

Abstract
Tuberculosis (TB) is the most lethal bacterial infectious disease worldwide. It is notoriously difficult to treat, requiring a cocktail of antibiotics administered over many months. The dense, waxy outer membrane of the TB-causing agent, Mycobacterium tuberculosis (Mtb), acts as a formidable barrier against uptake of antibiotics. Subsequently, enzymes involved in maintaining the integrity of the Mtb cell wall are promising drug targets. Recently, we demonstrated that Mtb lacking malic enzyme (MEZ) has altered cell wall lipid composition and attenuated uptake by macrophages. These results suggest that MEZ contributes to lipid biosynthesis by providing reductants in the form of NAD(P)H. Here, we present the X-ray crystal structure of MEZ to 3.6 Å. We use biochemical assays to demonstrate MEZ is dimeric in solution and to evaluate the effects of pH and allosteric regulators on its kinetics and thermal stability. To assess the interactions between MEZ and its substrate malate and cofactors, Mn2+ and NAD(P)+, we ran a series of molecular dynamics (MD) simulations. First, the MD analysis corroborates our empirical observations that MEZ is unusually flexible, which persists even with the addition of substrate and cofactors. Second, the MD simulations reveal that dimeric MEZ subunits alternate between open and closed states, and that MEZ can stably bind its NAD(P)+ cofactor in multiple conformations, including an inactive, compact NAD+ form. Together the structure of MEZ and insights from its dynamics can be harnessed to inform the design of MEZ inhibitors that target Mtb and not human malic enzyme homologs.
Keywords: malic enzyme, lipid biosynthesis, anaplerotic node, structure, NAD(P)H
Graphical Abstract
This shows that the eukaryotic large-subunit malic enzymes (MEs) which have the GER-motif are tetrameric and the prokaryotic MEs with a non-GER motif are dimeric including Mtb. The distinguishing GER and non-GER motif forms the basis for a potential compact NAD+ adduct that may be used for structure-based drug design that would target Mtb and not human ME.
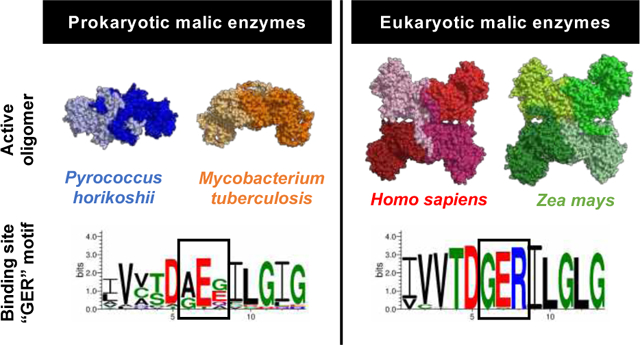
Tuberculosis (TB) is the leading cause of infectious death globally with 1.5 million TB-related deaths and 1.1 million new infections in 2018 alone1. This global pandemic is fueled by the emergence of drug-resistant strains of the causative agent Mycobacterium tuberculosis (Mtb) as well as a catastrophic synergy with HIV. TB treatment involves a cocktail of antibiotics which need to be taken for a minimum of 6 months2. Non-compliance is common and has contributed to the rapid increase of multi- and extensively-drug resistant Mtb strains. New, less toxic anti-TB therapeutics with shorter treatment regimens are urgently needed.
In a recent study we demonstrated that the enzymes at the anaplerotic node of central carbon metabolism are essential for Mtb intracellular survival in macrophages and therefore are potential therapeutic targets3. The anaplerotic node consists of enzymes that connect the pathways of glycolysis, gluconeogenesis and the tricarboxylic acid (TCA) cycle. These proteins regulate the flux distribution between anabolism, catabolism and energy supply to the cell, while replenishing the intermediates of the TCA cycle (Figure 1A). One of the enzymes in this node is malic enzyme (ME), which can reversibly convert malate and NAD(P)+ to pyruvate, CO2 and NAD(P)H (Figure 1B). Our previous study suggests that Mtb ME (MEZ) has a role in lipid biosynthesis (forward direction as a L-malate decarboxylase) and a minor back-up role in CO2-dependent anaplerosis (reverse reductive pyruvate carboxylation reaction)3, Figure 1. Although we showed that MEZ is not required for growth on gluconeogenic or glycolytic substrates, the MtbΔmez deletion strain had impaired macrophage invasion3. Additionally, the MtbΔmez colonies had a glossy and viscous morphology, which has been observed previously for Mtb mutants that lack cell-wall lipid transporters and a trehalose dimycolate esterase4–7, suggesting that the cell envelope lipid composition had been altered. Indeed, the MtbΔmez strain had a buildup of apolar free fatty acids and mycolic acids as compared to wildtype Mtb, which resulted in an accompanying decrease in the levels of cell-wall bound mycolates in total lipid extractions3. These results suggest that a major role of Mtb MEZ is as a L-malate decarboxylase (forward direction reaction), to produce NAD(P)H for lipid biosynthesis.
Figure 1. The PEP-pyruvate-oxaloacetate node or anaplerotic (ANA) node of metabolism in Mtb.

A The larger arrows point in the predicted physiological direction. Abbreviations: PCA, pyruvate carboxylase; MEZ, malic enzyme, PCK, PEP carboxykinase; PPDK, phosphate pyruvate dikinase, MDH, malate dehydrogenase. B The enzyme reaction of MEZ.
It has been shown that mez is part of the phoPR regulon, a two-component system that regulates genes essential for the biosynthesis of complex lipids, which supports the role of MEZ in lipid metabolism8. Specifically, the MtbΔphoP mutant, similar to MtbΔmez, results in a structurally altered cell envelope composition providing further evidence that MEZ supports NAD(P)H production for the synthesis of complex lipids. The connection between MEs and lipid biosynthesis has been observed in other species9–11. Studies of eukaryotic oleaginous microorganisms, with an emphasis on algae and yeast, demonstrate that MEs produce NAD(P)H for lipid biosynthesis including fatty-acid rich storage compounds such as triacylglycerol (TAG)10. In Streptomyces coelicolor, it has been shown that deletion of the NAD+- and NADP+-dependent ME expressing genes results in the decreased production of TAG9. Moreover, the overexpression of ME in Rhodococcus jostii during growth on glucose promoted an increased NADP+-dependent ME activity resulting in a 2-fold increase in fatty acid biosynthesis11. Together these studies support that MEZ plays a major role in the production of reducing agent, NAD(P)H, required for Mtb lipid biosynthesis and virulence. Therefore, it is compelling to consider MEZ as a potential candidate for structure-based anti-TB drug design.
The biological importance of MEs is reflected in its ubiquity amongst many species, including eukaryotes, prokaryotes and archaea. It follows that there is a large, diverse group of X-ray crystal structures of NADP+- and NAD+-dependent MEs in the Protein Data Bank (PDB) ranging from high eukaryotes to prokaryotes12–14. Characterized MEs typically adopt dimeric or tetrameric assemblies with subunits (i.e. monomers) ranging from 40–50 kDa for small subunit prokaryotic MEs to ~60 kDa for large subunit MEs found predominately in eukaryotes and some prokaryotes (Figure S1). To-date, there have been three distinct structural classes of MEs reported: small subunit, large subunit and chimeric (the subset of chimeric MEs will not be further discussed). The structures of eukaryotic large subunit MEs consist of a tetrameric assembly comprised of a “dimer of dimers”, as previously described15 (Figure S1B). Each monomer has four distinct domains (A-D), similar to Figure 2A. Within higher eukaryotes, the presence of a C-terminal extension in Domain D facilitates the tetramerization of two dimers14 (Figure S1B). By contrast, in plant MEs, dimer tetramerization is facilitated by an N-terminal extension in Domain A12. The other distinct class consists of prokaryotic small subunit MEs, which are dimeric and have a minimal Domain A and no Domain D. For these small subunit MEs assembly of the dimer is required for full formation of the active site, wherein each monomer subunit contributes a Lys and a Tyr to the active site, both of which are required for catalysis13. By comparison, large subunit ME proteins have fully formed active sites, independent of dimerization. Alignment of representative prokaryote and eukaryote large subunit ME sequences highlight the presence of an extended N- and/or C-terminal extension among the large subunit MEs that is unique to eukaryotes and is required for tetramer formation (Figures S1 and S2B). In all structural classes of MEs reported, Domains B and C have conserved motifs and a high degree of structural similarity, where Domain B is the divalent metal binding domain and Domain C is the canonical NAD(P)H-binding Rossman-like fold domain (Figure 2A). A large body of biochemical data exists for MEs, particularly for higher eukaryotic and plant MEs, where ME mechanism of action, catalytic residues and activators and inhibitors have been well studied12,15,16. This has been particularly well-reviewed for higher eukaryotes by Tong et al.14. In contrast, there is much less known about the biochemistry of large subunit prokaryotic MEs, apart from E. coli MEs17.
Figure 2. Structural Domains and Oligomeric State of MEZ.

A. Structural cartoon of the MEZ monomer with the domains colored. B. Structural cartoon of dimeric MEZ with domains colored as in A. C. Gel filtration chromatogram of MEZ to identify its oligomeric state. His-tagged MEZ was run on a S200 HiLoad 16/600 column at 0.5mL/min and compared with protein standards. The predominant MEZ peak (Peak 1) is the dimeric species (expected MW is 124 kDa), which runs at an approximate size of 120 kDa. SDS-PAGE analysis of peak B reveals a contaminant protein, and peak C is probably imidazole. Peak A is likely higher order MEZ aggregates. D. Residues located at the MEZ dimeric interface from each monomer are depicted in grey and pink sphere representation. The AEA151 motif is indicated in yellow spheres.
The X-ray crystal structure of MEZ is of paramount importance to understanding the enzyme’s biochemistry and assessing its suitability as an anti-TB drug target. In this study, we solved the structure of MEZ in its apo form and determined that MEZ, even though it is a member of the large subunit ME family, is dimeric. Previously, we demonstrated that MEZ utilizes NAD+ or NADP+ as its cofactor3. To shed light on the divalent metal, substrate and cofactor binding modes, we carried out molecular dynamics (MD) simulations on the docked MEZ dimer structure. These simulations highlighted several unique factors about MEZ; (1) the requirement of divalent metal for malate binding; (2) the overall inherent structural flexibility of MEZ compared to other ME structures; and (3) the most stable MEZ dimer appears to form a mixed open:closed conformation in the presence of Mn2+, malate and NAD(P)+. Finally, we highlight sequence, structural and cofactor binding mode differences between MEZ and human ME, which can be harnessed as a template for MEZ structure-based inhibitor design. Altogether, this work contributes significantly to the biochemical and structural understanding of MEZ and establishes the potential of this enzyme as an anti-TB drug target.
Results
Structure of Mtb malic enzyme (MEZ)
MEZ is a dimeric in crystalline and solution forms
The structure of MEZ was solved to 3.6 Å resolution in space group P21 (Table 1), with an Rwork/Rfree of 30.5/ 32.6 (Table 1). The MEZ X-ray crystal structure has four subunits in the asymmetric unit, where the two dimers have a similar assembly to other large subunit ME dimers (Figures S1B & 2B). Whilst the two dimers form a few interactions at their interface, this crystallographic MEZ tetrameric state does not resemble any previously observed large subunit eukaryote ME tetramer assembly and is unlikely to be biologically relevant, Figure S2C12,14. As MEZ is a large subunit ME, it was surprising that MEZ crystallized as a dimer. However, sequence analysis revealed that MEZ lacks the N- or C-terminal extension that facilitates tetramer formation in plant and higher eukaryotic MEs, respectively (Figures S1 & S2B). To confirm the oligomeric state of MEZ in solution, we carried out size exclusion chromatography (SEC) and the results confirm that MEZ is dimeric in solution at pH 7.0 (Figure 2C).
Table 1.
Data collection and refinement statistics for MEZ crystal structure
| MEZ | |
|---|---|
| Data collection | |
| Space group | P 21 |
| Cell dimensions | |
| a, b, c (Å) | 94.5, 143.2, 117.6 |
| α, β, γ (°) | 90, 109.8, 90 |
| Resolution (Å) | 46.0–3.6 (3.7–3.6)a |
| Rmergeb | 0.14 (0.78) |
| CC1/2c | 0.98 (0.49) |
| I / σI | 4.43 (1.06) |
| Completeness (%) | 99.7 (99.5) |
| Redundancy | 2.0 (2.0) |
| Refinement | |
| Resolution (Å) | 46.0–3.6 (3.7–3.6) |
| Total reflections | 68476 (6833) |
| Unique reflections | 34246 (3417) |
| Rwork / Rfreed | 30.5/32.6 |
| Ramachandran favored (%) | 90.0 |
| Ramachandran outliers (%) | 0.60 |
| No. atoms | |
| Protein | 13482 |
| Ligands | 30 |
| Water | 100 |
| B-factors (Å2) | |
| Protein | 74.2 |
| Ligands | 59.1 |
| Water | 37.1 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.84 |
| PDB-ID | 6URF |
Values within parentheses refer to the highest resolution shell.
Rmerge = ∑∑|Ihkl—Ihkl(j) |/∑Ihkl, where Ihkl(j) is observed intensity and Ihkl is the final average value of intensity.
CC1/2 = , where is the variance of the average intensities and is the average of the variances of the averaged (merged) intensities.
Rwork = ∑||Fobs| - |Fcalc||/∑|Fobs| and Rfree = ∑||Fobs| - |Fcalc||/∑|Fobs|, where all reflections belong to a test set of 5% data randomly selected in Phenix.
Structure of MEZ and a comparison with other large subunit ME structures
Like other large subunit MEs14,18, the MEZ monomer is composed of four domains (Figure 2A). Domain A (residues 1–113) is composed of five α-helices (α1-α5, Table S1 and Figure S2A), and predominately mediates the dimer interface (Figures 2B&D). Domain B (residues 114–265, and 455–525) has an internal β-sheet composed of 5 parallel β-strands (β1/β5/β2/β6/β7). The rest of domain B is helical: α6-α12 and α20-α22. Domain C is formed by residues 266–454 and bears the Rossmann-like fold of ME proteins. The MEZ Rossmann-like fold has a 5- or 6-stranded parallel β-sheet (β9/β8/β10/β11/β12 in subunits B-D and also β14* in subunit A) and following the strand β12 is a conserved β-hairpin composed of strands β13 and β14. The β-sheet is enclosed by seven α-helices: α13-α19. Lastly, domain D (residues 526–545) is the C-terminus tail of MEZ and is largely unstructured except for a final α-helix, α23 (Figures 2A and S2A&B)
The four subunits of MEZ in the asymmetric unit are similar. Dimer 1 is comprised of subunits A and B, while Dimer 2 is formed by subunits C and D. Dimer 1 and Dimer 2 superimpose with a root mean square deviation (rmsd) of 1.58 Å over all atoms. Moreover, the subunits also align closely, with rmsds ranging between 0.82 – 1.53 Å between subunits.
Although the four subunits are similar, there are some noteworthy differences. There is variability in secondary structure elements between individual subunits (Table S1). For example, α7 is only present in subunit B, α17 is present in every subunit except B, and the final α23 is only present in subunits A and C. Finally, subunit C has an additional helix before α1, which aligns to an α-helix in human ME (PDB: 1QR6)15. Moreover, there is also variability in β-strands between subunits. Large subunit ME proteins have a conserved β-hairpin (β3/β4) that is partially observed in MEZ; electron density is observed for this β-hairpin in subunit C, partially observed in subunits B and D, and strikingly, no electron density is observed for β3/β4 in subunit A. Additionally, for all the subunits, we observed mobility in β-strands within the Rossmann-like fold β-sheet, particularly mobile is β14*, which can extend and participate in a β-hairpin (β13/β14). Finally, in the NAD(P)H binding site of subunits A, B and C, several entire residues are not modeled (Table S1) due to an absence of observable electron density, and for all subunits, several sidechains were left unmodelled for similar reasons. Finally, there is a loop region that has been left partially unmodelled due to missing electron density between β9 and α17 in all subunits (Table S1). These differences between subunits underlie the overall flexibility that is observed in the MEZ structure.
The MEZ structure captured an apo-state, which is unsurprising considering the divalent metal, NAD(P)H and substrate-binding sites are unoccupied. Notably, compared to other ME structures in the PDB, MEZ appears to be the least ordered as (1) many secondary structure elements are random coils or have no observable electron density in the MEZ structure compared to others and (2) MEZ appears to be relatively flexible as considerable efforts were made to improve the diffraction quality of MEZ crystals to no avail.
Dimerization interface of MEZ
The dimer interface, similar to those seen in other large subunit ME proteins (Figure S1B), is facilitated predominately by hydrophobic interactions and some H-bonds between Domain A from each monomer (Figure 2B&D). The surface areas buried in Dimer 1 and Dimer 2 are 2453 and 2405 Å2, respectively, where the monomers have a surface area of 23730–24105 Å2. At the interface between the two dimers in the MEZ structure asymmetric unit there are few contacts and very little surface area buried, 377 and 396 Å2 for Subunits A & D and B & C (Figure S2C), respectively, suggesting that the tetramer in the asymmetric unit is a consequence of crystal packing rather than MEZ forming a stable tetramer in solution. As mentioned above, the oligomeric state of MEZ in solution was confirmed to be dimeric at pH 7.0 (Figure 2C).
MEZ kinetics data
MEZ kinetics show cooperativity with malate and NAD+
We previously showed that, although MEZ was able to catalyze the reverse reductive pyruvate carboxylation, this enzyme clearly has a strong preference for the forward gluconeogenic direction3, Figure 1B. To inform the structural studies, we performed a more detailed kinetic characterization of purified MEZ at two different pHs, Table 2. At pH 7.4 the initial rates of MEZ-catalyzed L-malate decarboxylation measured at various concentrations of malate demonstrated sigmoidal kinetics irrespective of the co-factor used, which implies positive cooperative malate binding and has been described for other MEs (Figure S3)19,20. Although the Hill coefficients were similar when either cofactor was used, the K0.5 (substrate concentration at which half maximal velocity/half-saturation is achieved) for malate (NAD+) was approximately 50% lower than the K0.5 for malate (NADP+). The cooperative effect of malate binding was enhanced at pH 6.6 in the presence of NAD+ but was abolished with NADP+ as the cofactor. Furthermore, due to enhanced malate cooperativity at low pH, we also observe an increase in catalytic efficiency of MEZ with NAD+ at pH 6.6. The results also suggest that binding of NAD+ occurs in a positive cooperative fashion whereas the binding of NADP+ follows Michaelis-Menten kinetics. Cooperativity would enable MEZ to produce step-like rate responses to changes in malate and NAD+ concentration, and thereby respond rapidly to changing cellular conditions. We confirmed that MEZ preferred NAD+ over NADP+, with a K0.5 value for NAD+ nearly 3 times lower than the Km for NADP+ at pH 7.4 (Table 2).
Table 2.
Kinetic constants for MEZ
| Substrate/cofactor (pH) | Km or K0.5 (mM) | Vmax (mmol min−1 μg −1) | Vmax/ Km or K0.5 (min−1μg−1) | Hill coefficient |
|---|---|---|---|---|
| Gluconeogenic reaction | ||||
| (L-malate decarboxylation) | ||||
| aL-malate with constant NAD+ (pH 6.6) | 3.9±0.2b | 63.2±0.2 | 16.1 | 1.6–2.2 |
| aL-malate with constant NADP+ (pH 6.6) | 16.6±1.3 | 44.0±1.0 | 2.7 | n/ad |
| aL-malate with constant NAD+ (pH 7.4) | 18.7±0.5b | 127.3±1.8 | 6.8 | 2.1–2.7 |
| aL-malate with constant NADP+ (pH 7.4) | 36.9±2.2b | 48.27±2.3 | 1.3 | 2.2–2.6 |
| cConstant L-malate with NAD+ (pH 6.6) | 0.5±0.4b | 16.±0.4 | 33.8 | 1.4–1.6 |
| cConstant L-malate with NADP+ (pH 6.6) | 0.8±0.1 | 58.7± 4.8 | 77.2 | n/a |
| cConstant L-malate with NAD+ (pH 7.4) | 0.5±0.03b | 16.8±0.5 | 33.7 | 1.4–1.6 |
| cConstant L-malate with NADP+ (pH 7.4) | 1.4±0.02 | 21.1±0.2 | 14.8 | n/a |
Kinetic values are given as averages ± SEM.
L-malate (0.25 – 100 mM) and NAD(P)+ is 1.6 mM
Kinetic for these reactions are sigmoidal and the reported values are K0.5 values
NAD(P)+ (0.05 – 1.6 mM) and [L-malate] is 100 mM
n/a is not applicable
MEZ kinetics show pH dependence and allosteric regulation
Previous studies have shown that several metabolic intermediates can act as allosteric regulators of ME21,22. When NAD+ is the MEZ cofactor, the addition of fumarate and succinate activated the forward malate decarboxylation reaction to 361% and 290% of the unmodified activity, respectively, and aspartate induced a smaller increase in activity, Figure 3A. Like E. coli ME, oxaloacetate inhibited MEZ activity to 61%17, Figure 3A. Finally, pyruvate, glutamate and acetate did not significantly modify the MEZ activity even at high concentrations (data not shown).
Figure 3. Biophysical and Biochemical Analyses of MEZ.

A. Regulatory properties of recombinant MEZ. MEZ forward activity using NAD+ as a cofactor was measured at pH 7.4 in the absence or presence of each effector whilst the malate concentration was maintained at 4 mM. Values are expressed as percentages of the activity measured in the control assay containing no effector. B. Thermal stability (Tm) of MEZ measured at various pHs with different small molecules (2mM) using DSF. All conditions included 2mM MnCl2. Experiments were performed at least in triplicate.
To evaluate the effects of pH and potential allosteric regulators on the oligomeric assembly of MEZ, we performed SEC analysis at pH 6.0 and pH 7.4 after incubation of MEZ with 100-fold molar excess of fumarate, succinate and aspartate. In all conditions studied, MEZ remained dimeric (data not shown) as observed for MEZ alone (Figure 2C).
We further examined the impact of ligand binding and pH on the thermal stability of MEZ. Using differential scanning fluorimetry (DSF), we determined the melting temperature (Tm) of MEZ in the presence of 10 mM MnCl2 with 200-fold molar excess of NAD(P)+, malate, or potential allosteric activators at various pHs (6.0, 6.5, 7.0 and 7.5). The addition of cofactors or substrate had minimal impact on the Tm of MEZ, with the greatest shift (~1.7°C) occurring at pH 7.5 between apo MEZ and MEZ in the presence of NAD+ (Figure 3B). While incubation with potential ME allosteric activators had little effect, there is a clear trend between MEZ thermal stability and pH. At low pH (6.0 & 6.5), the Tms are comparable, ranging from 51.0–52.5°C; at higher pH (7.0 & 7.5), the measured Tms are lower, ranging from 45.5–49°C. MEZ is stabilized in a more acidic environment and this increased stability coincides with elevated activity in the kinetic experiments (Table 2). Thus, MEZ appears to be partially regulated by pH: destabilized and less active at higher pH, and more stable and active at lower pH. Similarly, the activity of photosynthetic MEs are regulated by pH12,14.
MEZ MD simulations with divalent metal, malate and NAD(P)H
Divalent metal coordinating and active site residues are largely conserved across MEZ and other large subunit MEs
All MEs studied thus far, require binding of either Mn2+ or Mg2+ in the active site to reinforce the structural integrity of the enzyme and to aid in substrate, intermediate and product binding during catalysis23. The divalent metal binding site is highly conserved among both prokaryotic and eukaryotic MEs and comprises two Asp residues and one Glu residue: this conservation is also observed in MEZ and consists of Glu240, Asp241, and Asp264 (Figure S2B). Additional critical catalytic residues in other MEs studied are Lys169 and Tyr96 (numbering for MEZ)14. In eukaryotes, an Arg at position 165 in human mitochondrial ME (position 151 in MEZ) is also critical for catalysis, and its modification disrupts malate binding without disrupting cofactor binding14. While this Arg is highly conserved among large subunit eukaryotic species, it is less conserved among large subunit prokaryotes (Figure S2B). The substitution of Arg for Ala in this position for MEZ (Ala151) distinguishes it from all other structurally resolved large subunit MEs.
MEZ requires divalent metal for stable malate binding
We solved the X-ray crystal structure of MEZ in its apo conformation. We attempted to soak the apo-MEZ crystals with high concentrations and to co-crystallize MEZ with malate, MgCl2 or MnCl2, NAD(P)H and allosteric effectors. However, crystals showed no improvement in resolution and no observable electron density for any small molecules. To fully understand the structure of MEZ in the presence of its substrate, divalent metal and cofactor, we manually modelled these components into the apo MEZ structure referencing the small molecule binding modes observed in the human mitochondrial ME structure (PDB: 1PJ2). We used Mn2+ as the divalent metal as MEZ has a preference for Mn2+ compared to Mg2+ 3. After docking, we energy-minimized the structural complexes with Amber24 before performing MD simulations. Because metals often present challenges in parameterization of classical MD simulations, we initially simulated MEZ in the presence of malate and either NAD+ or NADP+ only. However, in the absence of Mn2+, we observed that malate binding is unstable in the active site (Figure S4) and dissociates within the first 10 ns of simulation.
After minimizing and equilibrating the structure of MEZ with Mn2+, malate and NAD(P)+ bound, we observe that within subunit A, the metal ion, malate and NAD(P)+ molecules, are not significantly displaced, and malate is stabilized by both the divalent metal and the nicotinamide group of NAD(P)+. However, within subunit B, we observed a greater degree of drift of the divalent metal and small molecules (Figure S5A). In the presence of NAD+, subunit A retains contacts along the entire length of the dinucleotide, whereas the contacts of the adenine base with MEZ are lost within subunit B (Figure S5B). The same is observed for NADP+, where the contacts of MEZ chain A with NADP+ form along the length of molecule, and in contrast, there are no contacts between chain B and the adenine base, Figure S5C. Even though the ribose 2’-phosphate forms no contacts with MEZ in chain B, NADP+ in chain B is only slightly displaced from NADP+ in chain A (Figure S5).
MEZ is conformationally flexible
To explore the NAD(P)+ binding modes, examine the potential cooperative dynamics between subunits within the dimer, and provide insight into the specific interactions that facilitate use of either NAD+ or NADP+ for catalysis, we ran MD simulations of MEZ with malate, NAD+ or NADP+, and Mn2+.
To evaluate and compare the overall flexibility of MEZ in its apo (open) and holo (closed) forms, we computed the root mean squared fluctuation (RMSF) of the αC atom for each residue and averaged the values over the five simulation repeats (Figure S6A). For domains A, B1 and B2 the RMSF values generally range between 1 and 3 Å and these regions are relatively stable compared to the rest of the protein chain. Domain D, and especially domain C, exhibit the greatest instability; residues in Domain D have RMSF values ranging from 1.5–3.5 Å while domain C has the broadest range from 1–7 Å for the apo MEZ simulation. When compared to RMSF values of other systems, domains C and D are particularly flexible; RMSF values computed from simulations of the highly compact and stable lysozyme are below 1 Å25, while dimers of Amyloid β(1–40), a highly mobile and flexible protein, range from 1–6 Å26. Notably, simulations of Zea mays ME showed only modest structural flexibility when compared to MEZ, with computed RMSF values from 1–2.7 Å over all four domains27. Interestingly, though binding might typically be expected to modestly stabilize fluctuations the addition of malate, Mn2+ and NAD(P)+ to the simulations had no impact on the overall stability of the MEZ protein backbone, apart from some reduction in computed αC RMSF values for residues between Gln355 and Ala430 (Figure S6B). This is consistent with our empirical observations that soaking or co-crystallizing substrates, cofactors and metals with MEZ failed to improve the diffraction quality of our crystals.
Fluctuations between open and closed active site states
Previous studies of MEZ homologs suggest that MEs can adopt open and closed states [13, 31]. In our simulations described above, we also observe expansion and contraction of the MEZ active site, which was tracked by recording the distance between αC atoms of Pro244 and Ala300 on two neighboring helices, α12 and α15, respectively (Figure 4). Here, we evaluated the relative stability of cofactor binding in neighboring subunits and whether there is an interdependence of open and closed states within the dimer.
Figure 4. Conformational Dynamics of MEZ Active Site.

A & B. Snapshots from MD simulations of MEZ with Mn2+, malate, and NAD+. A The distance between Pro244 and Ala300 is depicted with a dashed line and labeled in Å for the closed form and the open form. B. Superimposition of the open and closed form of NAD+-MEZ subunits from A. C. & E. The distance between the Pro244 and Ala300 are plotted for the five 100 ns simulation replicates. C. is MEZ with NAD+, malate, and Mn2+, and E. is MEZ with NADP+, malate, and Mn2+. Each simulation replicate represents an independent simulation for which the starting velocities have been randomized and are unique to each replicate. All subunits in all simulations are initiated from the closed form. The * denotes simulations where the NAD(P)+ cofactors and malate substrate remain stably bound in the active site of both subunits. D. & F. Represent the dimer with one monomer in the open conformation and the other in the closed form. An example of snapshots of the dimer with NAD(P)+ cofactors, malate and Mn2+ at the end of MD simulation 5 & 3 shown in panels C & E, respectively. In D, one monomer in the closed conformation (grey cartoon) and the other in the open conformation (pink cartoon) with malate (cyan) and NAP+ (yellow) in sticks and Mn2+ (pink, sphere), and the AEA151 motif is highlighted in yellow spheres. In F, one monomer in the closed conformation (grey cartoon) and the other in the open conformation (blue cartoon) with malate (cyan) and NAP+ (salmon) in sticks and Mn2+ (pink, sphere), and the AEA151 motif is highlighted in yellow spheres.
Among the five simulations of MEZ in the presence of Mn2+, malate and NAD(P)+, the subunits adopt both closed and open states (all 5 simulations are shown in Figures 4C&E), with subunits in some simulations remaining stably in one state or the other, but in other cases, multiple transitions occur. In simulations where the cofactor NADP+ remains stably bound with the nicotinamide group near the malate and Mn2+ (Simulations 1, 3, 5, Figures 4E&F), the subunits stably adopt opposite forms or in the case of Simulation 5 appear to rapidly fluctuate between the open and closed states. In the case of MEZ with Mn2+, malate and NAD+, the subunits also transition between open and closed states, Figures 4A–D. Further, for simulations where the NAD+ cofactor is stably bound in both subunits (Simulations 3, 5, Figure 4C), the subunits within the dimer adopt opposite open-closed forms.
From the MD simulations, we can draw general conclusions regarding the global dynamics of the protein in association with its substrate and cofactors. All simulations showed a fluctuation between the open and closed forms. Furthermore, in simulations with NAD(P)+, malate and Mn2+, where active sites from both subunits are occupied by cofactor, substrate and divalent cation, the data suggest that within the dimer, the closed ‘active’ form is only stabilized when the neighboring subunit stably adopts an open conformation, even though the two active sites within the dimer are not in close proximity to one another (Figures 4D&F). These results are further supported by the kinetic data above, which suggest that MEZ is allosterically regulated by the substrate malate, Table 2.
NAD(P)+ co-factor binding modes
To identify the residues that participate in cofactor binding and that specifically facilitate the binding of both NAD+ and NADP+, we selected the simulations with the most stable cofactor binding for both MEZ subunits and used the final simulation frame as the reference for analysis. To compare relative stability amongst simulations, we computed the average RMSF for each cofactor atom in both subunits, with the most stable conformations being the lowest average RMSF. For MEZ with Mn2+, malate and NAD+, we selected Simulation 3 (Figure 4C); while for MEZ with Mn2+, malate and NADP+, we selected Simulation 1 (Figure 4E).
MEZ has two NAD+ binding modes: an elongated and a collapsed form
With Mn2+, malate and NAD+, we observe two stable conformations of MEZ: one elongated and the other collapsed (Figure 5A). Indeed, multiple conformations of NAD(H) have been observed and described previously in crystallographic, solution NMR, and MD studies25,26,28,29. Specifically, the collapsed form is commonly observed for unbound NAD(H) in solution30, but has also been observed in holo structures, including in the exo-site of human mitochondrial ME (PDB ID:1PJ3, 1PJL)31,32. To identify additional examples of enzymes in complex with the collapsed form of NAD+, we retrieved all instances of NAD+ molecules deposited in the PDB (a total of 4,163 sets of coordinates) and computed Tanimoto scores with OpenEye Scientific’s Shape Toolkit using the collapsed form we observe in our MEZ simulations as the reference structure. The three highest scoring NAD+ molecules (with the most similar conformation to the collapsed NAD+ in MEZ) were observed in Apylsia California ADP ribosyl cyclase, which cyclizes NAD+ into cyclic-ADP-ribose (PDB ID: 3ZWM)33, as well as prokaryotic CBS domain proteins (PDB ID: 2RC3, 4FRY), whereby the domain regulates enzyme and transport activities in response to adenosyl group binding.
Figure 5. Putative NAD(P)+ Binding Modes in MEZ after MD simulations.

A. Bound cofactor conformations. Representative conformations of the NAD+ (yellow) and NADP+ (salmon) binding modes in simulations of MEZ with Mn2+ and malate. The conformations were selected by taking the final frame of the simulation trajectories where the cofactors were most stably bound. The elongated form of NAD+ represents an active conformation while the collapsed form of NAD+ occupies the binding site in an inactive form. For NADP+ simulations, the cofactor occupies a single semi-collapsed, active form with the nicotinamide oriented near the malate substrate. B. Putative NAD+ binding modes in dimeric MEZ. In simulations of MEZ (white) with NAD+ (yellow), Mn2+ (magenta), and malate (cyan), the NAD+ cofactor adopts two distinct binding modes. In Chain A, the NAD+ extends along the pocket in an elongated conformation. The nicotinamide interacts with malate while making polar contacts with many MEZ residues. In Chain B, the NAD+ cofactor is folded over in a collapsed conformation and occupies a much more limited space of the MEZ binding pocket, with the nicotinamide interacting with MEZ residues, Mn2+ and malate substrate, and thus this likely represents an inactive bound conformation. C. Putative NADP+ binding mode in MEZ for Chain A (closed) and Chain B (open). In simulations of MEZ with NADP+ (salmon), Mn2+, and malate, NADP+ makes several polar interactions with residues in the MEZ binding pocket and with malate. In both the open and closed forms, the NADP+ 2’-phosphate group is stabilized by hydrogen bonds to an Arg sidechain.
Here we suggest that the collapsed form of NAD+ in complex with MEZ represents an inactive, albeit stable conformation; the nicotinamide group is oriented away from the malate substrate and Mn2+ cation, and instead makes polar contacts with the backbone of Glu150 (Figure 5B, chain B). This form is also stabilized by polar contacts with malate, Asp264, Arg414, and the backbone of Leu153 and Asp241. As noted previously, human mitochondrial ME (PDB: 1PJ3) as well as all other structurally resolved large subunit MEs, have a conserved ‘GER165 (Glu163-Arg165)’ motif in the active site, corresponding to residues 149–151 in MEZ (Figure S2B). In MEZ, this motif is substituted by residues ‘AEA151’. Given that the collapsed form of NAD+ is proximal to Ala151, it is possible that the presence of an Arg residue at this position could interrupt or perturb the stability of the collapsed conformation in the MEZ active site (Figure 6). We speculate that this collapsed form of NAD+ may be unique among non-’GER’ containing large subunit MEs. The elongated conformation of NAD+ appears to adopt a catalytically relevant orientation (Figure 5B, chain A), with the nicotinamide group oriented toward the malate and Mn2+ cation. This active form is stabilized by several polar interactions with the malate substrate and sidechains of Arg343, Asp330, Thr383, Ser409, Arg414 as well as the backbone of Gly301 and Asn410 (Figure 5B), and an additional interaction of Arg331 sidechain with the aromatic ring of the adenine base.
Figure 6. A significant difference between MEZ and human ME.
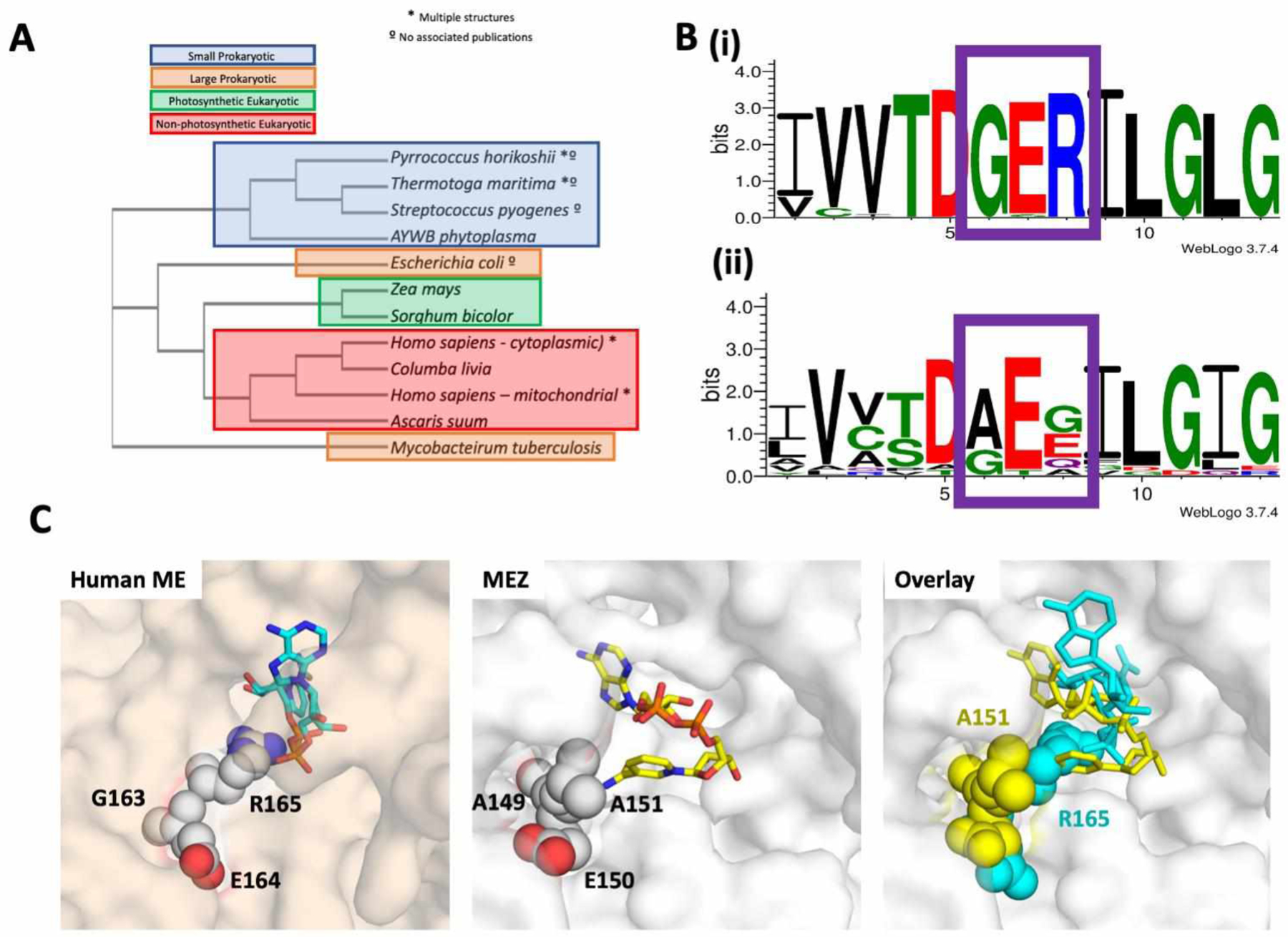
A. Phylogenetic tree of ME structures including MEZ. There are over 20 structures of malic enzymes (ME) in the Protein Data Bank, representing 12 isoforms from 11 distinct species. A sequence-based phylogenetic tree of these MEs was generated using PDB IDs: 1WW8, 1VL6, 2HAE, 2A9F, 5CEE, 6AGS, 5OU5, 6C7N, 3WJA, 1GQ2, 1PJ2, 1O0S, 6URF using Clustal Omega58. MEZ represents a distinct fork in the structural phylogenetic tree for large subunit MEs. B. Alignments comparing the amino acids around the conserved GER-motif (boxed in purple) in eukaryotic MEs and the equivalent region in Gram-positive MEs reveals that, (i) while the eukaryotic MEs GER-motif is highly conserved, (ii) Mtb and other Gram-positive organisms lack the GER-motif and instead possess a (A/G)E(E/G/Q/A) motif. Alignments were generated in WebLogo 3.7.459. Organisms included in this analysis are listed in the supplemental material. C. Surface representations of the human ME (PDB 1PJ2, wheat) and MEZ (white). Right panel: Human ME active site showing the GER165 (spheres) motif interacting with NAD+ (cyan, stick). Middle panel: MEZ active site showing the AEA151 motif (spheres) interacting with the compact form of NAD+ (yellow, stick). Left panels: Overlay of the human ME and MEZ structures with the human ME GER165 motif and NAD+ in yellow and the MEZ AEA151 motif and NAD+ in cyan.
MEZ has one NADP+ binding mode: a semi-collapsed form
By comparison, in simulations of MEZ with Mn2+, malate and NADP+, the cofactor adopts a semi-collapsed form (Figure 5A) which appears to be well oriented for catalysis with the nicotinamide group located near the malate substrate and Mn2+ cation (Figure 5C). NADP+ makes several polar contacts with malate as well as sidechain and backbone atoms of the surrounding residues. The ligand is stabilized by interactions with Arg248, Thr268, Thr299, Ala300, Arg331, and Ser382. Notably in both the open and closed forms, the 2’-phosphate group on the ribose of NADP+ (absent in NAD+) is stabilized by a hydrogen bond to an Arg sidechain (Figure 5C). Tanimoto analysis was performed using the semi-collapsed structure of NADP+ as the reference structure with all structures of NADP+ in the PDB (3164 sets of coordinates). Interestingly, the three top-scoring NADP+ molecules are found in structures of Mtb nicotinamide-mononucleotide adenylyltransferase (NadD) (PDB ID: 4S1O) and Mycobacterium abscessus NadD (PDB ID: 4YMI & 5VIR). These results suggest that the semi-collapsed NADP+ form observed in MEZ MD simulations is biologically relevant.
Discussion
Many bacterial species have multiple copies of genes encoding for MEs16,17,34, which underscores this enzyme’s physiological importance in central carbon metabolism as well as in the provision of NAD(P)H for anabolic processes such as fatty acid biosynthesis. Several eukaryotes also possess multiple ME isoforms, which are often distinct from each other in their cofactor preference. For example, humans possess three MEs; NADP-dependent ME is found in the cytosol while the other two are localized to the mitochondria14. By contrast, Mtb and other mycobacteria are reliant on only one ME and as such require flexible mechanisms of regulation in order to satisfy metabolic requirements.
Unlike other characterized large subunit MEs, MEZ forms a stable dimer
In general, large subunit eukaryotic MEs are observed as either a stable tetramer or in a dimer-tetramer equilibrium, wherein substrate binding or allosteric activators drive the formation of the higher-order tetrameric state12,35,36. Structural stabilization of these tetramers is facilitated by the presence of a C- or N-terminal extension12,14, where mutations in the C-terminal extensions of pigeon ME and human cytosolic ME disrupt formation of the tetramer assembly37,38. In photosynthetic eukaryotes, the N-terminal extension is long (50–60 residues) when compared to the moderately extended C-termini of non-photosynthetic eukaryotes (20–30 residues), Figure S2B. Structurally, both these N- and C-termini extensions of one dimer interact with the adjacent dimer stabilizing the tetramer interface (Figure S1). In non-photosynthetic eukaryotes, the C-terminus extends up and away from the dimer along the middle of the solvent facing region of the adjacent dimer, while in photosynthetic eukaryotes, the N-terminus forms a β-strand that interacts with the neighboring dimers’ N-terminus to form a pair of antiparallel β-sheets at the vertex of the tetramer interface (Figure S1B). The only large subunit prokaryote ME in the PDB and the closest structural homolog to MEZ, E. coli ME, is also dimeric in assembly (PDB: 6AGS). The E. coli ME dimer superimposes well with the MEZ dimer (rmsd 2.20 Å over all atoms) and has a similar dimer interface (Figure 2D). Compared to MEZ, E. coli ME has modest 17-residue and 7-residue extensions at the N- and C-termini, respectively (Figure S2B). Notably, in the E. coli ME dimer, the first and last β-strand from alternate monomers form two 2-stranded β-sheets, in contrast to the MEZ structure, where the N- and C-termini of all monomers are random coils apart from a C-terminal single-turn helix (Figure 2A). Both prokaryotic large subunit MEs, E. coli ME and MEZ, crystallized as a dimer; however SEC and biochemical data suggest E. coli ME assembles as a tetramer17.
MEZ may be partially regulated by pH
The regulation of MEs by pH has been studied in other organisms. In eukaryotic species, it has been observed that acidic pH drives the dissociation of the tetramer/dimer equilibrium toward dimeric and monomeric species. Disruption of tetramer formation has been linked to substrate inhibition of ME by malate at a pH of less than 7.022,39,40. Photosynthetic C4-ME is inhibited by malate in a pH dependent manner when the stromal pH drops to 7.0–7.5 from pH 8.012. The molecular basis of this inhibition was shown to be related to the destabilization of the higher order oligomeric state12. In our DSF studies, we observe that MEZ is stabilized at lower pH (Figure 3B); however, by SEC the oligomeric state persists as a dimer at both pH 7.5 (Figure 2C) and pH 6.0 (data not shown). This may have biological relevance as mez is part of the phoPR regulon, which is upregulated at low pH8. In support of this, we show biochemically that the catalytic efficiency of MEZ improves at lower pH (Table 2).
MEZ is allosterically regulated by fumarate and succinate
Our biochemical analysis of MEZ suggests that like many eukaryotic MEs, MEZ operates with cooperative kinetics with respect to malate18,19. In the case of human mitochondrial and Ascaris suum MEs, fumarate acts as an allosteric activator and supplants the cooperative kinetic effects of malate binding21,41. For human mitochondrial ME, the fumarate binds at two symmetric allosteric sites at the dimer interface; these two sites are coordinated by Arg67 and Arg91 residues from each subunit. While our MEZ structure has no ligands bound at these sites, the structurally corresponding residues, Arg51 and Arg75 are organized in a similar configuration at the dimer interface, suggesting that binding of ligands at the two symmetric allosteric sites may also disrupt cooperative binding of malate in MEZ. Further, our kinetic data show that decarboxylating activity is enhanced in the presence of fumarate and succinate, and inhibited by oxaloacetate (Figure 3A). Finally, MEZ activity is enhanced by fumarate more than succinate, which likely stems from higher energetic favorability in binding the more rigid fumarate due to reduced entropic costs or from the fact that fumarate is more easily deionized at low pH in comparison to succinate.
Most of the characterized MEs are allosterically controlled. Indeed studies of the homologous E. coli ME demonstrated allosteric upregulation by aspartate and downregulation by oxaloacetate17. Metabolically it is advantageous that fumarate and succinate, both products of the TCA cycle immediately upstream of malate, regulate MEZ (Figure 1A). Similarly, oxaloacetate inhibits MEZ to prevent a futile cycle occurring between pyruvate and oxaloacetate17.
Our kinetic data indicated that MEZ activity is cooperative with respect to NAD+ but not NADP+. MD simulations results for dimeric MEZ with NAD(P)+, malate and Mn2+ also suggest that the closed ‘active’ form is most stable when the neighboring subunit adopts its open conformation. Furthermore, with the cofactor NADP+ present, the most stable open-closed dimeric form is when NADP+ adopts a semi-collapsed, catalytically active form in both subunits (Figure 5C). As binding modes are similar for both subunits, MEZ activity is unlikely to be cooperative for NADP+. In contrast, in the presence of NAD+, the most stable open-closed dimeric form exists when NAD+ adopts two conformational forms, a catalytically active elongated form in one subunit and an inactive collapsed form in the other (Figure 5B). These results are further supported by the kinetic data, which shows that MEZ activity is cooperative with respect to NAD+ and not NADP+ as a cofactor.
The plasticity of MEZ: its biological relevance and druggability
Generally speaking, it has been observed that structural protein disorder is associated with either 1) increased promiscuity and/or 2) a disorder-to-order transition upon protein-ligand interactions, or 3) a disorder-to-order transition upon protein-protein interactions34. Given that we do not observe a decrease in disorder with our simulations nor an increase in MEZ thermal stability in the presence of cofactors and substrate (Figure 3B), there is little evidence to support a disorder-to-order transition upon ligand binding. However, we do not know whether MEZ forms protein-protein interactions that facilitate a transition to a more ordered state. A recent study demonstrated that cytosolic human ME1 activates and forms a stable hetero-complex with 6-phosphogluconate dehydrogenase (6GPD), which notably also generates NADPH to potentially maintain redox homeostasis42. Mtb has an analogous coenzyme, F420-dependent 6-phosphogluconate dehydrogenase (FGD1), which generates reduced coenzyme F420. While FGD1 is structurally distinct from human 6GPD43, MEZ may interact with FGD1 or have other protein-interaction partners. Ultimately, we posit that the structural plasticity of MEZ facilitates cofactor or substrate promiscuity and allosteric regulation. These features allow MEZ to generate reductants for lipid biosynthesis and the maintenance of redox homeostasis rapidly in response to metabolic and environmental stressors, which Mtb experiences in the human host.
Due to its role in lipid biosynthesis3, MEZ presents a potential anti-TB drug target. Similarly, human MEs have also been identified as potential targets for developing anti-cancer therapeutics44–46. Notably, a phylogenetic tree of all available large subunit ME structures in the PDB demonstrates that MEZ is evolutionarily distinct from other MEs (Figure 6A). And importantly, there are distinct structural differences between MEZ and the human ME homologs. In our MD simulations, we observe that the NAD+ cofactor can adopt multiple stable binding modes (Figure 5A). For the compact NAD+ MEZ-bound form, the ribose group near the NAD+ adenine motif forms polar contacts with the Mn2+ ion and malate substrate, but the nicotinamide group is oriented away from the active site and thus, is not suitably positioned for catalysis (Figure 5B, chain B). In order to accommodate this orientation, MEZ adopts a conformation wherein the β-hairpin turn (β3/β4) of residues 148–151 containing part of the AEA151 motif is displaced from the binding pocket as its backbone makes interactions with the compact NAD+ molecule (Figure 5B, chain B and Figure 6C, middle). Notably, in other ME structures such as human mitochondrial ME, rather than the AEA151 motif there is GER165, and residues at the apex of this β-hairpin form a helix that is flanked by the Arg165 residue that anchors this turn to the ME active site by forming polar contacts with malate and NAD+ (PDB ID: 1PJ2) (Figure 6C, left). In MEZ, Arg165 is substituted for an alanine (Ala151), and as the β-hairpin no longer possesses a positively charged sidechain to coordinate the malate substrate, the β-hairpin has greater flexibility. Indeed, this flexibility is observed in the apo MEZ structure as the residues that form the β-hairpin are random coil in Chain A, but an ordered β-hairpin in chains B, C & D (Table S1).
We suspect that the stable binding of NAD+ in a compact form is unique to MEZ and other prokaryotic MEs that lack the conserved GER165 motif (Figure 6B & S2B). Interestingly, this compact NAD+ conformation is similar to cyclic-ADP-ribose33. As MEZ can accommodate the compact form due to the structural differences with human MEs (Figure 6C, right), an inactive cyclized NAD derivative may therefore be an excellent starting point to design Mtb specific inhibitors of MEZ. The role of MEZ in the Mtb life cycle and its unique structural qualities presents an appealing opportunity for the design of selective inhibitors, and warrants further exploration.
Methods
MEZ purification
MEZ was purified as described3. Briefly a pNIC28-BSA4 plasmid containing the MEZ gene with an N-terminal His-tag was transformed into BL21(DE3) cells and grown in LB media containing 50 μg/ml kanamycin at 37°C. Once the cells reached 0.6–0.8 OD, expression was induced with 1mM IPTG and the temperature was reduced to 18 °C for approximately 18 hours. Cells were harvested by centrifugation for 30 minutes at 5000 rpm. Pellets were washed with STE buffer (10 mM Tris-HCl, pH8, 100 mM NaCl, 1 mM EDTA) and stored at −20°C. Cell pellets were resuspended in Buffer A (50 mM Tris pH 7.6, 500 mM NaCl, 50 mM Imidazole, 0.14 mM BME (β-mercaptoethanol), 5mM MgCl2, 4% glycerol) containing lysozyme and 1 mM PMSF (phenylmethylsulfonyl fluoride). Following sonication and centrifugation to remove debris, the cell lysate was loaded onto a Ni2+-bound His-Trap column and washed with several column volumes of Buffer A, followed by a high salt wash (three column volumes of Buffer A with 2M NaCl). The MEZ was eluted with a gradient of Buffer A and Buffer A containing 500mM Imidazole. MEZ containing fractions were identified via SDS-PAGE analysis and collected and pooled. For DSF and size exclusion chromatography, MEZ was dialyzed into Buffer A overnight at 4°C.
MEZ crystallization
Prior to crystallization studies, MEZ was concentrated and dialyzed (Amicon, 30 kDa MWCO) into 50mM trisodium Citrate-citric acid, 100mM NaCl, pH 6.0, per the results of a pre-crystallization DSF buffer screen performed using the Meltdown program47, wherein MEZ exhibited a trend of greater thermal stability at lower pH (pHs 5.5–10 were tested). Notably, prior attempts at crystallization (before identification of an optimal protein buffer and pH), yielded few diffracting crystals and none were reproducible. Crystallization conditions were identified via a collection of Hampton Research sparse matrix crystal screens. Crystal hits were optimized to produce diffraction quality crystals. Notably, protein concentration and drop sizes were all varied. Cryo-conditions crystal seeding, in-situ proteolysis, dehydration, additive screens and the addition of small organic compounds including alanine, glycine, tartronic acid, malate and cofactors NADH and NADPH (2–10mM) via soaking and co-crystallization were all explored. Of the more than 2000 crystals that were harvested, most diffracted no better than 8.0 Å and just two crystals diffracted to 4.0 Å or better.
The crystal condition that gave the highest resolution diffracting crystal was 0.2M NaCl, 0.1M HEPES, pH 7.5, 2mM alanine, 12% PEG 8000 and the crystal was cryo-frozen in 20% glycerol. The diffraction data set was collected at 70K to a resolution of 3.6 Å at ALS and was indexed and integrated in XDS48, and scaled in Aimless49. A solution was generated via molecular replacement using Phaser50, utilizing a dimer search model generated by iTasser51 from the structure of human ME (PDB ID: 2AW5, chains B&C). The model was refined in Coot and phenix.refine50,52.
MEZ Enzyme Assays
MEZ enzyme activity was assayed as described3. Briefly, MEZ enzyme activity was measured at 30 °C by spectrophotometrically measuring the NAD(P)H formation at 340 nm. The reaction mixture contained: 100 mM Tris, 100 mM malate, 0.5 mM MnCl2, 1.6 mM NAD(P)+ at pH 7.4 or pH 6.6. The kinetic constants were determined by varying the concentration of malate (0.25 – 100 mM) or NAD(P)+ (0.05–1.6 mM) while leaving other components at saturation concentrations. The kinetic constants are the means of three-six measurement and were calculated by fitting the initial rates to the Michaelis-Menten Equation (V = Vmax*[S]/(Km + [S]) and allosteric sigmoidal enzyme kinetics (V = Vmax*[S]h/(K0.5h + [S]h), where h is the Hill coefficient by non-linear regression using GraphPad Prism 7.04. When different compounds were tested as potential inhibitors or activators of enzyme activity, MEZ forward activities (using NAD+ as a cofactor) were measured at pH 7.4 in the absence or presence of 0.5, 1.0, or 2.0 mM of each effector (oxaloacetate, succinate, fumarate, pyruvate, aspartate, glutamate or acetate) whilst the malate concentration was maintained at 4 mM, approximately one-fifth of the K0.5, malate value. The results are presented as the percentages of activity in the presence of the effectors in relation to the activity measured in the absence of the metabolites (100%).
Protein Thermal Stability Measurements
Protein thermal stability was determined using differential scanning fluorimetry (DSF). For DSF measurements, proteins were incubated with 25 or 40 μM SYPRO orange dye in 20 mM sodium phosphate (pH 7.4), 150 mM NaCl. Samples were heated from 25 °C to 96 °C at 1 °C/min using an Mx3005P qPCR machine (Agilent Technologies). The dye was excited at 492 nm, and fluorescence emission was monitored at 610 nm. Melting curves were obtained in duplicate, and each experiment was conducted independently three times. Melting temperatures (Tms) were determined using nonlinear regression to determine melting-curve inflection points.
Size exclusion chromatography
Size exclusion chromatography for MEZ was run on a S200 HiLoad 16/600 column at 0.5mL/min with protein standards from Bio-Rad to determine the oligomeric state of MEZ in solution. MEZ was run in its apo form, or with the addition of 100 molar excess of fumarate, succinate and alanine, with 2mM of the small molecule in the running buffer.
Molecular Dynamics Simulations
To prepare MEZ for simulations, a dimer of the most complete chain B was generated by aligning a duplicate copy of chain B with its partner, chain A, in PyMol (DeLano Scientific, CA) and submitting the B/B dimer to pdbfixer53 to model in missing atoms, residues and loops using default protonation states at pH 7.0. Malate, NAD+ and NADP+ were manually docked in PyMol so as to mimic binding modes observed in MEZ’s closest structural homolog by sequence, human mitochondrial ME (PDB ID: 1PJ2).
Cofactors NAD+ and NADP+ were prepared using previously optimized Amber parameters54. The Mn2+ metal site parameters were built using Amber’s MCPB.py program55. Malate and Mn2+ were prepared using antechamber from AmberTools1824 with GAFF version 1.7 and AM1-BCC charges. In the MCPB.py input file, the terminal oxygens of residues Asp241 and Asp264 were listed as having bonded pairs with the Mn2+ ion and the malate ligand was identified as a non-amino acid included in the metal complex. Geometry optimization and force constant calculations were performed with input files generated by MCPB.py using Gaussian09. Output parameters were evaluated with parmed56 and satisfied the criteria outlined in the MCPB.py tutorial.
Following this, each system was solvated using tleap from AmberTools1824 with a 10 Å rectangular box of TIP3P water, extending from the surface of the protein to the box edge with sodium ions added to neutralize the charge of the system. Minimization proceeded using sander from Amber14 with steepest descents running for 20000 steps, followed by heating from 100 to 300 K with a constant volume for 25000 time steps of 2 fs. Equilibration continued using sander for 500000 time steps of 2 fs under constant pressure with positional restraints initially applied on all non-water atoms at 50 kcal/mol/Å2 and progressively increased in increments of 5 kcal/mol/Å2 over ten 50000-step segments. The resulting topology and coordinate files for each system were used as inputs for further MD simulations. Production simulations were executed in OpenMM 7.1.057 using a Langevin integrator with a 2 fs time step and a friction coefficient of 10 ps‒1. For each of four systems (MEZ only, MEZ/malate/NAD+, MEZ/Mn2+/malate/NAD+, and MEZ/Mn2+/malate/NADP+), five 100 ns simulations were initiated with random starting velocities.
Molecular Dynamics Analysis
Analysis of trajectory data was performed using MDTraj 1.9.3. In order to systematically track the stability of malate in the binding site, we computed the distance between C1 atom of malate and the αC atom of Asn455 for the duration of each simulation. Similarly, the relative positioning of NAD(P)+ in relation to the active site was tracked by recording the distance between the C4 atom of the nicotinamide group of NAD(P)+, which is reduced to form NAD(P)H, and the αC atom of Asn455. To quantify differences in the conformational stability of MEZ alone or in complex with malate and NAD(P)+, we computed and averaged the RMSF values of each αC atom in the protein backbone for all simulations and plotted them as shown in (Figure S6). Visual inspection of the trajectories revealed open and closed states for the MEZ subunits; in order to assign the relative state, we tracked the relative proximity of two neighboring helices by computing the distance between αC atoms of Pro244 and Ala300 (Figure 4). For analysis of NAD(P)+ binding modes, we identified one simulation among the five replicates of MEZ/Mn2+/malate/NAD+ and MEZ/Mn2+/malate/NADP+ for which the cofactor was most stable. Sustained binding of the cofactor in the active site of MEZ was assessed by computing the distance between the NAD(P)+ carbon atom at position 4 of the nicotinamide group and the αC atom of a stable residue in the binding pocket, Asn455. Relative stability of the cofactor amongst the replicates was imputed by comparing the RMSF for each atom in the NAD+ or NADP+ ligand; the final frame was used to examine the contacts between the protein and each cofactor.
Supplementary Material
Acknowledgements
We would like to thank Tom Mendum for useful discussions and Tom Poulos and Jose Amaya for critical reading of the manuscript. We thank the Advanced Light Source at Berkeley National Laboratories (ALS) and the Stanford Synchrotron Radiation Lightsource (SSRL) for their invaluable help in data collection, and The Triton Shared Computing Cluster (TCSS) at the San Diego Supercomputing Center (SSDC) for their computing support.
Funding
C.W.G. thanks the National Institutes of Health (NIH) for financial support (P01-AI095208), K.H.B. thanks the NIH for support from a predoctoral training grant (T32GM108561), D.L.M. appreciates financial support from the NIH (R01-GM108889), and B.J.C. was funded through a UCI Chancellor’s Postdoctoral Fellowship. D.J.V.B. is grateful to the Medical Research Council for financial support (MR/K01224X/1).
Abbreviations Used
- 6GPD
6-phosphogluconate dehydrogenase
- ADP
adenosine diphosphate
- BME
β-mercaptoethanol
- DSF
differential scanning fluorimetry
- FGD1
F420-dependent 6-phosphogluconate dehydrogenase
- HIV
human immunodeficiency virus
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- kDa
kilodalton
- MD
molecular dynamics
- ME
malic enzyme
- MEZ
Mycobacterium tuberculosis malic enzyme
- Mtb
Mycobacterium tuberculosis
- NAD(P)+ / NAD(P)H
nicotinamide adenine dinucleotide (phosphate)
- NadD
mononucleotide adenylyltransferase
- NMR
nuclear magnetic resonance
- PMSF
phenylmethylsulfonyl fluoride
- PDB
protein data bank
- rmsd
root mean square deviation
- RMSF
root mean square fluctuation
- SEC
size exclusion chromatography
- TAG
triacylglycerol
- TB
Tuberculosis
- TCA
tricarboxylic acid cycle
Footnotes
Supporting Information
Within the support information is an extended Figure legend for Figure 6B. Additionally, there is a supporting table (Table S1) and supporting figures (Figures S1-S6). Finally, there is a supporting reference. This information is available free of charge on the ACS Publications website.
Accession Numbers
Mtb ME (MEZ): P9WK25 PDB ID code: 6URF. Authors will release the atomic coordinates and experimental data upon article publication.
References
- (1).The World Health Organization. Global tuberculosis report 2020, https://www.who.int/publications/i/item/9789240013131 (10/14/2020).
- (2).Nahid P; Dorman SE; Alipanah N; Barry PM; Brozek JL; Cattamanchi A; Chaisson LH; Chaisson RE; Daley CL; Grzemska M; Higashi JM; Ho CS; Hopewell PC; Keshavjee SA; Lienhardt C; Menzies R; Merrifield C; Narita M; O’Brien R; Peloquin CA; Raftery A; Saukkonen J; Schaaf HS; Sotgiu G; Starke JR; Migliori GB; Vernon A Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin Infect Dis 2016, 63, e147–e195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Basu P; Sandhu N; Bhatt A; Singh A; Balhana R; Gobe I; Crowhurst NA; Mendum TA; Gao L; Ward JL; Beale MH; McFadden J; Beste DJV The anaplerotic node is essential for the intracellular survival of Mycobacterium tuberculosis. J Biol Chem 2018, 293, 5695–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fay A; Czudnochowski N; Rock JM; Johnson JR; Krogan NJ; Rosenberg O; Glickman MS Two Accessory Proteins Govern MmpL3 Mycolic Acid Transport in Mycobacteria. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ojha AK; Trivelli X; Guerardel Y; Kremer L; Hatfull GF Enzymatic hydrolysis of trehalose dimycolate releases free mycolic acids during mycobacterial growth in biofilms. J Biol Chem 2010, 285, 17380–17389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Owens CP; Chim N; Graves AB; Harmston CA; Iniguez A; Contreras H; Liptak M; Goulding CW The Mycobacterium tuberculosis Secreted Protein, Rv0203, Transfers Heme to Membrane Proteins, Mycobacterial membrane protein Large 3 (MmpL3) and MmpL11. J Biol Chem 2013, 288, 21714–21728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pacheco SA; Hsu FF; Powers KM; Purdy GE MmpL11 protein transports mycolic acid-containing lipids to the mycobacterial cell wall and contributes to biofilm formation in Mycobacterium smegmatis. J Biol Chem 2013, 288, 24213–24222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Baker JJ; Johnson BK; Abramovitch RB Slow growth of Mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol Microbiol 2014, 94, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rodriguez E; Navone L; Casati P; Gramajo H Impact of malic enzymes on antibiotic and triacylglycerol production in Streptomyces coelicolor. Appl Environ Microbiol 2012, 78, 4571–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ratledge C The role of malic enzyme as the provider of NADPH in oleaginous microorganisms: a reappraisal and unsolved problems. Biotechnol Lett 2014, 36, 1557–1568. [DOI] [PubMed] [Google Scholar]
- (11).Hernandez MA; Alvarez HM Increasing lipid production using an NADP(+)-dependent malic enzyme from Rhodococcus jostii. Microbiology 2019, 165, 4–14. [DOI] [PubMed] [Google Scholar]
- (12).Alvarez CE; Bovdilova A; Hoppner A; Wolff CC; Saigo M; Trajtenberg F; Zhang T; Buschiazzo A; Nagel-Steger L; Drincovich MF; Lercher MJ; Maurino VG Molecular adaptations of NADP-malic enzyme for its function in C4 photosynthesis in grasses. Nat Plants 2019, 5, 755–765. [DOI] [PubMed] [Google Scholar]
- (13).Alvarez CE; Trajtenberg F; Larrieux N; Saigo M; Golic A; Andreo CS; Hogenhout SA; Mussi MA; Drincovich MF; Buschiazzo A The crystal structure of the malic enzyme from Candidatus phytoplasma reveals the minimal structural determinants for a malic enzyme. Acta Crystallogr D Struct Biol 2018, 74, 332–340. [DOI] [PubMed] [Google Scholar]
- (14).Chang GG; Tong L Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry 2003, 42, 12721–12733. [DOI] [PubMed] [Google Scholar]
- (15).Xu Y; Bhargava G; Wu H; Loeber G; Tong L Crystal structure of human mitochondrial NAD(P)+-dependent malic enzyme: a new class of oxidative decarboxylases. Structure 1999, 7, R877–889. [PubMed] [Google Scholar]
- (16).Bukato G; Kochan Z; Swierczynski J Different regulatory properties of the cytosolic and mitochondrial forms of malic enzyme isolated from human brain. Int J Biochem Cell Biol 1995, 27, 1003–1008. [DOI] [PubMed] [Google Scholar]
- (17).Bologna FP; Andreo CS; Drincovich MF Escherichia coli malic enzymes: two isoforms with substantial differences in kinetic properties, metabolic regulation, and structure. J Bacteriol 2007, 189, 5937–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Coleman DE; Rao GS; Goldsmith EJ; Cook PF; Harris BG Crystal structure of the malic enzyme from Ascaris suum complexed with nicotinamide adenine dinucleotide at 2.3 A resolution. Biochemistry 2002, 41, 6928–6938. [DOI] [PubMed] [Google Scholar]
- (19).Voegele RT; Mitsch MJ; Finan TM Characterization of two members of a novel malic enzyme class. Biochim Biophys Acta 1999, 1432, 275–285. [DOI] [PubMed] [Google Scholar]
- (20).Hsieh JY; Liu GY; Chang GG; Hung HC Determinants of the dual cofactor specificity and substrate cooperativity of the human mitochondrial NAD(P)+-dependent malic enzyme: functional roles of glutamine 362. J Biol Chem 2006, 281, 23237–23245. [DOI] [PubMed] [Google Scholar]
- (21).Karsten WE; Pais JE; Rao GS; Harris BG; Cook PF Ascaris suum NAD-malic enzyme is activated by L-malate and fumarate binding to separate allosteric sites. Biochemistry 2003, 42, 9712–9721. [DOI] [PubMed] [Google Scholar]
- (22).Hsieh JY; Shih WT; Kuo YH; Liu GY; Hung HC Functional Roles of Metabolic Intermediates in Regulating the Human Mitochondrial NAD(P)(+)-Dependent Malic Enzyme. Sci Rep 2019, 9, 9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chang HC; Chou WY; Chang GG Effect of metal binding on the structural stability of pigeon liver malic enzyme. J Biol Chem 2002, 277, 4663–4671. [DOI] [PubMed] [Google Scholar]
- (24).Case DA; Cerutti DS; Cheatham TE III; Darden TA; Duke RE; Giese TJ; Gohlke H; Goetz AW; Greene D; Homeyer N; Izadi S; Janowski P; Kaus, A. J; Kovalenko TSL, LeGrand S, Li P, Luchko T, Luo R, Madej B, Merz KM, Monard G, Needham P, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Salomon-Ferrer R, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, York DM and Kollman PA AmberTools 17. 2017. [Google Scholar]
- (25).Borkotoky S; Murali A A computational assessment of pH-dependent differential interaction of T7 lysozyme with T7 RNA polymerase. BMC Struct Biol 2017, 17, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Watts CR; Gregory AJ; Frisbie CP; Lovas S Structural properties of amyloid beta(1–40) dimer explored by replica exchange molecular dynamics simulations. Proteins 2017, 85, 1024–1045. [DOI] [PubMed] [Google Scholar]
- (27).Bovdilova A; Alexandre BM; Hoppner A; Luis IM; Alvarez CE; Bickel D; Gohlke H; Decker C; Nagel-Steger L; Alseekh S; Fernie AR; Drincovich MF; Abreu IA; Maurino VG Posttranslational Modification of the NADP-Malic Enzyme Involved in C4 Photosynthesis Modulates the Enzymatic Activity during the Day. Plant Cell 2019, 31, 2525–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Costa MG; Benetti-Barbosa TG; Desdouits N; Blondel A; Bisch PM; Pascutti PG; Batista PR Impact of M36I polymorphism on the interaction of HIV-1 protease with its substrates: insights from molecular dynamics. BMC Genomics 2014, 15 Suppl 7, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Parthasarathy R; Fridey SM Conformational variability of NAD+ in the free and bound states: a nicotinamide sandwich in NAD+ crystals. Science 1984, 226, 969–971. [DOI] [PubMed] [Google Scholar]
- (30).Guillot B; Lecomte C; Cousson A; Scherf C; Jelsch C High-resolution neutron structure of nicotinamide adenine dinucleotide. Acta Crystallogr D Biol Crystallogr 2001, 57, 981–989. [DOI] [PubMed] [Google Scholar]
- (31).Tao X; Yang Z; Tong L Crystal structures of substrate complexes of malic enzyme and insights into the catalytic mechanism. Structure 2003, 11, 1141–1150. [DOI] [PubMed] [Google Scholar]
- (32).Yang Z; Batra R; Floyd DL; Hung HC; Chang GG; Tong L Potent and competitive inhibition of malic enzymes by lanthanide ions. Biochem Biophys Res Commun 2000, 274, 440–444. [DOI] [PubMed] [Google Scholar]
- (33).Kotaka M; Graeff R; Chen Z; Zhang LH; Lee HC; Hao Q Structural studies of intermediates along the cyclization pathway of Aplysia ADP-ribosyl cyclase. J Mol Biol 2012, 415, 514–526. [DOI] [PubMed] [Google Scholar]
- (34).Lerondel G; Doan T; Zamboni N; Sauer U; Aymerich S YtsJ has the major physiological role of the four paralogous malic enzyme isoforms in Bacillus subtilis. J Bacteriol 2006, 188, 4727–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hsieh JY; Chen SH; Hung HC Functional roles of the tetramer organization of malic enzyme. J Biol Chem 2009, 284, 18096–18105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Edwards GE; Andreo CS NADP-malic enzyme from plants. Phytochemistry 1992, 31, 1845–1857. [DOI] [PubMed] [Google Scholar]
- (37).Chang HC; Chang GG Involvement of single residue tryptophan 548 in the quaternary structural stability of pigeon cytosolic malic enzyme. J Biol Chem 2003, 278, 23996–24002. [DOI] [PubMed] [Google Scholar]
- (38).Hsieh JY; Li SY; Chen MC; Yang PC; Chen HY; Chan NL; Liu JH; Hung HC Structural characteristics of the nonallosteric human cytosolic malic enzyme. Biochim Biophys Acta 2014, 1844, 1773–1783. [DOI] [PubMed] [Google Scholar]
- (39).Murugan S; Hung HC Biophysical characterization of the dimer and tetramer interface interactions of the human cytosolic malic enzyme. PLoS One 2012, 7, e50143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Chang GG; Huang TM; Chang TC Reversible dissociation of the catalytically active subunits of pigeon liver malic enzyme. Biochem J 1988, 254, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hsieh JY; Liu JH; Yang PC; Lin CL; Liu GY; Hung HC Fumarate analogs act as allosteric inhibitors of the human mitochondrial NAD(P)+-dependent malic enzyme. PLoS One 2014, 9, e98385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yao P; Sun H; Xu C; Chen T; Zou B; Jiang P; Du W Evidence for a direct cross-talk between malic enzyme and the pentose phosphate pathway via structural interactions. J Biol Chem 2017, 292, 17113–17120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bashiri G; Squire CJ; Moreland NJ; Baker EN Crystal structures of F420-dependent glucose-6-phosphate dehydrogenase FGD1 involved in the activation of the anti-tuberculosis drug candidate PA-824 reveal the basis of coenzyme and substrate binding. J Biol Chem 2008, 283, 17531–17541. [DOI] [PubMed] [Google Scholar]
- (44).Hsieh JY; Li SY; Tsai WC; Liu JH; Lin CL; Liu GY; Hung HC A small-molecule inhibitor suppresses the tumor-associated mitochondrial NAD(P)+-dependent malic enzyme (ME2) and induces cellular senescence. Oncotarget 2015, 6, 20084–20098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sarfraz I; Rasul A; Hussain G; Hussain SM; Ahmad M; Nageen B; Jabeen F; Selamoglu Z; Ali M Malic enzyme 2 as a potential therapeutic drug target for cancer. IUBMB Life 2018, 70, 1076–1083. [DOI] [PubMed] [Google Scholar]
- (46).Wen Y; Xu L; Chen FL; Gao J; Li JY; Hu LH; Li J Discovery of a novel inhibitor of NAD(P)(+)-dependent malic enzyme (ME2) by high-throughput screening. Acta Pharmacol Sin 2014, 35, 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Rosa N; Ristic M; Seabrook SA; Lovell D; Lucent D; Newman J Meltdown: A Tool to Help in the Interpretation of Thermal Melt Curves Acquired by Differential Scanning Fluorimetry. J Biomol Screen 2015, 20, 898–905. [DOI] [PubMed] [Google Scholar]
- (48).Kabsch W Xds. Acta Crystallogr D Biol Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Evans PR; Murshudov GN How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yang J; Zhang Y I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 2015, 43, W174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (53).Eastman P; Friedrichs MS; Chodera JD; Radmer RJ; Bruns CM; Ku JP; Beauchamp KA; Lane TJ; Wang LP; Shukla D; Tye T; Houston M; Stich T; Klein C; Shirts MR; Pande VS OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J Chem Theory Comput 2013, 9, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Ryde U Molecular dynamics simulations of alcohol dehydrogenase with a four- or five-coordinate catalytic zinc ion. Proteins 1995, 21, 40–56. [DOI] [PubMed] [Google Scholar]
- (55).Li P; Merz KM Jr. MCPB.py: A Python Based Metal Center Parameter Builder. J Chem Inf Model 2016, 56, 599–604. [DOI] [PubMed] [Google Scholar]
- (56).Swails J; Zhu T; He X; Case DA AFNMR: automated fragmentation quantum mechanical calculation of NMR chemical shifts for biomolecules. J Biomol NMR 2015, 63, 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol 2017, 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sievers F; Higgins DG Clustal Omega for making accurate alignments of many protein sequences. Protein Sci 2018, 27, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Crooks GE; Hon G; Chandonia JM; Brenner SE WebLogo: a sequence logo generator. Genome Res 2004, 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.