

Abstract
Alpha-1 antitrypsin (AAT) has protective functions in animal islet transplantation models. While the therapeutic effect of AAT therapy is currently being tested in clinical trials, we investigated the mechanism of AAT protection in a clinically relevant marginal intrahepatic human islet transplantation model. In recipients receiving islets and AAT, 68.9% (20/29) reached normoglycemia, compared to 35.7% (10/28) in those receiving islets only, at 60 days posttransplant (PT). AAT-treated mice had lower serum levels of inflammatory cytokines immediately PT. Reduced M1 macrophages were observed in livers of AAT-treated recipients compared to controls as evidenced by flow cytometry and RNA-seq transcriptional profiling analysis. In vitro AAT suppressed IFN-γ-induced M1 macrophage activation/polarization via suppression of STAT1 phosphorylation and iNOS production. AAT inhibits macrophage activation induced by cytokines or dying islets, and consequently leads to islet cell survival. In a macrophage depletion mouse model, the presence of M1 macrophages in the liver contributed to graft death. AAT, through suppressing macrophage activation, protected transplanted islets from death and dysfunction in the human islet and NOD-SCID mouse model. The protective effect of AAT was confirmed in a major mismatch allogeneic islet transplantation model. Taken together, AAT suppresses liver macrophage activation that contributes to graft survival after transplantation.
Keywords: basic (laboratory) research/science, diabetes, graft survival, islet transplantation, islets of Langerhans, macrophage/monocyte biology: activation, translational research/science
1. INTRODUCTION
Islet/β cell transplantation offers a therapeutic option for the treatment of type 1 diabetes. However, even in syngeneic islet transplantation models, up to 60% of islet cells die immediately posttransplant (PT) as a result of innate inflammatory responses.1,2 Understanding the mechanisms and suppressing these responses may reduce early islet cell death and enhance the efficacy of islet transplantation.
Alpha-1 antitrypsin (AAT) is an acute phase reactant and serine protease inhibitor that inhibits both protease and non-protease enzymes, including neutrophil elastase, cathepsin G, and others.3 Plasma AAT levels and activity are lower in diabetic patients than in non-diabetic controls. Therefore, AAT deficiency may be associated with diabetes.4 AAT exerts anti-inflammatory effects via suppression of cytokine production, complement activation, and immune cell infiltration.5,6 In pancreatic β cells, AAT functions as an anti-apoptotic and anti-inflammatory agent.7–9 In murine models of diabetes, a single injection of AAT to non-obese diabetic (NOD) mice reduced the intensity of insulitis, increased β cell mass, promoted β cell regeneration, and prevented the onset of diabetes via modulating T regulatory cells (Tregs).8 AAT protected mouse and non-human primate islet grafts from failure by reducing islet cell death, promoting vascularization,9–11 and suppressing inflammation.12 The effects of AAT therapy in chronic pancreatitis patients undergoing islet autotransplantation are currently being evaluated in clinical trials by our center and others.
Macrophages are one of the major cells of the innate immune system.13 They can be divided into a variety of tissue-specific resident cells and marrow-derived circulating monocytes that travel to inflammatory sites via the bloodstream.14 Resident macrophages in specific organs, such as the liver, participate in tissue repair, immune surveillance, and homeostasis. Both resident and inflammatory macrophages can be activated and become classically activated macrophages (M1) or alternatively activated (M2) macrophages. M1 macrophages can be activated by cytokines, including interferon (IFN)-γ and lipopolysaccharide (LPS), and exert proinflammatory and host defense functions via secretion of TNF-α, inducible nitric oxide (iNOS), and superoxide anions. In contrast, M2 macrophages can be activated by interleukin (IL)-4 and IL-13, and contribute to tissue repair and resolution of inflammation, by secretion of IL-10, IL-1 receptor α, and TGF-β.15 While the stimuli for macrophage polarization into the classical M1 and alternative M2 pathways are likely much more complex and less bipolar, the paradigm is useful to study therapeutics that affect macrophage function.
In patients, islets are infused into the liver via the portal vein. Activated liver resident macrophages (Kupffer cells) and inflammatory macrophages secrete proinflammatory cytokines and contribute to islet cell death after transplantation. In the meantime, dying β cells also produce iNOS and high mobility group box 1 (HGMB) that attract more macrophages to the liver and enhance inflammation and β cell death. We have shown that AAT treatment to the recipients protects mouse islets from β cell death after transplantation in the syngeneic islet transplantation model.12,16 However, whether AAT suppresses macrophage activation/M1 polarization as a mechanism for reduced islet death and better function remains largely unknown. In this study, we assess whether macrophages are the direct cellular targets of AAT and whether treatment with AAT protects human islets from cell death and dysfunction via modulating macrophage activation.
2. MATERIALS AND METHODS
2.1. Animals
Male NOD-SCID, BALB/c, and C57BL/6 mice at 6–8 weeks of age (The Jackson Laboratory) were used in the study. All procedures and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina.
2.2. Islet transplantation
Diabetes was induced in NOD-SCID or C57BL/6 mice using streptozotocin (STZ) as previously described.12 Human islets were from Georgetown University. Donor information and islet characteristics are listed in Table S1. In human islet transplantation, 85 islet equivalent number (IEQ) per gram of mouse body weight were transplanted via portal vein into the liver of the NOD-SCID mice. In the allogeneic islet transplantation model, BALB/c islets (400–450) were transplanted under the kidney capsule of the diabetic C57BL/6 mice. Non-fasting blood glucose levels were measured every 2 or 3 days until 60 days PT and mice with blood glucose levels <200 mg/dl were considered normoglycemic.
2.3. AAT injection
Recipient mice were given intraperitoneal human AAT (Prolastin C, Grifols) at 2 or 4 mg/mouse every 2 days through 12 days PT. Mice receiving vehicle at the same schedules were used as controls.
2.4. Intravenous glucose tolerance test (IVGTT) and human C-peptide levels
Mice fasted overnight were injected with a glucose solution (1 g/kg body weight, i.v.). A small aliquot of blood/serum was obtained at 0, 15, 30, 60, 90, and 120 min for glucose and human C-peptide measurements using a Freestyle glucometer and the Human C-peptide ELISA kit (ALPCO).
2.5. Serum cytokine levels
Blood samples were collected before (day 0) and at 1, 3, and 7 days PT. The presence of 23 mouse cytokines in serum was quantified using the Bio-Plex Pro Mouse Cytokine 23-plex Assay kit (Bio-Rad), following the manufacturer's instructions.
2.6. Liver macrophage isolation
Livers were perfused with an enzyme solution containing 1 mg/ mL of collagenase type IV (GIBCO); 1 mg/mL Pronase E (Millipore), 20 μg/mL of DNase (Sigma-Aldrich), and 3 mmol/L of CaCl2 in HBSS (Sigma-Aldrich) at 3 ml/min for 20 min as previously described.17,18 The liver was gently removed, minced, and filtered. Cells were washed multiple times and cultured in DMEM medium with 10% fetal bovine serum, 1% penicillin, and streptomycin in 5% CO2 at 37°C. Floating cells were removed at 2 hours after cell plating and macrophages were allowed to continually attach to the plate overnight before testing.
2.7. Flow cytometry
Macrophages were harvested, washed with PBS, and fixed with 2% paraformaldehyde. Cells were incubated with respective antibodies or their corresponding isotype controls for 1 hour in the dark on ice. Expression of cellular markers was analyzed on the BD LSRFortessa X20 flow Cytometer. Data were analyzed using the FlowJo software.
2.8. RT-PCR and transcriptional profiling analysis of liver macrophages
Total RNA was extracted and mRNA levels of genes were analyzed by real-time (RT)-PCR using standard methods as reported previously.12 For RNA-seq analysis of liver macrophages, RNA libraries were constructed using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB, #E7760S/L). Directional mRNA-seq was conducted using the Illumina HiSeq 4000 system, using the paired-end 150 reads option. RNA-seq paired-end reads were aligned to the 10-mm mouse reference genome (GRCm38 release 89) using the ultrafast universal RNA-seq aligner (STAR v2.5.3).19 Genewise counts were assessed with HTSeq-count v.0.8.0.20 DESeq2 v1.16.1 was used to compute regularized log 2 levels of gene expression and estimate differentially expressed genes.21 Between-group differences above a two-fold change with a false discovery rate-adjusted P-value (padj) < .05 were considered differentially expressed. g: Profiler (https://biit.cs.ut.ee/gprofiler/gost) was used for pathway and Gene Ontology (GO) analysis.
2.9. Measurements of islet cell apoptosis
Raw264.7 cells were treated by IFN-γ at 100 ng/mL for 24 hours. Human islets were co-cultured with the Raw cells in the presence or absence of AAT at 0.5 mg/mL for another 24 hours. Islets were removed and embedded in OCT and sectioned. Islet cell death was identified using the fluorescein In Situ Cell Death Detection kit (Roche Diagnostics). Cell death was confirmed using an apoptosis ELISA kit.
2.10. Macrophage activation when co-cultured with cytokine-treated dying islets in vitro
Human islets were treated with cytokines (IFN-γ [100 ng/mL], IL-1β [5 ng/mL], and TNF-α [100 ng/mL]) or vehicles for 24 hours, and cell death was measured by the apoptosis ELISA kit. Raw264.7 cells were co-cultured with vehicle- or cytokine-treated islets for 24 hours in the presence or absence of 0.5 mg/mL AAT. mRNA expressions of CD11c, IL-6, iNOS, and TNF-α were measured by RT-PCR analysis.
2.11. Western blot
Raw264.7 cells were lysed using protein lysis buffer, and protein concentration was measured using the spectrometer with analysis by immunoblotting using antibodies against p-STAT1 and STAT1. Beta-actin expression was detected using the anti-β-actin antibody and used as the endogenous control. Protein expression was quantified using the ImageJ software.
2.12. Islet transplantation into macrophage-depleted recipients
Clodronate-containing liposomes (lip-clod, Liposoma, 50 mg/kg) were given to recipients after STZ injection and before human islet transplantation as previously described.22,23 Liver tissue was collected 24 hours post liposome administration to confirm macrophage depletion by RT-PCR analysis and immunohistochemistry staining for F4/80 expression. Human islets alone (control group), islets with IFN-γ-treated Raw264.7 cells (1 × 106, M1 macrophages, IFN-γ group), or islets with Raw264.7 cells treated with IFN-γ in the presence of AAT (AAT group) were transplanted into the livers of the macrophage-depleted diabetic NOD-SCID mice. Nonfasting blood glucose levels of mice were measured daily until 21 days PT.
2.13. Statistical analysis
Percentage of mice reaching normoglycemia was analyzed and plotted using Kaplan-Meier survival statistics compared by the log-rank test. Data are expressed as mean ± standard error of the mean (SEM). Differences between groups were compared for statistical significance by ANOVA with Tukey-Kramer post hoc test, Student's t test, or Z test; P < .05 denoted significance.
3. RESULTS
3.1. ATT improves the survival and function of islets transplanted into the STZ-induced diabetic mice
We showed in our previous study that AAT treatment (2 mg/mouse) to the islet transplant recipient prolonged the survival of mouse islet grafts in a syngeneic intrahepatic islet transplantation model in C57BL/6 mice.12 To explore the potential clinical application, we tested the protective effects of AAT on graft survival and function when human islets were transplanted into the immunodeficient NOD-SCID mice that had been rendered diabetic (two consecutive blood glucose levels >350 mg/dl) by STZ treatment. In this model, when 400–450 human islets were transplanted, only 35.7% (10/28) of recipients in the control group achieved normoglycemia at day 60. In contrast, 46.7% (7/15) of recipients receiving AAT at 2 mg/ mouse (p = .5 vs. control by log-rank test) and 68.9% (20/29) of recipients receiving AAT at 4 mg/mouse groups achieved normoglycemia (p = .009 vs. control) at day 60 PT (Figure 1A). Average days needed to reach normoglycemia were 6.1 ± 2.9 days in control, and 3.1 ± 0.6 days in the AAT 4 mg/mouse group (Figure 1B). Based on these data, we used 4 mg/mouse AAT treatment in all the following studies.
FIGURE 1.

AAT treatment improves islet graft survival and function. (A–F) Data collected from human islets transplanted into NOD-SCID mouse model. (A) Percentage of recipients reaching normoglycemia in mice receiving AAT at 2 mg/mouse (n = 15), 4 mg/mouse (n = 29), or vehicle controls (CTR, n = 28). (B) Days to reach normoglycemia in islet transplant mice receiving AAT (4 mg/mouse) or vehicle. (C,D) Blood glucose levels during an intravenous glucose tolerance test (IVGTT) performed in AAT-treated and CTR mice at 7 and 28 days posttransplant. Inset, area under the curve of the blood glucose levels during an IVGTT. (E) Serum human C-peptide levels in recipients as measured by ELISA (n = 8 in each group). (F) Body weight changes in CTR or AAT group (n = 15–20 in each group). Data shown are mean ± SEM. *P < .05 by Student'sttest. (G–I) Data collected from the BALB/c to C57BL/6 islet transplantation model. (G) Percentage of mice with primary nonfunction in AAT-treated (n = 10) and control mice (n = 10). (H) Average nonfasting blood glucose levels of control and recipient mice (n = 10 in each group). (I) Percentage of allograft survival in AAT and control mice. *P < .05 by Z test
AAT-treated mice had better islet function as shown by lower blood glucose levels and significantly smaller glucose area under the curve during the IVGTT (Figure 1C,D, and insets) and higher basal and glucose-stimulated human C-peptide levels in serum (Figure 1E) at 7- and 28-days PT. The levels of mouse C-peptide in both groups were undetectable (not shown), suggesting that normoglycemia was maintained by the transplanted human islets. Besides, AAT-treated recipients exhibited better physical condition and faster recovery of body weight loss postsurgery (Figure 1F).
We next tested the protective effect of AAT in a major mismatch allogeneic islet transplantation model in which BALB/c islets (H-2b) were transplanted into diabetic C57BL/6 H-2d recipients. In this model, primary islet function occurred in only 50% (5/10) of control recipients when 400–450 islets were transplanted. In contrast, 90% (9/10) of the AAT-treated recipients showed primary function (p = .02, Z test, Figure 1G). The average blood glucose levels in AAT-treated mice were lower than mice in the control group during the follow-up period (until day 60, Figure 1H). Also, in mice that demonstrated primary graft function, AAT islets survived much longer (surviving 8, 10, 12, 16, 16, 21, 60, 60, and 60 days, median: 16 days) than controls (surviving 4, 8, 12, 21, and 60 days, median 12 days) (Figure 1I). These data further confirmed the protective effect of AAT on the primary function of transplanted islets.
3.2. AAT reduces serum proinflammatory cytokine levels PT
Nonspecific inflammatory responses lead to islet graft loss in both autologous and allogeneic islet transplantation settings PT.24To test whether AAT suppressed inflammation post intrahepatic islet transplantation, we measured cytokine secretion profiles in serum samples collected from AAT and the control NOD-SCID recipients before and at days 1, 3, and 7 post human islet transplantation using the Bio-Plex pro cytokine 23-plex assay kit. Most cytokine levels were elevated in control mice at day 1 PT. Presence of proinflammatory cytokines, including IL-1α, IL-1β, IL-6, IFN-γ, TNF-α, granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and others, was significantly reduced in serum from AAT-treated recipients compared to controls at day 1 PT (Figure 2A). Meanwhile, inflammatory-related chemokines, including MIP-1α, MIP-1β, KC, eotaxin, and MCP-1, were also significantly reduced in AAT-treated recipients compared with control at day 1 PT (Figure 2A). Most of the elevated cytokine levels declined to basal levels by day 7. Additionally, AAT-treated mice had higher levels of IL-4 and IL-10, and two anti-inflammatory cytokines, at day 3, although there was a lower IL-10 expression in the AAT group at day 1 (Figure 2B). Our data showed that AAT inhibits serum proinflammatory cytokines while increased anti-inflammatory cytokine levels in NOD-SCID mice receiving human islet transplantation.
FIGURE 2.
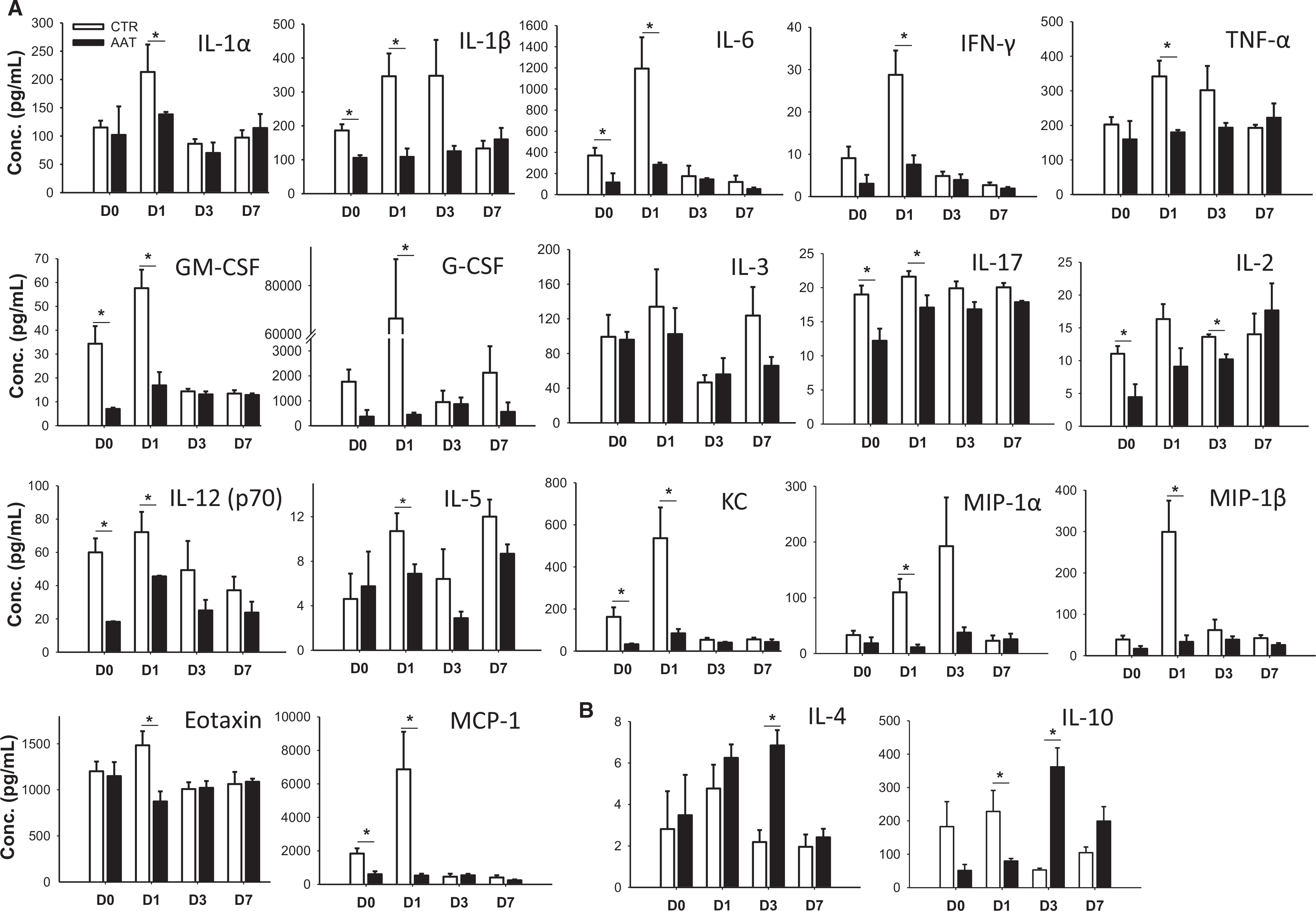
Cytokine levels in AAT or control recipients after transplantation. (A) Serum cytokines and (B) IL-4 and IL-10 levels from AAT or CTR mice at 0, 1, 3, and 7 days posttransplant measured by the Bio-Plex Pro Cytokine 23-Plex Assay kit. Data are shown as mean ± SEM, n = 3–4 in each time point; *P < .05 by Student's t test
3.3. AAT suppresses liver macrophage activation
Kupffer cells and infiltrating macrophages play central roles in the inflammatory responses within the liver after intrahepatic islet transplantation. Graft survival was improved when macrophage activation was inhibited.25 To determine whether AAT suppressed liver macrophage activation in our model, we assessed M1 and M2 macrophage phenotypes in relation to total F4/80+ liver macrophages at day 1 PT in the NOD-SCID recipients. Although AAT-treated recipients had similar total numbers of F4/80+ macrophages compared to controls (58.4 ± 3.1% vs. 60.0 ± 6.7%), they had significantly fewer activated M1 macrophages (F4/80+CD11c+CD206−, 20.4 ± 4.1% vs. 36.8 ± 2.7%; p = .02) and increased M2 macrophages (F4/80+CD11c−CD206+, 14.0 ± 0.6% vs. 3.8 ± 0.3%, p = .01) at 1-day PT (Figure 3A,B). The phenotypes of macrophages were confirmed by reduced mRNA expressions of M1 marker genes, including CD11c, iNOS, and TNF-α, and increased M2 marker genes, including CD206, IL-10, and Arginase 1 (Arg1), in the AAT group compared to controls (Figure 3C). These data suggest that treatment with AAT suppressed M1 and favored M2 macrophage polarization in the livers of NOD-SCID mice bearing human islet grafts.
FIGURE 3.
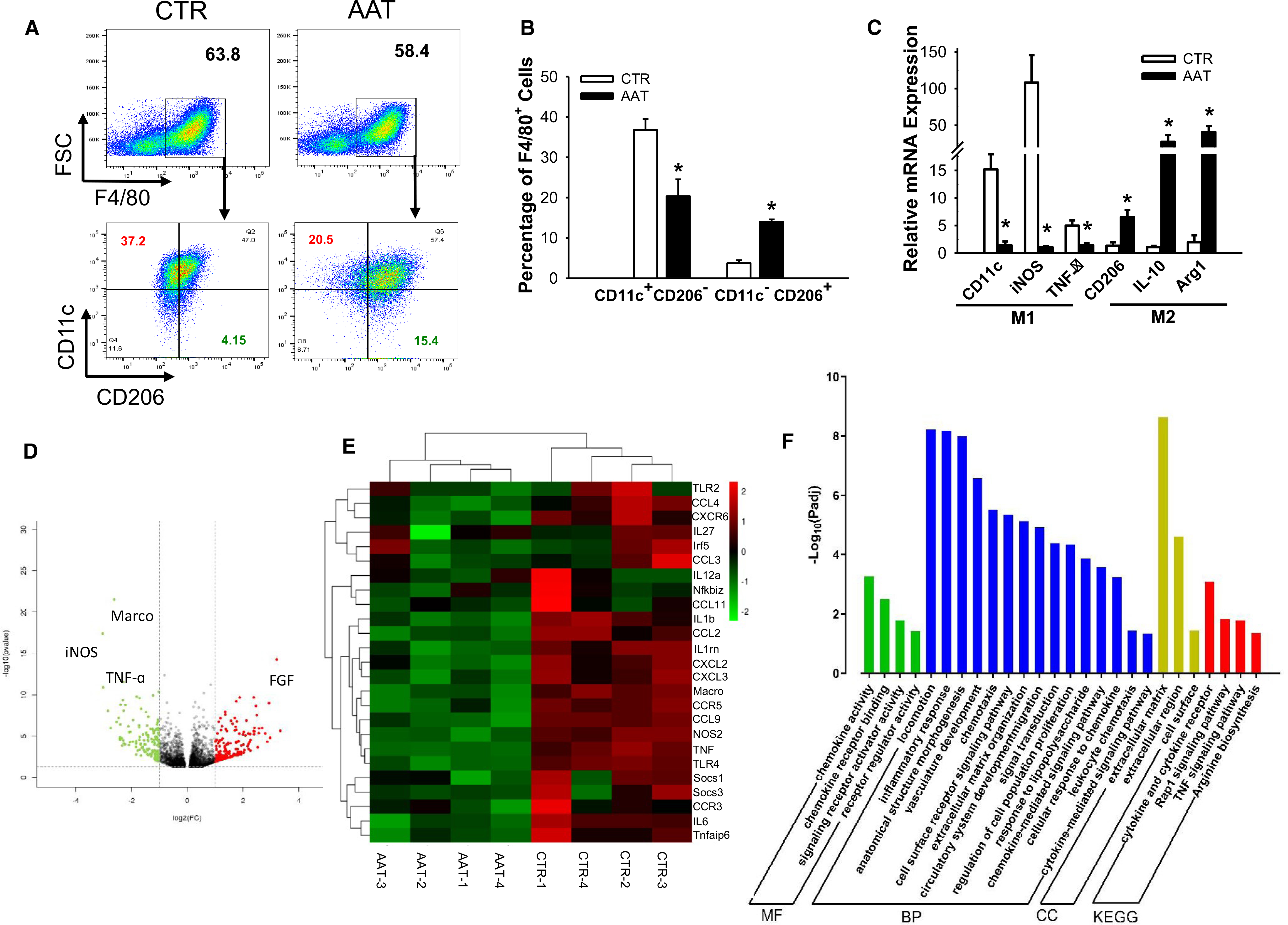
Liver macrophage transcription profiles from AAT-treated mice show reduced M1 percentages at 1-day PT. (A) Flow cytometric plots showing expression of F4/80 (upper panel), CD11c, and CD206 (lower panels) in liver macrophages harvested from AAT or control mice. (B) Quantification of M1 (F4/80+CD11c+CD206−) and M2 (F4/80+CD11c−CD206+) macrophages in livers. (C) Relative mRNA expression of the M1 (CD11c, iNOS, and TNF-α, left panel) and M2 (CD206, IL-10, and Arg1, right panel) marker genes measured by RT-PCR. Data are presented as mean ± SEM; n = 6 in each group; *P < .05 by Student's t test. (D) Volcano plot of differentially expressed genes between control (CTR) and AAT-treated groups (AAT). (E) Heatmap of consensus M1 macrophage-related genes. Each column represents data from an individual AAT-treated or control mouse and each row represents a gene. Gene expression is shown by pseudocolor scale (−2 to 2), with red denoting high expression level and green denoting low expression level of a gene. (F) Pathway and Gene Ontology analysis of genes downregulated in AAT-treated mice macrophages. MF, molecular function; BP, biological process; CC, cellular component; KEGG, Kyoto Encyclopedia of Genes and Genomes
Transcriptional profiles of liver macrophages from control and AAT-treated mice at 1-day PT showed that 344 genes were differentially expressed with a fold change (FC) >2 and padj < .05 in the AAT-treated group compared with the control group (Figure S1). The genes most attenuated by AAT included M1 macrophage-related genes iNOS, Marco, and TNF-α (Figure 3D). Analysis of a broader set of M1 makers also indicated that AAT-treated recipients had reduced M1-related cytokine and chemokine gene expression than the control group (Figure 3E). These data suggest that AAT suppressed the expression of M1 macrophage-related genes that might have contributed to enhanced islet survival in our islet transplantation model. The GO and pathway and interactive network enrichment were performed to assess the functional outcomes of AAT-altered genes in macrophages. Clusters of inflammatory response, chemokine activity, and cytokine and cytokine receptor GO pathways were significantly affected by AAT treatment (Figure 3F).
3.4. AAT suppresses IFN-γ-induced Raw264.7 macrophage activation by suppression of STAT1 phosphorylation in vitro
To decipher whether AAT has a direct effect on macrophage activation, we treated Raw264.7 cells with IFN-γ and measured M1 marker gene expression in the presence and absence of 0.5 mg/ mL of AAT by RT-PCR. Expression of M1 marker genes, including CD11c, TNF-α, IL-6, and iNOS, at the transcriptional level was measured at 1, 3, 6, 24, and 36 hours after treatment with 100 ng/ mL of IFN-γ. Control cells treated with vehicle or AAT alone did not lead to any change of expression of all four genes. IFN-γ stimulation leads to increased expression of CD11c, TNF-α, IL-6, and iNOS in a time-dependent manner. Cells treated with AAT showed significantly less gene expression at 3–36 hours posttreatment (Figure 4A–D). In contrast, there was no difference in expression of most M2 macrophage-related genes in Raw264.7 cells treated with IL-4 with or without AAT (Figure S2).
FIGURE 4.
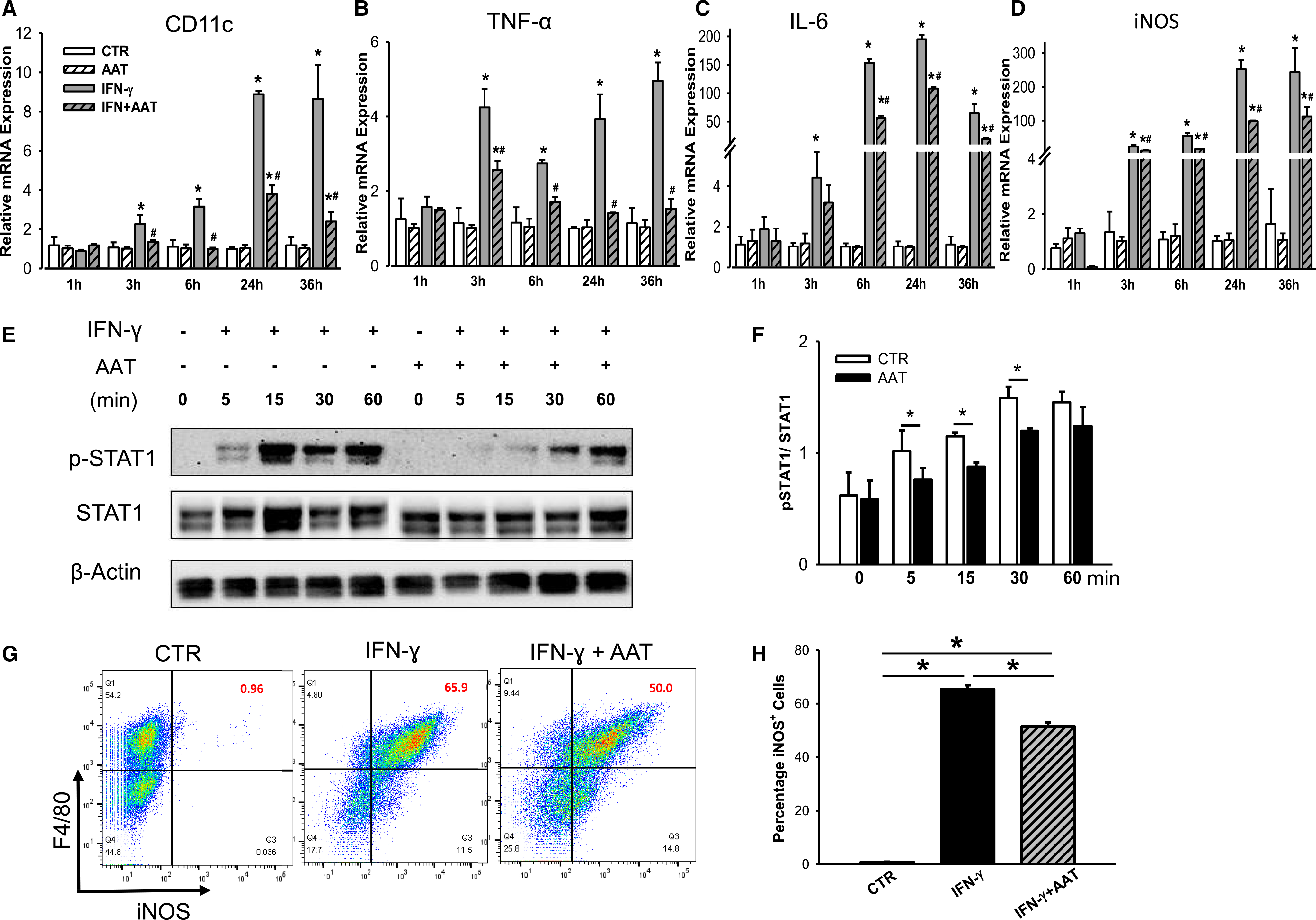
AAT suppresses IFN-γ-induced M1 macrophage activation by suppression of STAT1 phosphorylation in vitro. Expression of CD11c (A), TNF-α (B), IL-6 (C), and iNOS (D) in IFN-γ-treated Raw264.7 cells in the presence or absence of AAT at 1, 3, 6, 24, and 36 hoursafter treatment. CTR, nontreated Raw cell control; AAT, cells incubated with AAT (0.5 mg/mL) only; IFN, cells treated with IFN-γ (100 ng/mL) only; IFN+AAT, cells treated with IFN-γ in the presence of AAT. (A–D) Data shown are mean ± SEM from three to five individual experiments; differences were compared by one-way ANOVA test. *P < .05 vs. CTR; #P < .05 vs. IFN-γ treatment. (E) Protein expression of phosphorylation STAT1 (p-STAT1) and total STAT1 was measured in Raw264.7 cells stimulated with IFN-γ in the presence and absence of AAT by Western blot analysis. (F) Quantification of p-STAT1/STAT1. (G) Expression of M1 macrophage intracellular marker iNOS was measured by flow cytometry. (H) Quantification of iNOS+ cell population among F4/80+ macrophages. (E–H) Experiments were repeated three times; *P < .05 by Student's t test
To explore the molecular mechanism of AAT inhibition on IFN-γ-induced M1 activation, we measured the phosphorylation of STAT1, a key molecular mediator of M1 polarization,26 in Raw264.7 cells treated with IFN-γ in the presence or absence of AAT. In control cells, phosphorylation of STAT1 (Tyr701) was presented from 5 to 30 minutes after IFN-γ treatment. Addition of AAT significantly suppressed STAT1 phosphorylation (Figure 4E,F) and iNOS expression (Figure 4G,H). These results suggest that AAT inhibits M1 macrophage activation at least in part by suppressing IFN-γ-induced STAT1 phosphorylation and iNOS production.
3.5. AAT inhibits macrophage activation induced by cytokines or inflamed/dying islets and consequently leads to islet cell survival
The presence of activated macrophages is a characteristic feature that occurs in the destructive cascade of some islet cells after transplantation.27 This was confirmed in vitro when more islet cells underwent apoptosis when co-cultured with IFN-γ-activated Raw264.7 macrophages (Figure 5A). In contrast, there was significantly less islet cell death when islets were co-cultured with AAT pretreated, IFN-γ-treated Raw264.7 macrophages (Figure 5B,C). These data show that AAT suppresses macrophage activation and consequently reduced islet cell death.
FIGURE 5.

AAT protects islets cells from activated macrophage-induced apoptosis. (A) Human islet cell death after co-culture with IFN-γ-activated macrophages by TUNEL staining. Nuclei were stained with DAPI (blue), β-cells with the anti-insulin antibody (red), TUNEL+ cells are green. (B) Quantification of TUNEL+ cells within an islet; 15–20 islets were included in each group. (C) Islet cell death measured by the apoptosis ELISA kit. CTR, human islets; IFN, human islets co-cultured with IFN-γ-treated macrophages; IFN+AAT, human islets co-cultured with IFN-γ-treated macrophages in the presence of AAT. (D) Death of vehicle or cytokine-treated islets measured by cell apoptosis ELISA kit. (E) M1 marker gene expression in Raw264.7 cells measured at 24 hours after co-cultured with cytokine-treated islets. Vehicle, nontreated Raw264.7 cells; Cyto-Islet, cytokine (human IL-1β, TNF-α, and IFN-γ)-treated human islets; Cyto-Islet +AAT, cytokine-treated human islets in the presence of AAT. Data shown are mean ± SEM; *P < .05, CTR vs. Cyto-islets or Cyto-islets+AAT, and #P < .05 Cyto-islets vs. Cyto-islets+AAT, analyzed by one-way ANOVA
Meanwhile, inflamed/dying islets contribute to their demise releasing cytokines and chemokines that attract inflammatory macrophages to the liver and activate resident macrophages, which cause more islet death.28 To mimic the in vivo transplantation microenvironment, we treated human islets with cytokine cocktails, including IL-1β, IFN-γ, and TNF-α, and confirmed the presence of increased islet apoptosis (Figure 5D). We co-cultured dying or healthy islets with Raw264.7 cells for 24 hours in the presence and absence of AAT, and measured M1 marker gene expression in Raw264.7 cells. Co-culture with dying islets led to increased expression of CD11c, IL-6, iNOS, and TNF-α, and treatment with AAT suppressed the expression of those genes in macrophages (Figure 5E). These data show the AAT suppresses macrophage activation induced by inflamed/dying islets. Taken together, AAT suppresses macrophage activation induced by cytokines or dying islets, which subsequently leads to islet cell survival.
3.6. AAT suppresses M1 macrophage activation and contributes graft survival and function
To obtain direct evidence that macrophage activation plays an essential role in islet graft death and whether AAT can attenuate the detrimental effects of activated macrophages in vivo, we established a liver macrophage depletion diabetic mouse model by injecting diabetic NOD-SCID mice with lip-clod, and transplanted human islets with or without IFN-γ-treated Raw cells, and measured islet graft function (Figure 6A). Treatment with lip-clod effectively depleted F4/80 macrophages in mice livers as shown by liver tissue RT-PCR for F4/80 expression and immunohistochemistry staining (Figure 6B,C). At day 21 PT, all macrophage-depleted control recipients receiving human islets alone achieved normoglycemia (n = 6). In contrast, only 55.6% (5/9) of recipients receiving islets and IFN-γ-treated Raw cells achieved normoglycemia (n = 9, p = .001 vs. control, log-rank test), compared to 90% (9/10) of recipients receiving islets and AAT-treated IFN-γ Raw cells (p = .003 vs. IFN-γ group) (Figure 6D). Without macrophage depletion, about 35.7% (10/28) of diabetic NOD-SCID mice achieved normoglycemia at day 21. In addition, the AAT group showed significantly better graft function compared to the IFN-γ control group, as indicated by lower blood glucose levels and smaller glucose AUC during an IVGTT at 7 days PT (Figure 6E). Therefore, our data show that the presence of M1 macrophages in the liver contributed to the primary nonfunction of transplanted islets. Through suppression of macrophage activation, AAT protects transplanted islets from dysfunction.
FIGURE 6.
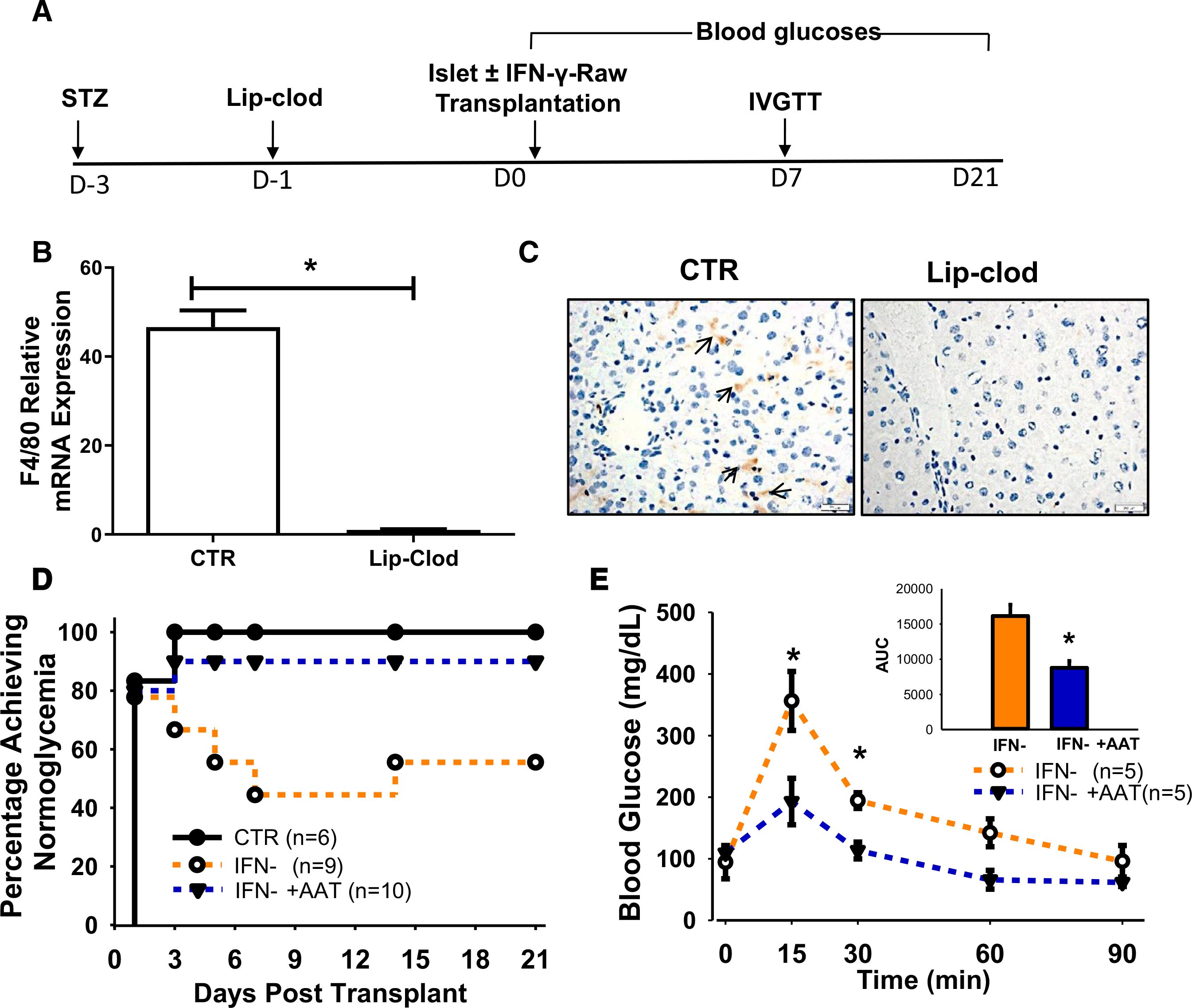
AAT suppresses macrophage activation and improves survival of transplanted human islets in macrophage-depleted diabetic NOD-SCID mice. (A) Study timeline of macrophage depletion and diabetes induction in the NOD-SCID mice. (B) Quantitation of F4/80 mRNA expression and immunostaining of F4/80+ macrophages in livers of control and lip-clod-treated mice at 24 hourspost lip-clod or PBS (CTR). (C) F4/80 staining in normal and macrophage-depleted livers. Arrows point to macrophages. (D) Percentages of recipients reaching normoglycemia after human islet transplantation in recipients receiving human islets alone (CTR), human islets with IFN-γ-treated Raw264.7 cells (IFN-γ), or human islets with IFN-γ-treated Raw264.7 cells in the presence of AAT (IFN-γ +AAT). (E) Blood glucose levels during the IVGTT performed at 7 days posttransplant. Inset, AUC of IVGTT. *P < .05, Student's t test
4. DISCUSSION
AAT therapy is a promising treatment that could improve islet graft survival after transplantation.8,10,12 AAT protects islet grafts via suppression of neutrophil infiltration and regulation of natural killer (NK) cell responses that contribute to islet graft acceptance.8,29–32 Whether AAT has a direct effect on macrophages, a major component of the innate immune response after human islet transplantation, is still unknown. In this study, we demonstrate for the first time, using a clinically relevant human islet transplantation model, that AAT protects islet graft survival at least in part via suppression of M1 macrophage polarization in both in vivo and in vitro systems. In vivo, we found that more mice receiving human islet grafts achieved normoglycemia when AAT was administered. Macrophages from AAT-treated recipients showed a distinct transcriptional profile with reduced M1 marker gene expression compared to cells from controls. In vitro, using Raw264.7 cells, we found that AAT suppressed IFN-γ-induced M1 macrophage polarization via inhibition of STAT1 phosphorylation. Importantly, by depletion of liver macrophages, we showed direct evidence that M1 macrophages in the liver contributed to primary nonfunction of transplanted islets.
Both macrophages and neutrophils contribute to inflammatory islet injury following islet transplantation into the liver.24,33,34 Multiple studies show that transient macrophage inhibition or inhibition of cytokine production prolonged islet allograft survival.25,35 For example, Kaufman and colleague showed that in a major mismatch islet transplantation model, primary nonfunction of transplanted islets was a result of a cell-mediated host-immune response dependent on macrophages or macrophage byproducts. Silica-induced macrophage depletion completely abolished primary nonfunction.36 In this study, we confirm those findings and introduce potential therapy to downregulate M1 macrophage polarization and cytokine-induced injury by treatment with AAT. Kupffer cells comprise 80% of the liver tissue macrophages in the human body. They contribute to injuries to other cells via secretion of free radicals and inflammatory cytokines, including IL-1, IL-6, TNF-α, IFN-γ, coagulation factors, complement, reactive oxygen, and nitrogen species.25,37 After islet transplantation, Kupffer cells can be activated by many factors including transplanted islets, injuries to the sinusoidal endothelium,25 the residual endotoxin derived from the islet isolation process,38 and dying acinar cells in the islet product.39 Furthermore, intraislet macrophages residing within the pancreatic islets, although low in numbers, may mediate islet injury.40 In addition, monocyte-derived inflammatory macrophages recruited by tissue injury can be activated and contribute to islet damage.41 However, whether the AAT effect was on Kupffer cells, intraislet macrophages, inflammatory macrophages, or all of these cannot be distinguished in this study.
Recent studies show that human AAT dose protocols and routes of administration impact the protective effects of AAT therapy for islet-related injuries.42 We found that infusion of 4 mg/mouse of AAT achieved better protection to human islet grafts compared to the lower dose of 2 mg/mouse. Our experience shows that the dose of human AAT needs to be optimized in different transplantation situations. For example, in the syngeneic islet transplantation model using C57BL/6 mice, treatment of recipients with seven doses of AAT at 2 mg/mouse every other day led to 60% of recipients reaching normoglycemia,12 while in this study when human islets were transplanted into NOD-SCID mice, a dose of 4 mg/mLwas needed to achieve similar protection. The requirement of different doses for different transplantation settings is not fully explained, although higher doses of AAT may be needed to suppress complement activation associated with human (xenoislet) transplantation. The body weight of each mouse averaged 25 g, and the approximate dose of AAT in our model was 160 mg/kg every 2 days. Studies have shown that AAT up to 240 mg/ kg (about 6 mg/mouse for a mouse of 25 g) was safe in animals and patients.29 Therefore, higher doses or different dosing schedules of AAT might be needed to achieve better protection in human islets.
It is worth noting that mice receiving AAT survived significantly longer with quicker recovery after surgery than vehicle-treated controls. Continuous AAT infusion in mice led to the death of mice in certain mouse species due to generation of antibodies;29 however, we found that AAT injection was safe in the C57BL/6 and the NOD-SCID mice. Although our studies were focused on the impact of AAT on macrophage function, we observed that AAT-treated mice showed lower levels of serum cytokines and chemokines from day 0 (about 2–4 hours after AAT injection and before islet transplantation). Treatment with STZ can induce cell apoptosis and cause multiple injuries to liver, cardiac, and adipose tissues.43,44 Since AAT has a protective effect on many tissues by downregulation of inflammatory factors,7,45,46 the better survival of transplanted islets and better ter survival of treated mice may not be entirely macrophage specific.
Therefore, we believe that AAT exerted its protective effects at least in part through suppression of macrophage activation. In a previous study, islet cells co-cultured with activated macrophages were lysed within 15 hours in vitro,47 which is consistent with our data that the co-culture of islets with IFN-γ-stimulated macrophages led to increased β-cell death in vitro.
In addition, this study focuses on early events following human islet transplantation into the immunodeficient recipients, since early islet cell death is a major issue hindering the success of islet transplantation.1 While it is not a focus of this study, it is important to assess the long-term protective effects of AAT in immunocompetent mice, since notable chronic decline in islet function has been observed in both autologous and allogeneic islet transplantations.48,49
5. CONCLUSIONS
We identified a novel mechanism that explains the protective effects of AAT on human islet graft survival after intrahepatic islet transplantation, which was mediated by suppression of macrophage activation/polarization. These data provide strong evidence that AAT treatment has the potential to be used to enhance human islet graft survival and function in clinical islet transplantation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Xian C Li, Peixiang Lan, and Don Rockey for sharing protocols for isolation of liver macrophages. We thank the Reeves Family for their generous donation. We thank Dr. Mateusz Tomczyk for sharing the protocol for macrophage depletion. We thank Ms. Jennifer Lee for critical language editing. This study was supported in part by the National Institutes of Health (DK105183, DK120394, and DK118529) and the Department of Veterans Affairs (VA-ORD BLR&D Merit I01BX004536). All procedures and protocols were approved by the Institutional Animal Care and Use Committee (IACUC, No 170117) at the Medical University of South Carolina.
Funding information
National Institutes of Health, Grant/ Award Number: DK105183, DK120394 and DK118529; Department of Veterans Affairs, Grant/Award Number: I01BX004536; Institutional Animal Care and Use Committee, Grant/Award Number: 170117; Medical University of South Carolina
Abbreviations:
- AAT
alpha-1 antitrypsin
- FC
fold change
- IFN-γ
interferon-gamma
- IL
interleukin
- iNOS
inducible nitric oxide
- IVGTT
intravenous glucose tolerance test
- M1
classically activated macrophages
- M2
alternatively activated macrophages
- PT
posttransplant
- TNF-α
tumor necrosis factor-alpha
Footnotes
DISCLOSURE
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. Dr. Strange has grants paid to the Medical University of South Carolina by Grifols and consults for Grifols, outside of the current project. The other authors have no conflict of interest to disclose.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study were included in this published article and its additional files.
REFERENCES
- 1.Biarnes M, Montolio M, Nacher V, Raurell M, Soler J, Montanya E. Beta-cell death and mass in syngeneically transplanted islets exposed to short- and long-term hyperglycemia. Diabetes. 2002;51(1):66–72. [DOI] [PubMed] [Google Scholar]
- 2.Alejandro R, Cutfield RG, Shienvold FL, et al. Natural history of intrahepatic canine islet cell autografts. J Clin Invest. 1986;78(5):1339–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breit SN, Wakefield D, Robinson JP, Luckhurst E, Clark P, Penny R. The role of alpha 1-antitrypsin deficiency in the pathogenesis of immune disorders. Clin Immunol Immunopathol. 1985;35(3):363–380. [DOI] [PubMed] [Google Scholar]
- 4.Sandstrom CS, Ohlsson B, Melander O, Westin U, Mahadeva R, Janciauskiene S. An association between Type 2 diabetes and alpha-antitrypsin deficiency. Diabet Med. 2008;25(11):1370–1373. [DOI] [PubMed] [Google Scholar]
- 5.Ehlers MR. Immune-modulating effects of alpha-1 antitrypsin. Biol Chem. 2014;395(10):1187–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger M, Liu M, Uknis ME, Koulmanda M. Alpha-1-antitrypsin in cell and organ transplantation. Am J Transplant. 2018;18(7):1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang B, Lu Y, Campbell-Thompson M, et al. Alpha1-antitrypsin protects beta-cells from apoptosis. Diabetes. 2007;56(5):1316–1323. [DOI] [PubMed] [Google Scholar]
- 8.Koulmanda M, Bhasin M, Hoffman L, et al. Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci USA. 2008;105(42):16242–16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis EC, Shapiro L, Bowers OJ, Dinarello CA. Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci USA. 2005;102(34):12153–12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis EC, Mizrahi M, Toledano M, et al. alpha1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci USA. 2008;105(42):16236–16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koulmanda M, Sampathkumar RS, Bhasin M, et al. Prevention of nonimmunologic loss of transplanted islets in monkeys. Am J Transplant. 2014;14(7):1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Sun Z, Gou W, et al. alpha-1 antitrypsin enhances islet engraftment by suppression of instant blood-mediated inflammatory reaction. Diabetes. 2017;66(4):970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu Q, Yang H, Shi Q. Macrophages and bone inflammation. J Orthop Translat. 2017;10:86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon S, Pluddemann A, Martinez EF. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262(1):36–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Gou W, Kim DS, Strange C, Wang H. Clathrin-mediated endocytosis of alpha-1 antitrypsin is essential for its protective function in islet cell survival. Theranostics. 2019;9(13):3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smedsrod B, Pertoft H. Preparation of pure hepatocytes and reticuloendothelial cells in high yield from a single rat liver by means of Percoll centrifugation and selective adherence. J Leukoc Biol. 1985;38(2):213–230. [DOI] [PubMed] [Google Scholar]
- 18.Zeng WQ, Zhang JQ, Li Y, Yang K, Chen YP, Liu ZJ. A new method to isolate and culture rat kupffer cells. PLoS One. 2013;8(8):e70832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weisser SB, van Rooijen N, Sly LM. Depletion and reconstitution of macrophages in mice. J Vis Exp. 2012;66:4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Rooijen N, Hendrikx E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol Biol. 2010;605:189–203. [DOI] [PubMed] [Google Scholar]
- 24.Barshes NR, Wyllie S, Goss JA. Inflammation-mediated dysfunction and apoptosis in pancreatic islet transplantation: implications for intrahepatic grafts. J Leukoc Biol. 2005;77(5):587–597. [DOI] [PubMed] [Google Scholar]
- 25.Bottino R, Fernandez LA, Ricordi C, et al. Transplantation of allogeneic islets of Langerhans in the rat liver: effects of macrophage depletion on graft survival and microenvironment activation. Diabetes. 1998;47(3):316–323. [DOI] [PubMed] [Google Scholar]
- 26.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang X, Moore DJ, Ketchum RJ, et al. Resolving the conundrum of islet transplantation by linking metabolic dysregulation, inflammation, and immune regulation. Endocr Rev. 2008;29(5):603–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sigrist S, Oberholzer J, Bohbot A, et al. Activation of human macrophages by allogeneic islets preparations: inhibition by AOP-RANTES and heparinoids. Immunology. 2004;111(4):416–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashkenazi E, Baranovski BM, Shahaf G, Lewis EC. Pancreatic islet xenograft survival in mice is extended by a combination of alpha-1-antitrypsin and single-dose anti-CD4/CD8 therapy. PLoS One. 2013;8(5):e63625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleixo-Lima G, Ventura H, Medini M, Bar L, Strauss P, Lewis EC. Mechanistic evidence in support of alpha1-antitrypsin as a therapeutic approach for type 1 diabetes. J Diabetes Sci Technol. 2014;8(6):1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koulmanda M, Bhasin M, Fan Z, et al. Alpha 1-antitrypsin reduces inflammation and enhances mouse pancreatic islet transplant survival. Proc Natl Acad Sci USA. 2012;109(38):15443–15448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonigk D, Al-Omari M, Maegel L, et al. Anti-inflammatory and immunomodulatory properties of alpha1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci USA. 2013;110(37):15007–15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guttman O, Yossef R, Freixo-Lima G, Rider P, Porgador A, Lewis EC. alpha1-Antitrypsin modifies general NK cell interactions with dendritic cells and specific interactions with islet beta-cells in favor of protection from autoimmune diabetes. Immunology. 2014;144(3):530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang K, Weaver JD, Li Y, Chen X, Liang J, Stabler CL. Local release of dexamethasone from macroporous scaffolds accelerates islet transplant engraftment by promotion of anti-inflammatory M2 macrophages. Biomaterials. 2017;114:71–81. [DOI] [PubMed] [Google Scholar]
- 35.Tran PO, Gleason CE, Robertson RP. Inhibition of interleukin-1beta-induced COX-2 and EP3 gene expression by sodium salicylate enhances pancreatic islet beta-cell function. Diabetes. 2002;51(6):1772–1778. [DOI] [PubMed] [Google Scholar]
- 36.Kaufman DB, Platt JL, Rabe FL, Dunn DL, Bach FH, Sutherland DE. Differential roles of Mac-1+ cells, and CD4+ and CD8+ T lymphocytes in primary nonfunction and classic rejection of islet allografts. J Exp Med. 1990;172(1):291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Decker K. Biologically active products of stimulated liver macrophages (Kupffer cells). Eur J Biochem. 1990;192(2):245–261. [DOI] [PubMed] [Google Scholar]
- 38.Vargas F, Vives-Pi M, Somoza N, et al. Endotoxin contamination may be responsible for the unexplained failure of human pancreatic islet transplantation. Transplantation. 1998;65(5):722–727. [DOI] [PubMed] [Google Scholar]
- 39.Gray DW, Sutton R, McShane P, Peters M, Morris PJ. Exocrine contamination impairs implantation of pancreatic islets transplanted beneath the kidney capsule. J Surg Res. 1988;45(5):432–442. [DOI] [PubMed] [Google Scholar]
- 40.Carrero JA, McCarthy DP, Ferris ST, et al. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc Natl Acad Sci USA. 2017;114(48):E1041 8–E10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Groot M, Schuurs TA, Keizer PP, Fekken S, Leuvenink HG, van Schilfgaarde R. Response of encapsulated rat pancreatic islets to hypoxia. Cell Transplant. 2003;12(8):867–875. [PubMed] [Google Scholar]
- 42.Baranovski BM, Ozeri E, Shahaf G, et al. Exploration of alpha1-antitrypsin treatment protocol for islet transplantation: dosing plan and route of administration. J Pharmacol Exp Ther. 2016;359(3):482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valentovic MA, Alejandro N, Betts Carpenter A, Brown PI, Ramos K. Streptozotocin (STZ) diabetes enhances benzo(alpha) pyrene induced renal injury in Sprague Dawley rats. Toxicol Lett. 2006;164(3):214–220. [DOI] [PubMed] [Google Scholar]
- 44.Eleazu CO, Eleazu KC, Chukwuma S, Essien UN. Review of the mechanism of cell death resulting from streptozotocin challenge in experimental animals, its practical use and potential risk to humans. J Diabetes Metab Disord. 2013;12(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jedicke N, Struever N, Aggrawal N, et al. alpha-1-antitrypsin inhibits acute liver failure in mice. Hepatology. 2014;59(6):2299–2308. [DOI] [PubMed] [Google Scholar]
- 46.Lee S, Lee Y, Hong K, et al. Effect of recombinant alpha1-antitrypsin Fc-fused (AAT-Fc)protein on the inhibition of inflammatory cytokine production and streptozotocin-induced diabetes. Mol Med. 2013;19:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burkart V, Kolb H. Protection of islet cells from inflammatory cell death in vitro. Clin Exp Immunol. 1993;93(2):273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Webb MA, Illouz SC, Pollard CA, et al. Islet auto transplantation following total pancreatectomy: a long-term assessment of graft function. Pancreas. 2008;37(3):282–287. [DOI] [PubMed] [Google Scholar]
- 49.Ryan EA, Paty BW, Senior PA, et al. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54(7):2060–2069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study were included in this published article and its additional files.