

Abstract
Protein cages (viral and non-viral) found in nature have evolved for a variety of purposes and are found in all kingdoms of life. The main functions of these nanoscale compartments are the protection and delivery of nucleic acids e.g. virus capsids, or the enrichment and sequestration of metabolons e.g. bacterial microcompartments. This review focuses on recent developments of protein cages for use in immunotherapy and therapeutic delivery. In doing so, we highlight the unique ways in which protein cages have informed on fundamental principles governing bio-nano interactions. With the enormous existing design space among naturally occurring protein cages, there is still much to learn from studying them as biomimetic particles.
Introduction
Protein cages are complex macromolecular aggregates, generated from the precise self-assembly of repeated protein subunits. There is a vast diversity of protein cages found in nature including examples comprised of just tens of proteins to thousands. Within this expansive variability in size and shape of protein cages, individual examples are geometrically constrained and generally comprised of a defined number of subunits. As such, they are highly suited to high-resolution bio-physical characterisation such as X-ray crystallography and cryo-electron microscopy, which in many cases provides atomic-level structural detail.1,2 As molecular components for nanomedical devices for the delivery of antigens and therapeutic molecules, self-assembled protein cages display numerous advantages, including the possibility of rational and geometrically defined surface modifications with molecular precision. In addition, protein cages are biocompatible, they are synthesised using standard biotechnology manufacturing processes and they are capable of carrying heterologous cargos via a range of native and heterologous encapsidation strategies. The majority of engineered protein cages are derived from virus capsids including plant viruses, mammalian viruses and bacteriophages. Although bacterial nanoscale compartments such as encapsulins,3,4 ferritin,5,6 and compartments based on enzyme complexes such as lumazine synthase and the E2 subunit of pyruvate dehydrogenase,7–9 have all been extensively investigated as naturally occurring protein cage biomimetic vehicles for biomedical applications.10,11 Furthermore, the computational design of protein cages from non-cage forming proteins is emerging as a source of novel assemblies based on the geometries of protein particles found in nature.12–15 An exciting recent area is the rational engineering16,17 and/or directed laboratory evolution18–23 of cage-forming proteins that is expanding the geometric constraints of existing components for the assembly of protein cages as biomimetic particles.
The variety of protein cage shapes and sizes is illustrated by the range of viruses and virus-like particles (VLPs) used in bio-nanotechnology, from the small isometric plant viruses (~25–30 nm) to the larger protein shells (~80–100 nm) and filamentous particles (up to ~900 nm long) of various bacteriophage.24 The available variation in nanoscale geometry, combined with high individual structural fidelity and particle homogeneity has facilitated their use in investigating the molecular details of bio-nano interactions as discussed below (Fig. 1). For example, the study of virus-derived particles interacting with the immune system is defining the size range and shape of the most effective immunogens; probing interactions with cellular receptors that mediate uptake has shown the importance of surface ligand density in biomimetic design; and analysis of the biodistribution of virus-derived particles is demonstrating the importance of size and shape in circulation and cell interactions. Not only have they proved useful in elucidating these concepts, protein cages are also, therefore, useful in the design of biomimetic vehicles that take advantage of this fundamental knowledge in bio-nano interactions. Here, we review the latest developments in the use of protein cages as immunotherapeutics and as targeted delivery vehicles, with a focus on virus-derived particles.
Fig. 1.
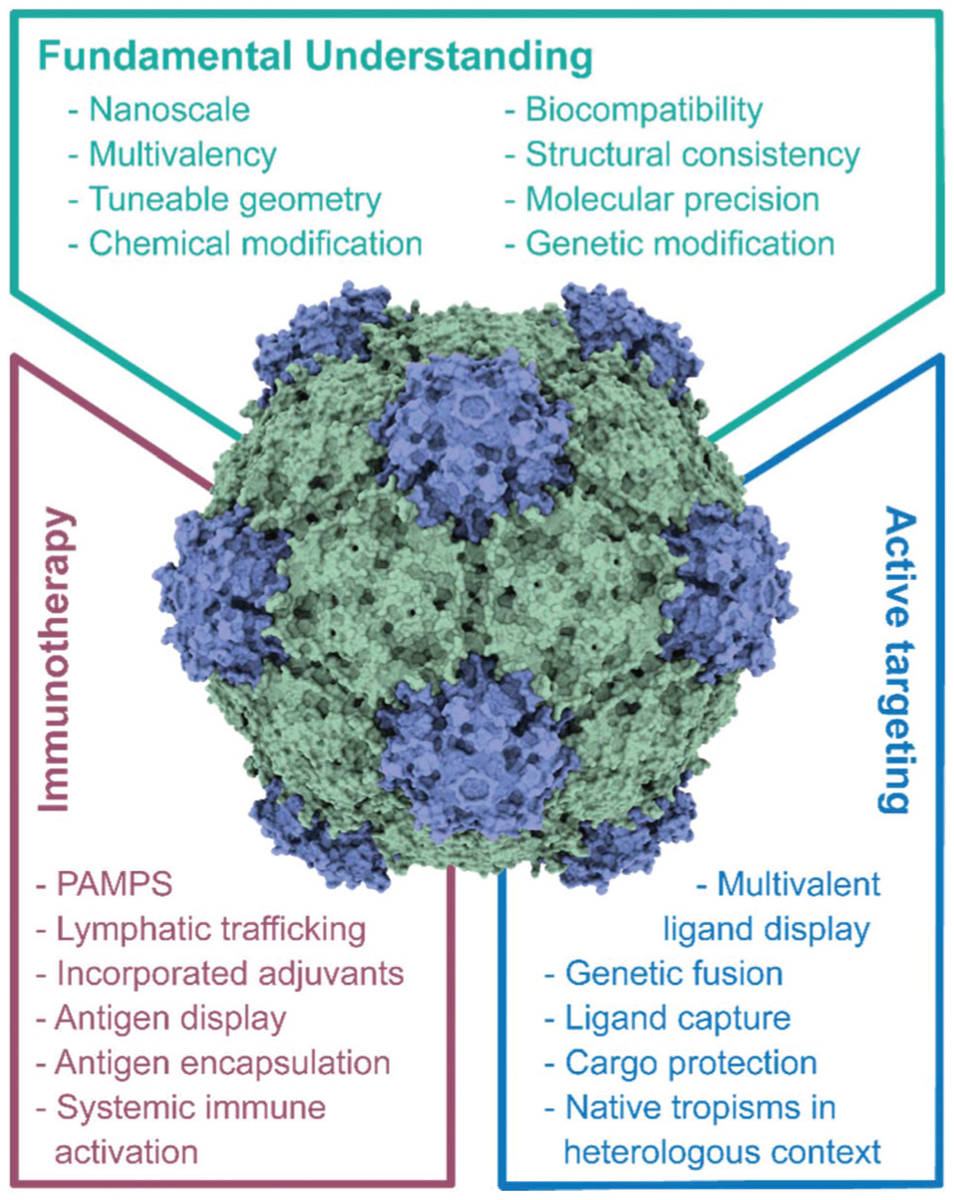
The use of protein cages as biomimetic vehicles. Summary of the salient characteristics of protein cages that have enabled improved knowledge of the molecular details of bio-nano interactions (green). Key properties and features of protein cages that are driving their use as prophylactic and therapeutic vaccines (blue). Key properties and features of protein cages that enable their development as cell-type specific, active targeting vehicles (purple). PAMPs, pathogen-associated molecular patterns.
Stimulating the immune system
The earliest report on the use of VLPs – recombinant virus capsids, as virus mimetics are as vaccines.25 Whether icosahedral or filamentous, the geometry and multivalency of VLPs present pathogen-associated molecular patterns (PAMPs) that trigger innate immune recognition through pathogen-associated molecular recognition receptors (PRRs), most commonly toll-like receptors (TLRs).26 While the proteinaceous capsids are recognized by TLR227 (and likely other PRRs), VLPs may also be engineered to contain RNA and also signal through TLR7/8.28,29 And of course, opportunity exists to co-deliver immune-stimulatory molecules, such as oligodeoxynucleotides (CpGs) serving as TLR9 ligands.30 These immune-stimulatory properties in combination with their size, generally ranging from 20–200 nm for isometric particles, make them ideal candidates for vaccine delivery to the draining lymph nodes and priming interactions with antigen-presenting cells (APCs). It has been demonstrated that intact particles are required to achieve efficient transport and APC processing,31 but it is also emerging that the shape of the protein particle has a significant impact with small (~30 nm) spherical particles found to promote greater APC uptake and processing than filamentous particles.32 Clinical examples of VLP vaccines include those against Human papilloma virus such as GlaxoSmithKline Biologicals’ Cervarix® or Merck’s Gardasil®. Advances in synthetic biology and improved understanding of the engineering of expression systems is fuelling the development pipeline with new vaccine candidates. In early development stages is a plant-produced polio vaccine;33 and poised to enter the market, is Medicago’s plant-based quadrivalent recombinant VLP influenza vaccine.34
Besides the development of non-infectious VLPs as vaccines against the cognate virus, heterologous VLP display technologies have long been established.25 Here, desired epitopes are displayed on a VLP platform. Various engineering design concepts have been developed to achieve epitope display: epitopes can be fused to the coat proteins via genetic engineering, chemical conjugation as well as enzymatic ligation, together enabling the display of peptides, proteins as well as small molecules and carbohydrates. Disease targets include infectious disease, autoimmune disease, addiction, and cancer. The concepts have been widely reported in the literature and we refer the reader to the following reviews and references therein.25,35–38 The particular advantages of using multivalent protein cages as immunogen carriers are highlighted in the following examples. Small molecules such as nicotine and carbohydrates are generally poorly immunogens. Chemical coupling through orthogonal chemistries and display on VLP carriers, however, has been demonstrated to effectively prime humoral and cellular immunity against small molecules. In the case of nicotine, a vaccine has been proposed to help with nicotine addiction;39 and carbohydrate vaccines are candidates for therapeutic cancer vaccines.40 Diverse chemistries and genetic approaches have been developed to display peptide epitopes on the surface of protein cages, while the internal cavity of some VLPs and encapsulins also offers the potential of encapsulating immunogens.41,42 This approach was demonstrated by encapsulating internal influenza proteins into VLPs that stimulated a strong cytotoxic T cell response.42 Combining encapsulation of T cell epitopes with surface-displayed B-cell epitopes thus presents the possibility of creating a truly biomimetic nanostructure that would be capable of stimulating a balanced immune response.
In these applications, the protein cage serves multiple functions: (i) fusion to a large stable structure protects the epitope from premature degradation; (ii) based on their size, VLPs and other protein cages deliver epitopes to the draining lymph nodes and target professional APCs.31 The multivalent protein cage not only serves to deliver the epitope, but also serves as an adjuvant: signalling through PRRs as described above will send immune-stimulatory signals and leads to efficient processing of the delivered epitopes.26 Moreover, as shown with VLPs at least, the carrier’s own epitopes serve as co-stimulatory molecules and processing through the endolysosomal pathways allows for cross-presentation boosting humoral and cellular immunity.43 Although potentially problematic if the immune response is principally targeted against the carrier, a heterologous prime-boost concept was recently presented that made use of immune-orthogonal carriers. Here, the same epitope (tumour-associated antigen human epidermal growth factor receptor 2, HER2) was delivered through repeat administrations using distinct plant VLP carriers for the initial prime and subsequent boosts. Enhanced efficacy of the heterologous delivery of the antigen was achieved by focusing the immune response on the HER2 target and therefore delaying tumour onset after prophylactic vaccination.44
Lastly, recent research has focused on the use of VLPs for in situ vaccine applications. Here the VLP is directly administered into an identified tumour where the immune-stimulatory properties of VLPs activate innate immunity. When successful the process recruits and activates macrophages, neutrophils, and natural killer cells that phagocytose and lyse tumour cells providing a source of antigen to be processed by the innate immune cells; engagement of APCs then also triggers adaptive immune activation. Therefore, this local therapy stimulates systemic anti-tumour immunity and immune memory to protect from outgrowth of metastatic disease and recurrence. Using VLPs and virus particles from Cowpea mosaic virus, efficacy has been demonstrated in mouse models of glioma, ovarian cancer, breast cancer, colon cancer, and melanoma.45,46 Ongoing trials in companion dogs with melanoma indicate that the potent antitumour efficacy of VLPs can be replicated in these animals.47–49 Other VLPs considered as in situ vaccines include M13 bacteriophage,50 Potato virus X,51 Tobacco mosaic virus,52 and Papaya mosaic virus.53 As a regulatory proof-of-concept for this class of therapeutic, already on the market is Amgen’s IMLYGIC® an oncolytic virotherapy engineered to produce cytokines that recruit immune cells to process antigens released from the lysed tumour cells.
Evading the immune system and delivery
Viruses and their VLPs, whether of mammalian, plant or bacteriophage origin, are immunogenic because of their shared properties, principally size and multivalency. Nevertheless, we are beginning to understand that different protein cages are immunogenic to varying degrees. The immunogenicity may also be viewed as a double-edged sword: when protein cages are engineered as vectors for drug and gene delivery, we must overcome the immunogenic properties to avoid premature clearance and/or immunotoxicity. Some approaches borrow strategies from the nanotechnology field, where polymer-coatings are applied to confer stealth properties. The most common example is PEGylation, where poly(ethylene glycol) (PEG) is covalently attached to the VLP; this has long been explored for viral gene delivery.54 Another strategy is to render viruses less immunogenic by genetically removing ‘hotspots’ of antibody recognition, thus enabling viral gene delivery vectors to escape pre-existing immunity.55 An extension of this approach is the use of a broad panel of immune-orthogonal variants of a given protein cage for repeated administration, which is often required during a therapeutic treatment regimen or, as described above, for prime-boost immunisation strategies. The sequential use of Adeno-associated virus coat protein variants of gene delivery vectors successfully evaded adaptive immune response in a preclinical model.56
On the other hand, the nanoscale morphology and, in particular, the multivalency of protein shells also provides opportunity when it comes to engineering cell interactions and the delivery of therapeutic proteins and nucleic acids or imaging agents. The study of virus–cell interactions has shown that there is a close relationship between the valency of attachment and infectious uptake.57,58 Viruses carefully control the residence time and mobility at the cell surface via receptor clustering59,60 in order to balance attachment with the lateral mobility needed to find, or induce, permissive endocytic sites.57 Indeed, assembly of individual subunits into VLPs is required for efficient uptake in vitro.61 Work on ligand-conjugated virus particles has shown that uptake is also influenced by aspect ratio and symmetry of ligand distribution.62 The importance of the symmetry and local density of ligand display in determining interactions with tumour receptors has also been elucidated using RGD63 and IL-4R targeting peptide bunches64 using ferritin cages. In both genetic and chemical conjugations, the distance of the targeting ligands from the surface of the protein cage plays an important role in targeting efficiency and subsequently cellular uptake.65 In addition to informing what parameters are useful for cell uptake, chemically consistent virus-derived particles with varying aspect ratios have also been used to show how this parameter influences biodistribution and intratumoural diffusion.66,67
Leveraging on the multivalent display of ligands on the surface of non-viral protein cages, active targeting to specific cell types has been reported. Recent attempts include targeting of cell receptors on pancreatic cancer using neurophilin-1 binding peptides conjugated to heat shock protein particles,68 hepatocellular carcinoma GPRC78 receptor targeting using SP94 peptides on both encapsulains69 and ferritin,70 and heparan sulfate proteoglycan targeting of glioma71 by GKRK peptides displayed on ferritin cages. The sophisticated designs are departures from the traditional fibronectin-derived RGD peptides72 that indiscriminately bind to cell membranes or by relying on the natural binding of ferritin cages to transferrin receptors.73,74 Targeting moieties are introduced to the surface of the protein cages by genetic fusion, biological or chemical conjugations. Peptides are typically fused to the protein cage subunits and self-assembly of the subunits provides precise spatial control on the arrangements of the targeting ligands on the surface of the protein cages. In addition to peptides, small molecules such as hyaluronic acid75 and folic acid76 have been chemically conjugated to ferritin and the pyruvate dehydrogenase E2 multi-enzyme complex to target CD44 lung cancer and cervical cancer cells, respectively. Recent examples of immunotherapy using pyruvate dehydrogenase E2 particles show that the display of CpG and non-CpG DNA increases targeting to and uptake of particles by APCs.77 Co-delivery of melanoma-derived peptide gp100 and CpG in this way boosted T-cell response and increase animal survival rate by 40%.78 Biomacromolecules such as antibody or modified versions, such as single chain variable fragments, have also been displayed using chemical conjugation79 or by displaying peptides that bind the constant fragment of antibody, capturing the ligand for display.80 Similarly, biological conjugation using SpyTag/SpyCatcher on the surface of encapsulins81 and SortaseA enzyme to modify E2 particles82 have been shown to impart multiple functionalities such as receptor targeting and temperature-dependent phase transitions.
In addition to the presentation of functional moieties, exploiting the cargo carrying capacity of protein cages is an alternative strategy for evading the immune system. For example, the delivery of RNA can be particularly challenging due to its recognition by the immune system and protein cages such as Cowpea chlorotic mottle virus may be used to shield and deliver these sensitive molecules.83 This platform is also used to efficiently deliver small interfering RNAs (siRNAs) to mammalian cells.84,85 In addition to nucleic acids, protein cages have been used to encapsidate therapeutic enzymes such as cytochrome P450s.86 Encapsidation strategies for protein cargo loading is becoming increasingly refined from direct fusion to the structural proteins,87 to engineered electrostatic interactions,88,89 to interactions between capsid proteins of viruses or encapsulins with minimal peptides derived from their native internal proteins.61,90–93 The studies reporting these strategies inform on the characteristics of the protein cage and, in turn, increase the opportunities for protein cages to be used in biomimetic therapeutic contexts. The potential is especially attractive when combined with external functionalisation,94 though for active targeting this potential has yet to be reached.
Passive targeting and native tropism in heterologous contexts
Despite the various attempts to display targeting ligands on the surface of protein cages to impart active targeting in vitro, the in vivo efficacies are yet to be rigorously tested. Protein cages face similar challenges to other nanoparticle-based formulations, specifically liver and renal clearance. Despite these challenges, efforts to engineer protein cages as receptor-targeted cancer therapeutics is predicated on the expectation that they will passively accumulate in tumour tissues. The typical dimensions of protein cages and VLPs, generally below 500 nm, is well within the limit of enhanced permeation and retention (EPR) effect that results in selective accumulation due to the leaky vasculature found in tumours but not in normal tissues.95 Indeed, clearance by various filtering systems – hepatic, renal, lymphatic – are in general far more effective in terms of nanoparticle accumulation than engineered active, or receptor-mediated, targeting is in avoiding these. Perhaps then, the best use of naturally multivalent and precisely organised nanoparticles is to use native tropisms for delivery to specific tissues and cell types. Serotypes of Adeno-associated viruses are used in this way to favour gene delivery to specific tissues.96 Examples include some of the most effective CRISPR/Cas-mediated in vivo gene editing to date, in this case, in the liver.97
While structurally defined mammalian viruses generally have known cell surface receptors, it is likely that non-viral protein cages and those derived from non-mammalian viruses also hold considerable potential for active targeting without further modification. Indeed, tissue tropism of some plant viruses has been reported. For example the plant virus, Cowpea mosaic virus, binds to the mammalian protein vimentin98 and thus can be targeted to cancer99 and cardiovascular inflammation.100 While most protein-based nanoparticles, including plant viruses, have short circulation and fast tissue clearance, a recent study highlighted that Physalis mosaic virus-like particles enabled longitudinal imaging in a mouse model of prostate cancer, providing fluorescent and magnetic resonance contrast for 10 days.101 In another example, it was shown that potato virus X has a native tropism to B cells in mice.102 Therefore, while much effort has been placed on borrowing mechanisms to achieve tissue specificity from the mammalian viruses (e.g. RGD peptide ligands) possible intrinsic targeting mechanisms of plant viruses, bacteriophages and other protein cages remains largely unexplored.
Conclusions
Protein cages have been effectively used to investigate fundamental aspects of bio-nano interactions. As discussed here, advances range from elucidating how the structure of viruses stimulate the immune system, to probing the roles of symmetry, valency and density on receptor-mediated cellular uptake. The biocompatibility of proteins cages, in addition to the capacity for precise engineering, means that they also hold considerable potential in immunotherapy and cellular delivery. In an immunotherapy context, the nanoscale size and repetitive surface geometry of protein cages results in stimulatory interactions with, and targeting to, tissues and cells of the immune system. For targeted delivery requiring avoidance of the immune system, biocompatibility, structural consistency, and amenability to sophisticated engineering have enabled significant progress. However, the true potential of many protein cage platforms could be in their undiscovered targeting abilities.
Acknowledgements
N. F. S. is supported by the following agencies NSF (CAREER DMR-1841848), NIH (R01 HL137674, R21 EB024874, R01 CA224605, R01 CA202814, U01 CA218292) and ACS (128319-RSG-15-144-01-CDD). S. L. is partially supported by NTU-Northwestern Institute for Nanomedicine. F. S. acknowledges the support of the CSIRO Synthetic Biology Future Science Platform.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.de Ruiter MV, Klem R, Luque D, Cornelissen JJLM and Castón JR, Nanoscale, 2019, 11, 4130–4146. [DOI] [PubMed] [Google Scholar]
- 2.Heddle JG, Chakraborti S and Iwasaki K, Curr. Opin. Struct. Biol, 2017, 43, 148–155. [DOI] [PubMed] [Google Scholar]
- 3.Giessen TW and Silver PA, Nat. Microbiol, 2017, 2, 17029. [DOI] [PubMed] [Google Scholar]
- 4.Giessen TW, Curr. Opin. Chem. Biol, 2016, 34, 1–10. [DOI] [PubMed] [Google Scholar]
- 5.Khoshnejad M, Parhiz H, Shuvaev VV, Dmochowski IJ and Muzykantov VR, J. Controlled Release, 2018, 282, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Gao H, Zhang Y, Liu G, Niu G and Chen X, Front. Chem. Sci. Eng, 2017, 11, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seebeck FP, Woycechowsky KJ, Zhuang W, Rabe JP and Hilvert D, J. Am. Chem. Soc, 2006, 128, 4516–4517. [DOI] [PubMed] [Google Scholar]
- 8.Song Y, Kang YJ, Jung H, Kim H, Kang S and Cho H, Sci. Rep, 2015, 5, 15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalmau M, Lim S and Wang SW, Nano Lett, 2009, 9, 160–166. [DOI] [PubMed] [Google Scholar]
- 10.Bhaskar S and Lim S, NPG Asia Mater, 2017, 9, e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molino NM and Wang SW, Curr. Opin. Biotechnol, 2014, 28, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bale JB, Gonen S, Liu Y, Sheffler W, Ellis D, Thomas C, Cascio D, Yeates TO, Gonen T, King NP and Baker D, Science, 2016, 353, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsia Y, Bale JB, Gonen S, Shi D, Sheffler W, Fong KK, Nattermann U, Xu C, Huang P-S, Ravichandran R, Yi S, Davis TN, Gonen T, King NP and Baker D, Nature, 2016, 535, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Padilla JE, Colovos C and Yeates TO, Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 2217–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, André I, Gonen T, Yeates TO and Baker D, Science, 2012, 336, 1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azuma Y, Herger M and Hilvert D, J. Am. Chem. Soc, 2018, 140, 558–561. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki E, Böhringer D, van de Waterbeemd M, Leibundgut M, Zschoche R, Heck AJR, Ban N and Hilvert D, Nat. Commun, 2017, 8, 14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asensio MA, Morella NM, Jakobson CM, Hartman EC, Glasgow JE, Sankaran B, Zwart PH and Tullman-Ercek D, Nano Lett, 2016, 16, 5944–5950. [DOI] [PubMed] [Google Scholar]
- 19.Butterfield GL, Lajoie MJ, Gustafson HH, Sellers DL, Nattermann U, Ellis D, Bale JB, Ke S, Lenz GH, Yehdego A, Ravichandran R, Pun SH, King NP and Baker D, Nature, 2017, 552, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartman EC, Jakobson CM, Favor AH, Lobba MJ, Álvarez-Benedicto E, Francis MB and Tullman-Ercek D, Nat. Commun, 2018, 9, 1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terasaka N, Azuma Y and Hilvert D, Proc. Natl. Acad. Sci. U. S. A, 2018, 115, 5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wörsdörfer B, Woycechowsky KJ and Hilvert D, Science, 2011, 331, 589. [DOI] [PubMed] [Google Scholar]
- 23.Davidsson M, Wang G, Aldrin-Kirk P, Cardoso T, Nolbrant S, Hartnor M, Mudannayake J, Parmar M and Björklund T, Proc. Natl. Acad. Sci. U. S. A, 2019, 201910061, DOI: 10.1073/pnas.1910061116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen AM and Steinmetz NF, Chem. Soc. Rev, 2016, 45, 4074–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohsen MO, Zha L, Cabral-Miranda G and Bachmann MF, Semin. Immunol, 2017, 34, 123–132. [DOI] [PubMed] [Google Scholar]
- 26.Bachmann MF and Jennings GT, Nat. Rev. Immunol, 2010, 10, 787–796. [DOI] [PubMed] [Google Scholar]
- 27.Shepardson KM, Schwarz B, Larson K, Morton RV, Avera J, McCoy K, Caffrey A, Harmsen A, Douglas T and Rynda-Apple A, mBio, 2017, 8, e01356–17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Albakri MM, Veliz FA, Fiering SN, Steinmetz NF and Sieg SF, Immunology, 2019, 159, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carignan D, Herblot S, Laliberte-Gagne ME, Bolduc M, Duval M, Savard P and Leclerc D, Nanomedicine, 2018, 14, 2317–2327. [DOI] [PubMed] [Google Scholar]
- 30.Storni T, Ruedl C, Schwarz K, Schwendener RA, Renner WA and Bachmann MF, J. Immunol, 2004, 172, 1777–1785. [DOI] [PubMed] [Google Scholar]
- 31.Link A, Zabel F, Schnetzler Y, Titz A, Brombacher F and Bachmann MF, J. Immunol, 2012, 188, 3724–3733. [DOI] [PubMed] [Google Scholar]
- 32.Shukla S, Myers JT, Woods SE, Gong X, Czapar AE, Commandeur U, Huang AY, Levine AD and Steinmetz NF, Biomaterials, 2017, 121, 15–27. [DOI] [PubMed] [Google Scholar]
- 33.Marsian J, Fox H, Bahar MW, Kotecha A, Fry EE, Stuart DI, Macadam AJ, Rowlands DJ and Lomonossoff GP, Nat. Commun, 2017, 8, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pillet S, Couillard J, Trepanier S, Poulin JF, Yassine-Diab B, Guy B, Ward BJ and Landry N, PLoS One, 2019, 14, e0216533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee KL, Twyman RM, Fiering S and Steinmetz NF, WIREs Nanomed. Nanobiotechnol, 2016, 8, 554–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hefferon KL, Vaccines, 2018, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohsen MO, Speiser DE, Knuth A and Bachmann MF, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol, 2019, e1579, DOI: 10.1002/wnan.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charlton Hume HK, Vidigal J, Carrondo MJT, Middelberg APJ, Roldao A and Lua LHL, Biotechnol. Bioeng, 2019, 116, 919–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maurer P, Jennings GT, Willers J, Rohner F, Lindman Y, Roubicek K, Renner WA, Muller P and Bachmann MF, Eur. J. Immunol, 2005, 35, 2031–2040. [DOI] [PubMed] [Google Scholar]
- 40.Yin Z, Chowdhury S, McKay C, Baniel C, Wright WS, Bentley P, Kaczanowska K, Gildersleeve JC, Finn MG, BenMohamed L and Huang X, ACS Chem. Biol, 2015, 10, 2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lagoutte P, Mignon C, Stadthagen G, Potisopon S, Donnat S, Mast J, Lugari A and Werle B, Vaccine, 2018, 36, 3622–3628. [DOI] [PubMed] [Google Scholar]
- 42.Patterson DP, Rynda-Apple A, Harmsen AL, Harmsen AG and Douglas T, ACS Nano, 2013, 7, 3036–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storni T and Bachmann MF, J. Immunol, 2004, 172, 6129–6135. [DOI] [PubMed] [Google Scholar]
- 44.Cai H, Shukla S, Wang C, Masarapu H and Steinmetz NF, J. Am. Chem. Soc, 2019, 141, 6509–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF and Fiering S, Nat. Nanotechnol, 2016, 11, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kerstetter-Fogle A, Shukla S, Wang C, Beiss V, Harris PLR, Sloan AE and Steinmetz NF, Cancers, 2019, 11, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoopes PJ, Mazur CM, Osterberg B, Song A, Gladstone DJ, Steinmetz NF, Veliz FA, Bursey AA, Wagner RJ and Fiering SN, Proc. SPIE-Int. Soc. Opt. Eng, 2017, 10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoopes PJ, Wagner RJ, Duval K, Kang K, Gladstone DJ, Moodie KL, Crary-Burney M, Ariaspulido H, Veliz FA, Steinmetz NF and Fiering SN, Mol. Pharm, 2018, 15, 3717–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoopes PJ, Moodie KL, Petryk AA, Petryk JD, Sechrist S, Gladstone DJ, Steinmetz NF, Veliz FA, Bursey AA, Wagner RJ, Rajan A, Dugat D, Crary-Burney M and Fiering SN, Proc. SPIE-Int. Soc. Opt. Eng, 2017, 10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eriksson F, Tsagozis P, Lundberg K, Parsa R, Mangsbo SM, Persson MA, Harris RA and Pisa P, J. Immunol, 2009, 182, 3105–3111. [DOI] [PubMed] [Google Scholar]
- 51.Lee KL, Murray AA, Le DHT, Sheen MR, Shukla S, Commandeur U, Fiering S and Steinmetz NF, Nano Lett, 2017, 17, 4019–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murray AA, Wang C, Fiering S and Steinmetz NF, Mol. Pharm, 2018, 15, 3700–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lebel ME, Chartrand K, Tarrab E, Savard P, Leclerc D and Lamarre A, Nano Lett, 2016, 16, 1826–1832. [DOI] [PubMed] [Google Scholar]
- 54.Wonganan P and Croyle MA, Viruses, 2010, 2, 468–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith JK and Agbandje-McKenna M, PLoS Pathog, 2018, 14, e1006929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moreno AM, Palmer N, Alemán F, Chen G, Pla A, Jiang N, Leong Chew W, Law M and Mali P, Nat. Biomed. Eng, 2019, 3, 806–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boulant S, Stanifer M and Lozach PY, Viruses, 2015, 7, 2794–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varga CM, Wickham TJ and Lauffenburger DA, Biotechnol. Bioeng, 2000, 70, 593–605. [DOI] [PubMed] [Google Scholar]
- 59.Cureton DK, Harbison CE, Cocucci E, Parrish CR and Kirchhausen T, J. Virol, 2012, 86, 5330–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Müller M, Lauster D, Wildenauer HHK, Herrmann A and Block S, Nano Lett, 2019, 19, 1875–1882. [DOI] [PubMed] [Google Scholar]
- 61.Dashti NH, Abidin RS and Sainsbury F, ACS Nano, 2018, 12, 4615–4623. [DOI] [PubMed] [Google Scholar]
- 62.Wen AM and Steinmetz NF, Adv. Healthcare Mater, 2014, 3, 1739–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang T, Zhu W, Li C, Zhang F, Gao D, Zhang ZP, Liang A, Zhang XE and Li F, Small, 2019, 15, e1904838. [DOI] [PubMed] [Google Scholar]
- 64.Jeon JO, Kim S, Choi E, Shin K, Cha K, So IS, Kim SJ, Jun E, Kim D, Ahn HJ, Lee BH, Lee SH and Kim IS, ACS Nano, 2013, 7, 7462–7471. [DOI] [PubMed] [Google Scholar]
- 65.Shewmake TA, Solis FJ, Gillies RJ and Caplan MR, Biomacromolecules, 2008, 9, 3057–3064. [DOI] [PubMed] [Google Scholar]
- 66.Chariou PL, Lee KL, Pokorski JK, Saidel GM and Steinmetz NF, J. Phys. Chem. B, 2016, 120, 6120–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shukla S, Eber FJ, Nagarajan AS, DiFranco NA, Schmidt N, Wen AM, Eiben S, Twyman RM, Wege C and Steinmetz NF, Adv. Healthcare Mater, 2015, 4, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murata M, Narahara S, Kawano T, Hamano N, Piao JS, Kang JH, Ohuchida K, Murakami T and Hashizume M, Mol. Pharm, 2015, 12, 1422–1430. [DOI] [PubMed] [Google Scholar]
- 69.Moon H, Lee J, Min J and Kang S, Biomacromolecules, 2014, 15, 3794–3801. [DOI] [PubMed] [Google Scholar]
- 70.Jiang B, Zhang R, Zhang J, Hou Y, Chen X, Zhou M, Tian X, Hao C, Fan K and Yan X, Theranostics, 2019, 9, 2167–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhai M, Wang Y, Zhang L, Liang M, Fu S, Cui L, Yang M, Gong W, Li Z, Yu L, Xie X, Yang C, Yang Y and Gao C, Drug Delivery, 2018, 25, 1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uchida M, Flenniken ML, Allen M, Willits DA, Crowley BE, Brumfield S, Willis AF, Jackiw L, Jutila M, Young MJ and Douglas T, J. Am. Chem. Soc, 2006, 128, 16626–16633. [DOI] [PubMed] [Google Scholar]
- 73.Bouzinab K, Thomas NR, Turyanska L, Gershkovich P, Weston N, Ashford MB and Bradshaw TD, Neuro-Oncology, 2019, 21, iv4. [Google Scholar]
- 74.Ravishankar S and Lim S, Nano Res, 2019, 12, 2925–2932. [Google Scholar]
- 75.Luo Y, Wang X, Du D and Lin Y, Biomater. Sci, 2015, 3, 1386–1394. [DOI] [PubMed] [Google Scholar]
- 76.Ren D, Kratz F and Wang SW, Biochem. Eng. J, 2014, 89, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Molino NM, Neek M, Tucker JA, Nelson EL and Wang SW, ACS Biomater. Sci. Eng, 2017, 3, 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Molino NM, Neek M, Tucker JA, Nelson EL and Wang SW, Biomaterials, 2016, 86, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buecheler JW, Howard CB, de Bakker CJ, Goodall S, Jones ML, Win T, Peng T, Tan CH, Chopra A, Mahler SM and Lim S, J. Chem. Technol. Biotechnol, 2015, 90, 1230–1236. [Google Scholar]
- 80.Kickhoefer VA, Han M, Raval-Fernandes S, Poderycki MJ, Moniz RJ, Vaccari D, Silvestry M, Stewart PL, Kelly KA and Rome LH, ACS Nano, 2009, 3, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bae Y, Kim GJ, Kim H, Park SG, Jung HS and Kang S, Biomacromolecules, 2018, 19, 2896–2904. [DOI] [PubMed] [Google Scholar]
- 82.Chen Q, Sun Q, Molino NM, Wang SW, Boder ET and Chen W, Chem. Commun, 2015, 51, 12107–12110. [DOI] [PubMed] [Google Scholar]
- 83.Azizgolshani O, Garmann RF, Cadena-Nava R, Knobler CM and Gelbart WM, Virology, 2013, 441, 12–17. [DOI] [PubMed] [Google Scholar]
- 84.Lam P and Steinmetz NF, Biomater. Sci, 2019, 7, 3138–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pretto C and van Hest JCM, Bioconjugate Chem, 2019, 30, 3069–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sánchez-Sánchez L, Tapia-Moreno A, Juarez-Moreno K, Patterson DP, Cadena-Nava RD, Douglas T and Vazquez-Duhalt R, J. Nanobiotechnol, 2015, 13, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brillault L, Jutras PV, Dashti N, Thuenemann EC, Morgan G, Lomonossoff GP, Landsberg MJ and Sainsbury F, ACS Nano, 2017, 11, 3476–3484. [DOI] [PubMed] [Google Scholar]
- 88.Brasch M, Putri RM, de Ruiter MV, Luque D, Koay MST, Castón JR and Cornelissen JJLM, J. Am. Chem. Soc, 2017, 139, 1512–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zschoche R and Hilvert D, J. Am. Chem. Soc, 2015, 137, 16121–16132. [DOI] [PubMed] [Google Scholar]
- 90.McCoy K, Selivanovitch E, Luque D, Lee B, Edwards E, Castón JR and Douglas T, Biomacromolecules, 2018, 19, 3738–3746. [DOI] [PubMed] [Google Scholar]
- 91.O’Neil A, Reichhardt C, Johnson B, Prevelige PE and Douglas T, Angew. Chem., Int. Ed, 2011, 50, 7425–7428. [DOI] [PubMed] [Google Scholar]
- 92.Putri RM, Allende-Ballestero C, Luque D, Klem R, Rousou K-A, Liu A, Traulsen CHH, Rurup WF, Koay MST, Castón JR and Cornelissen JJLM, ACS Nano, 2017, 11, 12796–12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rurup WF, Snijder J, Koay MST, Heck AJR and Cornelissen JJLM, J. Am. Chem. Soc, 2014, 136, 3828–3832. [DOI] [PubMed] [Google Scholar]
- 94.Sharma J, Uchida M, Miettinen HM and Douglas T, Nanoscale, 2017, 9, 10420–10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fang J, Nakamura H and Maeda H, Adv. Drug Delivery Rev, 2011, 63, 136–151. [DOI] [PubMed] [Google Scholar]
- 96.Srivastava A, Curr. Opin. Virol, 2016, 21, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA and Zhang F, Nature, 2015, 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G and Manchester M, PLoS Pathog, 2009, 5, e1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Steinmetz NF, Cho C-F, Ablack A, Lewis JD and Manchester M, Nanomedicine, 2011, 6, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Plummer EM, Thomas D, Destito G, Shriver LP and Manchester M, Nanomedicine, 2012, 7, 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hu H, Masarapu H, Gu Y, Zhang Y, Yu X and Steinmetz NF, ACS Appl. Mater. Interfaces, 2019, 11, 18213–18223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shukla S, Wen AM, Ayat NR, Commandeur U, Gopalkrishnan R, Broome A-M, Lozada KW, Keri RA and Steinmetz NF, Nanomedicine, 2014, 9, 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]