
Figure 2.
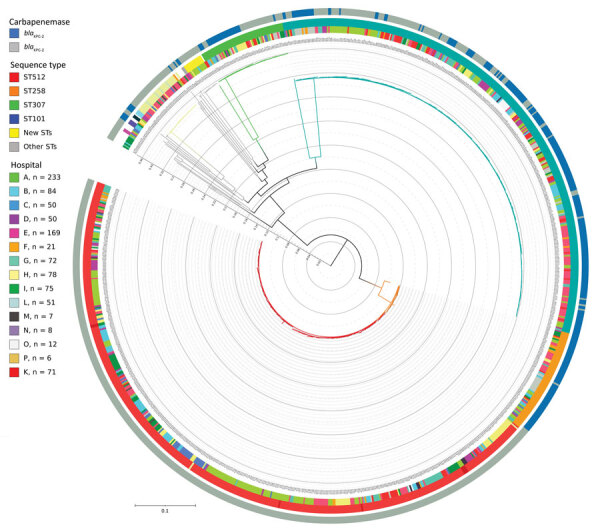
Phylogenetic tree of 989 Klebsiella pneumoniae genomes isolated at hospitals participating in the KPC-producing K. pneumoniae (KPC-Kp) study, Italy. The key shows the number of isolates included in the study provided by each center; 2 samples (1 from each from hospitals A and I) were excluded because the total quality of the assemblies was not sufficient to have high confidence in the SNPs called through all the genome (total coverage <30). Inner circle shows the KPC-Kp mechanism identified; middle circle shows hospitals from which strains were isolated; and the outer circle the shows identified STs. The whole genome core single-nucleotide polymorphisms (SNPs) were extracted from the 989 K. pneumoniae genome assemblies by using kSNP3.0 (https://sourceforge.net/projects/ksnp). Parametric maximum-likelihood estimation (general time-reversible plus gamma distribution plus invariable sites) analysis with 1,000 bootstrap estimates was used to infer the phylogeny. We used IQ-TREE (http://www.iqtree.org) to generate the tree and iTOL (https://itol.embl.de) to draw the tree. Major STs are represented by branch colors; ST512 and ST307 were the predominant STs. Major branches have bootstrap values >0.75 for branch support. Scale bar indicates nucleotide substitutions per site. KPC, Klebsiella pneumoniae–carbapenemase; ST, sequence type.