
Figure 3. Generating a consensus sequence.
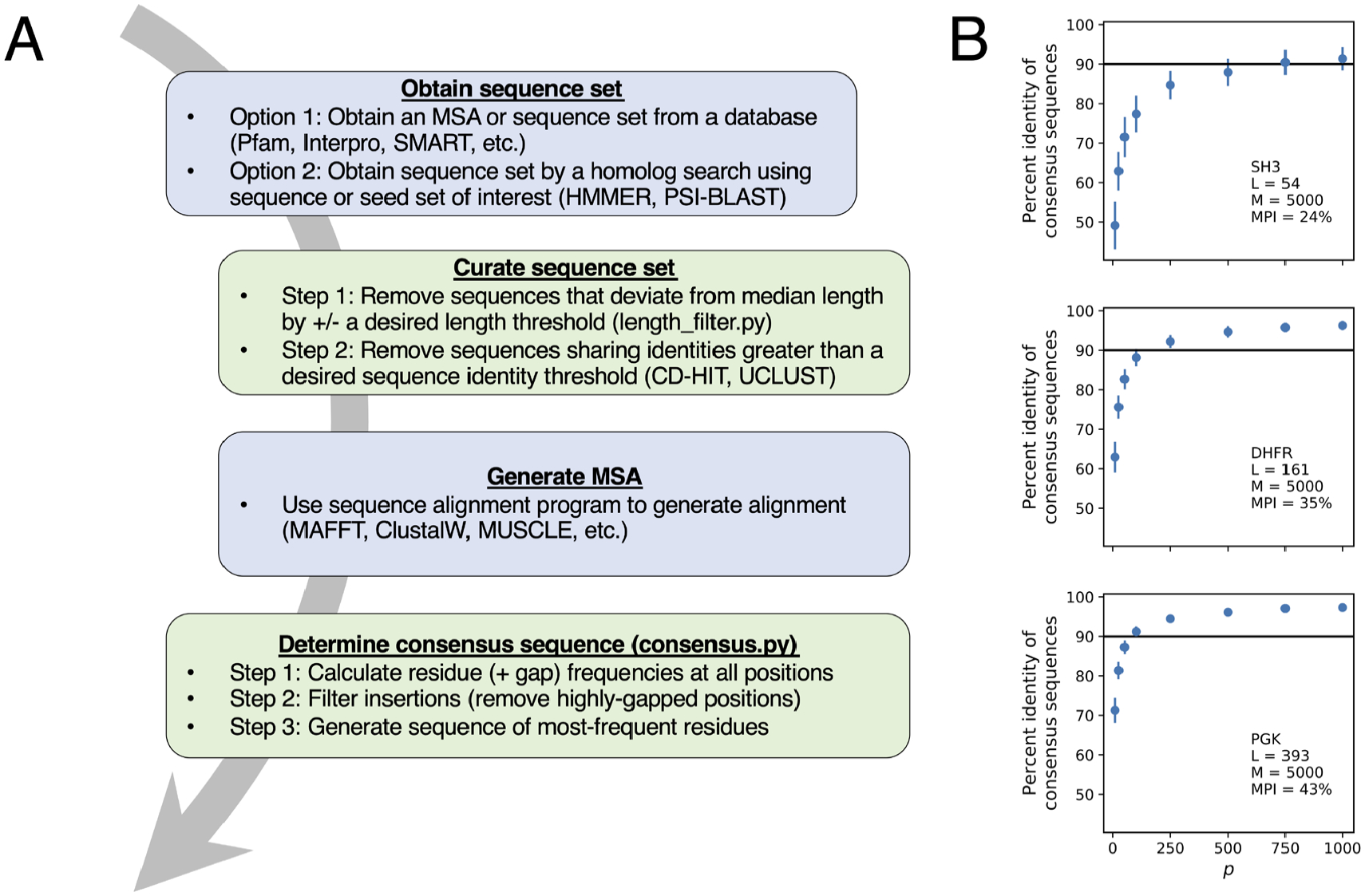
(A) Workflow for consensus sequence design. Major steps involve obtaining sequences, eliminating sequences with atypical length and high sequence identity, (re)aligning sequences, filtering gaps and insertions, and calculating residue frequencies. Examples of available databases and programs are highlighted where applicable. Green boxes contain steps that can be done using Python scripts noted in Section 3.3. (B) Identities among consensus sequences generated from random draws (sampling without replacement) of p sequences from a 5000 sequence MSA for SH3 domain (top), dihydrofolate reductase (middle), and phosphoglycerate kinase (bottom). Points represent the mean pairwise identity of 100 consensus sequences each obtained from random subsets of p sequences, and bars represent one standard deviation. Inset text indicates the length of each protein family (L), the number of sequences in each MSA (M), and the mean pairwise identity (MPI) of sequences in each 5,000 sequence MSA.