

Abstract
Cooperative bimetallic catalysis represents a fundamental approach in modern synthetic chemistry that addresses many challenges associated with the evolution of the field. Herein, we report bimetallic cooperative catalysis for the direct decarbonylative heteroarylation of ubiquitous carboxylic acids via acyl C–O/C–H coupling. This novel catalytic system exploits the cooperative action of a copper catalyst and a palladium catalyst under decarbonylative regimen, which enables for the highly chemoselective synthesis of important heterobiaryl motifs that enjoy a privileged role in the realm of organic synthesis through the coupling of carboxylic acids with heteroarenes in the absence of prefunctionalization or directing groups. This cooperative decarbonylative platform benefits from the direct use of common carboxylic acids and shows a remarkably broad substrate scope (>70 examples), including late-stage modification of pharmaceuticals and streamlined synthesis of bioactive agents. Extensive mechanistic and computational studies were conducted to gain insight into the mechanism of the bimetallic decarbonylative platform. The key step involves intersection of the two catalytic cycles via transmetallation of the copper-aryl species with the palladium(II) intermediate generated by oxidative addition/decarbonylation. We envision the versatile bimetallic cooperative decarbonylative framework will have an impact on planning synthetic approaches in cooperative catalysis, decarbonylative coupling and synthesis of biaryl motifs.
Keywords: bimetallic cooperative catalysis, heteroarylation, decarbonylation, carboxylic acids, C–O/C–H bond activation
Graphical Abstract

We report bimetallic cooperative catalysis for the direct decarbonylative heteroarylation of ubiquitous carboxylic acids via acyl C–O/C–H coupling. This novel catalytic system exploits the cooperative action of a copper catalyst and a palladium catalyst under decarbonylative regimen, which enables for the highly chemoselective synthesis of important heterobiaryl motifs that enjoy a privileged role in the realm of organic synthesis. This cooperative decarbonylative platform benefits from the direct use of ubiquitous carboxylic acids and shows a remarkably broad substrate scope (>70 examples), including late-stage modification of pharmaceuticals and streamlined synthesis of bioactive agents. The versatile bimetallic cooperative decarbonylative framework will have an impact on planning synthetic approaches in cooperative catalysis, decarbonylative coupling and synthesis of biaryl motifs.
Twitter: @Szostak_Group
Introduction
Heterocyclic compounds are molecular structures that contain at least two different elements in the ring, and are used as indispensable motifs across various facets of chemistry.[1] Heterocyclic chemistry is crucial in the preparation of new pharmaceutical agents, myriad of industrial products and polymers.[2,3] In comparison to the carbon-only backbone, heteroatoms engender the unique electronic and steric properties that have been particularly attractive in various fields of life sciences and technology, including pharmaceuticals, natural products, agrochemicals and functional materials.[4,5] It is noteworthy that a major percentage of commercial drugs relies on heterocyclic compounds (Figure 1A).[2a]
Figure 1.

Context of this work: cooperative decarbonylative heteroarylation of carboxylic acids via C–O/C–H coupling.
The widespread application of heterocycles as the backbone of a broad range of biologically active compounds has accelerated the demand for new technologies. Over the last decades, classic approaches for the synthesis of heterocycles have had a major impact on the advancement of organic chemistry (Figure 1B). Methods such as Paal-Knorr synthesis,[6,7] Hantzsch pyridine synthesis[8] and Fischer indole synthesis[9] are utilized on daily basis in the repertoire of practicing synthetic chemists. Modern academic and industrial studies on heterocyclic chemistry have been focused on the development of strategies for the synthesis of heterobiaryl motifs owing to the privileged nature of biaryls[4,5] and their pervasive presence in many pharmaceuticals, agrochemicals, natural products, organic materials and ligands for transition-metal-catalysis (Figure 1B).[10]
The most straightforward and efficient approach for the synthesis of heterobiaryls hinges upon transition-metal-catalyzed cross-coupling between haloarene/heteroarene or equivalent and metalated arene/heteroarene (Figure 1C, top).[4,5] Later, significant progress has been made by enabling direct C–H bond arylation of heteroarenes.[11] Carboxylic acid derivatives can be employed as electrophiles in the cross-coupling with heterocycles, including acyl fluorides,[12] acyl chlorides,[13] esters,[14] and amides[15] (Figure 1C, top).[16–20]
In the past years, the field of decarbonylative cross-coupling has experienced a rapid growth, in part owing to the high efficiency of generating aryl-metal intermediates.[21] Due to the undesired generation of activated starting materials in an additional synthetic step, the direct use of ubiquitous carboxylic acids has the major advantage over other precursors.[21] In recent years, elegant studies on decarboxylative cross-coupling of carboxylic acids have been disclosed, which employ carboxylic acids as nucleophilic coupling partners.[21–23] However, in many cases, steric and electronic effects limit the development and application of this platform, which is favored for electron-deficient or sterically-hindered carboxylic acids.[23] As a result, there is high demand for general and broadly applicable cross-coupling methods of carboxylic acids.
Decarbonylative cross-coupling of carboxylic acids can be traced to the work by de Vries in 1998 in the decarbonylative Heck reaction after conversion to symmetrical anhydrides,[24] followed by several studies demonstrating the utility of this attractive cross-coupling platform.[21–25] However, despite the distinct advantages of carboxylic acids as cross-coupling partners and the urgent demand for the efficient methods for the construction of valuable biaryl heterocycles, the decarbonylative synthesis of heterobiaryls from carboxylic acids is unknown.
We proposed that the palladium-catalyzed decarbonylative heteroarylation of carboxylic acids for the synthesis of heterobiaryls will offer a general and practical methodology for the synthesis of biaryl motifs from carboxylic acids, while at the same time exploiting the utility of the powerful decarbonylative activation mode of carboxylic acids (Figure 1C, bottom).
In the context of biaryl synthesis, rhodium catalysis has represented a particularly robust strategy for the construction of C–C bonds via direct C–H activation with the assistance of directing groups.[26–29] Rhodium-catalyzed decarbonylative arylation of carboxylic acids via C–H coupling with the assistance of directing groups has been reported (Figure 1D).[30,31] However, N-heterocyclic directing groups are necessary for coordination of the rhodium catalyst. Palladium and nickel catalysis have been considered as versatile catalytic systems for arylation reactions.[32–39] In 2019, we reported the palladium-catalyzed decarbonylative Suzuki-Miyaura cross-coupling of carboxylic acids with boronic acids as arylating reagents (Figure 1D).[40] While the protocol showed broad scope, prefunctionalization to form boronic acids is required for the cross-coupling.[41–43] With the continuous evolution of the field and the high demand for biaryl heterocycles, we anticipated that the direct implementation of heteroarenes as nucleophiles without prefunctionalization or directing groups in decarbonylative cross-coupling of carboxylic acids via C–O/C–H bond activation would allow for a broad range of heterobiaryls to be accesses from abundant carboxylic acid as electrophilic cross-coupling partners (Figure 1D, bottom).
As a critical advantage, the bimetallic cooperative catalysis was proposed via two catalytic cycles (Figure 1E): (i) a copper catalyst would first coordinate to the Lewis-basic group on heterocycle, leading to the heterocycle–Cu species by C–H activation; (ii) a palladium catalyst would insert into the acyl C–O bond of the carboxylic acids that has been activated in situ to give ArCOOPiv. The acyl–Pd–OPiv intermediate would next undergo decarbonylation to form Ar–Pd–OPiv intermediate. Finally, the two catalytic cycles would intersect during the transmetallation of the copper-aryl species with the palladium(II) intermediate to generate the final heterobiaryl product after reductive elimination. The transformation is distinct from previous studies by engaging cooperative cycle and decarbonylative acyl coupling of carboxylic acid anhydrides, which permits readily available carboxylic acids to be coupled with heteroarenes by direct deprotonation.
Noteworthy features of our study include: (1) direct use of ubiquitous carboxylic acids via robust C–O/C–H bond activations; (2) high chemoselectivity in the absence of prefunctionalization or directing groups; (3) bimetallic cooperative catalysis under decarbonylative regimen; (4) remarkably broad substrate scope, including late-stage modification of pharmaceuticals and streamlined synthesis of bioactive agents; (5) mechanistic studies providing key insights into the mechanism of the bimetallic decarbonylative catalysis platform.
Results and Discussion
The proposed palladium/copper catalyzed decarbonylative heteroarylation of carboxylic acids was examined using benzoxazole (1a) and benzoic acid (2a) as model substrates (see Supporting Information, SI). To our delight, after very extensive optimization, we identified the combination of Pd(OAc)2 (2.5 mol%), CuF2 (10 mol%) and dppb (5 mol%) in the presence of DMAP (1.5 equiv) and Piv2O (1.5 equiv) in dioxane at 160 °C as the optimum system to deliver the desired heterobiaryl product (3a) in 92% isolated yield (Figure 2).
Figure 2.
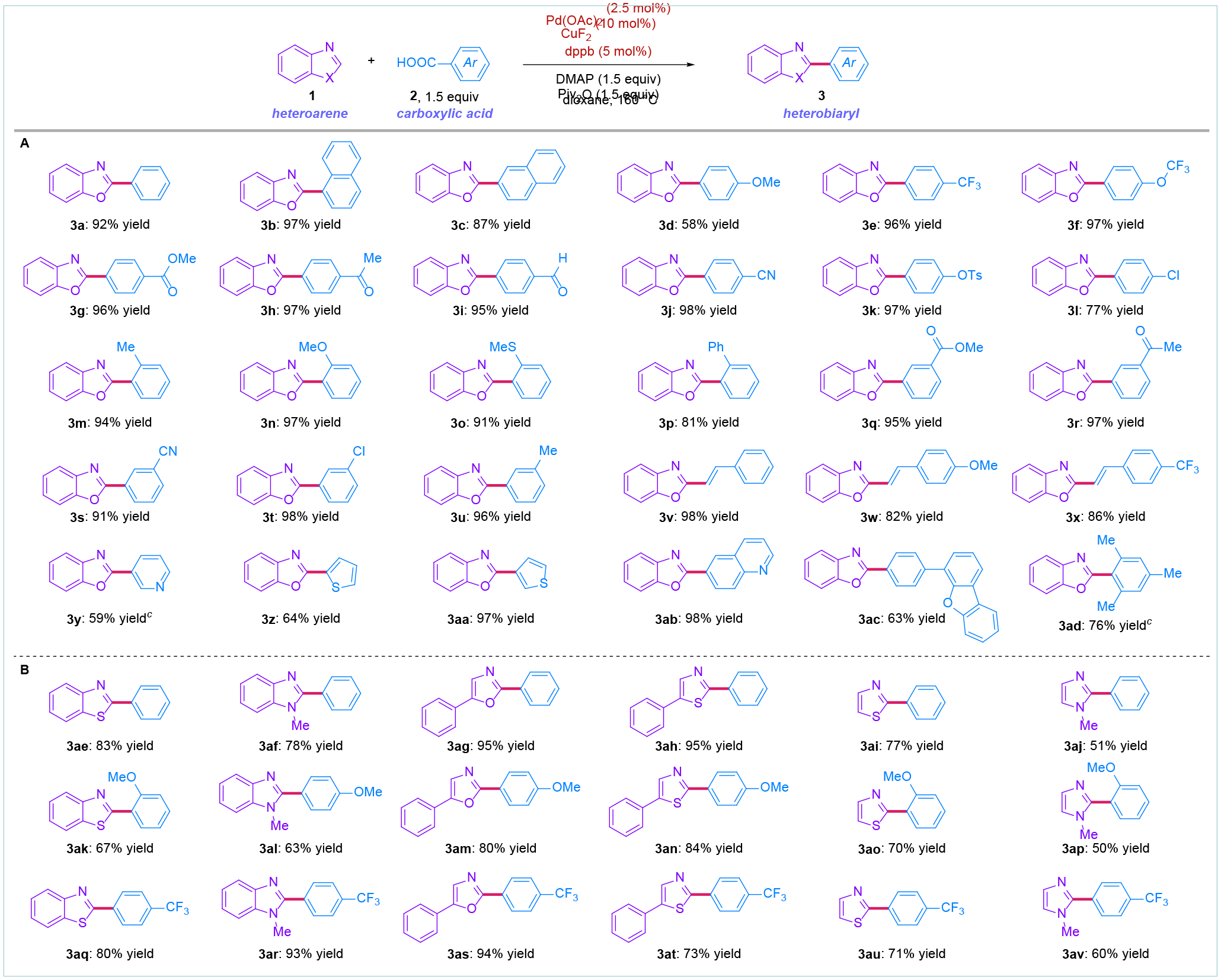
Scope of cooperative decarbonylative heteroarylation of carboxylic acids via C–O/C–H coupling. aConditions: Pd(OAc)2 (2.5 mol%), CuF2 (10 mol%), dppb (5 mol%), DMAP (1.5 equiv), piv2O (1.5 equiv). bPd(OAc)2 (2.5 mol%), CuF2 (10 mol%), dppb (5 mol%), DMAP (3.0 equiv), Piv2O (3.0 equiv). cPd(OAc)2 (5.0 mol%), CuF2 (20 mol%), dppb (10 mol%), DMAP (3.0 equiv), piv2O (3.0 equiv). See SI for details.
A summary of the optimization results is shown in the Supporting Information. Several optimization results are worth noting: (1) the use of either palladium or copper alone results in negligible conversion (<5%); (2) Dppb is the preferred ligand (96% yield); however, dppe (87%), dppp (72%), DPEPhos (61%) and dppf (93%) are also effective; (3) Among various copper salts examined, CuF2 is the preferred catalyst; however CuBr (61%), CuCl (63%), CuCN (58%), CuSO4 (43%), Cu(OAc)2 (56%) and Cu(OTf)2 (71%) also gave good to high yields; (4) The reaction is performed in the absence of an external inorganic base with pivalate acting as an internal base; (5) DMAP is the preferred Lewis base; however, the coupling is possible in the absence of DMAP (40% yield); (6) In all cases examined, the formation of heterobiaryl ketones (acyl coupling) was not observed, consistent with the high capacity of the cooperative system to facilitate decarbonylation.
With optimized conditions in hand, we next investigated the scope of carboxylic acids in this cooperative methodology for the synthesis of heterobiaryls using readily available carboxylic acids and heterocyclic substrates (Figure 2A). As shown, the scope of this approach is very broad, including excellent functional group tolerance to various sensitive electrophilic groups. Thus, various electron-neutral (3a-3c), electron-donating (3d) and electron-deficient carboxylic acids (3e-3f) underwent the coupling in excellent yields. It is noteworthy that electrophilic groups that would be problematic in classical addition of highly nucleophilic organomagnesium or organolithium organometallics, such as esters (3g), ketones (3h), aldehydes (3i) and nitriles (3j) could be readily employed. Remarkably, synthetic handles for further functionalization by cross-coupling, such as tosyl (3k) and chloride (3l) are well-compatible, resulting in 97% and 77% yields. Furthermore, sterically-hindered carboxylic acids (3m-3p) are well-tolerated. Moreover, unbiased substrates functionalized at the meta position, including esters (3q), ketones (3r), nitriles (3s), chlorides (3t) and alkyl groups (3u) are perfectly compatible in this cooperative transformation. Importantly, cinnamic acids (3v-3x) could also be used in this decarbonylative platform to deliver styryl-heterocyclic motifs in good to excellent yields. Likewise, this approach permits for the synthesis of the challenging bis-heterocylic biaryls directly from heterocycles and heterocyclic carboxylic acids (3y-3ab). Finally, this catalytic system can also accommodate the synthesis of hetero-terphenyls (3ac) and extremely hindered carboxylic acids, such as mesityl (3ad), demonstrating high level of compatibility in decarbonylative transfer via C–O/C–H activation.
To demonstrate the functional group compatibility of the heterocyclic component, we next investigated the scope of heterocycles amenable to this cooperative coupling (Figure 2B). To our delight, various five-membered heterocycles containing nitrogen, oxygen and sulfur, such as benzothiazoles (3ae, 3ak, 3aq), benzimidazoles (3af, 3al, 3ar), oxazoles (3ag, 3am, 3as), thiazoles (3ah-3ai, 3an-3ao, 3at-3au) and imidazoles (3aj, 3ap, 3av) are compatible with this method, offering direct access to various heterobiaryl motifs.
As a further demonstration of this strategy, we applied this cooperative decarbonylative coupling to the direct, late-stage functionalization of pharmaceuticals (Figure 3). The synthesis of heterobiaryls from (Adapalene, 3aw), (Bexarotene, 3ax), (Tamibarotene, 3ay), (Repaglinide, 3az), (Diflufenican, 3ba), (Probenecid, 3bb), (Febuxostat, 3bc), (Tocopherol, 3bd), (Menthol, 3be) and (Cholosterol, 3bf) using various heterocycles, such as benzoxazole (3be-3bf), benzimidazole (3bg), imidazole (3bh), 4-carboethoxyoxazole (3bi), oxadiazole (3bj), oxazole (3bk), benzothiazole (3bl), benzimidazole (3bm), thiazole (3bn), 4-carbo-tert-butoxythiazole (3bo) and 1,3,7-trimethylxanthine (3bp) demonstrates the excellent functional group tolerance of this cooperative platform and strongly emphasizes the synthetic utility of the coupling approach directly exploiting readily available carboxylic acids as electrophilic synthetic handles. At the present stage, electron-withdrawing substituents on the heterocycle component are well-tolerated (Adapalene, 3bi), while electron-donating groups lead to lower yields.
Figure 3.

Late-stage functionalization of pharmaceuticals via cooperative decarbonylative C–O/C–H coupling. aConditions: Pd(OAc)2 (2.5 mol%), CuF2 (10 mol%), dppb (5 mol%), DMAP (1.5 equiv), piv2O (1.5 equiv). See SI for details.
Moreover, considering the similarity of the acidic C–H bond character between heterocycles and polyfluorinated arenes,[44] we investigated the decarbonylative arylation of pentafluorobenzene using carboxylic acids as electrophiles (Figure 4A). To our delight, a range of carboxylic acid substrates could be converted to the polyfluorinated biaryl or styrenyl products without modification of the reaction conditions. To demonstrate the utility of this robust cooperative decarbonylative platform, we performed a series of sequential transformations deploying the carboxylic acid functional group as a directing handle (Figure 4B–D). Thus, the ortho-C–H arylation directed by the carboxylic acid group,[45] followed by cooperative decarbonylative heteroarylation under standard reaction conditions delivered ortho-heteroterphenyls, which are used as heat storage agents (Figure 4B). Next, traceless toluene oxidation/cooperative heterobiaryl coupling delivered the para-heteroterphenyl product exploiting an alkyl group as a carboxylic acid equivalent in a formal C–C bond activation (Figure 4C).[46] Furthermore, electrophilic meta-C–H functionalization of benzoic acids is readily achieved by SNAr, which enables for orthogonal sequential couplings on the benzoic acid template as exemplified in the Suzuki–Miyaura cross-coupling/heterobiaryl coupling under standard conditions (Figure 4D).[47]
Figure 4.

Synthetic applications of cooperative decarbonylative heteroarylation of carboxylic acids via C–O/C–H coupling.
To illustrate the utility of this cooperative decarbonylative method in the synthesis of biologically active agents, we applied this protocol to the synthesis of hetero-terphenyl 3bh, a selective COX-2 inhibitor with the activity comparable to that of Celecoxib (Figure 4E, IC50 = 0.67).[48] Thus, the Suzuki-Miyaura coupling followed by cooperative decarbonylative heteroarylation permits for a convergent approach to introduce the heterocyclic biaryl scaffold without modification of the reaction conditions. Furthermore, the cooperative decarbonylative heteroarylation of 4-oxo-4H-chromene-2-carboxylic acid delivered the bis-heterobiaryl chromenone derivative 3bi, a class of bis-heterocycles that are used for the diagnosis of amyloid related disease as specific PET imaging probes (Figure 4F).[49]
Considering the remarkably broad scope and prospective applications of the cooperative decarbonylative coupling of carboxylic acids, we sought to gain insight into the reaction mechanism of this intriguing process. Consequently, we conducted a series of stoichiometric and DFT studies (vide infra). Stoichiometric studies are summarized in Figure 5.
Figure 5.

Mechanistic studies in cooperative decarbonylative heteroarylation of carboxylic acids via C–O/C–H coupling.
(1) To rule out the pathway by ketone decarbonylation,[50] we prepared heterobiaryl ketone 3a’ and subjected this substrate to the standard coupling conditions, resulting in close to quantitative recovery of starting material; the decarbonylative product was not formed under the reaction conditions (<2%) (Figure 5A). This experiment is also consistent with decarbonylation prior to transmetallation (vide infra).
(2) To gain insight into the proposed transmetallation step, we prepared 2-benzoxazolyl copper,[12] and subjected this compound to the coupling conditions in the presence of benzoic acid, Piv2O and a palladium catalyst, which resulted in the formation of the desired heterobiaryl product in 97% yield (Figure 5B). This experiment is consistent with copper to palladium transmetallation in the reaction mechanism.
(3) To investigate the effect of copper catalyst, we performed the coupling using well-defined [Cu(IPr)Cl] and [Cu(dppb)I] catalysts, which resulted in 51% and 30% yield of the heterobiaryl product (Figure 5C). These experiments indicate that well-defined sources of copper could catalyze the reaction, consistent with heterocycle C–H activation by the copper catalyst.[11a,44]
(4) To investigate the active species formed from the carboxylic acid, we prepared ArCO2Piv as the proposed intermediate and subjected this substrate to the reaction conditions, resulting in 90% yield of the heterobiaryl product (Figure 5D). This experiment is consistent with activation of the carboxylic acid to give ArCO2Piv as the active intermediate.
(5) Next, in order to gain insight into the copper catalyst, we performed the coupling using CuCl and CuCl2 under the standard conditions (Figure 5E). The reaction resulted in a similar conversion (CuCl: 63%; CuCl2: 66%) demonstrating that copper catalysts at both I and II oxidation state are suitable.
(6) To investigate the observed selectivity in the coupling, we performed a series of stoichiometric competition experiments (Figure 5F–H). Thus, competition experiments between different heterocycles revealed the following order of reactivity: benzoxazole > oxazole > benzothiazole > benzimidazole > imidazole (Figure 5F–G), which is consistent with the relative C–H bond acidity (benzoxazole, pKa = 24.8; oxazole, pKa = 27.7; benzothiazole, pKa = 27.8; thiazole, pKa = 29.4; N-Me-benzimidazole, pKa = 32.1; N-Me-imidazole, pKa = 33.7).[44] Competition experiments between differently substituted benzoic acids revealed that electron-deficient carboxylic acids are inherently more reactive than their electron-rich counterparts, while sterically-hindered benzoic acids are more reactive than unsubstituted benzoic acids (Figure 5H). The increased steric demand of acyl-metal complexes favors decarbonylation[21] (vide infra).
Computational studies.
To further understand this cooperative decarbonylative coupling, we performed DFT calculations on the catalytic cycle. The DFT-computed free energy profile of the operative pathway is shown in Figure 6A. From the anhydride-coordinated complex int1, the oxidative addition via TS2 cleaves the acyl C–O bond and generates the acylpalladium(II) intermediate int3. Subsequent decarbonylation via TS4 leads to the CO-coordinated species int5, and CO dissociates to generate the arylpalladium(II) intermediate int6. int6 complexes with the arylcopper intermediate int7 to form the bimetallic complex int8. This complexation from int6 to int8 is endergonic by 11.3 kcal/mol because the 15.1 kcal/mol endergonicity of the arylcopper generation (int15 to int7) is included; the energy reference for the arylcopper intermediate int7 is the stable LCuF(oxazole) species int15. From int8, the transmetallation via TS9 leads to the biarylpalladium(II) intermediate int10. int10 dissociates the copper(I)pivalate int11, and subsequent C–C reductive elimination via TS13 produces the product-coordinated complex int14. int14 eventually liberates the cross-coupling product and regenerates the catalytic active species int1. Based on the computed free energy profile, the on-cycle resting state is the acylpalladium(II) intermediate int3. The rate-determining step is the transmetallation via TS9, which requires an overall barrier of 28.7 kcal/mol comparing with the resting state int3.
Figure 6.

DFT calculations of cooperative decarbonylative heteroarylation of carboxylic acids via C–O/C–H coupling.
The DFT-computed free energy changes of the arylcopper(I) int7 generation is presented in Figure 6A. From the LCuF(oxazole) species int15, the out-sphere deprotonation by pivalate anion occurs through TS16, which generates the anionic arylcopper intermediate int17. Int17 has the coordinated pivalic acid quenched by DMAP, and the anionic arylcopper(I)F species dissociates the fluoride anion to produce the arylcopper(I) int7. This fluoride dissociation is necessary to provide the required coordination site of the arylcopper species int7 in the complexation with arylpalladium intermediate int6. Our calculations suggest that the generation of int7 is quite facile, requiring a 14.9 kcal/mol barrier for the out-sphere deprotonation via TS16. This indicates that the Cu-catalyzed C–H bond activation is not rate-determining in the cooperative catalysis. We also considered a number of alternative pathways for the arylcopper(I) int7 generation, which are summarized in Figure 6B. The out-sphere and inner-sphere deprotonations with copper(I)pivalate are both less favorable via TS20 and TS23 respectively. On the basis of mechanistic and DFT studies we proposed the catalytic cycle shown in Figure 7.
Figure 7.

Proposed catalytic cycle.
Conclusion
In conclusion, we have reported the bimetallic cooperative system for the direct decarbonylative heteroarylation of aryl carboxylic acids via C–O/C–H bond activation. Thus, a cooperative action of palladium and copper catalysts under decarbonylative regimen permits for the synthesis of privileged heterobiaryls with broad scope of substrates and excellent functional group tolerance. The prospective impact of this synthetic platform was showcased in the late-stage modification of pharmaceuticals and streamlined synthesis of bioactive motifs, directly exploiting the pervasive presence of the carboxylic acid moiety in drugs and common synthetic motifs. Extensive mechanistic studies and DFT provided insight into the catalytic mechanism, whereby the two catalytic cycles merge via transmetallation of the copper-aryl species with the palladium(II) intermediate. The power of this bimetallic decarbonylative coupling is reflected by efficient couplings across a range of heteroarens and benzoic acids as electro-philes that are beyond decarboxylative generation of aryl nucleophiles from carboxylic acids. Considering the great advantage of carboxylic acids as electrophilic components in cross-coupling reactions,[51] this method should have broader implications for the development of decarbonylative couplings and the rapid synthesis of heterobiaryl building blocks. Further studies on cooperative decarbonylative catalysis are currently underway and will be reported in due course.
Supplementary Material
Acknowledgements
We thank the NIH (1R35GM133326, M.S.), the NSF (CAREER CHE-1650766, M.S.), Rutgers University (M.S.), NSFC (21702182 and 21873081, X.H.), Fundamental Research Funds for the Central Universities (X.H.), and Zhejiang University (X.H.) for generous financial support. The Bruker 500 MHz spectrometer used in this study was supported by the NSF-MRI (CHE-1229030). Additional support was provided by the Rutgers Grad-uate School in the form of Dean’s Dissertation Fellowship (C.L.). Calculations were performed on the high-performance computing system at the Department of Chemistry, Zhejiang University.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Joule JA, Mills K, Heterocyclic Chemistry; Wiley-Blackwell: Oxford, 2013; [Google Scholar]; b) Noel S, Cadet S, Gras E, Hureau C, Chem. Soc. Rev 2013, 42, 7747–7762; [DOI] [PubMed] [Google Scholar]; c) Jin Z, Nat. Prod. Rep 2013, 30, 869–915; [DOI] [PubMed] [Google Scholar]; d) Palmer DC, Oxazoles: Synthesis, Reactions and Spectroscopy, Part A; John Wiley & Sons: Hoboken, 2003; [Google Scholar]; e) Palmer DC, Oxazoles: Synthesis, Reactions and Spectroscopy, Part B; John Wiley & Sons: Hoboken, 2004; [Google Scholar]; f) Luca LD, Curr. Med. Chem 2006, 13, 1–23. [PubMed] [Google Scholar]; g) Grimmett MR, Imidazole and Benzimidazole Synthesis; Academic Press: London, 1997. [Google Scholar]
- [2].a) McGrath NA, Brichacek M, Njardarson JT, J. Chem. Educ 2010, 87, 1348–1349; [Google Scholar]; b) Cabrele C, Reiser O, J. Org. Chem 2016, 81, 10109–10125; [DOI] [PubMed] [Google Scholar]; c) Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW, Chem. Soc. Rev 2016, 45, 546–576; [DOI] [PubMed] [Google Scholar]; d) Blakemore DC, Castro L, Churcher I, Rees DC, Thomas AW, Wilson DM, Wood A, Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- [3].a) Roughley SD, Jordan AM, J. Med. Chem 2011, 54, 3451–3479; [DOI] [PubMed] [Google Scholar]; b) Brown DG, Boström J, J. Med. Chem 2016, 59, 4443–4458; [DOI] [PubMed] [Google Scholar]; c) Vitaku E, Smith DT, Njardarson JT, J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- [4].For reviews, see:; a) Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem. Int. Ed 2005, 44, 4442–4489; [DOI] [PubMed] [Google Scholar]; b) Hassan J, Sevignon M, Gozzi C, Schulz E, Lemaire M, Chem. Rev 2002, 102, 1359–1470; [DOI] [PubMed] [Google Scholar]; c) Alberico D, Scott ME, Lautens M, Chem. Rev 2007, 107, 174–238. [DOI] [PubMed] [Google Scholar]
- [5].For reviews on direct C–H functionalization of heteroaromatic compounds, see:; a) Campeau L-C, Fagnou K, Chem. Commun 2006, 1253–1264; [DOI] [PubMed] [Google Scholar]; b) Colby DA, Bergman RG, Ellman JA, Chem. Rev 2010, 110, 624–655; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ackermann L, Vicente R, Kapdi AR, Angew. Chem. Int. Ed 2009, 48, 9792–9826; [DOI] [PubMed] [Google Scholar]; d) Guo XX, Gu DW, Wu Z, Zhang W, Chem. Rev 2015, 115, 1622–1651. [DOI] [PubMed] [Google Scholar]
- [6].Paal C, Ber. Dtsch. Chem. Ges 1884, 17, 2756–2767. [Google Scholar]
- [7].Knorr L, Ber. Dtsch. Chem. Ges 1884, 17, 2863–2870. [Google Scholar]
- [8].Hantzsch A, Chem. Ber 1881, 14, 1637–1638. [Google Scholar]
- [9].Fischer E, Jourdan F, Ber. Dtsch. Chem. Ges 1883, 16, 2241–2245. [Google Scholar]
- [10].For a recent study, see:; Lee S, Park SB, Org. Lett 2009, 11, 5214–5217. [DOI] [PubMed] [Google Scholar]
- [11].a) Huang J, Chan J, Chen Y, Borths CJ, Baucom KD, Larsen RD, Faul MM, J. Am. Chem. Soc 2010, 132, 3674–3675; [DOI] [PubMed] [Google Scholar]; b) Canivet J, Yamaguchi J, Ban I, Itami K, Org. Lett 2009, 11, 1733–1736; [DOI] [PubMed] [Google Scholar]; c) Muto K, Yamaguchi J, Itami K, J. Am. Chem. Soc 2012, 134, 169–172. [DOI] [PubMed] [Google Scholar]
- [12].For heteroarylation of acyl fluorides, see:; Ogiwara Y, Iino Y, Sakai N, Chem. Eur. J 2019, 25, 6513–6516. [DOI] [PubMed] [Google Scholar]
- [13].For heteroarylation of acyl chlorides, see:; Harn NK, Gramer CJ, Anderson BA, Tetrahedron. Lett 1995, 36, 9453–9456. [Google Scholar]
- [14].For decarbonylative heteroarylation of esters, see:; a) Amaike K, Muto K, Yamaguchi J, Itami K, J. Am. Chem. Soc 2012, 134, 13573–13576; [DOI] [PubMed] [Google Scholar]; b) Meng L, Kamada Y, Muto K, Yamaguchi J, Itami K, Angew. Chem. Int. Ed 2013, 52, 10048–10051. [DOI] [PubMed] [Google Scholar]
- [15].For decarbonylative heteroarylation of amides, see:; Zhou P-X, Shi S, Wang J, Zhang Y, Li C, Ge C, Org. Chem. Front 2019, 6, 1942–1947. [Google Scholar]
- [16].For reviews on cross-coupling of amides, see:; a) Liu C, Szostak M, Chem. Eur. J 2017, 23, 7157–7173; [DOI] [PubMed] [Google Scholar]; b) Li G, Ma S, Szostak M, Trends Chem. 2020, 2, 914–928. [Google Scholar]
- [17].a) Liu C, Meng G, Liu Y, Liu R, Lalancette R, Szostak R, Szostak M, Org. Lett 2016, 18, 4194–4197; [DOI] [PubMed] [Google Scholar]; b) Liu C, Liu Y, Liu R, Lalancette R, Szostak R, Szostak M, Org. Lett 2016, 18, 4194–4197; [DOI] [PubMed] [Google Scholar]; c) Liu C, Li G, Shi S, Meng G, Lalancette R, Szostak R, Szostak M, ACS Catal. 2018, 8, 9131–9139; [Google Scholar]; d) Liu C, Shi S, Liu Y, Liu R, Lalancette R, Szostak R, Szostak M, Org. Lett 2018, 20, 7771–7774; [DOI] [PubMed] [Google Scholar]; e) Liu C, Lalancette R, Szostak R, Szostak M, Org. Lett 2019, 21, 7976–7981. [DOI] [PubMed] [Google Scholar]
- [18].Meng G, Szostak M, Angew. Chem. Int. Ed 2015, 54, 14518–14522. [DOI] [PubMed] [Google Scholar]
- [19].Shi S, Meng G, Szostak M, Angew. Chem. Int. Ed 2016, 55, 6959–6963. [DOI] [PubMed] [Google Scholar]
- [20].Liu C, Szostak M, Angew. Chem. Int. Ed 2017, 56, 12718–12722. [DOI] [PubMed] [Google Scholar]
- [21].For reviews on decarbonylative cross-coupling, see:; a) Lu H, Yu TY, Xu PF, Wei H, Chem. Rev 2020, doi: 10.1021/acs.chemrev.0c00153; [DOI] [PubMed] [Google Scholar]; b) Dzik WI, Lange PP, Gooßen LJ, Chem. Sci 2012, 3, 2671–2678; [Google Scholar]; For reviews on decarboxylative cross-coupling of carboxylic acids, see:; c) Goossen LJ, Rodriguez N, Goossen K, Angew. Chem. Int. Ed 2008, 47, 3100–3120; [DOI] [PubMed] [Google Scholar]; d) Rodriguez N, Goossen LJ, Chem. Soc. Rev 2011, 40, 5030–5048; [DOI] [PubMed] [Google Scholar]; e) Wei Y, Hu P, Zhang M, Su W, Chem. Rev 2017, 117, 8864–8907. [DOI] [PubMed] [Google Scholar]
- [22].For decarboxylative heteroarylation, see:; a) Zhang F, Greaney MF, Angew. Chem. Int. Ed 2010, 49, 2768–2771; [DOI] [PubMed] [Google Scholar]; b) Hu P, Zhang M, Jie X, Su W, Angew. Chem. Int. Ed 2012, 51, 227–231; [DOI] [PubMed] [Google Scholar]; c) Zhang Y, Zhao H, Zhang M, Su W, Angew. Chem. Int. Ed 2015, 54, 3817–3821; [DOI] [PubMed] [Google Scholar]; d) Kan J, Huang S, Lin J, Zhang M, Su W, Angew. Chem. Int. Ed 2015, 54, 2199–2203. [DOI] [PubMed] [Google Scholar]
- [23].For recent studies on decarboxylative heteroarylation, see:; a) Crovak RA, Hoover JM, J. Am. Chem. Soc 2018, 140, 2434–2437; [DOI] [PubMed] [Google Scholar]; b) Chen L, Ju L, Bustin KA, Hoover JM, Chem. Commun 2015, 51, 15059–15062. [DOI] [PubMed] [Google Scholar]
- [24].Stephan MS, Teunissen AJJM, Verzijl GKM, de Vries JG, Angew. Chem. Int. Ed 1998, 37, 662–664. [DOI] [PubMed] [Google Scholar]
- [25].a) Zhang X, Jordan F, Szostak M, Org. Chem. Front 2018, 5, 2515–2521; [Google Scholar]; b) Goossen LJ, Paetzod J, Angew. Chem. Int. Ed 2002, 41, 1237–1241; [DOI] [PubMed] [Google Scholar]; c) Goossen LJ, Rodriguez N, Chem. Commun 2004, 40, 724–725; [DOI] [PubMed] [Google Scholar]; d) Kraus GA, Riley S, Synthesis 2012, 44, 3003–3005; [Google Scholar]; e) Liu Y, Kim KE, Herbert MB, Fedorov A, Grubbs RH, Stoltz BM, Adv. Synth. Catal 2014, 356, 130–136; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) John A, Hogan LT, Hillmyer MA, Tolman WB, Chem. Commun 2015, 51, 2731–2733. [DOI] [PubMed] [Google Scholar]
- [26].Fagnou K, Lautens M, Chem. Rev 2003, 103, 169–196. [DOI] [PubMed] [Google Scholar]
- [27].Jang YJ, Larin EM, Lautens M, Angew. Chem. Int. Ed 2017, 56, 11927–11930. [DOI] [PubMed] [Google Scholar]
- [28].Sidera M, Fletcher SP, Nat. Chem 2015, 7, 935–939. [DOI] [PubMed] [Google Scholar]
- [29].Goetzke FW, Mortimore M, Fletcher Angew SP. Chem. Int. Ed 2019, 58, 12128–12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pan F, Lei Z-Q, Wang H, Li H, Sun J, Shi Z-J, Angew. Chem. Int. Ed 2013, 52, 2063–2067. [DOI] [PubMed] [Google Scholar]
- [31].Lei Z-Q, Pan F, Li H, Li Y, Zhang X-S, Chen K, Wang H, Li Y-X, Sun J, Shi Z-J, J. Am. Chem. Soc 2015, 137, 5012–5020. [DOI] [PubMed] [Google Scholar]
- [32].For palladium-catalyzed hydroarylation, see:; Vasquez AM, Gurak JA, Joe CL, Cherney EC, Engle KM, J. Am. Chem. Soc 2020, 142, 10477–10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].For nickel-catalyzed 1,2-diarylation, see:; Derosa J, Kleinmans R, Tran VT, Karunananda MK, Wisniewski SR, Eastgate MD, Engle KM, J. Am. Chem. Soc 2018, 140, 17878–17883. [DOI] [PubMed] [Google Scholar]
- [34].For Co/Ni-catalyzed hydroarylation, see:; Shevick SL, Obradors C, Shenvi RA, J. Am. Chem. Soc 2018, 140, 12056–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].For Fe/Ni-catalyzed hydroarylation, see:; Green SA, Vasquez-Cespedes S, Shenvi RA, J. Am. Chem. Soc 2018, 140, 11317–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].For Ni-catalyzed alkylarylation, see:; Shekhar KC, Dhungana RK, Shrestha B, Thapa S, Khanal N, Basnet P, Lebrun RW, Giri R, J. Am. Chem. Soc 2018, 140, 9801–9805. [DOI] [PubMed] [Google Scholar]
- [37].For Ni-catalyzed difunctionalization, see:; Shrestha B, Basnet P, Dhungana RK, Shekhar KC, Thapa S, Sears JM, Giri R, J. Am. Chem. Soc 2017, 139, 10653–10656. [DOI] [PubMed] [Google Scholar]
- [38].For Ni-catalyzed arylboration, see:; Sardini SR, Lambright AL, Trammel GL, Omer HM, Liu P, Brown MK, J. Am. Chem. Soc 2019, 141, 9391–9400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].For Ni-catalyzed diarylation, see:; Gao P, Chen L-A, Brown MK, J. Am. Chem. Soc 2018, 140, 10653–10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu C, Ji CL, Qin ZX, Hong X, Szostak M, iScience 2019, 19, 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu C, Ji CL, Hong X, Szostak M, Angew. Chem. Int. Ed 2018, 57, 16721–16726. [DOI] [PubMed] [Google Scholar]
- [42].Liu C, Qin ZX, Ji CL, Hong X, Szostak M, Chem. Sci 2019, 10, 5736–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu C, Ji CL, Zhou T, Hong X, Szostak M, Org. Lett 2019, 21, 9256–9261. [DOI] [PubMed] [Google Scholar]
- [44].Boogaerts IIF, Fortman GC, Furst MRL, Cazin CSJ, Nolan SP, Angew. Chem. Int. Ed 2010, 49, 8674–8677. [DOI] [PubMed] [Google Scholar]
- [45].De Sarkar S, Liu WP, Kozushkov SL, Ackermann L, Adv. Synth. Catal 2014, 356, 1461–1479. [Google Scholar]
- [46].Ni J, Li J, Fan Z, Zhang A, Org. Lett 2016, 18, 5960–5963. [DOI] [PubMed] [Google Scholar]
- [47] <b/>(a).Mortier J Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds; Wiley: Hoboken, 2016; [Google Scholar]; b) Kraszkiewicz L, Sosnowski M, Skulski L, Synthesis 2006, 7, 1195–1199. [Google Scholar]
- [48].a) Seth K, Garg SK, Kumar R, Purohit P, Meena VS, Goyal R, Banerjee UC, Chakraborti AK, ACS Med. Chem. Lett 2014, 5, 512–516; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Barder TE, Walker SD, Martinelli JR, Buchwald SL, J. Am. Chem. Soc 2005, 127, 4685–4696. [DOI] [PubMed] [Google Scholar]
- [49].Fuchigami T; Nakayama M; Yoshida S; Katayama F; Makaie M WO 2019168170, March 1, 2019. [Google Scholar]
- [50].Ji CL, Hong X, J. Am. Chem. Soc 2017, 139, 15522–15529. [DOI] [PubMed] [Google Scholar]
- [51].For further pertinent studies on decarboxylative coupling of carboxylic acids, see:; a) Patra T, Maiti D, Chem. Eur. J 2017, 23, 7382–7401; [DOI] [PubMed] [Google Scholar]; b) Agasti S, Pal T, Achar TK, Maiti S, Pal D, Mandal S, Daud K, Lahiri GK, Maiti D, Angew. Chem. Int. Ed 2019, 58, 11039–11043; [DOI] [PubMed] [Google Scholar]; c) Agasti S, Maity S, Szabo KJ, Maiti D, Adv. Synth. Catal 2015, 357, 2331–2338; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Patra T, Nandi S, Sahoo SK, Maiti D, Chem. Commun 2016, 52, 1432–1435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.