
Figure 1. Extracting a consensus sequence from a multiple sequence alignment.
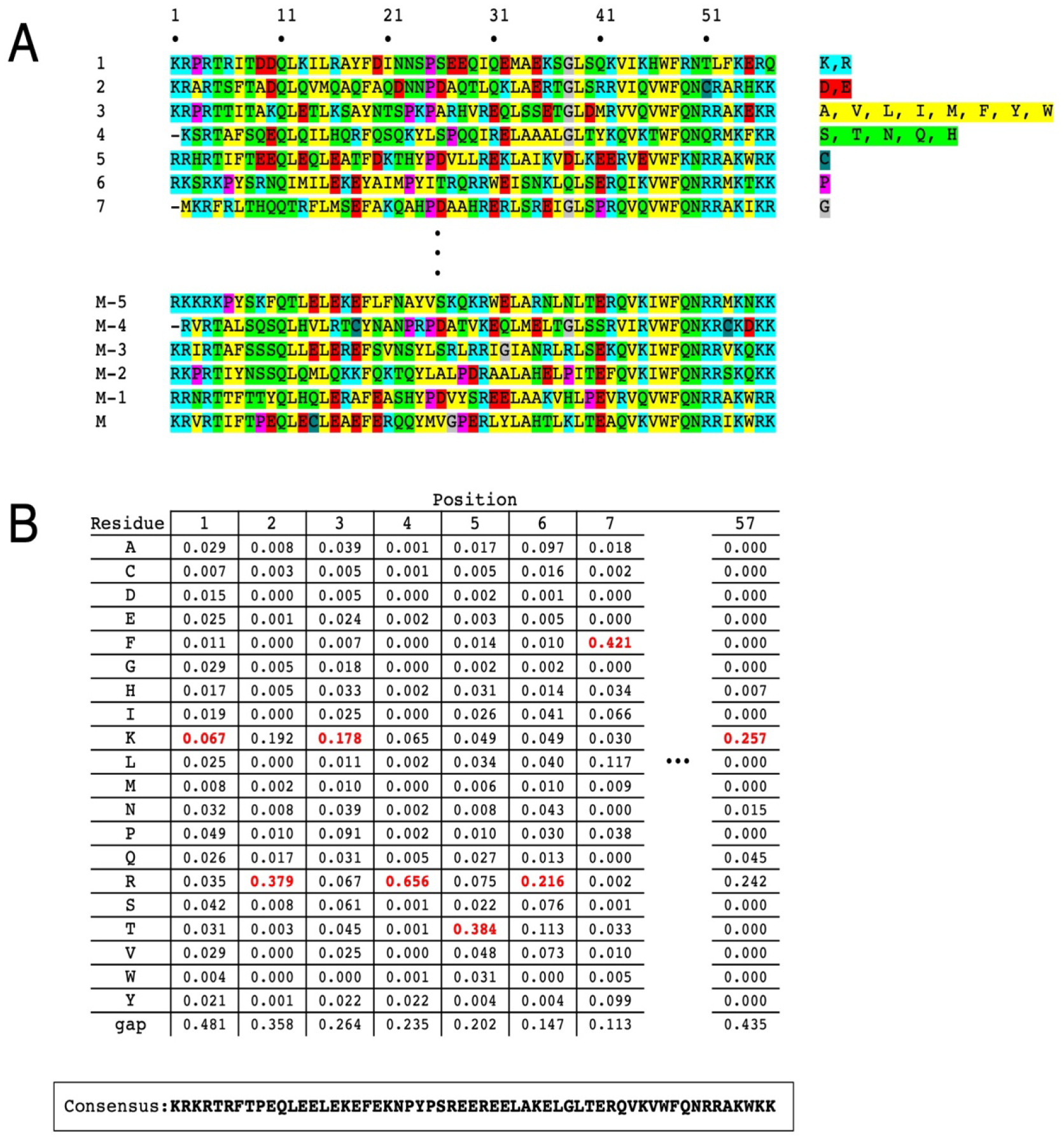
(A) An alignment of M=4,571 homeodomain sequences, each of length L=57 residues. Residues with similar (or unique) chemical features are highlighted to illustrate conservation. Positions with a gap in more than half the sequences are eliminated from the alignment. From such an alignment, the frequency of each type of residue (including remaining “gap residues”) is determined. (B) The consensus sequence (bottom) is obtained by taking the residue with the highest frequency (excluding gaps, see Section 3) at each position (red).