

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder consisting of progressive loss of motor neurons. TDP‐43 has been identified as a component of ubiquitin‐immunoreactive inclusions of motor neurons in ALS. We focused on the diffuse cytoplasmic TDP‐43 immunoreactivity in ALS neurons, and quantitatively assessed it in comparison with skein/round TDP‐43 and ubiquitin immunostaining in motor neurons of 30 sporadic ALS cases. The percentage of spinal motor neurons with cytoplasmic TDP‐43 immunoreactivity was higher than that of ubiquitin‐immunoreactive ones. The percentage of TDP‐43‐positive motor neurons was independent of neuron counts in anterior horns, while the percentage of ubiquitinated neurons was inversely correlated. Aiming to define the cytosolic localization of TDP‐43, the immunoblot analysis of spinal cord and frontal cortex showed that full‐length TDP‐43, the 45 kDa form and ubiquitinated TDP‐43 are found in the soluble inclusion‐free fraction. The present data suggest that delocalization, accumulation and ubiquitination of TDP‐43 in the cytoplasm of motor neurons are early dysfunctions in the cascade of the events leading to motor neuron degeneration in ALS, preceding the formation of insoluble inclusion bodies. Being cytoplasmic accumulation an ongoing event during the course of the illness, a therapeutic approach to this incurable disease can be envisaged.
Keywords: ALS, biochemistry, cell counting, immunohistochemistry, TDP‐43, ubiquitin
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive degenerative disorder in which the selective death of motor neurons in the brain and spinal cord leads to paralysis of voluntary muscles. Most cases are sporadic; less than 10% are inherited. Although excessive excitatory tone, protein misfolding, alterations in axonal transport, dysfunction of energy production and calcium metabolism have been described (30), the underlying pathological events of motor neuron death remain unclear.
The neuropathological hallmark of ALS is the intracytoplasmic accumulation of ubiquitin‐tagged proteins in motor neurons, appearing as round or skein‐like inclusions. Recently, TDP‐43 has been identified as a component of these inclusions in ALS 2, 29, 37), as well as in frontotemporal lobar degeneration (FTLD) with and without motor neuron disease 7, 9, 29). TDP‐43 has been shown to be a major disease protein in ALS (20), a view strengthened by the finding of TDP‐43 gene mutations in familial and sporadic ALS cases 13, 18, 35, 42), and of direct links between gene mutation and TDP‐43 neuropathology in ALS (40). The mechanisms underlying the segregation of TDP‐43 into cytoplasmic ubiquitinated aggregates and the implication of TDP‐43 in the pathogenesis of neurodegeneration remain unknown.
TDP‐43 and ubiquitin colocalize within the pathological cytoplasmic skein/round inclusions (23); however, the spectrum of TDP‐43 immunoreactivity appears to be wider than that of ubiquitin, and includes diffuse cytoplasmic immunoreactivity and diffuse punctate cytoplasmic staining in ALS 2, 12, 27, 33, 36) and in sporadic FTLD with motor neuron disease (4). The distinct cytopathological profile consisting of a cleared nucleus and diffuse cytoplasmic TDP‐43 immunostaining may represent an early stage of inclusion formation and has been considered “pre‐inclusions”4, 7, 11, 12, 27).
A pathological form of TDP‐43 has been found biochemically in sarkosyl‐insoluble fractions from ALS samples seemingly corresponding to the insoluble inclusion bodies 2, 23, 29, 36). The biochemical features of the diffuse cytoplasmic TDP‐43 seen by immunohistochemistry are so far not known.
The aims of the present study were to assess the quantitative relation between the two immunohistochemical patterns (diffuse staining and skein/round inclusion staining) of TDP‐43 in ALS, and to analyze biochemically TDP‐43 and its ubiquitination status in the cytosolic, inclusion‐free fraction from ALS samples. In an effort to correlate immunohistochemistry and disease progression, the number of immunopositive neurons was evaluated in relation to the number of neuron counts in anterior horns of ALS.
MATERIALS AND METHODS
Patients
Thirty patients with sporadic ALS underwent complete autopsy, including the brain and spinal cord. Nervous tissue was processed with formalin–paraffin. Specimens of frontal cortex and spinal cord from three sporadic ALS cases and three neuropathologically normal individuals matched for age were stored at −80°C for biochemical analysis.
The ALS patients were seen in a motor neuron diseases clinic. All patients had the diagnosis of definite ALS at death, according to the revised El Escorial criteria (5). They ranged in age from 41 to 74 years, and the median age was 61 years at death (9 women and 21 men). The duration of illness ranged from 5 to 60 months with a median duration of 30 months. The site of onset was bulbar in 10 patients and limbs in 20 patients. Two patients had overt dementia meeting Neary criteria (28) for clinical frontotemporal dementia: a 43‐year‐old male patient with behavioral and cognitive changes preceding the motor symptoms by 6 years, and a 40 year‐old female patient with behavioral abnormalities presenting during the follow‐up of her motor symptoms. A detailed neuropsychological testing for the evaluation of frontal type impairment was not performed.
Immunohistochemistry
Serial 5 µm thick paraffin sections from the cervical and lumbar spinal cord, frontal cortex, temporal cortex and hippocampus were immunostained by a monoclonal antibody to ubiquitin (clone 1510, Chemicon, Temecula, Calif.; 1:200), a polyclonal antibody to TDP‐43 (ProteinTech Group, Chicago, Ill.; 1:400) and a monoclonal anti‐GFAP (clone 6F2, Dako, Glostrup, Denmark; 1:100) The slides were pretreated by microwave to enhance immunoreactivity. The immune reaction was visualized by Strep‐ABC Vectastain® kit (Vector Laboratories, Burlingame, CA, USA) using diaminobenzidine as the chromogen. In parallel, TDP‐43 immunostaining was visualized by EnVision™ + Dual Link System‐HRP (Dako) in order to check and improve accuracy and consistency.
Quantitation of immunolabeling
The number of motor neurons containing ubiquitin‐immunopositive inclusions and TDP‐43 immunostaining were counted in three distinct slides 100 µm apart of lumbar spinal cord of each patient. Neurons were identified by the presence of nucleus and/or nucleolus, or by the large GFAP‐negative cell body in adjacent section. The percentage of immunopositive neurons out of the total number of motor neurons seen in each slide was recorded. The percentage of ubiquitin‐ and TDP‐43‐positive neurons was also counted in 200 hippocampal dentate granule neurons in the most densely labeled areas of the immunopositive cases.
Colocalization
The double immunofluorescence study was performed on selected sections using a rabbit polyclonal antibody against TDP‐43 (1:200) and a mouse monoclonal antibody against ubiquitin (clone 1510; 1:200). The secondary antibodies were polyclonal rabbit anti‐mouse TRIC (Dako; 1:100) and polyclonal swine anti‐rabbit FITC (Dako; 1:100). Slides were viewed with a Zeiss Axio‐Imager microscope (Zeiss, Göttingen, Germany).
Biochemical analysis
Reagents
TDP‐43 polyclonal antibody (BC001487) was from Protein Tech Group; glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) (sc‐25778) polyclonal antibody and secondary antibodies conjugated to HRP were from Santa‐Cruz, Heidelberg, Germany; ubiquitin polyclonal antibody (U5379) and protease (P8340) and phosphatase (P2850) inhibitor cocktails were from Sigma‐Aldrich, Milan, Italy; lamin B1 monoclonal antibody (33‐2000) was from Zymed Laboratories, San Francisco, Calif.; histone H4 antibody (2592) was from Cell Signaling Technology (CELBIO, Milan, Italy); polyvinylidene difluoride (PVDF) membranes were from GE Healthcare, Milan, Italy; and protein molecular markers (161‐0373) and RC DC protein assay kit were from BioRad, Milan, Italy. All other chemicals were analytical grade from Sigma‐Aldrich.
Tissue fractionation
Frozen specimens of the spinal cord and frontal cortex were homogenized in isotonic medium, hereafter defined as low salt buffer (LS‐B), consisting of 20 mM Tris (pH 7.2), 5 mM EDTA, 10% sucrose, 1 µL/mL of protease inhibitor cocktail and 2 µL/mL of phosphatase inhibitor cocktail. The homogenate was centrifuged at low speed (1.300 × g) for 20 minutes at 4°C, a speed sufficient to prevent nuclear lysis and to give rise to a supernatant and a pellet (pellet A) referred to include intact nuclei, unbroken cells and cellular debris 25, 38). Pellet A was washed in LS‐B and pelleted again at low speed. The supernatant was centrifuged at high speed (100.000 × g) for 120 minutes at 4°C and resolved in an additional pellet (pellet B) and in a clear soluble fraction. Pellet B was washed in LS‐B and sedimented again at high speed. The soluble fraction was centrifuged again at high speed for 60 minutes without no additional pellet; on this basis, this fraction was defined cytosolic fraction free from insoluble material (CS‐F).
Pellets A and B were separately suspended in a high salt buffer (HS buffer) consisting of 10 mM Tris–HCl (pH 7.5), 1 mM EGTA, 10% sucrose, 0.8 M NaCl, 1 µL/mL of protease inhibitor cocktail and 2 µL/mL of phosphatase inhibitor cocktail, added to an equal volume of 2% Triton X‐100 in HS buffer. Aliquots of the suspended pellets A and B were characterized by Western immunoblotting by using antibodies to the nuclear envelope marker lamin B1 and the nuclear matrix or nucleoplasm marker histone H4. The analysis revealed a strong immunopositivity of both pellets for the two markers (Figure 1). For this reason, pellets A and B were combined and processed by sequential fractionations 15, 16), finally giving rise to a sarkosyl‐insoluble pellet (SarkI‐P).
Figure 1.
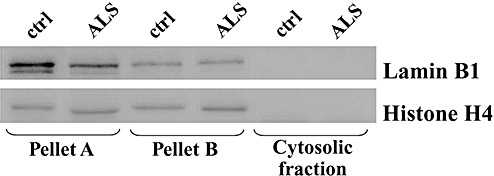
Characterization of pellets A and B, and cytosolic fraction from samples of amyotrophic lateral sclerosis spinal cord (ALS) and controls (ctrl) by using the nuclear‐specific markers lamin B1 and histone H4. Pellets A and B are positive to the nuclear markers; the cytosolic fraction [cytosolic fraction free from insoluble material (CS‐F)] is negative.
The soluble fraction CS‐F was characterized by Western immunoblotting by using lamin B1, histone H4 and the cytoplasmic marker GAPDH.
At the end of these procedures, the soluble fraction (CS‐F) and the SarkI‐P were separately added with the urea buffer A, consisting of 50 mM Tris (pH 6.8), 5% (w/v) SDS, 4 M deionized urea, 2% (v/v) β‐mercaptoethanol (39), incubated for 30 minutes at room temperature, centrifuged at 18 000 × g for 30 minutes, aliquoted and finally stored frozen at −70°C. Before freezing, all preparations were evaluated for protein content using the RC DC protein assay kit.
Western immunoblotting
Frozen samples of CS‐F and SarkI‐P were added with urea buffer B consisting of 50 mM Tris (pH 6.8), 2% glycerol, 5% (w/v) SDS, 4 M deionized urea, 40 mM dithiothreitol, 0.05% (w/v) bromophenol blue (39), incubated for 2 h at room temperature, centrifuged at 18 000 × g for 10 minutes and then subjected to SDS–PAGE on 10% or 12% (w/v) polyacrylamide gels and electroblotted on PVDF membranes (3). Blotted proteins were immunostained with antibodies to TDP‐43 and ubiquitin. As a control for equal protein loading, the antibody to GAPDH was used. Blots are representative of three or more independent experiments.
RESULTS
Immunohistochemistry
Spinal cord
No immunostaining for ubiquitin was observed in motor neurons of control cases, while a granular nuclear staining for TDP‐43 was present. Skein‐like or round intracytoplasmic ubiquitin‐ and TDP43‐immunoreactive deposits were found in motor neurons of ALS spinal cords. (Figure 2A,B). Other motor neurons showed a diffuse TDP‐43 immunostaining instead of the skein/round cytoplasmic aggregates (Figure 2C,D,E,G,H,I); the diffuse pattern of TDP‐43 did not overlap with ubiquitin immunoreactivity (Figure 2E,F). Diffuse cytoplasmic TDP‐43 was found also in motor neurons of large size and endowed with Nissl substance (Figure 2H). The nuclei of the neurons with cytoplasmic TDP‐43 immunostaining were negative for TDP‐43. Rare TDP‐43‐positive glial inclusions were present in the anterior horns.
Figure 2.
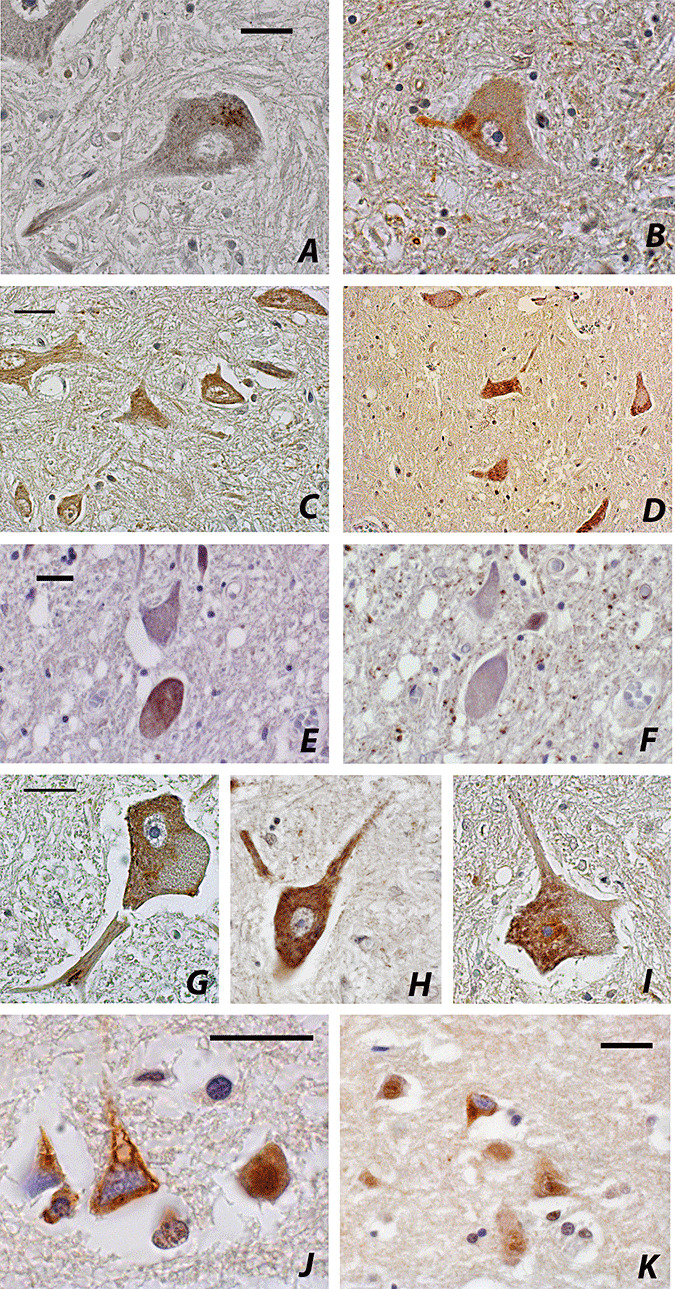
TDP‐43 and ubiquitin immunohistochemistry in sporadic ALS. (A) TDP‐43‐ and (B) ubiquitin‐positive skein‐like inclusion in spinal motor neurons; the nuclei of neurons are TDP‐43‐negative, DAB; (C), (D), (E), (G), (H), (I) Diffuse cytoplasmic TDP‐43 immunoreactivity in spinal motor neurons; (F) no ubiquitin staining in the TDP‐43‐positive neuron shown in (E), DAB; (J), (K) TDP‐43‐positive cytoplasm of neurons in the frontal cortex, DAB. Scale bars = 20 µm.
Double immunostaining showed an incomplete overlap of ubiquitin and TDP‐43 in some skein/round bodies, with some TDP‐43‐positive deposits partially overlapping with ubiquitin‐positive ones (Figure 3A–C).
Figure 3.
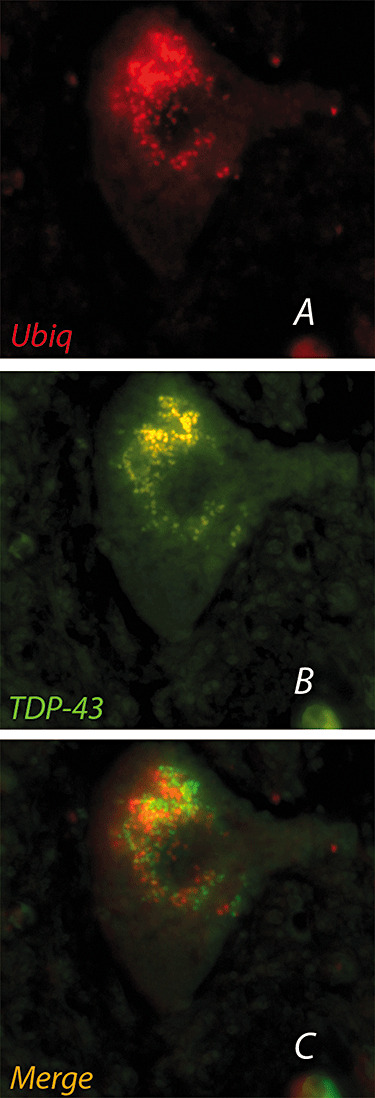
Double‐label immunofluorescence of cytoplasmic inclusions in an anterior horn motor neuron of a sporadic amyotrophic lateral sclerosis patient: TDP‐43 (A) and ubiquitin (B) do not completely overlap (C). A. Ubiquitin: TRIC, red. B. TDP‐43: FITC, green. C. Merged. ×1000.
Hippocampus, frontal cortex, temporal cortex
Cytoplasmic ubiquitin and TDP‐43 staining of hippocampal dentate neurons was found in six ALS patients, including the two ALS patients with dementia. Nuclear TDP‐43 staining was absent in the dentate neurons with cytoplasmic TDP‐43 staining.
In the remaining 24 cases, neither ubiquitin nor cytoplasmic TDP‐43 staining was observed in dentate neurons, while the nuclear immunoreactivity to the TDP‐43 antibody was a constant finding.
In the temporal and frontal cortex, TDP‐43 antibody decorated the neuronal nuclei; cytoplasmic immunostaining was observed in neurons of the frontal cortex in one ALS case only (Figure 2J,K).
Quantitative evaluation of immunostaining
Counting of TDP‐43‐positive neurons was performed in the slides revealed by the EnVision–Dual Link system which resulted to give the most consistent results. The mean number and percentage of TDP‐43‐ and ubiquitin‐positive motor neurons in the three slides of lumbar anterior horns and in the dentate gyrus of the hippocampus, as well as the mean number of motor neurons counted in the three slides of lumbar spinal cord, are reported in Table 1.
Table 1.
Quantitative immunohistochemistry of TDP‐43 and ubiquitin in 30 sporadic amyotrophic lateral sclerosis (ALS) patients.
| Anterior horns of lumbar spinal cord | Dentate gyrus | ||||||
|---|---|---|---|---|---|---|---|
| Cases | Neurons with diffuse citoplasmic TDP‐43 (%) | Neurons with skein/round TDP‐43 aggregates (%) | Neurons with TDP‐43 any type (%) | Neurons with skein/round ubiquitin aggregates (%) | Mean number of neurons in anterior horns | TDP‐ 43‐positive neurons (%) | Ubiquitin ‐positive neurons (%) |
| 1 sALS | 47 (74.61%) | 3 (4.76%) | 50 (79.37%) | 2 (3.17%) | 63 | ||
| 2 sALS | 51 (87.93%) | 4 (6.90%) | 55 (94.83%) | 6 (10.34%) | 58 | ||
| 3 sALS | 13 (24.07%) | 5 (9.26%) | 18 (33.33%) | 2 (3.70%) | 54 | ||
| 4 sALS | 6 (13.33%) | 9 (20.00%) | 15 (33.33%) | 10 (22.22%) | 45 | ||
| 5 sALS | 10 (25.00%) | 0 (0.00%) | 10 (25.00%) | 4 (10.00%) | 40 | ||
| 6 sALS | 0 (0.00%) | 5 (12.50%) | 5 (12.50%) | 2 (5.00%) | 40 | ||
| 7 sALS‐D | 4 (10.26%) | 6 (15.38%) | 10 (25.64%) | 8 (19.51%) | 40 | 23.08 | 35.15 |
| 8 sALS | 3 (7.90%) | 14 (36.84%) | 17 (44.74%) | 10 (26.32%) | 38 | ||
| 9 sALS | 19 (52.78%) | 0 (0.00%) | 19 (52.78%) | 2 (5.56%) | 36 | 6.08 | 5.88 |
| 10 sALS | 28 (65.12%) | 0 (0.00%) | 28 (65.12%) | 5 (19.23%) | 34.5 | ||
| 11 sALS | 6 (20.00%) | 1 (3.33%) | 7 (23.33%) | 4 (10.26%) | 34.5 | ||
| 12 sALS | 6 (18.75%) | 12 (37.50%) | 18 (56.25%) | 8 (25.00%) | 32 | ||
| 13 sALS | 14 (48.28%) | 0 (0.00%) | 14 (48.28%) | Na | 29 | 2.14 | 1.69 |
| 14 sALS | 24 (88.89%) | 0 (0.00%) | 24 (88.89%) | Na | 27 | ||
| 15 sALS | 1 (7.14%) | 2 (14.29%) | 3 (21.43%) | 5 (12.82%) | 26.5 | ||
| 16 sALS | 0 (0.00%) | 5 (16.00%) | 5 (16.00%) | 7 (28.00%) | 25 | ||
| 17 sALS | 1 (4.00%) | 1 (4.00%) | 2 (8.00%) | 5 (20.00%) | 25 | ||
| 18 sALS | 12 (46.16%) | 2 (7.69%) | 14 (53.85%) | 4 (17.39%) | 24.5 | ||
| 19 sALS | 8 (38.09%) | 4 (19.05%) | 12 (57.14%) | 6 (23.08%) | 23.5 | ||
| 20 sALS | 1 (4.35%) | 1 (4.35%) | 2 (8.70%) | 2 (8.70%) | 23 | ||
| 21 sALS | 3 (14.29%) | 7 (33.33%) | 10 (47.62%) | 12 (57.14%) | 21 | 6.33 | 4.85 |
| 22 sALS | 14 (58.34%) | 2 (8.33%) | 16 (66.67%) | 2 (13.33%) | 19.5 | ||
| 23 sALS‐D | 11 (36.67%) | 6 (20.00%) | 17 (56.67%) | 3 (33.33%) | 19.5 | 3.72 | 3.55 |
| 24 sALS | 16 (84.21%) | 0 (0.00%) | 16 (84.21%) | 1 (5.26%) | 19 | ||
| 25 sALS | 1 (6.66%) | 4 (26.67%) | 5 (33.33%) | 6 (33.33%) | 16.5 | ||
| 26 sALS | 6 (26.09%) | 3 (13.04%) | 9 (39.13%) | 2 (22.22%) | 16 | ||
| 27 sALS | 2 (18.18%) | 2 (18.18%) | 4 (36.36%) | 2 (18.18%) | 11 | ||
| 28 sALS | 0 (0.00%) | 2 (18.18%) | 2 (18.18%) | 6 (54.54%) | 11 | ||
| 29 sALS | 1 (7.70%) | 2 (15.38%) | 3 (23.08%) | 3 (33.33%) | 11 | 4.25 | 3.48 |
| 30 sALS | 4 (44.44%) | 0 (0.00%) | 4 (44.44%) | 2 (33.33%) | 7.5 | ||
na = not applicable.
The overall percentage of TDP‐43‐positive spinal motor neurons ranged from 8% to 94.83% (mean ± SD: 43.27 ± 23.91). The overall percentage of spinal motor neurons with TDP‐43‐positive skeins or round bodies was 0% to 37.5% (mean ± SD: 12.17 ± 11.08), and 0% to 88.89% (mean ± SD: 31.11 ± 27.93) of neurons had diffuse cytoplasmic immunostaining for TDP‐43. The overall percentage of spinal motor neurons with ubiquitin‐positive inclusions ranged from 3.17% to 57.14% (mean ± SD: 20.51 ± 13.81) (Table 2).
Table 2.
Percentage of TDP‐43‐ and ubiquitin‐positive motor neurons in anterior horns of 30 sporadic amyotrophic lateral sclerosis patients.
| Mean number (%) | Standard deviation (%) | Range (%) | |
|---|---|---|---|
| Neurons with diffuse cytoplasmic TDP‐43 | 31.11 | 27.93 | 0–88.89 |
| Neurons with skein/round TDP‐43 aggregates | 12.17 | 11.08 | 0–37.50 |
| Neurons with TDP‐43 any type | 43.27 | 23.91 | 8–94.83 |
| Neurons with skein/round ubiquitin aggregates | 20.51 | 13.81 | 3.17–57.14 |
After dividing the ALS cases into groups according to the mean number of motor neurons counted in the anterior horns, the percentage of neurons showing ubiquitin‐positive inclusions increased with the decreasing number of neurons. This was not the case for TDP‐43. The highest percentage of neurons with diffuse cytoplasmic TDP‐43 was counted in the anterior horns with the highest number of residual neurons. Conversely, the percentage decreased with the decreasing number of neurons, even if neurons with cytoplasmic TDP‐43 were frequent also in the anterior horns containing very few motor neurons (Figure 4). The percentage of neurons with skein‐like/round TDP‐43 inclusions did not show a relevant modification with the decreasing number of neurons. The percentage of TDP‐43‐positive skein/round inclusions was similar to that of the ubiquitin‐positive inclusions in the subgroups with more than 40 residual neurons. If the number of residual neurons was lower than 30, however, ubiquitin‐positive inclusions were more numerous than the TDP‐43‐positive ones (Table 1; Figure 4).
Figure 4.
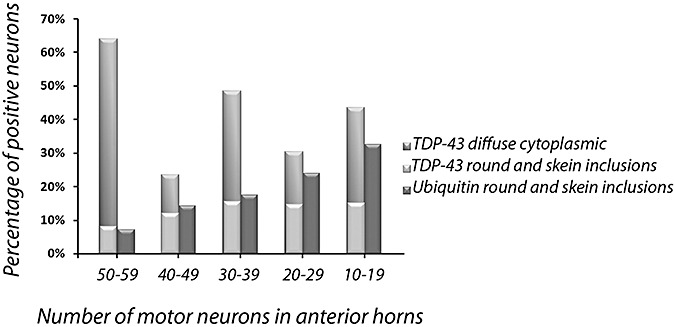
Percentage of TDP‐43‐ and ubiquitin‐positive spinal motor neurons by number of residual motor neurons in anterior horns of 30 sporadic amyotrophic lateral sclerosis patients.
In the dentate gyrus of the hippocampus, neurons with cytoplasmic ubiquitin‐ and TDP‐43‐positive inclusions ranged from 1.69% to 35.15%, and from 3.72% to 23.08%, respectively (Table 1). The possibility that some TDP‐43‐positive cells be glial cells cannot be excluded with certainty. Case by case, the percentage of ubiquitin‐immunopositive dentate neurons was similar to that of TDP‐43‐positive ones, with the exception of case #7, an ALS‐D patient affected by long‐lasting dementia, who had a higher percentage of ubiquitin‐ than TDP‐43‐positive neurons (Figure 5).
Figure 5.
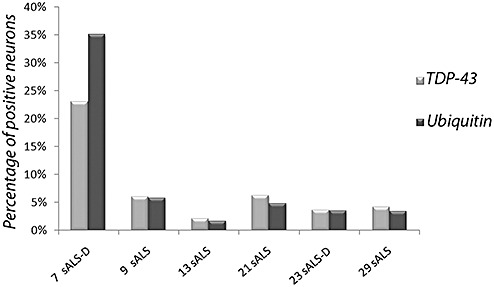
Percentage of TDP‐43‐ and ubiquitin‐ positive dentate neurons of hippocampus in the six immunopositive amyotrophic lateral sclerosis cases.
Biochemistry
The characterization showed that pellets A and B were strongly immunopositive to the two nuclear markers, while CS‐F resulted to be negative to the nuclear markers (Figure 1) and positive to the cytosolic marker GAPDH (Figure 7). On this basis, CS‐F resulted to be free of nuclear components, while pellets A and B included nuclear components in addition to the insoluble material. Therefore, CS‐F is representative of the soluble cytosolic compartment and does not include nuclei and nuclear debris. The results of the characterization prove that the storage of tissue specimens and the procedures by which subcellular fractionation was carried out did not cause any appreciable nuclear lysis. In addition, the absence of pellet after high‐speed centrifugation shows that CS‐F is free from insoluble bodies.
Figure 7.
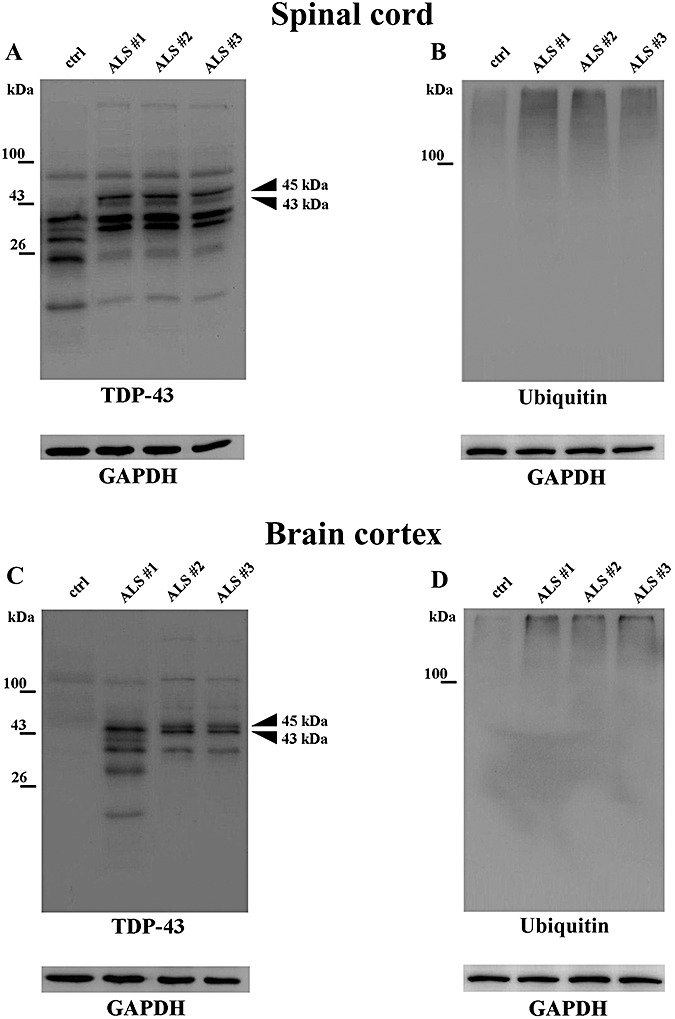
Immunoblotting of the cytosolic fractions (CS‐F) from samples of spinal cord (A,B) and cerebral cortex (C,D) of amyotrophic lateral sclerosis (ALS) and control patients (ctrl) with antibodies to TDP‐43 and ubiquitin. Arrowheads: 43 kDa, normal TDP‐43; 45 kDa: pathological form of TDP‐43. GAPDH immunoreactivity was used to confirm equal protein loading. The images are representative of three independent experiments performed on three patients and three controls.
In the SarkI‐P, derived from pellets A and B, the spectrum of TDP‐43 immunoreactivity in the blots from ALS spinal cord (Figure 6A) reproduced the previously reported one 2, 29, 36), highlighting a 43 kDa band accompanied by a 45 kDa band, a 28 kDa band and diffuse smeared staining including bands sized well above 43. In the blots of control samples, the 43 kDa band only was visible. These findings were reproduced in blots of SarkI‐P obtained from specimens of frontal cortex.
Figure 6.
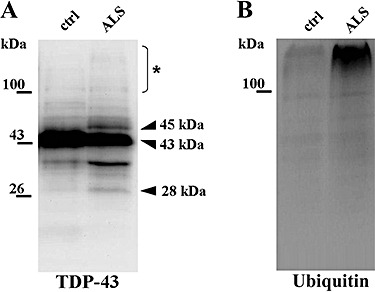
Immunoblotting of the sarkosyl‐insoluble fraction (SarkI‐P) from spinal cord specimens of amyotrophic lateral sclerosis patients (ALS) and control (ctrl) with antibodies to TDP‐43 (A) and ubiquitin (B). The 43 kDa band corresponds to full‐length TDP‐43; the 45 and 28 kDa bands are pathological TDP‐43 forms; the TDP‐43‐immunostained protein bands below 43 kDa are presumably cleavage products of TDP‐43 (29). (*) High‐molecular mass smear.
In CS‐F from ALS spinal cord, the antibody to TDP‐43 disclosed two immunoreactive bands at 43 and 45 kDa, being the latter darker than the former. By contrast, in the CS‐F blots of controls, the 43 kDa TDP‐43 band was barely detectable and the 45 kDa band was not visualized even after prolonged exposure to the antibody (Figure 7A). In both control and ALS immunoblots, a variety of TDP‐43 immunoreactive bands sized around 38 kDa and below were disclosed. A faint smear in the high‐molecular mass range was unveiled by the ubiquitin antibody in ALS and minimally in controls (Figure 7B).
In the CS‐F from frontal cortex specimens, a TDP‐43 immunoreactive band at 43 kDa, accompanied by a 45 kDa band was an exclusive feature of ALS cases (Figure 7C); a smear in the high‐molecular mass range was barely visible. A ubiquitin‐immunopositive wide smear localized well above 43 kDa was present in CS‐Fs from ALS and control specimens, being prominent in ALS (Figure 7D).
DISCUSSION
The novel finding of the present quantitative analysis is the prevalence of the diffuse cytoplasmic TDP‐43 as compared to the round/skein TDP‐43 and ubiquitin immunoreactivity in ALS‐affected spinal motor neurons. The diffuse cytoplasmic staining found by us in spinal motor neurons is reminiscent of the TDP‐43 immunopositive fine granules diffusely scattered in the neuronal cytoplasm in a number of brain areas of ALS patients 4, 27), in the hypoglossal nucleus of ALS (11) and in lower motor neurons of familial ALS with SOD1 gene mutation (32). Such immunostaining was considered as “pre‐inclusions.” The progressive changes in the TDP‐43 pathology going from diffuse/granular to skein/round inclusions have been described 27, 33, 36).
The diffuse cytoplasm localization of TDP‐43 in ALS motor neurons revealed by immunohistochemistry is confirmed by our biochemical analysis, highlighting a relevant 43 kDa TDP protein band, accompanied by a 45 kDa band, in the cytosolic inclusion‐free fraction of ALS samples (CS‐F). The banding pattern of CS‐F shares features with the pattern of the sarkosyl‐insoluble fraction reported in brain tissue from ALS and FTLD 2, 17, 23, 29, 36) and reproduced by us. The 45 kDa band is considered to be the phosphorylated form of full‐length TDP‐43 16, 29). The 28 kDa band frequently reported in immunoblots from ALS 17, 29, 36) was not evidenced in the CS‐F; this is possibly caused by the difficulty to detect these pathogenic C‐terminal fragments with the anti‐TDP‐43 antibody used. Consistently with the nuclear localization of normal TDP‐43, a 43 kDa band was not appreciable in the cytosolic fraction of control cortex and spinal cord, strengthening the value of our results.
A significant finding is the poor correlation between the detection of cytosolic TDP‐43 biochemically and the frequency of immunohistochemical cytoplasmic TDP‐43 reactivity in motor cortex of ALS. The ability to detect changes biochemically in areas without inclusions was reported for α‐synuclein in multiple system atrophy (10); it shows the existence of biochemically detected occult pathology in neurodegenerative diseases.
A number of additional novel dysfunctions were disclosed by the biochemical analysis. In the cytosolic fraction from ALS samples, both full‐length TDP‐43 and the 45 kDa form are accumulated and therefore independent of the inclusion bodies; these features were not so far demonstrated. In addition, TDP‐43 as a high‐molecular mass component tagged with various ubiquitin monomers is present not only in the fraction containing the inclusion bodies, but also in the cytosolic inclusion‐free fraction. Therefore, ubiquitinated TDP‐43 results to be not only a component of the insoluble inclusion bodies, but also a soluble cytosolic component in ALS. Immunohistochemistry is not able to reveal the diffuse ubiquitinated TDP‐43. Additional bands below 38 kDa were visible in both ALS and control blots; they have been considered to be proteolytic products of TDP‐43 (29).
TDP‐43 is a highly conserved nuclear protein that acts as a transcription repressor and activator of exon skipping (6), and is involved in nuclear bodies (26). The redistribution of TDP‐43 in ALS may correspond to loss of nuclear function, resulting in transcriptional deregulation, aberrant messenger RNA splicing and disintegration of nuclear bodies. Reduced stabilization of mRNA coding for the low‐molecular weight isoform of neurofilaments may also take place (36). Any of these defects can play a role in the pathogenesis of ALS. Loss of TDP‐43 nuclear function is consistent with either abnormal subcellular distribution or abnormal structural properties.
The events underlying the redistribution of TDP‐43 in ALS remain unclear. The abnormal accumulation of TDP‐43 as a native protein in the cytosolic fraction is, in itself, predictive of a defective import–export process. The cause can be either impairment of the transport mechanisms at the nuclear envelop or posttranslational events that lead to anomalous/incorrect folding of the protein. Either TDP‐43 gene mutations or dysfunctions in the folding machinery that interferes with correct trafficking across the nuclear envelope can be responsible. In this respect, TDP‐43 gene mutations have recently been reported in sporadic and familial ALS 13, 18, 35, 40, 42). Redistribution and sequestration of TDP‐43 in insoluble aggregates in neurons have been reproduced in cultured cells by altering endogenous TDP‐43 nuclear trafficking and by expressing mutants with defective nuclear localization or nuclear export signals (41).
The biochemical finding of TDP‐43 in high‐molecular mass form and the correspondence of TDP‐43 and ubiquitin bands in parallel blots predict the existence of TDP‐43 in an anomalous structural conformation, because aberrant proteins are selective substrates of ubiquitination 14, 19). In ALS, TDP‐43 can be viewed both as a structurally abnormal protein and an abnormal cytosolic component, becoming a potential substrate for ubiquitination and/or segregation into insoluble aggregates.
The ubiquitin/proteasome system is implicated in the processing of proteins and plays also a role in a large array of functions essential for cellular homeostasis 14, 19, 31); therefore, ubiquitinated derivatives are normally found in control tissues and cells by biochemical methods, as in the blots of our control samples.
The progressive disappearance of motor neurons is the critical event in ALS. Evidence of different stages of the process, including the presence of normal‐appearing neurons is found in anterior horns (5). Therefore, the number of surviving neurons in anterior horns represents a surrogate marker of the stage of neurodegeneration and disease progression. Survival duration of individual ALS patients may not reflect the pathological stage, being survival influenced by site of disease presentation, intercurrent morbid events and supporting care in the advanced phases of illness. The trend of the percentage of TDP‐43‐positive neurons by the absolute number of surviving neurons in anterior horns, as well as the finding of cytoplasmic TDP‐43 accumulation in apparently healthy motor neurons, suggests that TDP‐43 redistribution is an early event in the pathogenic pathway leading to individual motor neuron death. In addition, the present results suggest that the delocalization of TDP‐43 is an ongoing event during the disease progression, involving progressively intact neurons, and preceding the deposition of insoluble inclusion bodies.
The trend of ubiquitin‐positive and TDP‐43‐positive skein/round inclusions indicates that the deposition of ubiquitinated bodies is a late event. The association between the high percentage of ubiquitinated neurons and the low number of residual neurons in anterior horns, previously reported by us (34; see Figure 1), suggests that ubiquitinated inclusions are the markers of the final stages of cell damage. This interpretation is concordant with the reported significant correlation between the presence of ubiquitin‐immunoreactive inclusions and absence of Nissl substance in anterior horn cells of ALS patients (21).
The persistence of ubiquitinated inclusions as tombstones in severely damaged motor neurons suggests an overloading of the ubiquitin–proteasome system by an excess of altered proteins. Abnormal accumulation of ubiquitinated derivatives is commonly regarded as a hallmark of proteasome proteolytic function inhibition 1, 8). TDP‐43 is probably a major protein undergoing ubiquitination in ALS; however, the identity of other abnormal proteins remains unknown. The partial overlap between TDP‐43 and ubiquitin staining of neuronal inclusions in ALS was reported also by others and considered as evidence that TDP‐43 is not the unique target of ubiquitination in ALS (33). Accumulation of ubiquitin‐tagged TDP‐43 contributes to the hampering of proteasome‐mediated protein degradation.
In dentate neurons of the hippocampus, TDP‐43 and ubiquitin immunoreactivity do quantitatively overlap, with the exception of one case with long‐lasting dementia before ALS onset. In dentate neurons, the abnormal compartmentalization of TDP‐43 is possibly accompanied, or followed soon by ubiquitination. Ubiquitin‐positive inclusions are found in dentate neurons of 20%–60% of sporadic ALS without a history of dementia (24), suggesting a spectrum of FTD and ALS with a common underlying pathology. In our series, dentate pathology was found in 14% of non‐demented ALS patients and in the two ALS patients with dementia. A formal neuropsychological evaluation of our ALS patients without frank dementia could have revealed frontal type impairment in additional ALS patients, as reported (22).
In conclusion, TDP‐43 immunoreactivity in sporadic ALS patients involves a larger number of spinal motor neurons than ubiquitin and has two main features: (i) diffuse cytoplasmic staining; and (ii) skein/round inclusions. The altered distribution of TDP‐43 in the cytoplasm of the upper and lower motor neurons is confirmed by our biochemical analysis, and is probably an early event of the pathogenic pathway that leads to motor neuron degeneration. The coexistence of native TDP‐43, hyperphosphorylated TDP‐43 and ubiquitinated TDP‐43 in the cytosol suggests that ubiquitination is a later event. Deleterious perturbation of nuclear trafficking and/or solubility of TDP‐43 may result in a loss of function at the nuclear level, and/or in toxic gain of function in the cytosolic compartment, both affecting neuron metabolic function and vitality. The redistribution of TDP‐43 seems to be an ongoing event during disease progression. If this is the case, counteracting this event may represent a rationale therapeutic approach to the incurable disease.
ACKNOWLEDGMENT
This study was supported by Compagnia di San Paolo, Turin, Italy, grant 2004.1424.
REFERENCES
- 1. André P, Groettrup M, Klenerman P, De Giuli R, Booth BL Jr, Cerundolo V et al (1998) An inhibitor of HIV‐1 protease modulates proteasome activity, antigen presentation, and t cell responses. Proc Natl Acad Sci USA 95:13120–13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H et al (2006) TDP‐43 is a component of ubiquitin‐positive inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. [DOI] [PubMed] [Google Scholar]
- 3. Bjerrum OJ, Schafer‐Nielsen C (1986) Buffer system and transfer parameters for semi‐dry electroblotting with a horizontal apparatus. In: Electrophoresis'68. Dunn MJ (ed.), pp. 315–327. VCH: Weinheim. [Google Scholar]
- 4. Brandmeir NJ, Geser F, Kwong LK, Zimmerman E, Qian J, Lee VM, Trojanowski JQ (2007) Severe subcortical TDP‐43 pathology in sporadic frontotemporal lobar degeneration with motor neuron disease. Acta Neuropathol 115:147–149. [DOI] [PubMed] [Google Scholar]
- 5. Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299. [DOI] [PubMed] [Google Scholar]
- 6. Buratti E, Dörk T, Zuccato E, Pagani F, Romano M, Baralle FE (2001) Nuclear factor TDP‐43 and SR proteins promote in vitro and in vivo CFTR exon skipping. EMBO J 20:1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ et al (2007) TDP‐43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ciechanover A (2006) The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology 66:S7–S19. [DOI] [PubMed] [Google Scholar]
- 9. Davidson Y, Kelley T, Mackenzie IR, Pickering‐Brown S, Du Plessis D, Neary D et al (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA‐binding protein, TDP‐43. Acta Neuropathol 113:521–533. [DOI] [PubMed] [Google Scholar]
- 10. Dickson DW, Liu W‐K, Hardy J, Farrer M, Metha N, Uitti R et al (1999) Widespread alterations of α‐synuclein in multiple system atrophy. Am J Pathol 155:1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dickson DW, Joseph KA, Amador Ortiz C (2007) TDP‐43 in differential diagnosis of motor neuron disorders. Acta Neuropathol 114:71–79. [DOI] [PubMed] [Google Scholar]
- 12. Fujita Y, Mizuno Y, Takamata M, Okamoto K (2008) Anterior horn cells with abnormal TDP‐43 immunoreactivities show fragmentation of the Golgi apparatus in ALS. J Neurol Sci 269:30–34. [DOI] [PubMed] [Google Scholar]
- 13. Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D et al (2008) TDP‐43 A315T mutation in familial motor neuron disease. Ann Neurol 63:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glickman MH, Ciechanover A (2002) The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82:373–428. [DOI] [PubMed] [Google Scholar]
- 15. Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T et al (2007) TDP‐43 is deposited in the Guam parkinsonism–dementia complex brains. Brain 130:1386–1394. [DOI] [PubMed] [Google Scholar]
- 16. Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y et al (2008) Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64: 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M et al (2008) Enrichment of C‐terminal fragments in TAR DNA‐binding protein‐43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 173:182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574. [DOI] [PubMed] [Google Scholar]
- 19. Kisselev AF, Goldberg AL (2001) Proteasome inhibitors: from research tools to drug candidates. Chem Biol 8:739–758. [DOI] [PubMed] [Google Scholar]
- 20. Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ (2007) TDP‐43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol 114:63–70. [DOI] [PubMed] [Google Scholar]
- 21. Leigh PN, Whitwell H, Garofalo O, Buller J, Swash M, Martin JE et al (1991) Ubiquitin‐immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution and specificity. Brain 114:775–788. [DOI] [PubMed] [Google Scholar]
- 22. Lomen‐Hoerth C (2004) Characterization of amyotrophic lateral sclerosis and frontotemporal dementia. Dement Geriatr Cogn Disord 17:337–341. [DOI] [PubMed] [Google Scholar]
- 23. Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ et al (2007) Pathological TDP‐43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434. [DOI] [PubMed] [Google Scholar]
- 24. Mackenzie IRA, Feldman H (2005) Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol 64:730–739. [DOI] [PubMed] [Google Scholar]
- 25. Matunis MJ (2006) Isolation and fractionation of rat liver nuclear envelopes and nuclear pore complexes. Methods 39:277–283. [DOI] [PubMed] [Google Scholar]
- 26. Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE (2005) Depletion of TDP‐43 overrides the need for exonic and intronic splicing enhancers in the human apoA‐II gene. Nucleic Acids Res 33:6000–6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mori F, Tanji K, Zhang HX, Nishihira Y, Tan CF, Takahashi H, Wakabayashi K (2008) Maturation process of TDP‐43‐positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 116:193–203. [DOI] [PubMed] [Google Scholar]
- 28. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S et al (1998) Frontotemporal lobar degeneration. A consensus on clinical diagnostic criteria. Neurology 51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 29. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133. [DOI] [PubMed] [Google Scholar]
- 30. Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7: 710–723. [DOI] [PubMed] [Google Scholar]
- 31. Princiotta MF, Schubert U, Chen W, Bennink JR, Myung J, Crews CM, Yewdell JW (2001) Cells adapted to the proteasome inhibitor 4‐hydroxy‐5‐iodo‐3‐nitrophenylacetyl‐Leu‐Leu‐leucinal‐vivyl sulfone require enzymatically active proteasomes for continued survival. Proc Natl Acad Sci USA 98:513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robertson J, Sanelli T, Xiao S, Yang W, Horne P, Hammond R et al (2007) Lack of TDP‐43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett 420:128–132. [DOI] [PubMed] [Google Scholar]
- 33. Sanelli T, Xiao S, Horne P, Bilbao J, Zinman L, Robertson J (2007) Evidence that TDP‐43 is not the major ubiquitinated target within the pathological inclusions of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 66:1147–1153. [DOI] [PubMed] [Google Scholar]
- 34. Schiffer D, Autilio‐Gambetti L, Chiò A, Gambetti P, Giordana MT, Gullotta F et al (1991) Ubiquitin in motor neuron disease: study at the light and electron microscope. J Neuropathol Exp Neurol 50: 463–473. [DOI] [PubMed] [Google Scholar]
- 35. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al (2008) TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra‐Lantz C, Shoesmith C (2007) TDP‐43 is a human low molecular weight neurofilament (hNFL) mRNA‐binding protein. Mol Cell Neurosci 35:320–327. [DOI] [PubMed] [Google Scholar]
- 37. Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A et al (2007) TDP‐43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113:535–542. [DOI] [PubMed] [Google Scholar]
- 38. Tata JR (1974) Isolation of nuclei from liver and other tissues. Methods Enzymol 31(Pt A):253–262. [DOI] [PubMed] [Google Scholar]
- 39. Taylor CM, Coetzee T, Pfeiffer SE (2002) Detergent‐insoluble glycosphingolipid/cholesterol microdomains of the myelin membrane. J Neurochem 81:993–1004. [DOI] [PubMed] [Google Scholar]
- 40. Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB et al (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP‐43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM (2008) Disturbances of nuclear and cytoplasmic TAR DNA binding protein (TDP‐43) induces disease‐like redistribution, sequestration and aggregate formation. J Biol Chem 283:13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A et al (2008) TDP‐43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 63:538–542. [DOI] [PubMed] [Google Scholar]