III and V of the prefrontal cortex in post-mortem samples of individuals with schizophrenia [36–41]. The markers used in these studies bind complex carbohydrates known as glycosaminoglycans (GAGs) on chondroitin sulfate proteoglycans (CSPGs), which include aggrecan, brevican, phosphacan, neurocan, and versican [65]. CSPGs are key components of PNNs, and their GAG chains have been shown to regulate neurite outgrowth and activity [66, 67]. Local enzymatic degradation of the carbohydrate chains of CSPGs in the ventral hippocampus of mice resulted in increased locomotor activity and dopamine system functioning, drawing parallels with some animal models and clinical symptoms of schizophrenia [68].
Transcripts for a number of ECM molecules showed decreased expression in pyramidal neurons of the superior temporal gyrus in schizophrenia cases, including aggrecan and versican [69]. Proteomic analysis of the anterior temporal lobe from individuals with schizophrenia revealed a reduction of the aggrecan protein core (AGC) as well as HALPN2, a protein that links and stabilizes CPSGs to other components of the ECM [70, 71]. An increase in phosphacan mRNA levels, also known as PTPRZ1, have been reported in dorsolateral prefrontal cortex of schizophrenia cases, and transgenic mice overexpressing this gene displayed altered glutamatergic, GABAergic, and dopaminergic signaling as well as behavioral abnormalities as well as behavioral phenotypes [72]. A common variant in the gene encoding neurocan (NCAN) has been investigated as a genetic risk factor for both schizophrenia and bipolar disorder and showed correlation with cortical folding and cognitive function [73–76]. The most recent and best powered GWAS confirm the association of this locus with both schizophrenia and bipolar disorder, though it is important to note this complex region contains ~20 genes, making it difficult to determine whether NCAN is the causal gene in this locus [13, 77].
In addition to CSPGs, glycoproteins found in PNNs have exhibited altered mRNA or protein expression in the brains of individuals with schizophrenia, including reelin, semaphorins, and integrins, though the specific glycans on these proteins were not analyzed [78–82]. Two families of metalloproteinases that regulate PNNs have also been studied in the context of schizophrenia, including matrix metalloproteinases (MMPs) and “a disintegrin and metalloproteases with a thrombospondin motif” (ADAMs/ADAMTS) [83, 84]. Several of these enzymes showed altered mRNA expression in pyramidal neurons of the superior temporal gyrus in disease samples including ADAMTS1, ADAMTS6, MMP16, MMP24, and MMP25, and protein levels of an additional matrix metalloproteinase, MMP9, were detected at elevated levels in blood samples from schizophrenia cases [69, 85, 86]. A common variant in MMP16 has been associated with schizophrenia through GWAS, and one member of the ADAMs family of enzymes ADAMTSL3 localizes to asignificant but again complex and ambiguous locus; however, we are unaware of any association between genetic variants in the subset of specific glycosyltransferases that synthesize GAGs on CSPGs and schizophrenia [13].
Differential expression of glycosylation enzymes
Gene expression studies of glycosylation enzymes in individuals with schizophrenia have identified differences across several pathways of glycan synthesis, suggesting that glycosylation as a whole may be dysregulated in the disease rather than just one or two specific synthetic pathways.
Studies of enzymes that add fucose residues to glycoconjugates in the superior temporal gyrus of schizophrenia cases revealed increased protein expression of POFUT2, an O-fucosyltransferase that attaches fucose directly to serine and threonine residues, and decreased expression of FUT8, the only α−1,6-fucosyltransferase which generates core-fucosylated N-glycans [42]. Measurement of core-fucosylated N-glycans using the carbohydrate-binding lectin AAL, which is specific for fucose, revealed reduced binding in disease samples, consistent with decreased FUT8 expression. Fut8 knockout mice exhibit decreased core-fucosylated N-glycans across various brain regions, including the hippocampus, and display hyperactivity, reduced social interaction, decreased working memory, and impaired pre-pulse inhibition – behavioral correlates observed in some cases of schizophrenia [87]. Common targets of POFUT2 include the ADAMTS family of metalloproteinases which rely on O-fucosylation for secretion into the extracellular matrix where they can remodel proteoglycans of perineuronal nets (PNNs) [84, 88, 89].
Protein levels of two N-acetylglucosaminyltransferases, MGAT4A and B3GNT8, differed in the dorsolateral prefrontal cortex of elderly individuals with schizophrenia compared to controls [43]. MGAT4A, one of two enzymes responsible for adding a fourth branch onto complex N-glycans, also showed changes in mRNA expression in parvalbumin-type interneurons isolated from layer 3 of the dorsolateral prefrontal cortex [90]. Protein levels of B3GNT8, which adds polylactosamine units to N-glycans, is upregulated in gliomas relative to normal brain tissue, but its role in neurologic and psychiatric disorders is unknown [91].
Finally, analysis of glycosylation gene expression using a custom RNA microarray in samples of prefrontal cortex identified differential expression of several enzymes, spanning multiple glycosylation pathways including N-linked, O-linked, and glycolipid metabolism [44].
Alterations of CSF N-glycans
In contrast to studying a particular glycoprotein, glycoenzyme, or glycan structure, glycomics analyses can be utilized to determine the relative abundance of individual or classes of N-glycans removed from host proteins using PNGase F. One such study analyzed the N-glycans from glycoproteins within CSF and serum of antipsychotic-naive individuals with schizophrenia and controls using high performance liquid chromatography [45]. The results revealed a general downregulation of certain classes of N-glycans in the CSF of cases, including bisected and sialylated structures, as well as gender-specific changes in serum protein N-glycans such as a two-fold increase in two complex N-glycans (A4G4S4 and A3FG3S3) in males with the disorder. Additional experiments to place these glycan changes in a biological and functional context are required, but the systematic analysis provides evidence that glycosylation is altered in the disease state and not strictly limited to substrates in the brain.
Discussion
Glycosylation encompasses a range of processes that are critical to normal brain development and neuronal function. In this review, we synthesize evidence suggesting broad dysregulation of glycobiology in schizophrenia. As the diagnostic criteria for schizophrenia are based on clinical phenomenology without consideration of biologically-defined markers of disease, our field is in dire need of validated and approachable disease mechanisms for the development of diagnostics and treatments [92, 93]. The profound clinical heterogeneity encompassed by the diagnosis of schizophrenia contributes to our difficultly in identifying such targets, and also likely accounts for some of the diverse glycosylation changes observed in the disease state.
Glycosylation changes observed in schizophrenia span several brain regions, cellular pathways, classes of proteins, and steps of the synthetic pathway. This suggests a general dysregulation of glycosylation as a feature of the disease state and does not implicate a specific synthetic step or cell type as being uniquely vulnerable. For example, glutamate receptor subunits displayed bidirectional changes of glycosylation in individuals with schizophrenia, with GluK2 having more high-mannose N-glycans and GluA2 having fewer. The excitatory amino acid transporters EAAT1 and EAAT2 exhibited no difference in high-mannose N-glycans, but their complex N-glycans were different in brain samples from cases of schizophrenia in post-mortem studies. Glycoproteins from multiple cell types showed differences of glycosylation in the disease state, again illustrated by the neuronal proteins GluA2 and GluK2 and the glia proteins EAAT1 and EAAT2. Glycosylation differences are not limited to N-glycosylation and are found in polysialylation, O-glycosylation, glycosphingolipid metabolism, and proteoglycans as well. Studies of glycosylation changes in schizophrenia stop short of determining the functional consequences of the observed differences, highlighting our relatively limited understanding of glycosylation in the nervous system despite the clear functional importance of this pathway in brain development [4]. Future studies of glycosylation enzymes dysregulated in schizophrenia, such as POFUT2, FUT8, MGAT4A, and B3GNT8, should include analyses of which glycoproteins are modified by these enzymes in the brain and how they change in the disease state, facilitating a better understanding of how these changes contribute to the disorder.
Aberrant glycosylation in schizophrenia may be attributed to several factors, including medications and environmental stressors associated with a lifetime of living with severe mental illness, as well as an individual’s genetic risk. To determine if glycosylation changes play a causal role in the development of the disorder or instead result from exposures in the disease state, such variables must be considered. One study of first-break, antipsychotic-naive individuals with schizophrenia found differences in individual CSF N-glycans between cases and controls, illustrating that medications alone are probably not sufficient to explain the altered glycan patterns observed. However, in post-mortem samples, prior antipsychotic medication use can have a profound effect on proteins and metabolites in schizophrenia [94]. Antipsychotic treatment alone can alter glycosylation in individuals with schizophrenia, as demonstrated by changes in N-glycans on serum glycoproteins detected after 6 weeks of olanzapine treatment [95]. Olanzapine or chlorpromazine has also been shown to increase levels of PSA-NCAM in the prefrontal cortex of healthy adult rodents, and chlorpromazine increased the surface expression of PSA-NCAM in human neuroblastoma cells, suggesting that these medications may impact localization or recycling of the polysialylated protein [96, 97]. Individuals with schizophrenia show decreased levels of PSA-NCAM in the brain compared to controls, raising the question of whether treatment may mitigate this reduction.
With regard to whether glycosylation changes might be secondary to exposures associated with a lifetime of severe mental illness, one post-mortem study compared levels of PSA-NCAM in the dorsolateral prefrontal cortex (PFC) in individuals with schizophrenia to individuals with bipolar disorder, major depressive disorder (MDD), and controls, finding a reduction only in cases of schizophrenia [35]. However, a more recent study of PNN levels in the dorsolateral prefrontal cortex found reductions in both bipolar disorder and schizophrenia samples compared to controls [41]. We are not aware of additional experiments analyzing glycosylation in the brains of individuals with MDD, but some studies have reported changes in the plasma or serum protein N-glycome in depression [98–100]. These results highlight the complexity and challenge of determining whether glycosylation changes are related to a particular disease, symptom, or exposure.
Recent GWAS of schizophrenia have identified risk variants near five genes encoding glycosylation enzymes, as well as several proteins critically regulated by glycosylation (Reviewed in related publication, Mealer et al, 2020, submitted for publication). The functional implications of these variants have yet to be studied, but the genetic association of glycosylation enzymes with schizophrenia supports the view that this pathway is involved in pathogenesis of the disorder. Further, work from our group and others has shown that the strongest coding variant from schizophrenia GWAS, a missense mutation in SLC39A8, lowers concentrations of a manganese, a critical co-factor for glycosyltransferases, and leads to altered glycosylation [101, 102].
In sum, there is a compelling argument from observational analyses of plasma, CSF and post-mortem brain samples that aberrant glycosylation is involved in the pathophysiology of schizophrenia, but many questions remain to be answered including the temporal relationship and connection to heritable variation. A recent review by Mueller and Meador-Woodruff describes schizophrenia-associated changes in glycosylation as well as an array of additional post-translational modifications, highlighting the importance and complexity of molecular alterations in disease [103]. Studies of glycosylation and schizophrenia have primarily focused on N-glycosylation due to the existence of well-developed and validated tools (namely enzymes that specifically remove N-glycans from proteins), leaving comprehensive assessment of protein O-glycosylation, glycolipids, and proteoglycans relatively unexplored in the disease state. Genetic associations with schizophrenia include enzymes which can act on N-glycans, O-glycans, glycolipids, and proteoglycans, demonstrating the need to include analyses of each modification in future research. Moving forward, experiments should consider the genetic variation related to schizophrenia, the diversity of glycosylation modifications, the contribution of environmental factors, and clinical heterogeneity in order to develop a more comprehensive understanding of how glycosylation is dysregulated in this disease.
Acknowledgements:
This work was supported by a foundation grant from the Stanley Center for Psychiatric Research at the Broad Institute of Harvard/MIT (awarded to RGM).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest: The authors have declared that no conflicts of interest exist.
References:
- 1.Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15:346–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng BG, Freeze HH. Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet TIG. 2018;34:466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang IJ, He M, Lam CT. Congenital disorders of glycosylation. Ann Transl Med. 2018;6:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurological aspects of human glycosylation disorders. Annu Rev Neurosci. 2015;38:105–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott H, Panin VM. The role of protein N-glycosylation in neural transmission. Glycobiology. 2014;24:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai G, Salonikidis PS, Fei J, Schwarz W, Schülein R, Reutter W, et al. The role of N-glycosylation in the stability, trafficking and GABA-uptake of GABA-transporter 1. Terminal N-glycans facilitate efficient GABA-uptake activity of the GABA transporter. FEBS J. 2005;272:1625–1638. [DOI] [PubMed] [Google Scholar]
- 7.Kandel MB, Yamamoto S, Midorikawa R, Morise J, Wakazono Y, Oka S, et al. N-glycosylation of the AMPA-type glutamate receptor regulates cell surface expression and tetramer formation affecting channel function. J Neurochem. 2018;147:730–747. [DOI] [PubMed] [Google Scholar]
- 8.Storey GP, Opitz-Araya X, Barria A. Molecular determinants controlling NMDA receptor synaptic incorporation. J Neurosci Off J Soc Neurosci. 2011;31:6311–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lichnerova K, Kaniakova M, Park SP, Skrenkova K, Wang Y-X, Petralia RS, et al. Two N-glycosylation Sites in the GluN1 Subunit Are Essential for Releasing N-methyl-d-aspartate (NMDA) Receptors from the Endoplasmic Reticulum. J Biol Chem. 2015;290:18379–18390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skrenkova K, Lee S, Lichnerova K, Kaniakova M, Hansikova H, Zapotocky M, et al. N-Glycosylation Regulates the Trafficking and Surface Mobility of GluN3A-Containing NMDA Receptors. Front Mol Neurosci. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Min C, Zheng M, Zhang X, Guo S, Kwon K-J, Shin CY, et al. N-linked Glycosylation on the N-terminus of the dopamine D2 and D3 receptors determines receptor association with specific microdomains in the plasma membrane. Biochim Biophys Acta. 2015;1853:41–51. [DOI] [PubMed] [Google Scholar]
- 12.Iqbal S, Ghanimi Fard M, Everest-Dass A, Packer NH, Parker LM. Understanding cellular glycan surfaces in the central nervous system. Biochem Soc Trans. 2018. 17 December 2018. 10.1042/BST20180330. [DOI] [PubMed] [Google Scholar]
- 13.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varki A Biological roles of glycans. Glycobiology. 2017;27:3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henrissat B, Surolia A, Stanley P. A Genomic View of Glycobiology. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. , editors. Essent. Glycobiol,. 3rd ed.Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [Google Scholar]
- 16.Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13:448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol. 2011;21:576–582. [DOI] [PubMed] [Google Scholar]
- 18.Stanley P, Taniguchi N, Aebi M. N-Glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. , editors. Essent. Glycobiol,. 3rd ed.Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [Google Scholar]
- 19.Joshi HJ, Narimatsu Y, Schjoldager KT, Tytgat HLP, Aebi M, Clausen H, et al. SnapShot: O-Glycosylation Pathways across Kingdoms. Cell. 2018;172:632–632.e2. [DOI] [PubMed] [Google Scholar]
- 20.Brockhausen I, Stanley P. O-GalNAc Glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. , editors. Essent. Glycobiol,. 3rd ed.Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [PubMed] [Google Scholar]
- 21.Haltiwanger RS, Wells L, Freeze HH, Stanley P. Other Classes of Eukaryotic Glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. , editors. Essent. Glycobiol,. 3rd ed.Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [PubMed] [Google Scholar]
- 22.Schnaar RL. The Biology of Gangliosides. Adv. Carbohydr. Chem. Biochem, vol. 76, Elsevier; 2019. p. 113–148. [DOI] [PubMed] [Google Scholar]
- 23.Schnaar RL, Kinoshita T. Glycosphingolipids. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. , editors. Essent. Glycobiol,. 3rd ed.Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [PubMed] [Google Scholar]
- 24.Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53:372–381. [DOI] [PubMed] [Google Scholar]
- 25.Lindahl U, Couchman J, Kimata K, Esko JD. Proteoglycans and Sulfated Glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. , editors. Essent. Glycobiol,. 3rd ed.Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [Google Scholar]
- 26.Rowlands D, Sugahara K, Kwok J. Glycosaminoglycans and Glycomimetics in the Central Nervous System. Molecules. 2015;20:3527–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen L, Lind-Thomsen A, Joshi HJ, Pedersen NB, Have CT, Kong Y, et al. A glycogene mutation map for discovery of diseases of glycosylation. Glycobiology. 2015;25:211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joshi HJ, Hansen L, Narimatsu Y, Freeze HH, Henrissat B, Bennett E, et al. Glycosyltransferase genes that cause monogenic congenital disorders of glycosylation are distinct from glycosyltransferase genes associated with complex diseases. Glycobiology. 2018;28:284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. , editors. Essentials of Glycobiology. 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [PubMed] [Google Scholar]
- 30.Tucholski J, Simmons MS, Pinner AL, Haroutunian V, McCullumsmith RE, Meador-Woodruff JH. Abnormal N-linked glycosylation of cortical AMPA receptor subunits in schizophrenia. Schizophr Res. 2013;146:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tucholski J, Simmons MS, Pinner AL, McMillan LD, Haroutunian V, Meador-Woodruff JH. N-linked glycosylation of cortical N-methyl-D-aspartate and kainate receptor subunits in schizophrenia. Neuroreport. 2013;24:688–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauer D, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal glycosylation of EAAT1 and EAAT2 in prefrontal cortex of elderly patients with schizophrenia. Schizophr Res. 2010;117:92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mueller TM, Haroutunian V, Meador-Woodruff JH. N-Glycosylation of GABAA receptor subunits is altered in Schizophrenia. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2014;39:528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbeau D, Liang JJ, Robitalille Y, Quirion R, Srivastava LK. Decreased expression of the embryonic form of the neural cell adhesion molecule in schizophrenic brains. Proc Natl Acad Sci U S A. 1995;92:2785–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilabert-Juan J, Varea E, Guirado R, Blasco-Ibáñez JM, Crespo C, Nácher J. Alterations in the expression of PSA-NCAM and synaptic proteins in the dorsolateral prefrontal cortex of psychiatric disorder patients. Neurosci Lett. 2012;530:97–102. [DOI] [PubMed] [Google Scholar]
- 36.Pantazopoulos H, Woo T-UW, Lim MP, Lange N, Berretta S. Extracellular matrix-glial abnormalities in the amygdala and entorhinal cortex of subjects diagnosed with schizophrenia. Arch Gen Psychiatry. 2010;67:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pantazopoulos H, Markota M, Jaquet F, Ghosh D, Wallin A, Santos A, et al. Aggrecan and chondroitin-6-sulfate abnormalities in schizophrenia and bipolar disorder: a postmortem study on the amygdala. Transl Psychiatry. 2015;5:e496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mauney SA, Athanas KM, Pantazopoulos H, Shaskan N, Passeri E, Berretta S, et al. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol Psychiatry. 2013;74:427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pantazopoulos H, Boyer-Boiteau A, Holbrook EH, Jang W, Hahn C-G, Arnold SE, et al. Proteoglycan abnormalities in olfactory epithelium tissue from subjects diagnosed with schizophrenia. Schizophr Res. 2013;150:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enwright JF, Sanapala S, Foglio A, Berry R, Fish KN, Lewis DA. Reduced Labeling of Parvalbumin Neurons and Perineuronal Nets in the Dorsolateral Prefrontal Cortex of Subjects with Schizophrenia. Neuropsychopharmacology. 2016;41:2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alcaide J, Guirado R, Crespo C, Blasco-Ibáñez JM, Varea E, Sanjuan J, et al. Alterations of perineuronal nets in the dorsolateral prefrontal cortex of neuropsychiatric patients. Int J Bipolar Disord. 2019;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mueller TM, Yates SD, Haroutunian V, Meador-Woodruff JH. Altered fucosyltransferase expression in the superior temporal gyrus of elderly patients with schizophrenia. Schizophr Res. 2017;182:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kippe JM, Mueller TM, Haroutunian V, Meador-Woodruff JH. Abnormal N-acetylglucosaminyltransferase expression in prefrontal cortex in schizophrenia. Schizophr Res. 2015;166:219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narayan S, Head SR, Gilmartin TJ, Dean B, Thomas EA. Evidence for disruption of sphingolipid metabolism in schizophrenia. J Neurosci Res. 2009;87:278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanta JL, Saldova R, Struwe WB, Byrne JC, Leweke FM, Rothermund M, et al. Identification of N-glycosylation changes in the CSF and serum in patients with schizophrenia. J Proteome Res. 2010;9:4476–4489. [DOI] [PubMed] [Google Scholar]
- 46.Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–179. [DOI] [PubMed] [Google Scholar]
- 47.Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol Oxf Engl. 2015;29:97–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buckner RL, DiNicola LM. The brain’s default network: updated anatomy, physiology and evolving insights. Nat Rev Neurosci. 2019;20:593–608. [DOI] [PubMed] [Google Scholar]
- 49.Hunt MJ, Kopell NJ, Traub RD, Whittington MA. Aberrant Network Activity in Schizophrenia. Trends Neurosci. 2017;40:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao R, Penzes P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr Mol Med. 2015;15:146–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maupin KA, Liden D, Haab BB. The fine specificity of mannose-binding and galactose-binding lectins revealed using outlier motif analysis of glycan array data. Glycobiology. 2012;22:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mueller TM, Remedies CE, Haroutunian V, Meador-Woodruff JH. Abnormal subcellular localization of GABAA receptor subunits in schizophrenia brain. Transl Psychiatry. 2015;5:e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schnaar RL, Gerardy-Schahn R, Hildebrandt H. Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol Rev. 2014;94:461–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seki T, Arai Y. Distribution and possible roles of the highly polysialylated neural cell adhesion molecule (NCAM-H) in the developing and adult central nervous system. Neurosci Res. 1993;17:265–290. [DOI] [PubMed] [Google Scholar]
- 56.Cox ET, Brennaman LH, Gable KL, Hamer RM, Glantz LA, Lamantia A-S, et al. Developmental regulation of neural cell adhesion molecule in human prefrontal cortex. Neuroscience. 2009;162:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson CP, Fujimoto I, Rutishauser U, Leckband DE. Direct Evidence That Neural Cell Adhesion Molecule (NCAM) Polysialylation Increases Intermembrane Repulsion and Abrogates Adhesion. J Biol Chem. 2005;280:137–145. [DOI] [PubMed] [Google Scholar]
- 58.Nakata D, Troy FA. Degree of Polymerization (DP) of Polysialic Acid (PolySia) on Neural Cell Adhesion Molecules (N-CAMs): DEVELOPMENT AND APPLICATION OF A NEW STRATEGY TO ACCURATELY DETERMINE THE DP OF polySIA CHAINS ON N-CAMS. J Biol Chem. 2005;280:38305–38316. [DOI] [PubMed] [Google Scholar]
- 59.Arai M, Yamada K, Toyota T, Obata N, Haga S, Yoshida Y, et al. Association between polymorphisms in the promoter region of the sialyltransferase 8B (SIAT8B) gene and schizophrenia. Biol Psychiatry. 2006;59:652–659. [DOI] [PubMed] [Google Scholar]
- 60.Tao R, Li C, Zheng Y, Qin W, Zhang J, Li X, et al. Positive association between SIAT8B and schizophrenia in the Chinese Han population. Schizophr Res. 2007;90:108–114. [DOI] [PubMed] [Google Scholar]
- 61.Isomura R, Kitajima K, Sato C. Structural and functional impairments of polysialic acid by a mutated polysialyltransferase found in schizophrenia. J Biol Chem. 2011;286:21535–21545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hildebrandt H, Mühlenhoff M, Oltmann-Norden I, Röckle I, Burkhardt H, Weinhold B, et al. Imbalance of neural cell adhesion molecule and polysialyltransferase alleles causes defective brain connectivity. Brain J Neurol. 2009;132:2831–2838. [DOI] [PubMed] [Google Scholar]
- 63.Berretta S, Pantazopoulos H, Markota M, Brown C, Batzianouli ET. Losing the sugar coating: potential impact of perineuronal net abnormalities on interneurons in schizophrenia. Schizophr Res. 2015;167:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bitanihirwe BKY, Mauney SA, Woo T-UW. Weaving a Net of Neurobiological Mechanisms in Schizophrenia and Unraveling the Underlying Pathophysiology. Biol Psychiatry. 2016;80:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bandtlow CE, Zimmermann DR. Proteoglycans in the Developing Brain: New Conceptual Insights for Old Proteins. Physiol Rev. 2000;80:1267–1290. [DOI] [PubMed] [Google Scholar]
- 66.Laabs TL, Wang H, Katagiri Y, McCann T, Fawcett JW, Geller HM. Inhibiting glycosaminoglycan chain polymerization decreases the inhibitory activity of astrocyte-derived chondroitin sulfate proteoglycans. J Neurosci Off J Soc Neurosci. 2007;27:14494–14501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foscarin S, Raha-Chowdhury R, Fawcett JW, Kwok JCF. Brain ageing changes proteoglycan sulfation, rendering perineuronal nets more inhibitory. Aging. 2017;9:1607–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shah A, Lodge DJ. A loss of hippocampal perineuronal nets produces deficits in dopamine system function: relevance to the positive symptoms of schizophrenia. Transl Psychiatry. 2013;3:e215–e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pietersen CY, Mauney SA, Kim SS, Lim MP, Rooney RJ, Goldstein JM, et al. Molecular profiles of pyramidal neurons in the superior temporal cortex in schizophrenia. J Neurogenet. 2014;28:53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martins-de-Souza D, Gattaz WF, Schmitt A, Rewerts C, Marangoni S, Novello JC, et al. Alterations in oligodendrocyte proteins, calcium homeostasis and new potential markers in schizophrenia anterior temporal lobe are revealed by shotgun proteome analysis. J Neural Transm. 2009;116:275–289. [DOI] [PubMed] [Google Scholar]
- 71.Wang Q, Wang C, Ji B, Zhou J, Yang C, Chen J. Hapln2 in Neurological Diseases and Its Potential as Therapeutic Target. Front Aging Neurosci. 2019;11:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takahashi N, Sakurai T, Bozdagi-Gunal O, Dorr NP, Moy J, Krug L, et al. Increased expression of receptor phosphotyrosine phosphatase-β/ζ is associated with molecular, cellular, behavioral and cognitive schizophrenia phenotypes. Transl Psychiatry. 2011;1:e8–e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mühleisen TW, Mattheisen M, Strohmaier J, Degenhardt F, Priebe L, Schultz CC, et al. Association between schizophrenia and common variation in neurocan (NCAN), a genetic risk factor for bipolar disorder. Schizophr Res. 2012;138:69–73. [DOI] [PubMed] [Google Scholar]
- 74.Raum H, Dietsche B, Nagels A, Witt SH, Rietschel M, Kircher T, et al. A genome-wide supported psychiatric risk variant in NCAN influences brain function and cognitive performance in healthy subjects: NCAN Genotype Influences Brain Functioning. Hum Brain Mapp. 2015;36:378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schultz CC, Mühleisen TW, Nenadic I, Koch K, Wagner G, Schachtzabel C, et al. Common variation in NCAN, a risk factor for bipolar disorder and schizophrenia, influences local cortical folding in schizophrenia. Psychol Med. 2014;44:811–820. [DOI] [PubMed] [Google Scholar]
- 76.Wang P, Cai J, Ni J, Zhang J, Tang W, Zhang C. The NCAN gene: schizophrenia susceptibility and cognitive dysfunction. Neuropsychiatr Dis Treat. 2016;Volume 12:2875–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Habl G, Schmitt A, Zink M, von Wilmsdorff M, Yeganeh-Doost P, Jatzko A, et al. Decreased reelin expression in the left prefrontal cortex (BA9) in chronic schizophrenia patients. Neuropsychobiology. 2012;66:57–62. [DOI] [PubMed] [Google Scholar]
- 79.Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A. 1998;95:15718–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eastwood SL, Law AJ, Everall IP, Harrison PJ. The axonal chemorepellant semaphorin 3A is increased in the cerebellum in schizophrenia and may contribute to its synaptic pathology. Mol Psychiatry. 2003;8:148–155. [DOI] [PubMed] [Google Scholar]
- 81.Walsh MT, Ryan M, Hillmann A, Condren R, Kenny D, Dinan T, et al. Elevated expression of integrin alpha(IIb) beta(IIIa) in drug-naïve, first-episode schizophrenic patients. Biol Psychiatry. 2002;52:874–879. [DOI] [PubMed] [Google Scholar]
- 82.Pantazopoulos H, Berretta S. In Sickness and in Health: Perineuronal Nets and Synaptic Plasticity in Psychiatric Disorders. Neural Plast. 2016;2016:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beroun A, Mitra S, Michaluk P, Pijet B, Stefaniuk M, Kaczmarek L. MMPs in learning and memory and neuropsychiatric disorders. Cell Mol Life Sci. 2019;76:3207–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mohamedi Y, Fontanil T, Cobo T, Cal S, Obaya AJ. New Insights into ADAMTS Metalloproteases in the Central Nervous System. Biomolecules. 2020;10:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Domenici E, Willé DR, Tozzi F, Prokopenko I, Miller S, McKeown A, et al. Plasma protein biomarkers for depression and schizophrenia by multi analyte profiling of case-control collections. PloS One. 2010;5:e9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamamori H, Hashimoto R, Ishima T, Kishi F, Yasuda Y, Ohi K, et al. Plasma levels of mature brain-derived neurotrophic factor (BDNF) and matrix metalloproteinase-9 (MMP-9) in treatment-resistant schizophrenia treated with clozapine. Neurosci Lett. 2013;556:37–41. [DOI] [PubMed] [Google Scholar]
- 87.Fukuda T, Hashimoto H, Okayasu N, Kameyama A, Onogi H, Nakagawasai O, et al. Alpha1,6-fucosyltransferase-deficient mice exhibit multiple behavioral abnormalities associated with a schizophrenia-like phenotype: importance of the balance between the dopamine and serotonin systems. J Biol Chem. 2011;286:18434–18443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Du J, Takeuchi H, Leonhard-Melief C, Shroyer KR, Dlugosz M, Haltiwanger RS, et al. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev Biol. 2010;346:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Berardinelli SJ, Haltiwanger RS. Analyzing the Effects of O-Fucosylation on Secretion of ADAMTS Proteins Using Cell-Based Assays. In: Apte SS, editor. ADAMTS Proteases, vol. 2043, New York, NY: Springer New York; 2020. p. 25–43. [DOI] [PubMed] [Google Scholar]
- 90.Enwright III JF, Huo Z, Arion D, Corradi JP, Tseng G, Lewis DA. Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia. Mol Psychiatry. 2018;23:1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu J, Shen L, Yang L, Hu S, Xu L, Wu S. High expression of β3GnT8 is associated with the metastatic potential of human glioma. Int J Mol Med. 2014;33:1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lawrie SM, O’Donovan MC, Saks E, Burns T, Lieberman JA. Improving classification of psychoses. Lancet Psychiatry. 2016;3:367–374. [DOI] [PubMed] [Google Scholar]
- 93.Lawrie SM, O’Donovan MC, Saks E, Burns T, Lieberman JA. Towards diagnostic markers for the psychoses. Lancet Psychiatry. 2016;3:375–385. [DOI] [PubMed] [Google Scholar]
- 94.Chan MK, Tsang TM, Harris LW, Guest PC, Holmes E, Bahn S. Evidence for disease and antipsychotic medication effects in post-mortem brain from schizophrenia patients. Mol Psychiatry. 2011;16:1189–1202. [DOI] [PubMed] [Google Scholar]
- 95.Telford JE, Bones J, McManus C, Saldova R, Manning G, Doherty M, et al. Antipsychotic treatment of acute paranoid schizophrenia patients with olanzapine results in altered glycosylation of serum glycoproteins. J Proteome Res. 2012;11:3743–3752. [DOI] [PubMed] [Google Scholar]
- 96.Frasca A, Fumagalli F, Ter Horst J, Racagni G, Murphy KJ, Riva MA. Olanzapine, but not haloperidol, enhances PSA-NCAM immunoreactivity in rat prefrontal cortex. Int J Neuropsychopharmacol. 2008;11:591–595. [DOI] [PubMed] [Google Scholar]
- 97.Abe C, Nishimura S, Mori A, Niimi Y, Yang Y, Hane M, et al. Chlorpromazine Increases the Expression of Polysialic Acid (PolySia) in Human Neuroblastoma Cells and Mouse Prefrontal Cortex. Int J Mol Sci. 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Park DI, Štambuk J, Razdorov G, Pučić-Baković M, Martins-de-Souza D, Lauc G, et al. Blood plasma/IgG N-glycome biosignatures associated with major depressive disorder symptom severity and the antidepressant response. Sci Rep. 2018;8:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boeck C, Pfister S, Bürkle A, Vanhooren V, Libert C, Salinas-Manrique J, et al. Alterations of the serum N-glycan profile in female patients with Major Depressive Disorder. J Affect Disord. 2018;234:139–147. [DOI] [PubMed] [Google Scholar]
- 100.Yamagata H, Uchida S, Matsuo K, Harada K, Kobayashi A, Nakashima M, et al. Altered plasma protein glycosylation in a mouse model of depression and in patients with major depression. J Affect Disord. 2017. 19 August 2017. 10.1016/j.jad.2017.08.057. [DOI] [PubMed] [Google Scholar]
- 101.Lin W, Vann DR, Doulias P-T, Wang T, Landesberg G, Li X, et al. Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. J Clin Invest. 2017;127:2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mealer RG, Jenkins BG, Chen C-Y, Daly MJ, Ge T, Lehoux S, et al. A schizophrenia risk locus alters brain metal transport and plasma glycosylation. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mueller TM, Meador-Woodruff JH. Post-translational protein modifications in schizophrenia. Npj Schizophr. 2020;6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]