

Abstract
Eukaryotic RNA polymerase II (Pol II) contains a tail‐like, intrinsically disordered carboxy‐terminal domain (CTD) comprised of heptad‐repeats, that functions in coordination of the transcription cycle and in coupling transcription to co‐transcriptional processes. The CTD repeat number varies between species and generally increases with genome size, but the reasons for this are unclear. Here, we show that shortening the CTD in human cells to half of its length does not generally change pre‐mRNA synthesis or processing in cells. However, CTD shortening decreases the duration of promoter‐proximal Pol II pausing, alters transcription of putative enhancer elements, and delays transcription activation after stimulation of the MAP kinase pathway. We suggest that a long CTD is required for efficient enhancer‐dependent recruitment of Pol II to target genes for their rapid activation.
Keywords: enhancer, gene expression, RNA polymerase II, transcription kinetics
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics
Characterization of a human RNAPII mutant lacking half of its disordered regulatory tail reveals a role for terminal repeat number in efficient enhancer‐dependent recruitment of Pol II.

Introduction
RNA polymerase II (Pol II) carries out transcription of protein‐coding genes and many non‐coding RNA species in eukaryotic cells (Cramer, 2019). The largest subunit of Pol II, RPB1, contains a unique C‐terminal domain (CTD) that consists of heptapeptide repeats with the consensus sequence YSPTSPS (Allison et al, 1985; Corden et al, 1985). The CTD forms a long and flexible, tail‐like extension from the Pol II body (Cramer et al, 2001). The CTD is essential in yeast, Drosophila, and mammalian cells (Allison et al, 1988; Bartolomei et al, 1988). In vitro, the CTD is dispensable for basal RNA synthesis (Corden, 2013), but required for activated transcription (Koleske et al, 1992).
A plethora of biochemical and genetic studies have demonstrated that the CTD functions in coordinating the transcription cycle with RNA processing and other nuclear events (Phatnani & Greenleaf, 2006; Chapman et al, 2008; Buratowski, 2009; Mayer et al, 2012; Hsin & Manley, 2012; Eick & Geyer, 2013; Zaborowska et al, 2016; Harlen & Churchman, 2017; Gerber et al, 2020). The CTD recruits factors for pre‐mRNA processing, in particular splicing (Fong & Bentley, 2001; David et al, 2011; Hsin & Manley, 2012). Factor binding to the CTD relies on interactions with short regions in the CTD comprising only 1–3 heptapeptide repeats (Meinhart et al, 2005).
The length of the CTD differs strongly between species. CTD length appears to scale with genome size and to correlate inversely with gene density (Stiller & Hall, 2002; Quintero‐Cadena et al, 2020). Genetic studies revealed the minimal number of CTD repeats that supports viability of various species (Bartolomei et al, 1988; West & Corden, 1995; Schwer et al, 2012; Lu et al, 2019). In the yeast Saccharomyces cerevisiae, only eight out of the 26 CTD repeats are required for viability, although 13 repeats are needed to overcome temperature sensitivity (Nonet & Young, 1989). The human CTD contains 52 repeats, but human cells expressing Pol II with only 25 CTD repeats grow normally (Boehning et al, 2018).
Based on available literature, it remains poorly understood why a certain CTD length is required for Pol II function in various species. CTD length correlates with the distance between gene promoters and their regulatory enhancer elements, suggesting that the CTD facilitates promoter‐enhancer contacts (Allen & Taatjes, 2015). Consistent with this model, CTD shortening can reduce expression of genes that are controlled by enhancers (Allison & Ingles, 1989; Scafe et al, 1990; Gerber et al, 1995; Aristizabal et al, 2013). Recent studies suggested that the CTD is involved in the formation of promoter‐enhancer contacts that rely on protein clustering driven by liquid–liquid phase separation (LLPS) (Sabari et al, 2018; Nair et al, 2019). Indeed, the CTD can undergo LLPS in vitro (Boehning et al, 2018) and can be incorporated into LLPS droplets of transcriptional activators (Kwon et al, 2014; Burke et al, 2015). The CTD is also important for Pol II clustering in vivo, as shortening of the human CTD from 52 to 25 repeats decreases the number and size of Pol II clusters in the human cell nucleus (Boehning et al, 2018).
Here, we systematically investigate the functional consequences of shortening the length of the CTD in human cells. We use a human cell line that expresses Pol II with a CTD truncated from 52 to 25 repeats (Boehning et al, 2018). Multiomics analysis of this cell line shows that CTD shortening does not generally alter RNA synthesis and processing, but that rapid RNA synthesis changes in response to an external signal are delayed and compromised. These results reveal that the normal CTD length is required for efficient transcription activation in human cells and are consistent with the model that the CTD is critical for rapid Pol II recruitment to genes upon their activation.
Results
CTD shortening hardly alters the human transcriptome
To investigate the functional consequences of CTD length shortening in human cells, we used U2OS osteosarcoma cells carrying an α‐amanitin‐resistant variant of RPB1 (Meininghaus et al, 2000) with 52 CTD repeats (“RPB1‐52R”, corresponding to wild‐type) or with 25 CTD repeats (“RPB1‐25R”) (Boehning et al, 2018). To characterize RNA metabolism genome‐wide, we subjected both cell lines to transient transcriptome analysis (TT‐seq) (Fig 1A and B, Materials and Methods). TT‐seq uses a short pulse of metabolic RNA labeling with 4‐thiouridine and subsequent sequencing of the labeled, newly synthesized RNA fragments (Schwalb et al, 2016). Together with standard RNA‐seq and with the use of kinetic modeling (Sun et al, 2012), TT‐seq can provide RNA synthesis and degradation rates.
Figure 1. CTD shortening hardly changes the transcriptome and RNA metabolism in human cells.
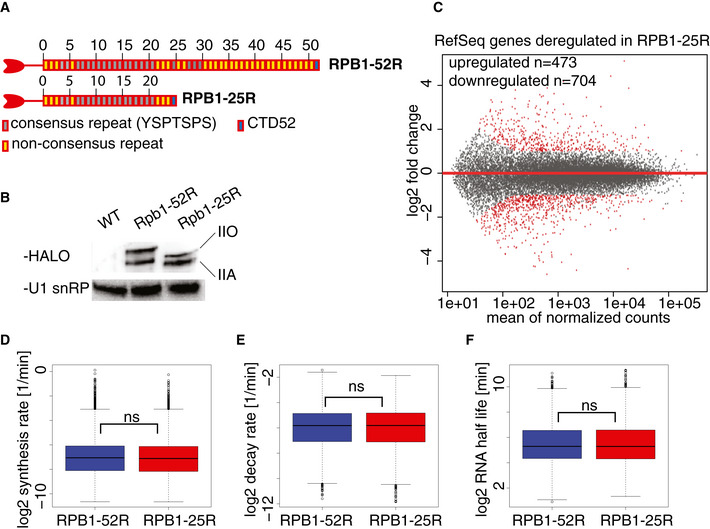
- Schematic representation of the CTD variants expressed in U2OS cells used in this study.
- Western blot verifying the expression of the CTD variants RPB1‐52R and RPB1‐25R in U2OS cells in whole cell extracts. U1 snRNP was used as a loading control.
- Changes in RNA synthesis upon CTD shortening. MA plot showing RNA synthesis changes in TT‐seq datasets upon CTD shortening in U2OS cells in steady state conditions. RPB1‐52R cells are used as a control, and the data were normalized using spike‐in counts. 16,214 expressed genes annotated in RefSeq were analyzed. Differentially expressed genes are in red. 473 genes were significantly upregulated (adjusted P value < 0.05, log2 fold change ≥ 1), and 704 genes were significantly downregulated (adjusted P value < 0.05, log2 fold change ≤ 1).
- Box plot of estimated RNA synthesis rates of expressed RefSeq transcripts (12,014 transcripts) based on TT‐seq and RNA‐seq datasets in RPB1‐52R and RPB1‐25R cells. Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range.
- Box plot of estimated RNA degradation rates of expressed RefSeq transcripts (12,014 transcripts) based on TT‐seq and RNA‐seq datasets in RPB1‐52R and RPB1‐25R cells. Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range.
- Box plot of estimated RNA half‐lives of expressed RefSeq transcripts (12,014 transcripts) based on TT‐seq and RNA‐seq datasets in RPB1‐52R and RPB1‐25R cells. Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range.
We collected TT‐seq and RNA‐seq data for two biological replicates in both RPB1‐52R and RPB1‐25R cells (Fig EV1A and B). We used RNA spike‐in probes to enable detection of global changes (Materials and Methods). We also confirmed that expression of the RPB1 mutation that confers α‐amanitin resistance does not alter RNA levels (Fig EV1C). Comparison of the data from the two different cell lines shows that CTD shortening does not lead to global changes in RNA levels for RefSeq genes (Fig 1C). Only ~ 7% of all expressed genes were differentially expressed (Padj < 0.05 with fold change > 2 (upregulated genes) or < 0.5 (downregulated genes)) (Fig 1C). Downregulated genes were enriched in factors regulating mesenchymal cell proliferation (Fig EV1D), in agreement with slightly reduced cell proliferation (Fig EV1E). We also did not observe changes in the RNA synthesis of transposable elements (Fig EV1F). Consistent with these findings, mRNA synthesis and degradation rates as well as mRNA half‐lives were also essentially unchanged (Fig 1D–F). In conclusion, CTD shortening does generally not alter the transcriptome or RNA metabolism in human cells.
Figure EV1. The transcriptome and RNA metabolism in human cells hardly change upon CTD shortening.
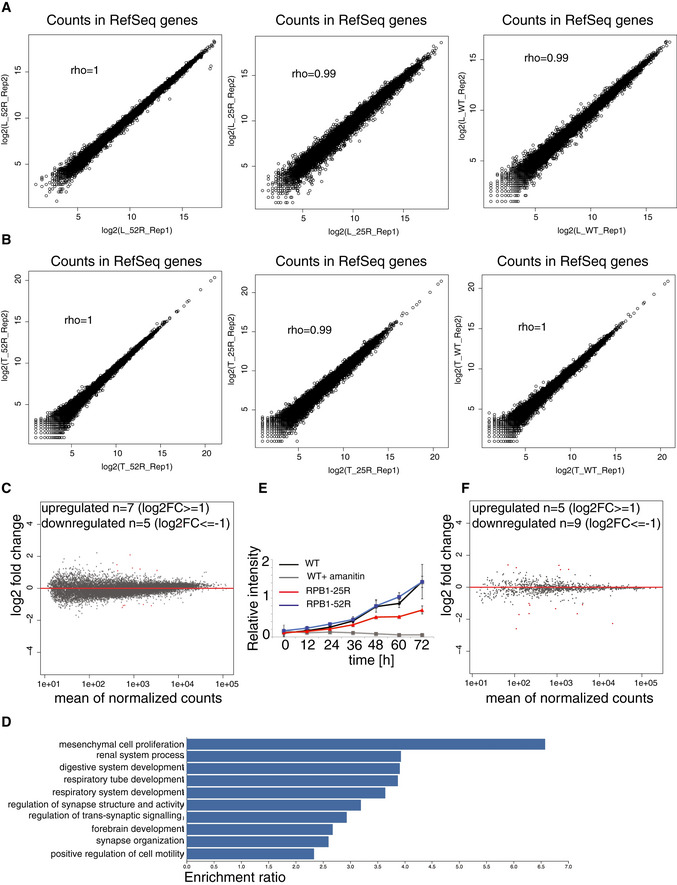
- Spearman correlation of read counts in RefSeq genes in biological replicates of TT‐seq experiments.
- Spearman correlation of read counts in RefSeq genes in biological replicates of total RNA‐ seq experiments.
- MA plot showing changes in RNA synthesis in TT‐seq datasets upon expression of the α‐amanitin‐resistant full‐length CTD variant in U2OS cells in steady state conditions. Wild‐type U2OS cells are used as a control, and the data were normalized using spike‐in counts. 16,214 expressed genes annotated in RefSeq were analyzed, 7 genes were significantly upregulated (adjusted P value < 0.05, log2 fold change ≥ 1, depicted in red) and 5 genes were significantly downregulated (adjusted P value < 0.05, log2 fold change ≤ 1, depicted in red).
- Gene ontology analysis of significantly downregulated genes (adjusted P value < 0.05, log2 fold change ≤ 1) upon CTD shortening. GO categories with FDR≤ 0.05 are shown.
- Proliferation curve generated for cells expressing the α‐amanitin‐resistant RPB1‐52R and RPB1‐25R CTD variants, as well as wild‐type U2OS cells. Error bars show standard deviation of 3 biological replicates. Statistical significance was estimated using two‐way ANOVA, followed by the Tukey multiple comparisons test. The difference in growth rate between RPB1‐52R and RPB1‐25R cells is significant (adjusted P value = 0.0014322).
- RNA synthesis of transposable elements in TT‐seq datasets upon expression RPB1‐25R CTD variant in U2OS cells in steady state conditions. Wild‐type U2OS cells are used as a control and the data were normalized using spike‐in counts. 1,026 expressed transposable elements were analyzed. Differentially expressed transposons are in red. Five were significantly upregulated (adjusted P value < 0.05, log2 fold change ≥ 1) and 9 were significantly downregulated (adjusted P value < 0.05, log2 fold change ≤ 1).
CTD shortening hardly affects pre‐mRNA splicing
In order to reveal possible consequences of CTD shortening for pre‐mRNA splicing kinetics, we used our TT‐seq datasets and quantified sequencing reads that were derived from unspliced transcripts (“unspliced reads”). These reads either span exon–intron junctions (5′ splice sites, 5′SSs) or intron–exon junctions (3′ splice sites, 3′SSs). To avoid confounding effects of alternative splicing, we used only major mRNA isoforms that constitute at least 70% of total RNA‐seq expression for a given gene (Materials and Methods). Using this criterion, we identified a total of 6,260 major RNA isoforms in RPB1‐52R and in RPB1‐25R cells, containing 24,393 5′SSs and 24,995 3′SSs. We then computed a “splicing ratio” that we defined as the ratio of spliced (exon–exon) reads over the sum of spliced and unspliced reads. CTD shortening did increase the splicing ratio only very slightly (Fig 2A), and this was independent of the position of the intron within the transcript (Fig 2B–D).
Figure 2. CTD shortening hardly alters pre‐mRNA splicing kinetics and mRNA isoforms.
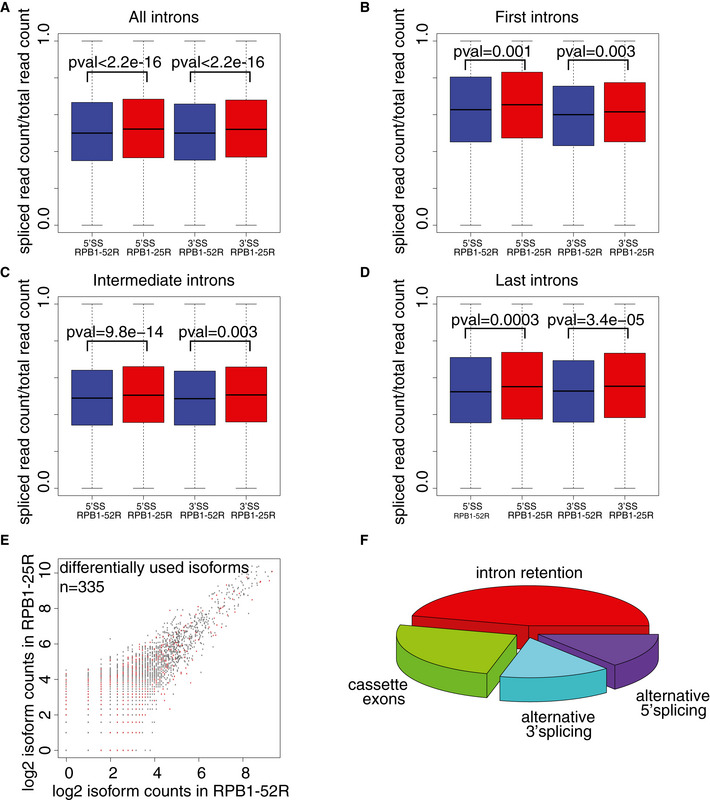
-
ABox plots showing ratios of spliced TT‐seq reads over total unspliced and spliced TT‐seq reads for all constitutive 5′SSs and 3′SSs detected in major RNA isoforms in RPB1‐52R and RPB1‐25R cells. P value was calculated using Mann–Whitney U‐test. Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
-
B–DBox plots showing ratios of spliced TT‐seq reads over total unspliced and spliced TT‐seq reads in RPB1‐52R and RPB1‐25R cells for all constitutive 5′SSs and 3′SSs detected in major RNA isoforms based on intron position in the transcript: (B) first intron junction, (C) intermediate intron junctions and (D) last intron junctions. P values were calculated using Mann–Whitney U‐test. Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range.
-
EDifferential mRNA isoform usage upon CTD shortening. Scatter plot showing read counts for mRNA isoforms detected in the chromatin fraction of RPB1‐52R and RPB1‐25R cells using long‐read sequencing. 47,416 isoforms were detected, RPB1‐52R condition was used as a control. 335 isoforms show differential expression (P value < 0.05, Fischer’s exact test).
-
FAlternative splicing events [in %] detected in 335 differentially expressed isoforms upon CTD shortening in U2OS cells.
To investigate whether CTD shortening affects alternative splicing (Cramer et al, 1999), we isolated chromatin‐associated RNA, which is enriched in nascent pre‐mRNAs (Bhatt et al, 2012). We directly sequenced these long RNAs with the Oxford Nanopore technology from both types of cells (Materials and Methods). In these data, only 335 out of 47,416 detected mRNA isoforms were differentially expressed (Fig 2E), and the majority of these showed intron retention (Fig 2F). Together, these results show that CTD shortening in human cells does hardly alter pre‐mRNA splicing and alternative splicing.
CTD shortening slightly alters promoter‐proximal pausing of Pol II
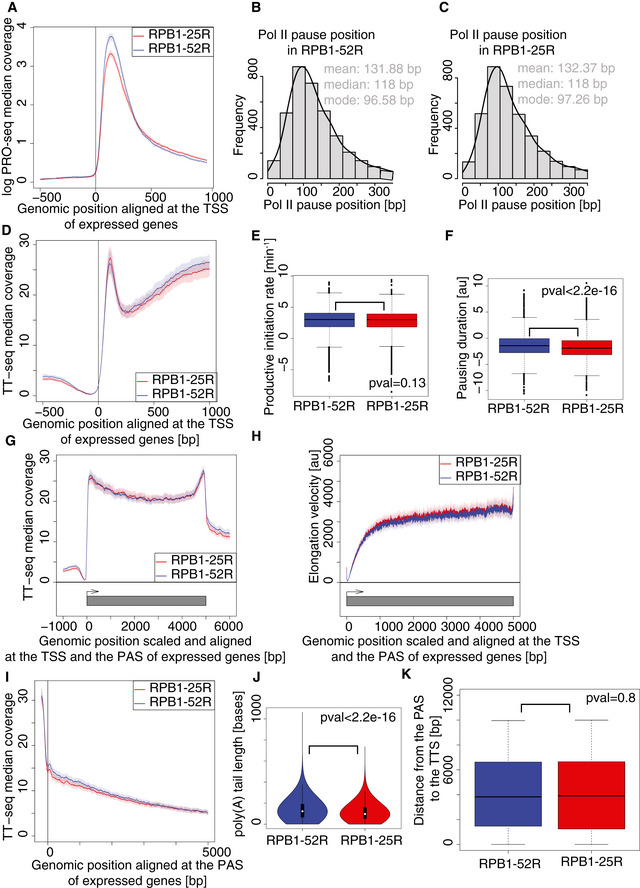
To investigate whether CTD shortening affects promoter‐proximal pausing, we used a previously developed multiomics approach that provides kinetic insights (Gressel et al, 2017, 2019). Briefly, this approach uses kinetic modeling to fit TT‐seq data and Pol II occupancy data and estimate the productive initiation frequency and the duration of Pol II pausing in the promoter‐proximal region. To map occupancy with transcriptionally engaged Pol II over the genome, we performed precision run‐on sequencing (PRO‐seq) (Mahat et al, 2016) in RPB1‐52R and RPB1‐25R cells for two biological replicates (Fig EV2A and B). Spike‐in probes were derived from Drosophila RNAs and used for quantification (Materials and Methods).
Figure EV2. CTD shortening leads to minor changes in Pol II pausing at steady state.
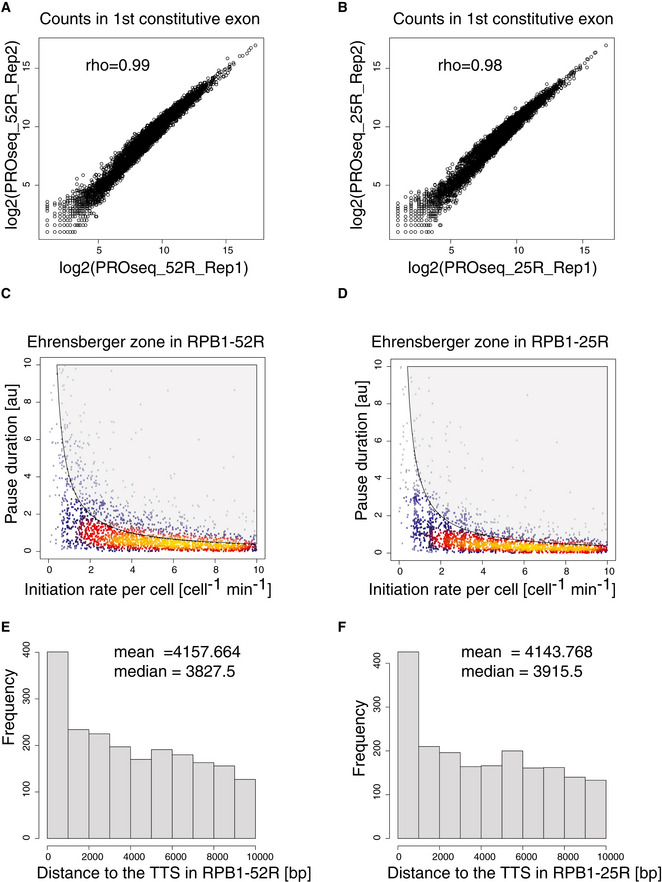
-
A, BSpearman correlation of read counts in the first constitutive exon of genes encoding major isoforms in biological replicates of PRO‐seq data.
-
C, DScatter plot of pausing duration (d) and productive initiation frequencies (I) in genes encoding major isoforms in RPB1‐52R and RPB1‐25R cells. The gray‐shaded area depicts impossible combinations of I and d according to published kinetic theory (Ehrensberger et al, 2013).
-
E, FHistogram of distances from the PAS to the transcription termination site (TTS) of genes encoding major isoforms.
Analysis of the PRO‐seq data showed that CTD shortening slightly decreased Pol II occupancy in the promoter‐proximal window downstream of the transcription start site (TSS) (Fig 3A), whereas the position of the Pol II occupancy peak remained unchanged (Fig 3B and C). To investigate the reasons for the decrease in Pol II occupancy, we calculated the productive transcription initiation frequency (I) and the duration of Pol II pausing (d) by combining TT‐seq with PRO‐seq occupancy data as described (Gressel et al, 2017). I was computed from TT‐seq coverage over non‐first constitutive exons, whereas d was obtained as a ratio of PRO‐seq signal over I in the promoter‐proximal window (see Materials and Methods). Whereas I was essentially unchanged, d was slightly decreased (Figs 3D–F, and EV2C and D). These results show that CTD shortening does not substantially alter Pol II pausing behavior under steady state conditions except for a slight decrease in pause duration.
Figure 3. CTD shortening leads to minor changes in Pol II pausing at steady state.
-
ACTD shortening leads to a decreased Pol II occupancy in promoter‐proximal region. Median coverage plot of PRO‐seq signal at genes encoding major isoforms. Solid lines represent median signal and shaded area refer to 95% bootstrap confidence intervals. Two independent biological replicates were analyzed.
-
B, CPol II pause position remains unchanged upon CTD shortening. Histogram of estimated positions of paused Pol II in RPB1‐52R and RPB1‐25R cells at genes encoding major isoforms with constitutive first exon longer than 100 bp.
-
DMedian coverage plot of TT‐seq signal at genes encoding major isoforms in RPB1‐52R and RPB1‐25R cells. Solid lines represent median signal and shaded area refer to 95% bootstrap confidence intervals. Two independent biological replicates were analyzed.
-
EBox plot showing productive transcription initiation rate in RPB1‐52R and RPB1‐25R cells for genes encoding major isoforms. P value = 0.13 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
-
FBox plot showing Pol II pausing duration in RPB1‐52R and RPB1‐25R cells for genes encoding major isoforms with constitutive first exon longer than 100 bp. P value < 2.2e‐16 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
-
GMedian scaled coverage plot of TT‐seq signal at genes encoding major isoforms. Solid lines represent median signal and shaded area refer to 95% bootstrap confidence intervals. Two independent biological replicates were analyzed.
-
HPol II velocity remains unchanged upon CTD shortening. Metagene profile of transcription elongation velocity at 4,480 genes longer than 10 kb encoding major isoforms. Solid lines represent median signal, and shaded area refer to 95% bootstrap confidence intervals.
-
IMedian coverage plot of TT‐seq signal centered at PAS at genes encoding major isoforms in RPB1‐52R and RPB1‐25R cells. Solid lines represent median signal, and shaded area refer to 95% bootstrap confidence intervals. Two independent biological replicates were analyzed.
-
JBox plot showing length of the poly(A) tail in RPB1‐52R and RPB1‐25R cells measured using Nanopore sequening data. P value < 2.2e‐16 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
-
KBox plot showing distance from the polyadenylation site (PAS) to the transcription termination site (TTS) in RPB1‐52R and RPB1‐25R cells for 1,958 genes encoding major isoforms. P value = 0.8 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range.
CTD shortening does not alter Pol II velocity and termination
TT‐seq metagene profiles over gene bodies were unaltered upon CTD shortening (Fig 3G), suggesting that Pol II elongation velocity is largely unchanged. To investigate this, we estimated Pol II velocity within expressed genes from our data as described previously (Gressel et al, 2017). We found that Pol II elongation velocity is unchanged upon CTD shortening (Fig 3H). We then plotted TT‐seq data around the polyadenylation site (PAS) and found no changes upon CTD shortening (Fig 3I). Poly‐A tail length as measured by Oxford Nanopore sequencing was not substantially altered either, indicating that 3′ end processing was normal (Fig 3J). We also called transcription termination sites downstream of the PAS as described (Schwalb et al, 2016) and also did not find any changes upon CTD shortening (Figs 3K and EV2E and F). In summary, these results indicate that Pol II elongation and termination do not show any obvious changes upon CTD shortening in human cells under steady state conditions.
CTD shortening reduces the number of transcribed putative enhancers
To investigate whether CTD shortening affects transcription of enhancers, we annotated putative enhancer RNAs (eRNAs) based on our TT‐seq data with the use of a previously established strategy (Schwalb et al, 2016; Michel et al, 2017; Zacher et al, 2017). We annotated 2,954 putative eRNAs in RBP1‐52R cells, but found only 1,779 enhancers in RBP1‐25R cells (Fig 4A). The lower number of annotated eRNAs in cells expressing Pol II with a shorter CTD did not arise from technical limitations because the TT‐seq samples were sequenced to the same depth of over 100 M reads (Table EV1). On average, putative eRNAs were also slightly shorter and showed slightly lower synthesis rates (Fig 4B and C). Putative enhancers were enriched in binding sites for members of the Activator Protein‐1 (AP‐1) transcription factor family, including FOS, JUN, and ATF (Fig EV3A). These factors function in bone development and regeneration (Ohta et al, 1991; Wagner, 2002) and in osteosarcoma tumorigenesis (Leaner et al, 2009), consistent with cell‐type specificity criteria for enhancers (Shlyueva et al, 2014; Heinz et al, 2015).
Figure 4. CTD shortening in human cells alters transcribed putative enhancers.
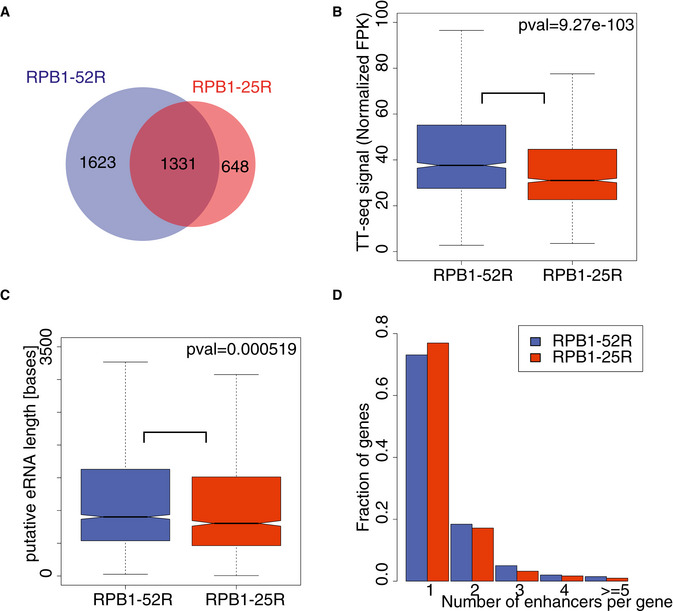
- Pie chart showing numbers of putative eRNAs annotated using TT‐seq data in RPB1‐52R and RPB1‐25R cells.
- Box plots showing RNA synthesis levels (RPKs) of putative eRNAs annotated using TT‐seq data in RPB1‐52R and RPB1‐25R cells. P value = 9.27e‐103 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
- Box plots showing lengths of putative eRNAs annotated using TT‐seq data in RPB1‐52R and RPB1‐25R cells. P value = 0.000519 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
- Histogram showing a distribution of number of putative eRNAs paired to genes in RPB1‐52R and RPB1‐25R cells.
Figure EV3. CTD shortening in human cells alters transcribed putative enhancers.
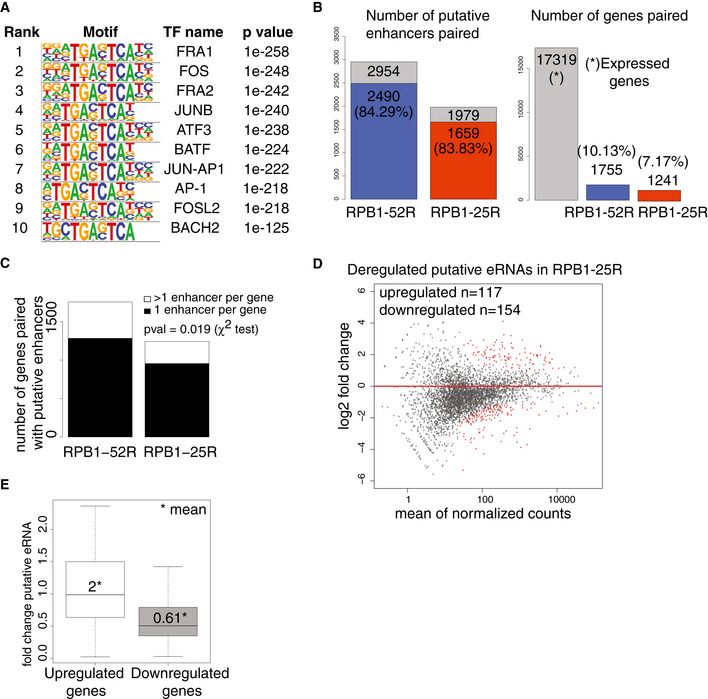
- Transcription factor‐binding sites enriches in the area of ± 50 bp from the TSS of putative eRNAs annotated using TT‐seq data in RPB1‐52R and RPB1‐25R cells. P values were determined by the hypergeometric test.
- Bar plots showing statistics of our enhancer‐promoter pairing strategy. The left bar plot shows a fraction of annotated putative enhancers that could be paired to promoters using our strategy. 2,490 putative enhancers in RPB1‐52R cells (out of all 2,954 enhancers annotated in RPB1‐52R cells) could be paired with promoters using our strategy (84.29% of all the putative enhancers we annotated in RPB1‐52R could be paired to promoters). In RPB1‐25R cells, 83.83% of annotated putative enhancers could be paired to promoters. The right panel shows a fraction of active promoters in U2OS cells that could be paired to putative enhancers using our strategy. There are 17,319 active promoters in U2OS cells (gray bar). In RPB1‐52R cells, 1,755 of active promoters could be paired with putative enhancers (which constitute 10.13% of all active promoters in RPB1‐52R cells). In the case of RPB1‐25R cells, 1,241 of all active promoters could be paired with putative enhancers (which constitutes 7.17% of all active promoters).
- Bar plot showing a number of putative enhancers paired per gene in RPB1‐52R and RPB1‐25R cells. P value = 0.019 (χ2 test).
- MA plot showing changes in putative eRNAs synthesis upon CTD shortening in U2OS cells in steady state conditions. RPB1‐52R cells are used as a control and the data were normalized using spike‐in counts. 4,335 putative eRNAs were analyzed, 117 putative eRNAs were significantly upregulated (adjusted P value < 0.05, log2 fold change ≥ 1, depicted in red) and 154 putative eRNAs were significantly downregulated (adjusted P value < 0.05, log2 fold change ≤ 1, depicted in red).
- Boxplot showing changes in putative eRNA synthesis if paired with genes that were significantly upregulated (adjusted P value < 0.05, log2 fold change ≥ 1) (left box) or significantly downregulated (adjusted P value < 0.05, log2 fold change ≤ 1) (right box) upon CTD shortening. Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
We then paired the obtained putative enhancers with their nearest transcriptionally active genes when their transcription start sites (TSS) were located within a maximum distance of 500 kb (Materials and Methods). Using this conservative approach, ~ 80% of annotated putative enhancers could be paired with at least one active gene in either cell line (Fig EV3B). Most active genes were paired with one putative enhancer (Figs 4D and EV3C). Genes that showed increased or decreased RNA synthesis upon CTD shortening generally also showed increased or decreased eRNA synthesis of their paired putative enhancers (Fig EV3D and E). These observations are consistent with the general finding that eRNA synthesis can serve as a proxy for enhancer activity (Melgar et al, 2011; Wu et al, 2014; Li et al, 2016). These results suggest that a fraction of putative enhancers change their transcription activity upon CTD shortening and that our pairing approach can recapitulate real functional interactions between target genes and their putative enhancers.
CTD shortening impairs transcriptional activation and enhancer transcription
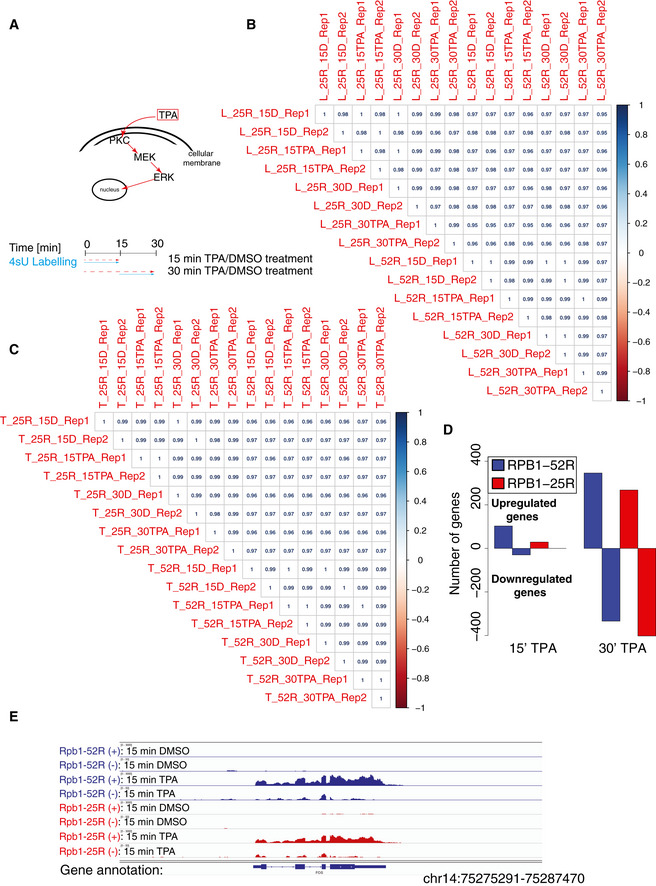
We next investigated whether CTD shortening influences signal‐induced transcription activation. We performed TT‐seq in RPB1‐52R and RPB1‐25R cells after their stimulation with 12‐O‐Tetradecanoylphorbol‐13‐acetate (TPA), a chemical that induces the MAP kinase signaling pathway (Kolb & Davis, 2004). Cells were stimulated with 200 nM TPA or DMSO (solvent control) for either 15 min or 30 min, and RNA synthesis was monitored by TT‐seq using a 4sU labeling time of 15 min (Fig EV4, EV5). We observed strongly reduced signal‐activated transcription induction in cells expressing Pol II with the shortened CTD. Upon 15 min of TPA stimulation, only 29 genes were significantly upregulated in RPB1‐25R cells in comparison with 113 genes in RPB1‐52R cells (Figs 5A and EV4D). Further, the genes that changed RNA synthesis in both cell lines at least twofold showed much lower response in cells with the shortened CTD (Figs 5B and EV4E). Among those genes were the well‐known immediate early genes FOS, JUNB, EGR1, and DUSP1 that encode regulatory factors (Herschman, 1991; Hargreaves et al, 2009). Upon 30 min of TPA stimulation, transcription activation was still impaired, albeit to a lower extent (Fig 5C and D).
Figure EV4. CTD shortening impairs transcription induction.
-
ASchematic representation of the experimental procedure used to study the effects of CTD shortening on TPA‐induced transcription.
-
B, CSpearman correlation of read counts in RefSeq genes of biological replicates in TT‐seq (B) and RNA‐seq (C) in RPB1‐52R and RPB1‐25R cells upon 15 and 30 min of TPA treatment (200 nM) and the respective DMSO controls.
-
DBar plot showing number of RefSeq genes that change RNA synthesis in TT‐seq datasets in RPB1‐52R and RPB1‐25R upon 15‐ and 30‐min treatment with TPA (200 nM). Genes showing adjusted P value < 0.05 and log2 fold change ≥ 1 were considered as significantly upregulated and genes showing adjusted P value < 0.05 and log2 fold change ≤ 1 were considered as significantly downregulated.
-
EExemplary genome browser view showing TT‐seq read coverage over FOS gene in RPB1‐52R and RPB1‐25R cells upon 15 min of TPA treatment (200 nM).
Figure EV5. CTD shortening delays transcription induction and changes transcription of putative enhancers.
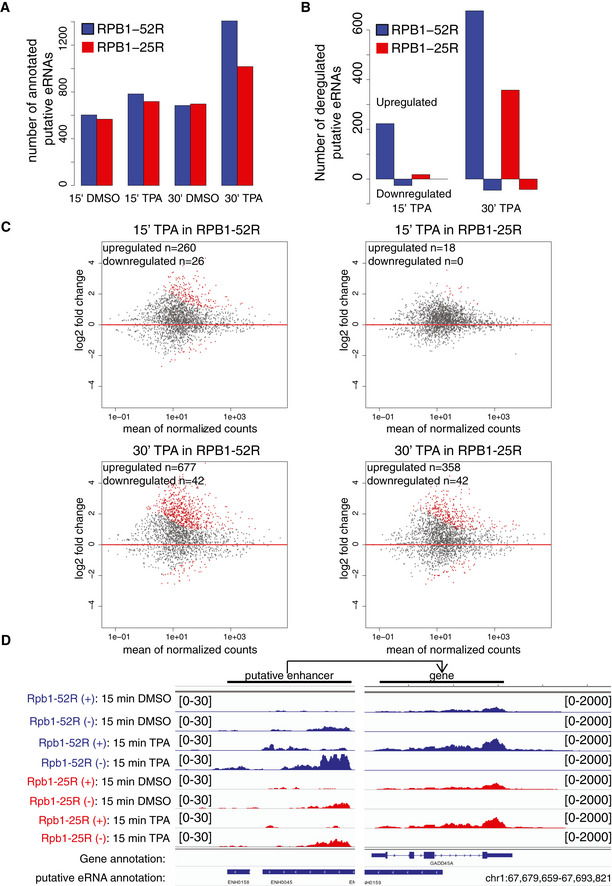
- Barplot showing number of annotated putative eRNAs upon 200 nM TPA or DMSO (solvent control) treatment in RPB1‐52R and RPB1‐25R cells.
- Bar plot showing number of differentially expressed putative eRNAs in TT‐seq datasets in RPB1‐52R and RPB1‐25R upon 15‐ and 30‐min treatment with TPA (200 nM). Putative eRNAs showing adjusted P value < 0.05 and log2 fold change ≥ 1 were considered as significantly upregulated and putative eRNAs showing adjusted P value < 0.05 and log2 fold change ≤ 1 were considered as significantly downregulated.
- MA plot showing changes in putative eRNAs synthesis in RPB1‐52R and RPB1‐25R cells upon 15 min or 30 min of TPA treatment. DMSO samples for respective cell lines were used as controls, and the data were normalized using library size normalization. Putative eRNAs showing adjusted P value < 0.05, log2 fold change ≥ 1, depicted in red) were considered significantly upregulated and putative eRNAs showing adjusted P value < 0.05, log2 fold change ≤ 1, depicted in red) were considered significantly downregulated.
- Exemplary genome browser view showing TT‐seq read coverage over GADD45 gene as well a region giving rise to a putative eRNA mapped to it in RPB1‐52R and RPB1‐25R cells upon 15 min of TPA treatment (200 nM).
Figure 5. CTD shortening impairs transcription induction.
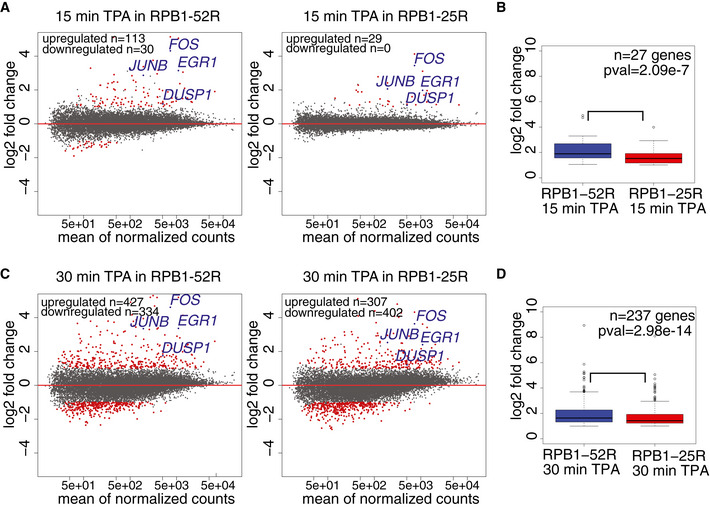
- Changes in RNA synthesis in response to 15 min of TPA treatment. MA plot showing RNA synthesis changes in TT‐seq datasets upon 15‐min treatment with TPA (200 nM) in RPB1‐52R and RPB1‐25R cells. DMSO treatment of the respective cell line was used as a control, and the data were normalized using library size normalization. 14,636 expressed genes annotated in RefSeq were analyzed. Differentially expressed genes are in red (adjusted P value < 0.05, log2 fold change ≥ 1 for upregulated genes and adjusted P value < 0.05, log2 fold change ≤ 1 for downregulated genes).
- Box plots showing log2 fold change of genes significantly upregulated upon 15‐min treatment with 200 nM TPA in RPB1‐52R and RPB1‐25R cells (adjusted P value < 0.05, log2 fold change ≥ 1). P value = 0.02257 (Mann–Whitney test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
- Changes in RNA synthesis in response to 30 min of TPA treatment. MA plot showing RNA synthesis changes in TT‐seq datasets upon 30 min treatment with TPA (200 nM) in RPB1‐52R and RPB1‐25R cells. DMSO treatment of the respective cell line was used as a control and the data were normalized using library size normalization. 14,636 expressed genes annotated in RefSeq were analyzed. Differentially expressed genes are in red (adjusted P value < 0.05, log2 fold change ≥ 1 for upregulated genes and adjusted P value < 0.05, log2 fold change ≤ 1 for downregulated genes)
- Box plots showing log2 fold change in RNA synthesis of genes significantly upregulated upon 30‐min treatment with 200 nM TPA in RPB1‐52R and RPB1‐25R cells (adjusted P value < 0.05, log2 fold change ≥ 1). P value = 0.001749 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
We next investigated whether the observed reduced transcription activation is accompanied by changes in enhancer transcription. We annotated putative enhancers based on their eRNA synthesis and paired them with nearby putative target genes using the same approach as above. We observed that the number of annotated putative enhancers was higher upon TPA stimulation compared with the DMSO control, showing that new putative enhancers became transcriptionally active (Figs 6A, and EV5A–C). In addition, TPA stimulation led to a larger fraction of genes pairing with more than one putative enhancer in RPB1‐52R cells compared with RPB1‐25R cells (Fig 6A). Also, putative enhancers paired with genes that were upregulated in both cell lines upon TPA induction showed higher eRNA synthesis in RPB1‐52R cells than in RPB1‐25R cells (Fig 6B–D). We conclude that CTD shortening impairs transcription activation of both, target genes and of their putative enhancers.
Figure 6. CTD shortening alters putative enhancers and delays transcription induction.
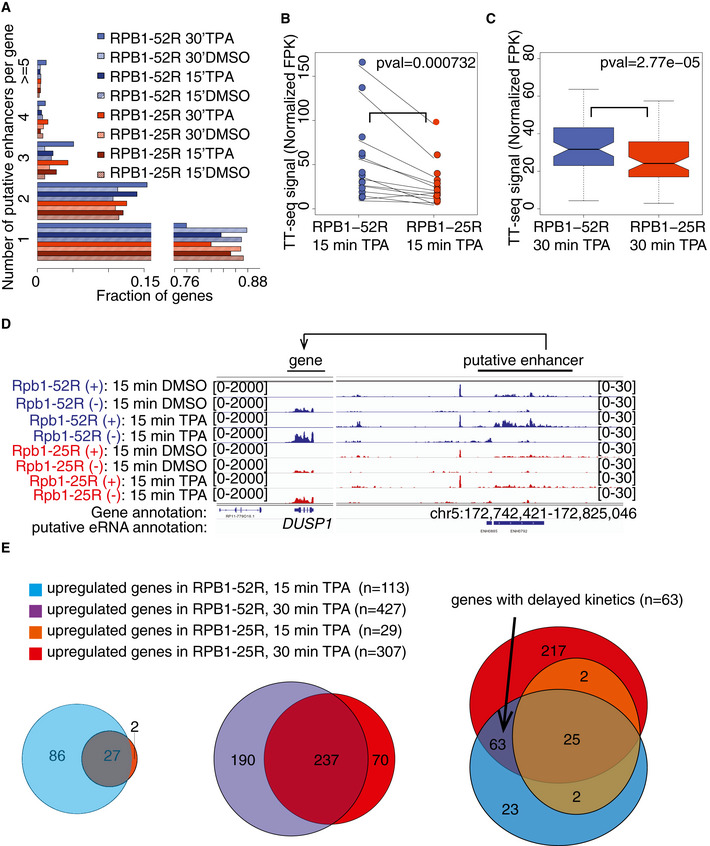
- Histogram showing the distribution of a number of putative eRNAs paired to genes in RPB1‐52R and RPB1‐25R cells upon 15 and 30 min TPA treatment (200 nM) as well as in the DMSO controls.
- Box plots showing RNA synthesis levels (RPK) of putative eRNAs paired with genes upregulated (adjusted P value < 0.05, log2 fold change ≥ 1) in RPB1‐52R and RPB1‐25R cells upon 15 min of TPA treatment (200 nM). P value = 0.000732 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
- Box plots showing RNA synthesis levels (RPK) of putative enhancer eRNAs paired with genes upregulated (adjusted P value < 0.05, log2 fold change ≥ 1) in RPB1‐52R and RPB1‐25R cells upon 30 min of TPA treatment (200 nM). P value = 2.77e‐5 (Mann–Whitney U‐test) Box limits are the first and third quartiles, the band inside the box is the median. The ends of the whiskers extend the box by 1.5 times the interquartile range. Two independent biological replicates were analyzed.
- Exemplary genome browser view showing TT‐seq read coverage over DUSP1 gene as well a region giving rise to a putative eRNA mapped to it in RPB1‐52R and RPB1‐25R cells upon 15 min of TPA treatment (200 nM).
- Venn diagrams showing overlaps between groups of genes upregulated upon 15 min TPA treatment in RPB1‐52R and RPB1‐25R cells (left panel), 30‐min TPA treatment in RPB1‐52R and RPB1‐25R cells (middle panel) as well as triple overlap between genes upregulated upon 15 min TPA in RPB1‐52R and RPB1‐25R cells with those upregulated upon 30 min TPA in RPB1‐25R cells (right panel). Genes showing adjusted P value < 0.05 and log2 fold change ≥ 1 were considered upregulated.
CTD shortening delays transcription activation
Our above analysis suggested that CTD shortening delays the natural gene induction during TPA stimulation, but does not generally change the identity of the target genes that are activated (Fig 6E). To investigate this, we identified 63 genes whose transcription was induced after 15 min of TPA stimulation in RPB1‐52R cells, but not in RPB1‐25R cells. We found that the same genes were induced in RPB1‐25R cells after 30 min of stimulation, showing that transcription activation was delayed, but that generally the same genes were targeted (Fig 6E, Table EV2). The same trend was observed for putative enhancers that were paired with these genes that showed delayed transcription activation (Fig EV5D). Overall, this indicates that CTD shortening delays signal‐induced transcription of target genes but does not generally alter which genes are induced.
Discussion
Here, we investigate how shortening of the Pol II CTD influences transcription kinetics in human cells. One important outcome of our comprehensive analysis is that CTD shortening hardly changes RNA metabolism in steady state. Based on available literature, this is not as surprising as it first appears. For example, release of Pol II from promoter‐proximal pausing involves phosphorylation of the linker region between the Pol II body and the CTD (Sdano et al, 2017; Vos et al, 2018a,b) and CTD shortening does not change the linker region, explaining why pausing behavior is not strongly altered. Also consistent with our work, a recent study using a different CTD truncation system reported no changes in Pol II pause release on selected single genes in human cells (Gerber & Roeder, 2020). Further, the recruitment of factors for co‐transcriptional RNA processing is not predicted to be defective upon CTD shortening because CTD‐binding proteins interact with only 1‐3 CTD repeats (Meinhart et al, 2005).
In addition, we show that CTD shortening delays transcription activation after cell stimulation, as exemplified by stimulation of the MAP kinase pathway. Stimulation of the MAP kinase pathway triggers a protein phosphorylation cascade that targets transcription factors and coactivators, modulating their interactions with target promoters and enhancers (Stevens et al, 2002; Yang et al, 2013; Klein et al, 2013). We therefore suggest that delayed transcription activation is caused by a defect in promoter‐enhancer communication that is enabled or facilitated by the CTD. Indeed, CTD truncation is known to reduce the expression of genes that are controlled by enhancers (L. A. Allison & Ingles, 1989; Scafe et al, 1990; Gerber et al, 1995; Aristizabal et al, 2013) and was recently shown to alter transcriptional bursting (Quintero‐Cadena et al, 2020). Factors that typically bind enhancer regions undergo liquid–liquid phase separation (LLPS) in vitro (Boija et al, 2018) and colocalize with Pol II in cells (Sabari et al, 2018; Cho et al, 2018). The CTD alone can undergo LLPS in vitro and can be recruited to nuclear condensates formed by transcription factors, and this behavior depends on CTD length (Kwon et al, 2014; Burke et al, 2015; Boehning et al, 2018). The length dependency is likely caused by a decreased valency of interactions when the CTD is shortened, as LLPS strongly depends on multivalent interactions (Hnisz et al, 2017). Taking these considerations together, we speculate that CTD shortening impairs enhancer‐ and LLPS‐dependent Pol II recruitment during cell stimulation, thereby delaying transcription activation.
This model of enhancer‐ and CTD length‐dependent Pol II recruitment may also explain why CTD length generally increases with genome size. Enhancers can be located tens or even hundreds of kilobases away from their target genes (Levine & Tjian, 2003). Cells with larger genomes therefore may rely on a longer CTD to efficiently recruit Pol II to target genes. In this respect, we note that CTD shortening not only alters CTD length but also its chemical nature because the distal part of the CTD, which is absent in our RPB1‐25R variant, contains heptapeptide repeat motifs that deviate from the consensus sequence at one or more amino acid positions (Dias et al, 2015; Simonti et al, 2015). It is known that such non‐consensus repeats can form hydrogels in vitro with the N‐terminal part of transcription factor TAF15 (Kwon et al, 2014), which raises the possibility that the distal CTD may interact with certain transcription factors. We therefore cannot exclude that some of the effects we observe here upon CTD shortening reflect in part a CTD function that is specific to the distal non‐consensus repeats.
In conclusion, our results show that the proximal half of the human CTD supports RNA synthesis and co‐transcriptional processes, whereas the full‐length CTD is required for rapid transcription activation. These observations are consistent with a prominent role of the CTD in enhancer‐dependent recruitment of Pol II to active genes, which is critical during transcription activation, when Pol II needs to be redistributed rapidly over the genome in the nucleus. Together, our results establish a crucial role of CTD length for efficient transcription dynamics in human cells.
Materials and Methods
Cell lines
We used previously reported human osteosarcoma U2OS cells (Boehning et al, 2018). RPB1‐52R cells stably express RPB1 containing an N‐terminal HALO tag, N792D mutation conferring α‐amanitin resistance, and 52 CTD repeats as in the wild‐type protein. RPB1‐25R mutant cells express only 25 repeats present in the wild‐type RPB1, which correspond to repeats 1–21 and 49–52. Cells were grown in DMEM(1X)+GlutaMAX‐I medium (Gibco, #21885‐025) with 10% FBS (Gibco, #10500‐064) in the presence 100 U/ml of Penicillin‐Streptomycin (Thermo Fisher, #15140122) in a humidified incubator with 5% CO2 at 37°C. Mutant cells were cultured with 1 μg/ml of α‐amanitin (Sigma‐Aldrich, #A2263). For stimulation experiments, cells were treated with TPA (Sigma‐Aldrich, #P1585) or DMSO (solvent control). DMSO was diluted 1:20,000 in the culturing medium to prevent its possible side effects on transcription (Verheijen et al, 2019). Drospohila Schneider‐2 cells were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, #ACC130). The cells were grown in Schneider's Insect Medium (VWR, #L0207‐500) with 10% FBS (Gibco, #10500‐064) at 25°C, protected from light. All cell lines were regularly tested for Mycoplasma contamination using PlasmoTest Mycoplasma Detection Kit (InvivoGen, #rep‐pt1).
Western blotting
1 × 106 cells were washed with ice‐cold PBS, centrifuged at 200 × g, 4°C and lysed with 100 μl of NuPAGE LDS Buffer (Invitrogen, #NP0007) supplemented with 0.4 mM (final concentration) of DTT and sonicated for 30 s with Ultrasonic Cleaner USC‐T instrument (VWR). The sample was diluted with 260 μl of water, and 30 μl were used for electrophoresis. Electrophoresis was performed using Mini‐Protean TGX gel (Bio‐Rad, #4561084) in 1× Tris/Glycin/SDS buffer (Bio‐Rad, #1610732) in the presence of size marker. The transfer was performed with Trans‐Blot Turbo system (Bio‐Rad, #1404156) in a Trans‐Blot Turbo instrument (Bio‐Rad) for 10 min using the high‐molecular weight program. After 1 h of incubation with 5% milk in PBS/0.1% Tween20, the membrane was probed overnight at 4°C with the following primary antibodies diluted in 1% milk/PBS/0.1% Tween20: α‐HALO (dilution 1:1,000, Promega, #G9211) or α‐U1 snRP (dilution 1:200, Santa Cruz, #sc‐39089). The membrane was washed three times with PBS/0.1% Tween, probed with HRP‐conjugated α‐mouse IgG (Abcam, #ab5870), developed using SuperSignal West Pico Plus Chemiluminescent Substrate (Thermo, #34580) and imaged with ChemoCam Imager ECL Type HR 16‐3200 (Intas).
Proliferation assay
Cell proliferation assay was performed using the CellTiter 96 AQueous One Solution Cell Proliferation Assay System (Promega, # G3582). U2OS cells were seeded in 96‐well plates (1,500 cells/well) in three technical replicates. Absorbance at 490 nm was measured with the Infinite M1000 Pro Tecan Plate Reader every 12 h for 72 h after 4 h of incubation with the MTS reagent. To determine the doubling time, background absorbance from the wells with medium only was subtracted from that of sample wells and background‐corrected mean absorbance value for each condition was determined. Doubling time was determined using the following formula: doubling time = 72 h*log2/log(background‐corrected mean absorbance at 72 h) − log(background‐corrected mean absorbance at 0 h). Estimated doubling times were 35 h for U2OS RPB1‐52R and 42 h for U2OS RPB1‐25R cells.
TT‐seq and RNA‐seq
TT‐seq was performed as described (Schwalb et al, 2016; Wachutka et al, 2019) with minor modifications. Specifically, two biological replicates of U2OS cells, 6 × 107 cells each were labeled for 15 min with 500 μM of 4‐thiouridine (4sU, Carbosynth, NT06186) at 37°C and 5% CO2. Each replicate was grown on 20 Petri dishes with the diameter of 15 cm. The cells on each plate were lysed with 5 ml of Qiazol Lysis Reagent (Qiagen, # 79306), and lysates belonging to a replicate were pooled and mixed with 60 μl of 6 ng/μl spike‐in mix prior to RNA isolation according to the manufacture’s protocol. Spike‐in sequences and their synthesis were as reported (Wachutka et al, 2019). 600 μg of RNA in batches of 200 μg of RNAs were sonicated to obtain fragments of < 6 kb using AFA micro tubes in Covaris S220 Focused‐ultrasonicator. 1 μg of each of the sonicated RNA from each replicate was kept for the preparation of total RNA (RNA‐seq). 4sU‐labeled RNA was purified from 200 μg batches of the fragmented RNAs. Biotinylation and purification of 4sU‐labeled RNAs was performed as described (Dölken et al, 2008; Wachutka et al, 2019). Libraries for sequencing were prepared using 100 ng of input RNA (for both, total and labeled RNA‐seq libraries) and TruSeq Stranded Total RNA Library Prep Gold (Illumina, #20020598), followed by sequencing with NextSeq500 (75 bases in paired‐end mode).
PRO‐seq
PRO‐seq was performed as described (Mahat et al, 2016; Jaeger et al, 2020) with minor modifications. Briefly, 5.4 × 107 U2OS cells and 0.6 × 107 of Schneider‐2 cells were used per replicate and two biological replicates were prepared. Nuclear run‐on reactions were performed in the presence of 25 nM of biotin‐11‐CTP (Jena Bioscience, #NU‐831‐BIOX) and 25 nM of biotin‐11‐UTP (Jena Bioscience, #NU‐821‐BIOX). cDNA generated from adapter‐ligated RNA was subject to 13 cycles of PCR in the final step. The sequencing library was size selected in two steps using RNAClean XP beads (Beckman Coulter, # A63987). The first step was performed with a ratio of beads:sample = 0.6:1 and the second step with a ratio of beads:sample = 1:1. Libraries were sequenced with NextSeq500 (42 bases in paired‐end mode).
Isolation of chromatin‐associated RNA and RNA long‐read sequencing
Isolation of chromatin‐associated RNA was performed as described (Conrad & Ørom, 2017) using 6 × 107 U2OS cells per replicate. One biological replicate was prepared. RNA isolated from the chromatin fraction was subject to purification of polyadenylated RNA using Dynabeads mRNA Purification kit (Thermo Fisher, #61006) according to manufacturer’s instructions. Two increase the purity of mRNA, two rounds of purification were performed for each sample. 500 ng of mRNA was converted into a sequencing library using Direct RNA Sequencing kit (Oxford Nanopore, #SQK‐RNA002) according to manufacturer’s instructions. Reverse transcription reaction step was omitted. The libraries were loaded onto previously primed SpotON flowcells and sequenced using MinION device (Oxford Nanopore).
Mapping and normalization of TT‐seq and RNA‐seq datasets
Paired‐end 75 bp reads were demultiplexed using 6 bp barcodes with bcl2fastq software (Illumina, v2.19.1). Mapping was performed using STAR (v2.6.1) (Dobin et al, 2013) and a hybrid genome containing Genome Reference Consortium version GRCh38 (hg38) of the human genome (from April 2014) and spike‐in sequences (Wachutka et al, 2019). The following parameters were set during mapping: ‐‐outFilterMismatchNmax 3 ‐‐outFilterMultimapScoreRange 0 ‐‐alignIntronMax 500000. Resulting bam files were filtered using SAMTOOLS (v1.9) (Li et al, 2009) to remove alignments with mapping quality lower than 7 (‐q 7). Only proper pairs (‐f2) were retained. Reads mapping to certain features were counted using HTSeq (v0.6.0) (Anders et al, 2015) using intersection‐strict mode and ‐‐stranded=reverse parameter. RefSeq genes considered expressed were defined as genes having showing log2 RPKM value equal or higher than −7 in TT‐seq datasets (for the longest transcript isoform annotated in RefSeq GRCh38) based on kernel density estimation plots of log2 RPKM values obtained for TT‐seq counts. Further steps of data analysis were performed using R/Bioconductor (R version 3.5.3) (R Development Core Team, 2011). TT‐seq datasets were normalized using RNA spike‐ins as previously described (Schwalb et al, 2016).
Sequencing depth σ for each sample j was calculated according to the following formula:
with read counts k for all available spike‐ins i in sample j for the RNA‐seq samples and for the labeled spike‐ins li in sample j for the TT‐seq samples. Cross‐contamination ε for each TT‐seq sample j was calculated with following formula:
Cross‐contamination was set to 1 for RNA‐seq samples. RNA synthesis and decay rates as well as RNA half‐lives were calculated as described previously (Schwalb et al, 2016; Michel et al, 2017). Differential gene expression analysis was performed using DESeq2 Bioconductor package (Love et al, 2014). TT‐seq coverage was calculated using coverage function from GRanges R Bioconductor package (Lawrence et al, 2013) and normalized using spike‐ins.
Gene ontology analysis
Gene ontology analysis was performed using the WebGestalt tool (Liao et al, 2019). Significantly deregulated genes determined from the TT‐seq datasets were analyzed with unchanged genes used as background. GO categories with FDR ≤ 0.05 are reported.
Determination of major RNA isoform
We used Salmon version 0.13.1 (Patro et al, 2017) in order to determine RNA‐seq datasets expressed major transcript isoforms per gene from GRCh38 RefSeq isoform annotation (ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh38_latest/refseq_identifiers/GRCh38_latest_rna.fna.gz). We retained the isoform that constitute at least 70% of total RNA‐seq expression for a given gene based on the mean value of Transcript Per Million (TPM) expression, which should reach the value of at least 0.5 TPM per sample. We removed overlapping genes from further analysis. This approach yielded 6260 major isoforms in RPB1‐52R and RPB1‐25R.
Calculation of splicing ratio
For the exon‐based splicing ratio analysis, we calculated reads overlapping a 4 bp window around the splice junction (2 bp located within the exon and 2 bp located within the intron) using findOverlaps function from the GenomicRanges Bioconductor R package (Lawrence et al, 2013). We limited our analysis to constitutive exons belonging to the major isoforms. Additionally, first exons had to be longer than 100 bp. We took into account only junctions that had more than 30 reads in the TT‐seq datasets. This approach yielded 2,295 of first 5′SSs, 2,713 of first 3′SSs, 2,415 of last 5′SSs, 2,424 of last 3′SSs, 19,683 of intermediate 5′SSs, and 19,858 of intermediate 3′SSs in RPB1‐52R and RPB1‐25R. “Unspliced” reads were defined as reads overlapping at least 3 bp with the 4 bp window around the splice junction. “Total” reads were defined as reads overlapping at least 2 bp with the 4 bp window around the splice junction. “Spliced reads” were calculated as difference between “total” reads and “spliced” reads. Splicing ratio was calculated by dividing “spliced” reads by “total” reads for each of the junction category. Statistical significance of observed differences was calculated using Mann–Whitney U‐test.
Estimation of RNA amount per cell
The RNA amount per cell was estimated using spike‐in counts as described (Gressel et al, 2017, 2019). For TT‐seq experiments, 6.8 × 107 RBP1‐52R cells and 5.6 × 107 RBP1‐25R cells were used. The number of spike‐in molecules per cell N [cell−1] for number of cells n was calculated according to the following formula:
with n × 10−3 g of spike‐ins m, n cells, the Avogadro number N A and the molar‐mass of the spike‐ins M calculated with the following equation:
where A n, U n, C n, and G n are the number of each respective nucleoside within each spike‐in. 159 refers to the molecular weight of a 5′ triphosphate. The conversion factor to RNA per cell k [cell−1] was calculated for all labeled spike‐ins i with the length L i as follows:
Initiation frequency estimation
Initiation frequency Ii for genes longer than 10 kb and encoding major isoforms in RPB1‐52R and RPB1‐25R was determined as described (Gressel et al, 2017, 2019) by fitting spike‐in normalized TT‐seq coverage on all non‐first constitutive exons in major isoforms for the labeling time t = 15 min to the following formula:
Our initiation frequency refers to the productive initiation frequency and does not take into account possible premature termination events.
Mapping of transcription termination site (TTS)
The TTSs of genes encoding major isoforms in RPB1‐52R and RPB1‐25R cells was determined as described (Schwalb et al, 2016). TT‐seq datasets were subject to variance stabilization transformation. TTS was determined in the window (potential termination window, PTM) of 10 kb downstream of the annotated PAS (polyadenylation site) for genes which did not overlap with the TSS of the next gene downstream. The selection was further restricted to transcripts with RPK value ≥ 100. The segmentation function from Bioconductor package tilingArray (Huber et al, 2006) was used to segment the PTM based on TT‐seq coverage drops of at least five times between two consecutive segments. The estimated TTS was determined as a border dividing two segments were the mean TT‐seq coverage dropped to 0 at least 500 bp downstream of the PAS. TTS was estimated for 1958 of expressed major isoforms in RPB1‐52R and RPB1‐25R cells.
PRO‐seq data analysis
PRO‐seq data were trimmed with cutadapt version 1.11 (Martin, 2011) using the following parameters: sample PROseq_25R_Rep1: cutadapt ‐a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCACACATCGATCT ‐A GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGT ‐m 20; sample PROseq_25R_Rep2: cutadapt ‐a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCACTGGTCAATCT ‐A GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGT ‐m 20; sample PROseq_52R_Rep1: cutadapt ‐a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCACGATCTGATCT ‐A GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGT ‐m 20; and sample PROseq_52R_Rep: cutadapt ‐a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCACGGACGGATCT ‐A GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGT ‐m 20 . The reads were mapped to the human genome (Genome Reference Consortium version GRCh38) of the human genome (from April 2014) and to the Drosophila melanogaster genome version BDGP6.28 with STAR (v2.6.1) (Dobin et al, 2013) using the following parameters: ‐‐outFilterMismatchNmax 3 ‐‐outFilterMultimapScoreRange 0 ‐‐alignIntronMax 500000.
Resulting bam files were filtered using SAMTOOLS (v1.9) (Li et al, 2009) to remove alignments with mapping quality lower than 7 (‐q 7). Only proper pairs (‐f2) were retained. In order to calculate normalization factors for PRO‐seq, HTSeq (v0.6.0) (Anders et al, 2015) with intersection‐strict mode and ‐‐stranded=reverse parameter was used to calculate reads mapping to D. melanogaster transcripts (Drosophila_melanogaster.BDGP6.28.100.gtf.gz from Ensembl.org) followed by calculation of size factors with DESeq2 Bioconductor package (Love et al, 2014). PRO‐seq sequencing Read1 mapping to the human genome was converted to reverse complement sequence and the last nucleotide incorporated by the Pol II was determined (Pol II position)—last base of the reversely complemented Read 1 for the “+” strand, and first base of the reversely complemented Read 1 for the “‐” strand. Coverage for Pol II position was calculated using coverage function from GRanges R Bioconductor package (Lawrence et al, 2013) and normalized using size factors determined with DESeq2 for D. melanogaster transcripts.
Calling of the Pol II paused sites
Pol II paused sites were determined as previously described (Gressel et al, 2017, 2019) for genes longer than 10 kb and encoding major isoforms. The paused sites were estimated in the window from the TSS to the end of the first exon. Only first exons longer than 100 bp were used. The paused site m was calculated using the maximization of the function ρi = maxmPim where ρi was a PRO‐seq normalized coverage value at least five times higher than the median of non‐negative normalized PRO‐seq coverage values Pim in the window.
Pol II pause duration calculation
Pause duration for genes longer than 10 kb and encoding major isoforms in RPB1‐52R and RPB1‐25R cells () in the pause window of ± 100 bp from the pausing site m was determined as described (Gressel et al, 2017, 2019) using initiation frequency () and normalized PRO‐seq coverage values () according to the following formula:
Elongation velocity estimation
Elongation velocity for genes longer than 10 kb and encoding major isoforms in RPB1‐52R and RPB1‐25R cells was calculated using the following formula:
For plotting, a smoothing approach was applied to the formula:
where wi is a weighting vector wi = (1, 2, 3, 4, 5, 6, 5, 4, 3, 2, 1).
Direct RNA Nanopore sequencing data mapping and analysis
Direct RNA Nanopore sequencing reads were obtained one replicate of RPB1‐52R cells and RPB1‐25R cells. Data processing was performed as previously described (Maier et al, 2020). FAST5 files were base‐called using Guppy 3.1.5 (Oxford Nanopore Technologies) with the following parameters: guppy_basecaller ‐i fast5 ‐s basecalled ‐‐num_callers 1 ‐‐cpu_threads_per_caller 12 ‐c rna_r9.4.1_70bps_hac.cfg ‐r ‐‐fast5_out ‐‐calib_detect ‐‐u_substitution on ‐q 0. Direct RNA nanopore sequencing reads were mapped with minimap2 2.10 (Li, 2016) to the GRCh38/hg38 genome assembly (Human Genome Reference Consortium) with the following parameters: minimap2 ‐ax splice ‐k14 ‐‐secondary=no. SAMtools 1.3.1 (Li et al, 2009) was used to quality filter SAM files, whereby alignments with MAPQ smaller than 20 (‐q 20) were skipped. Further data processing was carried out using the R/Bioconductor environment version 3.5.3 and Python version 3.6.10. Poly(A) tail length was estimated using nanopolish version 0.8.4 (Oxford Nanopore Technologies) “polya” algorithm. Isoform abundance was estimated using FLAIR (Tang et al, 2020) and Bedtools version 2.25.0 (Quinlan & Hall, 2010). Splice junctions were corrected using TT‐seq datasets with junctionsFromSam.py script.
Annotation of eRNAs
Actively transcribing enhancer RNA (eRNA) and other classes of transcription units (TUs) were predicted and annotated using coverage information from paired‐end TT‐seq data as described (Schwalb et al, 2016; Michel et al, 2017; preprint: Villamil et al, 2019). To reduce gaps in coverage within expressed transcripts and to increase detection sensitivity to transcripts that are weakly expressed, the human genome reference sequence (hg20/GRCh38) was first binned into 200‐bp sequences. Coverage was calculated as the count of TT‐seq fragment midpoints falling within each bin in a strand‐specific manner. Paired‐end reads with an inner mate distance greater than 500‐bp were discarded. The STAN R/Bioconductor package (Zacher et al, 2017) was used to implement a hidden Markov model with a Poisson log‐normal distribution to segment the binned genome into transcribed and untranscribed states. Contiguously transcribed bins and those separated by no more than 200‐bp were merged as individual TUs. To refine the start and end positions of predicted TUs, coverage data in a ± 400‐bp (2 bin) window around the original start and end positions were transformed into homoskedastic data using a variance stabilization transformation. The variance stabilized coverage was then converted to a stepwise function and the position of the boundary between two steps was designated as the start or end of the transcript.
Transcript classification was carried out by searching for strand‐specific overlaps with GENCODE‐annotated transcripts (release 31). Predicted TUs that have at least 50% reciprocal overlap with GENCODE‐annotated mRNAs and lncRNAs were classified as such. TUs that start < 1 kb downstream of these were discarded from further classification to account for downstream transcription run‐through past the cleavage site. TUs that are antisense to GENCODE‐annotated mRNA or lncRNA with transcription start sites (TSS) that are > 1 kb downstream of the mRNA or lncRNA’s TSS were classified as antisense RNA (asRNA). Those with TSS < 1 kb downstream of the mRNA/lncRNA TSS were classified as convergent RNA (conRNA), and those with TSS < 1 kb upstream of the mRNA/lncRNA TSS were classified as upstream antisense RNA (uaRNA). Remaining TUs, i.e., those with TSS > 1 kb away from any mRNA or lncRNA in either direction were classified as small intergenic non‐coding RNA (sincRNA). Enhancer states previously defined in 127 human cancer cell lines (NIH Roadmap Epigenomic Project) were used to identify eRNA from predicted TUs. asRNA and sincRNA with TSS within ± 500‐bp of Roadmap enhancer states were classified as putative eRNA. The eRNAs were further filtered using a minimum average expression cutoff of 40 fragments per kilobase (FPK) with at least 10 FPK in each replicate. These were further screened for bi‐directional transcription. Specifically, only eRNAs with another non‐coding TU on the opposite strand, starting within ± 600‐bp of its TSS, were included in the final eRNA annotation.
Promoter‐enhancer pairing
TT‐seq data were used to determine RefSeq‐annotated genes expressed under steady state and DMSO/TPA‐treated conditions. Each enhancer was paired to an expressed gene’s promoter by searching for the gene TSS on either strand that is nearest to the eRNA TSS within a maximum distance of ± 500 kb. Enhancer‐promoter pairs were validated by comparing the fold change in RNA synthesis across RPB1‐52R and RPB1‐25R of eRNAs and their paired genes. The average fold change in the RNA synthesis of eRNA paired to significantly upregulated (log2 fold change ≥ 1) or downregulated (log2 fold change ≤ −1) expressed genes (as determined by DESeq2, P value < 0.05) showed that eRNA synthesis changed in the same direction as their paired gene’s synthesis.
Transcription factor binding site analysis
Transcription factor binding site analysis was performed using HOMER (Heinz et al, 2010) in the window of ± 250 bp around the TSS of the annotated eRNAs in RPB1‐52R and RPB1‐25R. The following parameters of the findMotifsGenome.pl script were used: ‐size given ‐mask. P values were determined by the hypergeometric test.
Author contributions
Design, experiments, and bioinformatics analysis: AS. Bioinformatics analysis of putative enhancers: GV Advice on bioinformatics analysis: ML and BS. Cell lines: XD and CD‐D. Research design and supervision: PC. Manuscript preparation: AS and PC.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
The authors would like to thank Kerstin Maier and Petra Rus for sequencing and technical support, Sara Monteiro‐Martins for help in performing splicing analysis, members of the Cramer laboratory for insightful discussions and Martin Jäger for advice on the PRO‐seq experiment. A.S. was supported by an EMBO Long‐Term Postdoctoral Fellowship (ALTF‐529‐2015). P.C. was supported by the Deutsche Forschungsgemeinschaft (EXC 2067/1‐390729940) and the European Research Council Advanced Investigator Grant CHROMATRANS (grant agreement No 882357).
The EMBO Journal (2021) 40: e107015.
Data availability
RNA‐seq, TT‐seq, PRO‐seq, and Nanopore sequencing data are available under Gene Expression Omnibus GSE159092 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159092).
References
- Allen BL, Taatjes DJ (2015) The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol 16: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison LA, Moyle M, Shales M, James Ingles C (1985) Extensive homology among the largest subunits of eukaryotic and prokaryotic RNA polymerases. Cell 42: 599–610 [DOI] [PubMed] [Google Scholar]
- Allison LA, Wong JK, Fitzpatrick VD, Moyle M, Ingles CJ (1988) The C‐terminal domain of the largest subunit of RNA polymerase II of Saccharomyces cerevisiae, Drosophila melanogaster, and mammals: a conserved structure with an essential function. Mol Cell Biol 8: 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison LA, Ingles CJ (1989) Mutations in RNA polymerase II enhance or suppress mutations in GAL4. Proc Natl Acad Sci USA 86: 2794–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq‐A Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aristizabal MJ, Negri GL, Benschop JJ, Holstege FCP, Krogan NJ, Kobor MS (2013) High‐throughput genetic and gene expression analysis of the RNAPII‐CTD reveals unexpected connections to SRB10/CDK8. PLoS Genet 9: e1003758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomei MS, Halden NF, Cullen CR, Corden JL (1988) Genetic analysis of the repetitive carboxyl‐terminal domain of the largest subunit of mouse RNA polymerase II. Mol Cell Biol 8: 330–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DM, Pandya‐Jones A, Tong AJ, Barozzi I, Lissner MM, Natoli G, Black DL, Smale ST (2012) Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150: 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning M, Dugast‐Darzacq C, Rankovic M, Hansen AS, Yu T, Marie‐Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P et al (2018) RNA polymerase II clustering through carboxy‐terminal domain phase separation. Nat Struct Mol Biol 25: 833–840 [DOI] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM (2018) Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell 175: 1842–1855.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S (2009) Progression through the RNA polymerase II CTD Cycle. Mol Cell 36: 541–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke KA, Janke AM, Rhine CL, Fawzi NL (2015) Residue‐by‐residue view of in vitro FUS granules that bind the C‐terminal domain of RNA polymerase II. Mol Cell 60: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RD, Heidemann M, Hintermair C, Eick D (2008) Molecular evolution of the RNA polymerase II CTD. Trends Genet 24: 289–296 [DOI] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, Cisse II (2018) Mediator and RNA polymerase II clusters associate in transcription‐dependent condensates. Science 361: 412–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, Ørom UA (2017) Cellular fractionation and isolation of chromatin‐ associated RNA. Methods Mol. Biol. 1468: 1–9 [DOI] [PubMed] [Google Scholar]
- Corden JL, Cadena DL, Ahearn JM, Dahmus ME (1985) A unique structure at the carboxyl terminus of the largest subunit of eukaryotic RNA polymerase II. Proc Natl Acad Sci USA 82: 7934–7938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corden JL (2013) RNA polymerase II C‐terminal domain: tethering transcription to transcript and template. Chem Rev 113: 8423–8455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P, Cáceres JF, Cazalla D, Kadener S, Muro AF, Baralle FE, Kornblihtt AR (1999) Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell 4: 251–258 [DOI] [PubMed] [Google Scholar]
- Cramer P, Bushnell DA, Kornberg RD (2001) Structural basis of transcription: RNA polymerase II at 2.8 ångstrom resolution. Science 292: 1863–1876 [DOI] [PubMed] [Google Scholar]
- Cramer P (2019) Organization and regulation of gene transcription. Nature 573: 45–54 [DOI] [PubMed] [Google Scholar]
- David CJ, Boyne AR, Millhouse SR, Manley JL (2011) The RNA polymerase II C‐terminal domain promotes splicing activation through recruitment of a U2AF65‐Prp19 complex. Genes Dev 25: 972–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias JD, Rito T, Triglia ET, Kukalev A, Ferrai C, Chotalia M, Brookes E, Kimura H, Pombo A (2015) Methylation of RNA polymerase II non‐consensus Lysine residues marks early transcription in mammalian cells. Elife 4: e11215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölken L, Ruzsics Z, Rädle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P et al (2008) High‐resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrensberger AH, Kelly GP, Svejstrup JQ (2013) XMechanistic interpretation of promoter‐proximal peaks and RNAPII density maps. Cell 154: 713–715 [DOI] [PubMed] [Google Scholar]
- Eick D, Geyer M (2013) The RNA polymerase II carboxy‐terminal domain (CTD) code. Chem Rev 113: 8456–8490 [DOI] [PubMed] [Google Scholar]
- Fong N, Bentley DL (2001) Capping, splicing, and 3′ processing are independently stimulated by RNA polymerase II: Different functions for different segments of the CTD. Genes Dev 15: 1783–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber H, Hagmann M, Seipel K, Georgiev O, West MAL, Litingtungt Y, Schaffner W, Cordent JL (1995) RNA polymerase II C‐terminal domain required for enhancer‐driven transcription. Nature 374: 660–662 [DOI] [PubMed] [Google Scholar]
- Gerber A, Ito K, Chu CS, Roeder RG (2020) Gene‐specific control of tRNA expression by RNA polymerase II. Mol Cell 78: 765–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber A, Roeder RG (2020) The CTD is not essential for the post‐initiation control of RNA polymerase II activity. J Mol Biol 432: 5489–5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressel S, Schwalb B, Decker TM, Qin W, Leonhardt H, Eick D, Cramer P (2017) CDK9‐dependent RNA polymerase II pausing controls transcription initiation. Elife 6: e29736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressel S, Schwalb B, Cramer P (2019) The pause‐initiation limit restricts transcription activation in human cells. Nat Commun 10: 3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Horng T, Medzhitov R (2009) Control of inducible gene expression by signal‐dependent transcriptional elongation. Cell 138: 129–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Churchman LS (2017) The code and beyond: transcription regulation by the RNA polymerase II carboxy‐terminal domain. Nat Rev Mol Cell Biol 18: 263–273 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK (2015) The selection and function of cell type‐specific enhancers. Nat Rev Mol Cell Biol 16: 144–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschman HR (1991) Primary response genes induced by growth factors and tumor promoters. Annu Rev Biochem 60: 281–319 [DOI] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA (2017) A phase separation model for transcriptional control. Cell 169: 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin JP, Manley JL (2012) The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 26: 2119–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Toedling J, Steinmetz LM (2006) Transcript mapping with high‐density oligonucleotide tiling arrays. Bioinformatics 22: 1963–1970 [DOI] [PubMed] [Google Scholar]
- Jaeger MG, Schwalb B, Mackowiak SD, Velychko T, Hanzl A, Imrichova H, Brand M, Agerer B, Chorn S, Nabet B et al (2020) Selective mediator dependence of cell‐type‐specifying transcription. Nat Genet 52: 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Zaganjor E, Cobb MH (2013) Chromatin‐tethered MAPKs. Curr Opin Cell Biol 25: 272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb TM, Davis MA (2004) The tumor promoter 12‐O‐tetradecanoylphorbol 13‐acetate (TPA) provokes a prolonged morphologic response and ERK activation in Tsc2‐null renal tumor cells. Toxicol Sci 81: 233–242 [DOI] [PubMed] [Google Scholar]
- Koleske AJ, Buratowski S, Nonet M, Young RA (1992) A novel transcription factor reveals a functional link between the RNA polymerase II CTD and TFIID. Cell 69: 883–894 [DOI] [PubMed] [Google Scholar]
- Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL (2014) Phosphorylation‐regulated binding of RNA polymerase ii to fibrous polymers of low‐complexity domains. Cell 156: 374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaner VD, Chick JF, Donninger H, Linniola I, Mendoza A, Khanna C, Birrer MJ (2009) Inhibition of AP‐1 transcriptional activity blocks the migration, invasion, and experimental metastasis of murine osteosarcoma. Am J Pathol 174: 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Tjian R (2003) Transcription regulation and animal diversity. Nature 424: 147–151 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2016) Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32: 2103–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Rosenfeld MG (2016) Enhancers as non‐coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet 17: 207–223 [DOI] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B (2019) WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47: W199–W205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Portz B, Gilmour DS (2019) The C‐terminal domain of RNA polymerase II is a multivalent targeting sequence that supports drosophila development with only consensus heptads. Mol. Cell 73: 1232–1242.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, Lis JT (2016) Base‐pair‐resolution genome‐wide mapping of active RNA polymerases using precision nuclear run‐on (PRO‐seq). Nat Protoc 11: 1455–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier KC, Gressel S, Cramer P, Schwalb B (2020) Native molecule sequencing by nano‐ID reveals synthesis and stability of RNA isoforms. Genome Res 30: 1332–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17: 10 [Google Scholar]
- Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, Kremmer E, Eick D, Cramer P (2012) CTD Tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science 336: 1723–1725 [DOI] [PubMed] [Google Scholar]
- Meinhart A, Kamenski T, Hoeppner S, Baumli S, Cramer P (2005) A structural perspective of CTD function. Genes Dev 19: 1401–1415 [DOI] [PubMed] [Google Scholar]
- Meininghaus M, Chapman RD, Horndasch M, Eick D (2000) Conditional expression of RNA polymerase II in mammalian cells. Deletion of the carboxyl‐terminal domain of the large subunit affects early steps in transcription. J Biol Chem 275: 24375–24382 [DOI] [PubMed] [Google Scholar]
- Melgar MF, Collins FS, Sethupathy P (2011) Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol 12: R113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M, Demel C, Zacher B, Schwalb B, Krebs S, Blum H, Gagneur J, Cramer P (2017) TT‐seq captures enhancer landscapes immediately after T‐cell stimulation. Mol Syst Biol 13: 920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SJ, Yang L, Meluzzi D, Oh S, Yang F, Friedman MJ, Wang S, Suter T, Alshareedah I, Gamliel A et al (2019) Phase separation of ligand‐activated enhancers licenses cooperative chromosomal enhancer assembly. Nat Struct Mol Biol 26: 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Young RA (1989) Intragenic and extragenic suppressors of mutations in the heptapeptide repeat domain of Saccharomyces cerevisiae RNA polymerase II. Genetics 123: 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta S, Yamamuro T, Lee K, Okumura H, Kasai R, Hiraki Y, Ikeda T, Iwasaki R, Kikuchi H, Konishi J et al (1991) Fracture healing induces expression of the proto‐oncogene c‐fos in vivo Possible involvement of the Fos protein in osteoblastic differentiation. FEBS Lett 284: 42–45 [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods 14: 417–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatnani HP, Greenleaf AL (2006) Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev 20: 2922–2936 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero‐Cadena P, Lenstra TL, Sternberg PW (2020) RNA Pol II length and disorder enable cooperative scaling of transcriptional bursting. Mol Cell 79: 207–220.e8 [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2011) R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC et al (2018) Coactivator condensation at super‐enhancers links phase separation and gene control. Science 361: eaar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafe C, Chao D, Lopes J, Hirsch JP, Henry S, Young RA (1990) RNA polymerase II C‐terminal repeat influences response to transcriptional enhancer signals. Nature 347: 491–494 [DOI] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Hauf KF, Demel C, Tresch A, Gagneur J, Cramer P (2016) TT‐seq maps the human transient transcriptome. Science 352: 1225–1228 [DOI] [PubMed] [Google Scholar]
- Schwer B, Sanchez AM, Shuman S (2012) Punctuation and syntax of the RNA polymerase II CTD code in fission yeast. Proc Natl Acad Sci USA 109: 18024–18029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sdano MA, Fulcher JM, Palani S, Chandrasekharan MB, Parnell TJ, Whitby FG, Formosa T, Hill CP (2017) A novel SH2 recognition mechanism recruits Spt6 to the doubly phosphorylated RNA polymerase II linker at sites of transcription. Elife 6: e28723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A (2014) Transcriptional enhancers: from properties to genome‐wide predictions. Nat Rev Genet 15: 272–286 [DOI] [PubMed] [Google Scholar]
- Simonti CN, Pollard KS, Schröder S, He D, Bruneau BG, Ott M, Capra JA (2015) Evolution of lysine acetylation in the RNA polymerase II C‐terminal domain. BMC Evol Biol 15: 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JL, Cantin GT, Wang G, Shevchenko A, Shevchenko A, Berk AJ (2002) Transcription control by E1A and MAP kinase pathway via Sur2 mediator subunit. Science 296: 755–758 [DOI] [PubMed] [Google Scholar]
- Stiller JW, Hall BD (2002) Evolution of the RNA polymerase II C‐terminal domain. Proc Natl Acad Sci USA 99: 6091–6096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Larivière L, Maier KC, Seizl M, Tresch A, Cramer P (2012) Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 22: 1350–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AD, Soulette CM, van Baren MJ, Hart K, Hrabeta‐Robinson E, Wu CJ, Brooks AN (2020) Full‐length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11: 1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen M, Lienhard M, Schrooders Y, Clayton O, Nudischer R, Boerno S, Timmermann B, Selevsek N, Schlapbach R, Gmuender H et al (2019) DMSO induces drastic changes in human cellular processes and epigenetic landscape in vitro . Sci Rep 9: 4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villamil G, Wachutka L, Cramer P, Gagneur J, Schwalb B (2019) Transient transcriptome sequencing: computational pipeline to quantify genome‐wide RNA kinetic parameters and transcriptional enhancer activity. bioRxiv 10.1101/659912 [PREPRINT] [DOI] [Google Scholar]
- Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, Cramer P (2018a) Structure of activated transcription complex Pol II–DSIF–PAF–SPT6. Nature 560: 607–612 [DOI] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Urlaub H, Cramer P (2018b) Structure of paused transcription complex Pol II–DSIF–NELF. Nature 560: 601–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachutka L, Caizzi L, Gagneur J, Cramer P (2019) Global donor and acceptor splicing site kinetics in human cells. Elife 8: e45056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF (2002) Functions of AP1 (Fos/Jun) in bone development. Ann Rheumat Dis 61(Supplement 2): 40–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West ML, Corden JL (1995) Construction and analysis of yeast RNA polymerase II CTD deletion and substitution mutations. Genetics 140: 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Nord AS, Akiyama JA, Shoukry M, Afzal V, Rubin EM, Pennacchio LA, Visel A (2014) Tissue‐specific RNA expression marks distant‐acting developmental enhancers. PLoS Genet 10: e1004610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Sharrocks AD, Whitmarsh AJ (2013) MAP kinase signalling cascades and transcriptional regulation. Gene 513: 1–13 [DOI] [PubMed] [Google Scholar]
- Zaborowska J, Egloff S, Murphy S (2016) The pol II CTD: new twists in the tail. Nat Struct Mol Biol 23: 771–777 [DOI] [PubMed] [Google Scholar]
- Zacher B, Michel M, Schwalb B, Cramer P, Tresch A, Gagneur J (2017) Accurate promoter and enhancer identification in 127 ENCODE and roadmap epigenomics cell types and tissues by GenoSTAN. PLoS One 12: e0169249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Data Availability Statement
RNA‐seq, TT‐seq, PRO‐seq, and Nanopore sequencing data are available under Gene Expression Omnibus GSE159092 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159092).