

Microsporidia infection initiates a transcriptional response in parents, resulting in pathogen-resistant progeny.
Abstract
Parental infection can result in the production of offspring with enhanced immunity phenotypes. Critically, the mechanisms underlying inherited immunity are poorly understood. Here, we show that Caenorhabditis elegans infected with the intracellular microsporidian parasite N. parisii produce progeny that are resistant to microsporidia infection. We determine the kinetics of the response and show that intergenerational immunity prevents host-cell invasion by Nematocida parisii and enhances survival to the bacterial pathogen Pseudomonas aeruginosa. We demonstrate that immunity is induced by the parental transcriptional response to infection, which can be mimicked through maternal somatic depletion of PALS-22 and the retinoblastoma protein ortholog, LIN-35. We find that other biotic and abiotic stresses (viral infection and cadmium exposure) that induce a similar transcriptional response as microsporidia also induce immunity in progeny. Together, our results reveal how a parental transcriptional signal can be induced by distinct stimuli and protect offspring against multiple classes of pathogens.
INTRODUCTION
Animals have evolved diverse immune mechanisms to limit the negative impact of pathogens and parasites on host fitness. While immunological memory is typically considered a hallmark of the antibody-mediated adaptive immune system, memory of pathogen exposure has now been documented in animals lacking adaptive immunity. Although invertebrates only have innate immunity, at least 20 species, including insects, crustaceans, and mollusks, have now been shown to transfer protective immunity to their progeny following infection (1). Although this epigenetically inherited immunity can protect offspring against a variety of bacterial, fungal, and viral pathogens, it is largely unclear how immunity is induced. Several reports have described the deposition of bacterial cell-wall fragments in offspring following parental infection, as well as immune genes being up-regulated in both parents and their progeny (2, 3). Immune priming can be specific, whereby immunity is only active against the same strain of bacteria with which the parents were infected (4, 5). Conversely, it may be broad; for example, mealworm beetles primed with either fungi or a Gram-positive or Gram-negative bacteria induce immunity against Gram-positive pathogens (6). Although the effectors that provide immunity in the progeny are mostly unknown, antimicrobial peptides are often up-regulated in offspring (1, 2, 6).
Studies in the genetically tractable nematode Caenorhabditis elegans have enabled fundamental immune advances and shed light on many epigenetically inherited and stress-induced phenotypes (7–9). Hence, C. elegans has recently become a powerful model for the study of inherited innate immunity (10). In this host, antiviral immunity, mediated by the small RNA–mediated silencing of viral transcripts, was shown to last for at least three generations and be dependent on RNA interference (RNAi) pathways (11). Although there are conflicting reports about whether the natural Orsay virus can induce heritable immunity in C. elegans, injection with vesicular stomatitis virus was able to protect progeny against reinfection (12–14). Several studies have shown that learned bacterial avoidance can be transferred to progeny. In one case, heritable avoidance to Pseudomonas aeruginosa bacteria was dependent on RNAi pathways and a bacterial RNA used by C. elegans to inhibit host gene expression (15–17). Parental exposure to pathogenic bacteria can also protect offspring by increasing the likelihood of progeny adopting a stress-resistant dauer phenotype (18). Last, resistance can be induced by up-regulating immune genes in offspring. For example, intergenerational immunity to pathogenic Pseudomonas vranovensis is dependent on the induced expression of cysteine synthases (19).
Microsporidia are a large phylum of fungal-related parasites that infect most animal species (20). These pathogens can be lethal to immunocompromised humans and cause large economic losses by infecting agriculturally important animals such as honey bees, silk worms, and shrimp (21). Nematocida parisii is a natural microsporidia parasite that commonly infects C. elegans in the wild. Infection of C. elegans by N. parisii begins when microsporidia spores are ingested (22). Spores inside the intestinal lumen then fire a unique polar-tube structure that is used to transfer the cellular contents of the spore (sporoplasm), including the parasite’s genetic material, into the host intestinal cell (23). The pathogen then replicates intracellularly to form meronts and spreads from cell to cell by fusing the cells and forming syncytia (24). These meronts ultimately differentiate into mature spores, which exit infected cells nonlytically, and can result in the shedding of up to 200,000 spores during infection (22, 25). Microsporidia are pathogenic to C. elegans, resulting in reduced fecundity and ultimately death (24, 26).
Several mechanisms of innate immunity against microsporidia have been described in invertebrates. Microsporidia infection typically induces a strong host transcriptional response, which often includes up-regulation of a suite of different antimicrobial peptides (27). Although antimicrobial peptides are commonly up-regulated in other invertebrates, and C. elegans have many different families of these proteins, so far, only C-type lectins have been shown to be up-regulated upon N. parisii infection (28, 29). Instead, C. elegans has a recently identified stress/immune pathway called the “intracellular pathogen response” (IPR) that is induced upon infection by both microsporidia and Orsay virus (28, 30). The IPR includes up-regulation of ubiquitin adaptor proteins and is thought to be involved in clearing intracellular parasites (28, 31, 32).
In this study, we reveal robust resistance to microsporidia in the offspring of N. parisii–infected C. elegans. We show that inherited immunity markedly reduces host-cell invasion by N. parisii and also provides resistance to the bacterial pathogen P. aeruginosa. We find that immunity is conferred in a dose-dependent manner, lasts throughout development, and is maintained for a single generation. Using tissue-specific depletion or expression of negative regulators of the IPR (LIN-35 and PALS-22), we demonstrate that immunity in progeny is dependent on a parental transcriptional response and that this immunity is transferred from maternal somatic tissues to the progeny. The host transcriptional response and thus inherited immunity can be induced by both biotic and abiotic stresses that mimic the response to microsporidia. Together, these results provide insight into how inherited immune responses can be induced to protect offspring against pathogenic infection.
RESULTS
Parental infection by N. parisii confers immunity to C. elegans progeny in a dose-dependent manner
Infection of C. elegans with the natural microsporidian pathogen N. parisii delays development and affects worm fertility (23, 33). To quantify the effects of infection, we exposed early larval stage (L1) animals to varying doses of N. parisii spores. At 72 hours post-infection (hpi), animals were fixed and stained with the chitin-binding dye Direct Yellow 96 (DY96) (fig. S1, A and B). DY96 stains both microsporidia spores and worm embryos, allowing us to determine both parasite burden and host reproductive development. Worms infected with higher doses of spores exhibited greater parasite burden and a smaller body size at 72 hpi (fig. S1, C and D). Higher infection doses also correlated with a reduction in the percentage of gravid adults (animals carrying embryos) and a reduction in the number of embryos per animal (fig. S1, E and F). In agreement with these findings, infection of transgenic animals expressing a fluorescent protein in the germ line revealed germline deformities in worms exposed to high doses of spores (fig. S1G).
To determine the effects of parental infection on offspring, we infected parental (P0) generations at the L1 stage with a “very low,” “low,” or “moderate” dose of N. parisii that resulted in a ~10 to 50% reduction in the number of gravid adults (fig. S1E). At 72 hpi, infected adults were treated with a sodium hypochlorite solution to release the F1 embryos from adults and destroy microsporidia spores. As N. parisii infection is not vertically transmitted, resultant larvae are not infected with microsporidia (fig. S2) (26). F1 generations of synchronized L1s were then exposed to a high dose of spores. Imaging revealed a quantifiable reduction in the parasite burden of primed worms from infected parents, as compared to naïve worms from uninfected parents (Fig. 1, A and B). Immunity in F1 progeny was dose dependent; parents experiencing a higher infection burden gave rise to offspring that were more resistant to N. parisii. In agreement with our data for parasite burden, we saw that primed worms under infection conditions were larger and produced significantly more embryos than their naïve counterparts (Fig. 1, C to E). Together, these data reveal a robust and dose-dependent immunity phenotype in the offspring of N. parisii–infected worms.
Fig. 1. Parental infection by N. parisii confers immunity to the progeny of C. elegans.
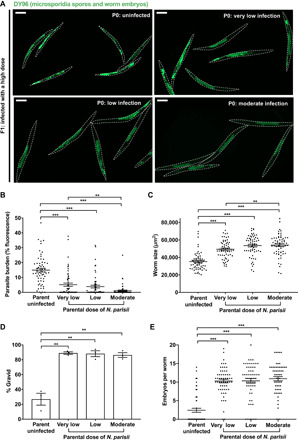
L1 stage N2 C. elegans were uninfected or exposed to varying concentrations of N. parisii spores (doses defined in Materials and Methods) for 72 hours. F1 L1 larvae were collected and infected with a high dose of N. parisii, fixed at 72 hpi, and stained with DY96. (A) Representative images of F1 populations stained with DY96, which binds to worm embryos and N. parisii spores. Scale bars, 200 μm. (B) Quantitation of DY96 fluorescent spores per worm. (C) Quantitation of worm area. (D) Percentage of worms that are gravid. (E) Number of embryos per worm. (B to E) Means ± SEM (horizontal bars) are shown. Data pooled from three independent experiments using n = 20 (B, C, and E) or n = 98 to 351 (D) worms per condition per experiment. The P values were determined by unpaired two-tailed Student’s t test. Significance with Bonferroni correction was defined as P < 0.0166. **P < 0.0033; ***P < 0.00033.
Primed worms showed no fitness advantage under noninfection conditions and were indeed smaller and contained fewer embryos than worms from uninfected parents (fig. S3). As infected worms are developmentally delayed, we tested the effects of parental developmental timing on offspring immunity. We did not observe any differences in infection outcomes of naïve progeny from parents of different ages, demonstrating that immunity is not conferred by delayed parental development (fig. S4).
Inherited immunity prevents microsporidia invasion events by restricting spores in the intestine
To resist microsporidia infection, animals may (i) prevent invasion of intestinal cells or (ii) destroy the invaded pathogen (23, 32). To determine by which mechanism inherited immunity confers resistance to N. parisii, we first performed invasion assays. Naïve or primed L1 worms were exposed to a high dose of spores and fixed at 30 min post-infection (mpi) or 3 hpi. To visualize invasion events in these animals, we performed fluorescence in situ hybridization (FISH) to detect N. parisii 18S ribosomal RNA in host intestinal cells, thus indicating the presence of intracellular sporoplasms. Our analysis revealed significantly fewer invasion events in primed animals at both time points, including an over 98% reduction in infectious events at 30 mpi (Fig. 2, A and B, and fig. S5A). To determine whether spores were present in the intestinal lumen, we also stained animals with DY96. Notably, we also observed far fewer spores in the guts of primed animals (Fig. 2, C and D, and fig. S5B).
Fig. 2. Inherited immunity prevents microsporidia invasion and P. aeruginosa colonization but not viral infection.
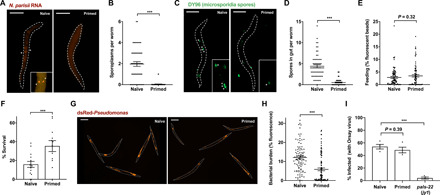
L1 stage N2 C. elegans were either not infected or infected with a low dose of N. parisii spores for 72 hpi. (A to D) F1 L1 larvae were exposed to a maximal dose of N. parisii, fixed at 30 mpi, and stained with DY96 (green), as well as a FISH probe to detect N. parisii 18S RNA (red). (A) Representative images of worms stained with FISH probe to detect invaded sporoplasms, marked by asterisks. Scale bars, 25 μm. (B) Number of sporoplasms per worm. Data pooled from three independent experiments using n = 16 to 20 worms per condition per experiment. (C) Representative images of worms stained with DY96 to detect spores, marked by asterisks, in the intestinal lumen. Scale bars, 25 μm. (D) Number of spores per worm. Data pooled from three independent experiments using n = 20 to 21 worms per condition per experiment. (E) F1 L1 larvae were fed fluorescent beads and fixed after 30 min. Quantitation of bead fluorescence per worm. Data pooled from three independent experiments using n = 24 to 30 worms per condition per experiment. (F) F1 L1 larvae were maintained on slow-killing plates with wild-type P. aeruginosa, and survival at 84 hpi was assessed. Data pooled from four independent experiments, each comprising two to four technical replicates, using n = 13 to 37 worms per condition per experiment. (G and H) F1 L1 larvae were maintained on slow-killing plates with dsRed–P. aeruginosa and fixed at 48 hpi. (G) Representative images of worm populations. Scale bars, 200 μm. (H) Quantitation of dsRed fluorescence per worm. Data pooled from three independent experiments using n = 21 to 47 worms per condition per experiment. (I) Naïve and primed F1 and pals-22 mutant L1 larvae were infected with Orsay virus, fixed at 16 hpi, and stained with FISH probe to detect Orsay virus RNA. Data pooled from four independent experiments with n = 52 to 158 worms per condition per experiment. Means ± SEM (horizontal bars) are shown. The P values were determined by unpaired two-tailed Student’s t test. (B, D to F, and H) Significance was defined as P < 0.05. ***P < 0.001. (I) Significance with Bonferroni correction was defined as follows: ***P < 0.0005.
To test whether a reduction of spores in our primed worms was a result of reduced feeding, we allowed naïve and primed L1 stage worms to feed on fluorescent beads. After 30 min or 3 hours, we fixed the populations and quantified fluorescence in individual animals. We failed to detect any reduction in feeding in primed worms at either time point (Fig. 2E and fig. S5C). At the 3-hour time point, primed animals actually fed significantly more than naïve worms, indicating that microsporidia invasion results in reduced feeding in C. elegans (fig. S5C).
We next tested whether a small lag in development of primed worms at the early L1 stage contributes to their immunity. Naïve and primed worms were exposed to spores immediately after initiating development or exposed to spores after 6 hours of growth on their typical Escherichia coli food source. The number of sporoplasms was quantified at 30 mpi, showing that after 6 hours of rest, primed worms still displayed significantly reduced microsporidia invasion (fig. S5D).
To test whether enhanced clearance of intracellular infection might also contribute to microsporidia resistance in primed animals, we performed pulse-chase experiments (23). Here, naïve and primed animals were maintained in the presence of high concentrations of N. parisii spores for 3 hours, before washing thoroughly to remove any microsporidia spores not inside the animals. The population was then split; half was immediately fixed to represent the initial infection, and the other half was maintained in the absence of spores until a 24-hour end point when they were fixed to assess clearance. Detection of sporoplasms using 18S RNA FISH and subsequent quantifications showed no evidence of intracellular pathogen clearance in the primed animals (fig. S5E). These data suggest that limiting invasion is the principal way by which inherited immunity provides protection against microsporidia.
To provide insight into the mechanisms underlying protection in primed animals, we next tested the specificity of the immune response. For this, we assayed resistance to the extracellular Gram-negative bacterial pathogen P. aeruginosa (strain PA14) using a well-established “slow killing” protocol (7). Here, naïve or N. parisii–primed animals were plated on wild-type PA14 at the L1 stage, and survival was assayed at 84 hpi. Primed worms were significantly less susceptible to P. aeruginosa infection than naïve animals (Fig. 2F). Slow killing by P. aeruginosa occurs as a result of bacterial accumulation in the gut (34). To visualize bacterial burden in our naïve and primed worms, we performed infection assays using dsRed-expressing PA14. At 48 hpi, we fixed and quantified fluorescence in our naïve and primed populations. In agreement with the data from survival assays, the bacterial burden in microsporidia-primed animals was significantly reduced (Fig. 2, G and H, and fig. S5F). A similar reduction in the bacterial burden of primed worms was observed using assays to quantify colony-forming units (CFUs) (fig. S5G). Together, these results suggest that a host intestinal factor may be protecting primed worms from infection by destroying microsporidia spores and other pathogenic microbes within the intestinal lumen.
To test whether inherited immunity could protect against all classes of pathogens, we tested the response of N. parisii–primed animals to the intracellular intestinal pathogen Orsay virus. Unexpectedly, FISH staining revealed that primed worms were similarly susceptible to Orsay virus as their naïve counterparts (Fig. 2I and fig. S5H). This result is particularly notable given the significant overlap between the transcriptional responses induced by Orsay virus and N. parisii, with both activating IPR genes (28). PALS-22 is a negative regulator of the IPR, and a loss-of-function mutation in pals-22 results in animals with a constitutively activated IPR that are resistant to viral and microsporidia infection but not to P. aeruginosa (Fig. 2I and fig. S5I) (31). Mutants deficient for pals-22 limit microsporidia infection by preventing invasion and clearing invaded parasites (fig. S5I). Thus, IPR-mediated immunity is distinct in several ways from the immunity found in primed animals.
Inherited immunity to N. parisii lasts a single generation and persists throughout development
We next sought to understand the kinetics of the inherited immune response to microsporidia infection. To determine whether immunity could be transmitted over multiple generations, we tested resistance to microsporidia in both the F1 and F2 progeny of N. parisii–infected worms. We observed resistance to microsporidia only in the F1 population, indicating that this response lasts for a single generation (Fig. 3, A and B, and fig. S6, A and B). To test the longevity of the inherited immune response within the F1 generation, we infected naïve and primed worms with N. parisii either at the L1 stage, 24 hours later at the L2/L3 stage, or 48 hours later at the L4 stage. After 30 mpi, worms were fixed and stained with a FISH probe to detect N. parisii, and the number of sporoplasms per individual was quantified. While immune-primed worms continued to show some resistance to infection at the L4 stage, resistance was strongest at the L1 stage of infection (Fig. 3C).
Fig. 3. Inherited immunity in N. parisii primed C. elegans lasts a single generation and is strongest in early larval stages.
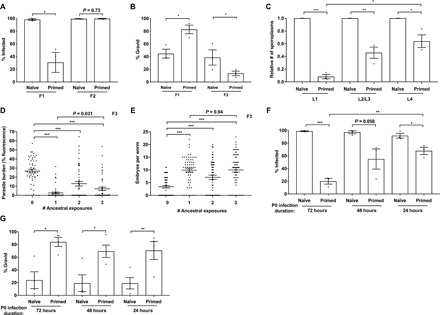
(A to C) L1 stage N2 C. elegans were either not infected or infected with a low dose of N. parisii spores for 72 hpi. (A and B) F1 embryo populations were then split and either tested for immunity or maintained under noninfection conditions for the collection and subsequent testing of F2 embryos. F1 and F2 L1 larvae were exposed to a high dose of N. parisii, fixed at 72 hpi, and stained with DY96. (A) Percentage of animals infected. (B) Percentage of worms that are gravid. (C) F1 larvae were challenged with a maximal dose of N. parisii spores at either the L1, L2/L3, or L4 stage; fixed at 30 mpi; and stained with N. parisii 18S RNA FISH probe. Number of sporoplasms per worm. Data pooled from three independent experiments using n = 16 to 20 (A and B) or n = 12 to 20 (C) worms per condition per experiment. (D and E) L1 stage N2 C. elegans (P0, F1, and F2) were infected with N. parisii for 72 hours, for either one, two, or three successive generations (see schematic in fig. S6C). F3 L1 larvae were exposed to a high dose of N. parisii, fixed at 72 hpi, and stained with DY96. (D) Quantitation of DY96 fluorescent spores per worm. (E) Number of embryos per worm. Data pooled from two independent experiments using n = 25 (D) or n = 30 (E) worms per condition per experiment. (F and G) N2 C. elegans were either not infected or infected with a low dose of N. parisii spores at the L1 stage (for 72 hours), L2/L3 stage (for 48 hours), or L4 stage (for 24 hours). F1 worms were infected with N. parisii as in (A). (F) Percentage of worms infected. Data pooled from three independent experiments using n = 100 to 197 worms per condition per experiment. (G) Percentage of worms that are gravid. Data pooled from four independent experiments using n = 100 to 197 worms per condition per experiment. Means ± SEM (horizontal bars) are shown. The P values were determined by unpaired two-tailed Student’s t test. (A, B, and G) Significance was defined as follows: *P < 0.05; **P < 0.01. (C and F) Significance with Bonferroni correction was defined as follows: *P < 0.025; **P < 0.005; ***P < 0.0005. (D and E) Significance with Bonferroni correction was defined as follows: ***P < 0.00033.
We next tested whether a greater ancestral history of infection might enhance immunity phenotypes and potentially enable the immune response to transmit over multiple generations. Animals were infected for one, two, or three sequential generations with N. parisii (P0s, F1s, and F2s). Susceptibility to microsporidia was assessed in F3 animals and in F4 animals following a single generation of rest without infection (fig. S6, C to E). We found that an increased history of ancestral infections did not enhance immunity phenotypes among the F3 populations (Fig. 3, D and E). Furthermore, resistance was seen exclusively in the F3 animals, and no resistance was observed in the F4 generations (fig. S6, F and G).
We next wanted to determine for how long a parent must be infected with N. parisii to transmit immunity to progeny. For this, parental generations were infected as previously at the L1 stage for 72 hours of total infection, or rested on their typical E. coli food source and subsequently infected as L2/L3s for 48 hours of total infection or as L4s for 24 hours of total infection. Infection assays on the resulting F1 progenies showed that parental worms infected only briefly as L4s were still able to confer immunity phenotypes to offspring, although to a lesser extent than animals from parents infected for longer periods (Fig. 3, F and G). As N. parisii takes over 48 hours to sporulate (24), these results indicate that the early stages of microsporidian infection alone (i.e., invasion and replication) are sufficient to induce immunity in progeny. Together, these data show that inherited immunity protects the F1 progeny from infected parents throughout development and supports a model in which the immediate parental environment is the most important factor in determining the immune competency of offspring.
The parental transcriptional response to infection triggers inherited immunity in offspring
N. parisii is an intestinal parasite that does not come into direct contact with the C. elegans germ line, and how information is transferred from the soma to the germ line is not known (fig. S7A). To explore this, we tested whether mutants defective in small-RNA inheritance and histone modification were able to transmit immunity phenotypes to offspring. We found that the offspring of N. parisii–infected mutants still became protected against infection (fig. S8) (19, 35). In addition, we found that the master immune regulator PMK-1 was not required for the induction of inherited immunity, consistent with this pathway not being involved in immunity to N. parisii (fig. S8, C and D) (26). These data indicate that the small RNA, histone modification, and PMK-1 pathways are not required for transmission of immunity.
To determine whether infection itself, or merely exposure to spores, is required for the transmission of inherited immunity from parent to progeny, we used heat-killed spores. Parent populations were either uninfected, exposed to live N. parisii, or exposed to heat-killed spores. Unlike live spores, heat-killed spores fail to induce the IPR, as demonstrated using transcriptional reporters for key IPR genes (fig. S7, B and C) (28). Consistent with a requirement for infection and induction of the IPR to initiate inherited immunity, the offspring of parents exposed to heat-killed spores showed no enhanced protection against N. parisii infection (Fig. 4, A and B).
Fig. 4. The parental transcriptional response to N. parisii triggers inherited immunity.
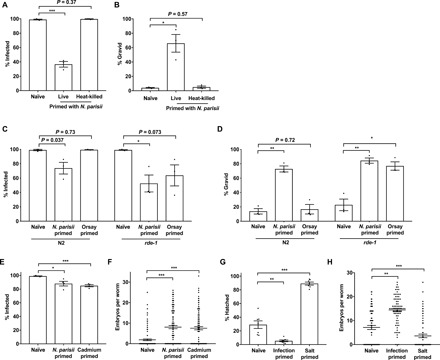
(A and B) L1 stage N2 C. elegans were either not infected or exposed to a low dose of live or heat-killed N. parisii spores for 72 hpi. F1 L1 larvae were exposed to a high dose of N. parisii, fixed at 72 hpi, and stained with DY96. (A) Percentage of worms infected. (B) Percentage of worms that are gravid. (C and D) L1 N2 and rde-1 mutants were not infected, infected with a low dose of N. parisii, or infected with Orsay virus for 72 hpi. F1 worms were infected with N. parisii as in (A). (C) Percentage of worms infected. (D) Percentage of worms that are gravid. (E and F) N2 C. elegans were either untreated or infected with a low dose of N. parisii spores or exposed to 50 mM cadmium from the L4 stage for 24 hours. F1 worms were infected with N. parisii as in (A). (E) Percentage of worms infected. (F) Number of embryos per worm. (G and H) L1 stage N2 C. elegans were either untreated, infected with a low dose of N. parisii spores on standard nematode growth media (NGM; 50 mM salt), or maintained on NGM containing 250 mM salt for 72 hours. (G) Percentage of F1 embryos hatching on 420 mM salt. (H) F1 worms were infected with N. parisii as in (A). Number of embryos per worm. (A to D) Data pooled from three independent experiments using n = 100 worms per condition per experiment. (E and F) Data pooled from four independent experiments using n = 100 worms (E) or n = 25 to 30 worms (F) per condition per experiment. (G) Data pooled from seven independent experiments using n = 100 to 215 worms per condition per experiment. (H) Data pooled from three independent experiments using n = 25 worms per condition per experiment. (A to H) Means ± SEM (horizontal bars) are shown. The P values were determined by unpaired two-tailed Student’s t test. Significance with Bonferroni correction was defined as follows: *P < 0.025; **P < 0.005; ***P < 0.0005.
We next tested whether other environmental conditions that induce a similar transcriptional response as microsporidia infection could also induce immunity in progeny. When analyzing mRNA sequencing data, we confirmed a previously noted similarity to the Orsay virus response and also noticed that the C. elegans response to N. parisii infection overlaps significantly with that of worms exposed to the heavy metal cadmium (fig. S7D).
To explore a role for the transcriptional response in inducing inherited immunity, we first performed N. parisii infection assays on the offspring of untreated parents or parents exposed to either N. parisii or Orsay virus. We saw that the Orsay-primed F1 progeny of loss-of-function rde-1 mutants, but not N2 worms, showed significantly reduced infection and improved fitness under microsporidia infection conditions, as compared to naïve controls (Fig. 4, C and D). This difference between the rde-1 genotype and N2 can be attributed to the increased susceptibility of rde-1 mutants to Orsay virus, allowing the P0s to have an enhanced viral response (fig. S7E) (36).
We next assayed the susceptibility of cadmium-primed offspring to N. parisii infection. Notably, resistance to microsporidia was observed in the offspring of parents exposed to cadmium (Fig. 4, E and F). In addition, cadmium-primed progeny exposed to P. aeruginosa had significantly lower bacterial burden than naïve animals, supporting a role for the transcriptional response in transmitting immunity against both these classes of pathogens (fig. S7F). In support of primed animals being specifically resistant to pathogen-induced stress rather than broadly resistant to all stresses, both infection-primed and cadmium-primed animals were less gravid than naïve worms when faced with a high concentration of cadmium (fig. S7G).
To test whether a stress that does not activate the IPR could induce inherited immunity, we grew parents under elevated salt conditions (fig. S7D) (37). While the salt-primed progeny of osmotically stressed parents were better able to cope with high-salt stress, these animals were significantly less fit than their naïve counterparts under N. parisii infection conditions (Fig. 4, G and H, and fig. S7H). Conversely, the offspring of N. parisii–infected animals were less viable than naïve animals under osmotic stress, further supporting a “cost” of priming (Fig. 4G). Together, these data support a role for the transcriptional response in transmitting inherited immunity to offspring and highlight the complex relationships that underlie responses to abiotic and biotic stresses across generations.
Progeny of animals with an artificially activated transcriptional response are resistant to infection
In addition to the previously characterized pals-22 mutant, we also observed that mutants of lin-35, the C. elegans ortholog of retinoblastoma protein (RB), induce a similar transcriptional response to microsporidia infection (fig. S7D) (38, 39). These mutants share extensive similarity with the transcriptional response to N. parisii, with 242 shared genes up-regulated at least twofold in both lin-35 and pals-22, as well as N. parisii–infected worms (Fig. 5). To determine whether activating this host response in the absence of infection or environmental stimuli would induce immunity, we performed infection assays in pals-22 and lin-35 mutants. In agreement with a role for the IPR in restricting infection, both pals-22 and lin-35 mutants had a significantly lower parasite burden than wild-type animals (Fig. 6A). However, pals-22 and lin-35 mutants have defects resulting in fewer embryos, even when grown under normal conditions. Thus, the number of embryos per worm in these mutants during infection conditions is similar to wild type (Fig. 6B).
Fig. 5. N. parisii infection induces many genes that are also up-regulated in both lin-35 and pals-22 mutants.
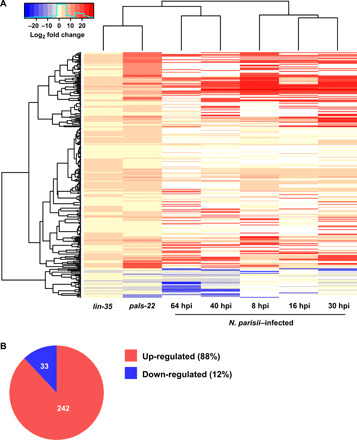
A shared transcriptional response was identified by determining genes that were either up- or down-regulated in both lin-35 and pals-22 mutants and at least one N. parisii infection time point. (A) Heatmap showing cluster analysis of the shared transcriptional response with the fold change of each cell corresponding to scale at the top. White cells in the heatmap represent genes not determined to be differentially expressed. (B) Fraction of shared genes that are either up- or down-regulated.
Fig. 6. Mutants that phenocopy the transcriptional response to infection transfer immunity to offspring through the maternal germ line.
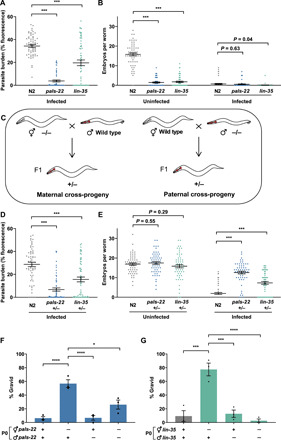
(A and B) L1 stage N2, pals-22(jy1), and lin-35(n745) worms were not infected or infected with a high dose of N. parisii, fixed at 72 hpi, and stained with DY96. (A) Quantitation of DY96 fluorescent spores per worm. (B) Number of embryos per worm. (A and B) Data pooled from three independent experiments using n = 20 worms per condition per experiment. (C) Schematic of mating to obtain maternal and paternal cross-progeny. Myo-2p::mCherry was used as a marker to distinguish cross-progeny from self-progeny. (D to G) P0 animals were allowed to mate for 24 hours. F1 worms were infected with N. parisii as in (A). (D) Quantitation of DY96 fluorescent spores per worm. (E) Number of embryos per worm. (D and E) Data pooled from three independent experiments using n = 20 worms per condition per experiment. Only hermaphrodite maternal cross-progeny were included in quantifications. (F and G) Percentage of worms that are gravid. Data pooled from three independent experiments using n = 13 to 67 worms per condition per experiment. Means ± SEM (horizontal bars) are shown. The P values were determined by ordinary one-way ANOVA with post hoc test . Significance was defined as follows: *P < 0.05; ***P < 0.001; ****P < 0.0001.
Next, to determine whether transcriptional response activation in these mutants could induce immunity in progeny, we performed mating assays. We first examined the cross-progeny of pals-22 and lin-35 mutant hermaphrodites mated with wild-type males to determine whether parents with an up-regulated transcriptional response generated resistant F1s (Fig. 6C). The heterozygous cross-progeny of both mutants produced a similar number of embryos in uninfected conditions as wild type, as the developmental timing and brood size are recessive traits of pals-22 and lin-35 mutants. When infected with N. parisii, cross-progeny have a lower pathogen load and produce more embryos than wild-type animals (Fig. 6, D and E). On average, 57% of pals-22 and 77% of lin-35 maternal cross-progeny became gravid under infection conditions, compared to less than 10% of wild-type animals (Fig. 6, F and G). In contrast, paternal cross-progeny, i.e., the offspring of wild-type hermaphrodites mated with males carrying either pals-22 or lin-35 mutations, do not exhibit improved reproductive fitness under infection conditions (Fig. 6, F and G). These results reveal that inherited immunity to microsporidia is maternally transferred and can be induced by transcriptional activation alone.
The signal for offspring resistance can originate in multiple somatic tissues
To identify the tissues required to induce the transcriptional response and thereby transmit inherited immunity, we used two methods. First, we infected the maternal cross-progeny of pals-22 mutants carrying a transgene for wild-type pals-22 under an endogenous promoter or under a promoter specific for a single-tissue type where pals-22 is typically expressed (30). While offspring of the pals-22 endogenous rescue strain were no longer resistant to infection, neuronal and hypodermal-specific rescue strains still produced more fit progeny but to a lesser extent than pals-22 mutants (Fig. 7, A and B). This indicates that a signal in the neuronal and hypodermal tissues may contribute to the transmission of inherited immunity but may not be crucial. The intestinal-specific rescue produced progeny that were less fit when infected than cross-progeny from pals-22 mutants, although this was not statistically significant, indicating that an intestinal signal could also contribute to transmission of immunity. The maternal cross-progeny of lin-35 mutants with wild-type lin-35 expressed under either an endogenous or intestine-specific promoter were no longer resistant to N. parisii, indicating that an intestinal signal was important for transmission of immunity in this case (Fig. 7, C and D). Furthermore, addition of lin-35 under the pie-1 germline promoter was seen to partially reduce fitness in infected offspring (Fig. 7, C and D).
Fig. 7. Induction of the IPR in somatic tissues induces inherited immunity.
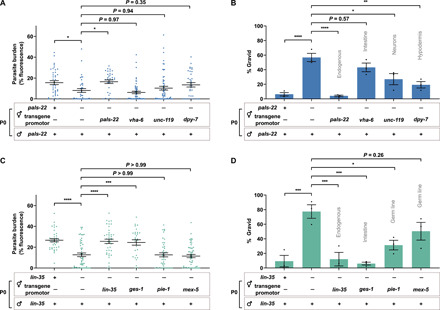
N2, pals-22, and lin-35 mutants with various rescue transgenes were allowed to mate for 24 hours. F1 L1 larvae were exposed to a high dose of N. parisii, fixed at 72 hpi, and stained with DY96. (A and C) Quantitation of DY96 fluorescent spores per worm. Data pooled from three independent experiments using n = 7 to 20 worms per condition per experiment. (B and D) Percentage of worms that are gravid. Data pooled from three independent experiments using n = 17 to 78 worms per condition per experiment. Tissues expressing the rescue transgenes are indicated on the graph. Only hermaphrodite maternal cross-progeny were included in quantifications. Means ± SEM (horizontal bars) are shown. The P values were determined by ordinary one-way ANOVA with post hoc test. Significance was defined as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
In a second approach, we inserted degron tags at endogenous pals-22 and lin-35 loci to take advantage of the auxin-inducible degradation system (40). We degraded either PALS-22 or LIN-35 in a tissue-specific manner in the parental generation only and assessed F1 progenies for resistance to microsporidia (fig. S9). Targeted degradation of PALS-22, and thus signal induction, in the adult intestine alone was sufficient to enhance immunity in progeny, while degradation in any other single tissue was unable to increase progeny fitness when infected (Fig. 8, A and B). In addition, degradation of LIN-35 in somatic tissues only was sufficient to confer resistance to offspring (Fig. 8, C and D). Together, these results suggest that induced expression of the IPR either in the adult intestine only or in multiple somatic tissues is sufficient to transmit inherited immunity to offspring.
Fig. 8. Somatic depletion of negative regulators of the IPR for a single generation induces inherited immunity.
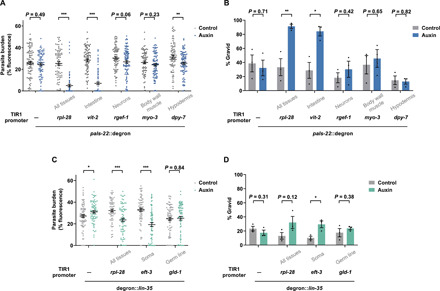
(A to D) P0 animals were grown on either control plates or plates containing 200 μM auxin from embryos to mediate degradation of PALS-22 or LIN-35 for 72 hours. F1 L1 larvae were exposed to a high dose of N. parisii, fixed at 72 hpi, and stained with DY96. (A and C) Quantitation of DY96 fluorescent spores per worm. Data pooled from three independent experiments using n = 19 to 39 worms per condition per experiment. (B and D) Percentage of worms that are gravid. Data pooled from three independent experiments using n = 100 to 214 worms per condition per experiment. Tissues where degradation was induced by auxin are indicated under the graphs. Means ± SEM (horizontal bars) are shown. The P values were determined by unpaired two-tailed Student’s t test. Significance was defined as follows: *P < 0.05; **P < 0.01; ***P < 0.001.
DISCUSSION
Inherited immunity is a nascent and rapidly growing field of research, with important consequences for our understanding of health and evolution. Multiple studies have now demonstrated that parental exposure to one pathogen can protect offspring against subsequent exposure to the same pathogen (1, 13, 19, 41). Several studies have also shown that parents exposed to a particular stress can produce progeny that are protected against the same stress and that abiotic stress can provide transgenerational resistance to pathogenic infection (5, 6, 42–44). Critically, it is not yet clear how the environment of the parent determines which stresses the offspring are protected against. Here, we show that inherited immunity to microsporidia can be activated by microsporidia and viral infection, as well as heavy metal stress, which all elicit a similar transcriptional response in the parents. Inherited immunity can also be activated through maternal somatic depletion of negative regulators of the transcriptional response. Activation of the transcriptional response within just a single generation in the intestine is sufficient to induce inherited immunity in offspring. Together, we have demonstrated that the signal for initiating inherited immunity against microsporidia is somatic induction of a shared maternal transcriptional stress response.
Inherited immunity phenotypes are often costly and can result in primed animals being more sensitive to other stresses (1, 45). Populations of C. elegans often develop within the same immediate environment, so parental infection is a good indicator of progeny exposure to microsporidia (46). Here, we find that progeny from microsporidia-infected parents are smaller, carry fewer embryos, and are more sensitive to osmotic and heavy metal stress. While inherited immunity, in response to microsporidia infection, lasts only a single generation, it can be induced in every generation, and this may be a strategy to limit fitness trade-offs.
Immunity to microsporidia in C. elegans is thought to function through activation of the IPR and associated pathogen clearance (28, 32). Up-regulation of the IPR results in immunity to both microsporidia and virus but not bacteria (31). Here, inherited immunity protects against microsporidia and bacteria but not virus. In addition, constant induction of the IPR genes in pals-22 and lin-35 mutants also greatly affects animal development, whereas offspring with activated inherited immunity have less severe growth defects (31, 47). Our results suggest that although induction of the IPR is sufficient to activate inherited immunity, inherited immunity and IPR-mediated immunity are separate immune responses. We also show that inhibition of the RB ortholog lin-35 provides immunity against microsporidia, both for the parents and their offspring. Although lin-35 has been implicated in stress responses and negative regulation of immune gene expression, this is the first example of lin-35 mutants providing pathogen resistance (48, 49). RB is evolutionarily conserved but, in mammals, acts as a positive regulator of antiviral immunity and immune cell development (50, 51).
Once the signal for inherited immunity is induced in the soma, the response must be transferred to developing progeny. Several inherited multigenerational responses that last for more than two generations are dependent on RNAi pathways (16, 52). Consistent with recent reports of responses that last only one or two generations being transmitted independent of these pathways, heritable immunity to microsporidia (which lasts a single generation) is not reliant on RNAi machinery (19, 35).
Being able to harness inherited immunity would provide a way to prevent infection of invertebrates (53). Inherited immunity can be induced without the pathogen itself, by molecules that activate an immune response, and thus, a similar approach could be used to combat microsporidia infections (54–56). Inherited immunity could be used to block infection of beneficial insects such as honey bees, and inhibition of this immunity could be used to improve the efficacy of microsporidia as biocontrol agents for locusts and other pests (57). Although this is the first report of inherited immunity to microsporidia, mosquitoes whose parents were infected with microsporidia contained significantly fewer malaria parasites. This suggests that manipulation of these pathways could also be used to prevent invertebrate vectors of human disease from transmitting infection (58, 59).
MATERIALS AND METHODS
Worm maintenance
C. elegans strains were maintained at 21°C on nematode growth media (NGM) plates seeded with 10× OP50-1 E. coli, as previously described (60). Strains used in this study are listed in table S1. For maintenance and infection assays, 10× concentrates of OP50-1 were prepared by growing cultures to saturation in lysogeny broth (LB) at 37°C for 16 to 18 hours. Populations were synchronized by washing worms off plates with M9 solution and bleaching with sodium hypochlorite/1 M NaOH until the embryos of gravid adults were released into solution. Eggs were washed three times with M9, resuspended in 5 ml of M9, and rotated at 21°C for 18 to 24 hours to allow embryos to hatch into L1s. For pelleting of live worms, animals were centrifuged in microcentrifuge tubes for 30 s at 1400g.
Construction of transgenic strains
For strains with tissue-specific expression of lin-35 in a lin-35 mutant background, MT10430 was crossed to DP38. The resulting lin-35(n745) I; unc-119(ed3) III double mutant was then crossed to YL398, YL402, YL409, and YL468 (47).
SapTrap was used to construct a repair template for introducing the auxin-inducible degron tag via CRISPR (61). Briefly, the ~500– to 600–base pair regions immediately upstream of the pals-22 start codon or downstream of the lin-35 stop codon were polymerase chain reaction–amplified from genomic N2 DNA to act as homology arms. These were cloned into the pDD379 backbone, along with guide RNA and other SapTrap-SEC kit plasmids (Addgene) (62). The repair construct and coinjection markers were microinjected into N2 worms, and hygromycin-resistant rollers were selected. The SEC was then excised via heat shock (63).
Strains carrying vit-2p::TIR1, rgef-1p::TIR1, myo-3p::TIR1, dpy-7p::TIR1, and rpl-28p::TIR1 were generated by MosSCI (64). Repair constructs were generated using Multisite LR Gateway cloning using pCFJ150 as LR entry vector and microinjected into strain EG6699.
All strains generated for this study were outcrossed to N2 at least three times before use. Plasmids and primers used in this study are listed in table S2.
Preparation of microsporidia spores
N. parisii spores were prepared as described previously (25). Briefly, microsporidia spores were used to infect large populations of C. elegans N2 worms. Infected worms were then harvested and mechanically disrupted using 1-mm-diameter zirconia beads (BioSpec Products Inc.), and the lysate was filtered through 5-μm filters (Millipore Sigma) to remove nematode debris. Spore preparations were assayed for bacterial growth, and those that were free of contaminating bacteria were stored at −80°C. Each assay was performed using spores of the same batch. In total, five batches were used in this study.
Microsporidia infection assays
Basic infection and priming assays
Synchronized populations of L1 worms were mixed with 1 ml of 10× OP50-1 alone as uninfected controls or 1 ml of 10× OP50-1 supplemented with N. parisii spores and plated on 10-cm NGM plates (doses defined in table S3 below). For priming assays, 2500 P0 animals were infected with a “low dose” of N. parisii such that >90% of animals were infected at 72 hpi, and >80% of animals were fertile to generate a sufficient yield of primed F1 embryos for subsequent experiments (fig. S1, C and E). At 72 hpi, worms were collected and washed three times in 1 ml of M9. To measure infection in the parental generation, 10% of P0 worms were fixed, and parasite burden and gravidity were assessed. The remaining 90% of worms were bleached to obtain naïve or primed embryos from uninfected or infected adults, respectively.
Following hatching in M9, 1000 naïve or primed F1 animals were mixed with 400 μl of 10× OP50-1 and challenged at the L1 stage on a 6-cm plate with a high dose of N. parisii. This resulted in ~10 to 20% of naïve animals being gravid, so that fitness increases or decreases would be easily detectable. At 72 hpi, worms were fixed and stained with DY96, and gravidity and infection were assessed by microscopy.
Assessment of parental age on inherited immunity
To test the effects of parental age on offspring immunity, 2500 P0 animals were either not infected or infected with a low dose of N. parisii at the L1 stage. At 64 and 72 hpi, P0 populations were bleached to release F1 embryos. Naïve and primed F1 animals were tested for inherited immunity as described in the “Basic infection and priming assays” section.
Assessment of parental infection duration on inherited immunity
To test how long animals must be infected to transmit immunity to offspring, 2500 P0 animals were either not infected or infected with a low dose of N. parisii at (i) the L1 stage, (ii) the L2/L3 stage after 24-hour rest on 10× OP50-1, or (iii) the L4 stage after 48-hour rest on 10× OP50-1. At 72 hours after L1, P0 populations were bleached to release F1 embryos. Naïve and primed F1 animals were tested for inherited immunity as described in the “Basic infection and priming assays” section.
Assessment of inherited immunity through development
To test inherited immunity throughout development, naïve or primed worms were obtained as described in the “Basic infection and priming assays” section. Next, 1000 naïve or primed animals were plated on 400 μl of 10× OP50-1 supplemented with a maximal dose of N. parisii spores at (i) the L1 stage, (ii) the L2/L3 stage after 24-hour rest on 10× OP50-1, or (iii) the L4 stage after 48-hour rest on 10× OP50-1. At 30 mpi, animals were fixed and stained with MicroB FISH probe to detect N. parisii 18S RNA, and the number of sporoplasms was quantified by microscopy.
To test the effects of small changes in development on inherited immunity, naïve or primed worms were obtained as described in the “Basic infection and priming assays” section. Next, 1000 naïve or primed L1 larvae were plated on 400 μl 10× OP50-1 supplemented with a maximal dose of N. parisii spores for their first exposure to food or after 6-hour rest on 10× OP50-1. At 30 mpi, animals were fixed and stained with MicroB FISH probe to detect N. parisii 18S RNA, and the number of sporoplasms was quantified by microscopy.
Assessment of spores in the gut and host cell invasion by N. parisii
To assay host cell invasion by microsporidia and the presence of spores in the gut, we conducted short infection assays (23). Naïve or primed F1 animals were obtained as described in the “Basic infection and priming assays” section. Next, 1000 naïve or primed animals were mixed with 4 μl of 10× OP50-1 and a maximal dose of N. parisii spores at the L1 stage and plated on NGM. At 30 mpi or 3 hpi, animals were fixed and stained with DY96 and MicroB FISH probe, and the number of spores or sporoplasms was quantified by microscopy.
Assessment of host clearance of N. parisii
To assay host clearance of microsporidia, we conducted pulse infection assays. For this, naïve or primed F1 animals were obtained as described in the “Basic infection and priming assays” section. Next, 2000 naïve or primed animals were mixed with 10× OP50-1 and a very high dose of N. parisii spores at the L1 stage and plated on NGM. At 3 hpi, populations were split and half of the animals were fixed. The remaining animals were maintained in the absence of spores until a 24-hour end point before fixing. Animals were stained with MicroB FISH probe, and the number of sporoplasms at each time point was quantified by microscopy.
Assessment of N. parisii–induced immunity over multiple generations
To assess immunity to microsporidia over multiple generations, we performed an ancestral infection assay (fig. S6C). Here, 2500 animals were infected at the L1 stage with N. parisii for one, two, or three successive generations (P0, F1, and F2). Each infection period lasted 72 hours before treating with sodium hypochlorite solution to obtain the next generation of embryos. F3 L1 populations were split and either subjected to infection testing or maintained under noninfection conditions for the collection and subsequent testing of F4 offspring. To test immunity, F3 and F4 larvae were exposed to a high dose of N. parisii at the L1 stage. At 72 hpi, F3 and F4 animals were fixed and stained with DY96 to visualize N. parisii spores.
Heat-killed spore infection assay
Synchronized populations of 2500 L1 worms were mixed with (i) 1 ml of 10× OP50-1 alone or 10× OP50-1 supplemented with a low dose of either (ii) live or (iii) heat-killed N. parisii spores and plated on NGM. For heat killing, live spores were treated at 65°C for 10 min. At 72 hpi, parental generations were bleached to obtain naïve embryos or embryos primed with heat-killed or live spores. Inherited immunity to N. parisii was tested as described in the “Basic infection and priming assays” section.
P. aeruginosa infection assays
Strains of wild-type or dsRed-expressing P. aeruginosa PA14 were used to assay survival and bacterial burden, respectively. Strains were grown in 3 ml of LB overnight from a single bacterial colony. A volume of 20 μl of bacterial culture was used to seed 3.5-cm slow-killing plates for survival assays (34). A volume of 50 μl of bacterial culture was used to seed 6-cm slow-killing plates for bacterial burden assays. To prevent C. elegans pathogen avoidance behavior (65), bacterial culture was spread to ensure that plates were fully covered with P. aeruginosa. Seeded plates were incubated for 24 hours at 37°C for growth of bacterial lawns and maintained for a further 24 hours at room temperature (RT) before infection assays. Naïve and primed worms were obtained as in the “Basic infection and priming assays” section. To assay survival, 20 to 30 naïve or primed L1 worms were transferred to 3.5-cm wild-type PA14-seeded plates and incubated at 25°C for 84 hours. Worms that failed to respond to pressure from a metal pick were considered nonviable.
To assay bacterial burden by fluorescence, 1000 naïve or primed L1 worms were plated on 6-cm dsRed PA14-seeded plates and incubated at 25°C for 48 hours. At 48 hpi, worms were collected from plates, washed three times in M9, and fixed, and bacterial burden was assessed by microscopy. Assays of bacterial burden by CFU counts were carried out in a similar manner to those described previously (66). Here, 1000 naïve or primed L1 worms were plated on 6-cm dsRed PA14-seeded plates and incubated at 25°C for 48 hours. At 48 hpi, three replicates of 10 worms were transferred to a 25 mM levamisole droplet on an NGM plate containing gentamicin (1 mg/ml) and ampicillin (1 mg/ml). Over ~30 min, worms were transferred to fresh droplets of levamisole on fresh plates three times to stop pharyngeal pumping and to kill and dilute PA14 stuck to the worm’s surface. Worms were then transferred to microcentrifuge tubes and washed three times in 1-ml sterile M9. Note that the final wash was saved and plated similar to worm lysates, to assess background CFUs. To lyse worms, 200-μl sterile phosphate-buffered saline (PBS) containing 1% Triton X-100 (Sigma-Aldrich) and an equal volume of 2.0-mm zirconia beads (BioSpec Products Inc.) were added to the washed nematodes before lysing with a cell disrupter for 3 min at 3000 rpm. Lysates were serially diluted in PBS, plated on LB plates, and incubated overnight at 37°C. Colonies were counted to determine the average CFU per nematode.
Orsay virus infection assays
Orsay virus filtrate was prepared as described previously (28). Briefly, plates of Orsay virus–infected animals were maintained until starvation. Virus shed by infected worms was collected by washing plates with M9, passing through 0.22-μm filters (Millipore Sigma), and stored at −80°C. For infections to test resistance to Orsay virus, naïve and primed worms were obtained as in the “Basic infection and priming assays” section. Next, 1000 naïve or primed L1s were mixed with 100 μl of 10× OP50-1 and 500 μl of the viral filtrate and then plated on 6-cm NGM. At 16 hpi, animals were fixed and FISH-stained to assess infection status.
To generate Orsay-primed animals, 2500 L1s were mixed with 1 ml of 10× OP50-1 and 500 μl of viral filtrate. At 72 hpi, animals were collected and bleached to obtain primed embryos. Inherited immunity to N. parisii was tested as described in the “Basic infection and priming assays” section.
Bead-feeding assay
Naïve or primed L1 worms were obtained as described in the “Basic infection and priming assays” section. Next, 1000 animals were placed on 6-cm plates in 400-μl total volume of M9 containing 10% (v/v) 10× OP50-1 and 4% (v/v) 0.2-μm green fluorescent polystyrene beads (Degradex, Phosphorex). Where spores were included for the 3-hour time point, a maximal dose of N. parisii spores was used. After 30 min or 3 hours, animals were fixed, and bead ingestion was assessed by microscopy.
Osmotic stress assays
Osmotic stress adaptation assays were performed as previously described (37). Synchronized populations of 2500 L1 worms were plated either (i) on standard NGM (50 mM salt) plates together with 1 ml of 10× OP50-1 alone or (ii) 10× OP50-1 supplemented with a low dose of N. parisii, or (iii) on NGM containing an elevated concentration of salt (250 mM) together with 1 ml of 10× OP50-1. At 80 hpi, parent populations were bleached to obtain naïve, infection-primed, or salt-primed embryos. To test resistance to osmotic stress, 1000 embryos were plated on NGM containing a high concentration of salt (420 mM). The percentage of embryos that had hatched by 48 hours was quantified using light microscopy. Inherited immunity to N. parisii was tested as described in the “Basic infection and priming assays” section.
Cadmium assays
We first confirmed that cadmium exposure induced IPR gene expression in C. elegans. This was determined by plating ERT054 and ERT071 fluorescent reporter worm strains on 50 mM calcium chloride and observing increased green fluorescent protein (GFP) expression in exposed worms, relative to unexposed controls. Synchronized populations of 2500 L4 worms were plated either (i) on standard NGM together with 1 ml of 10× OP50-1 alone or (ii) 1 ml of 10× OP50-1 supplemented with a low dose of N. parisii, or (iii) on NGM containing 50 mM cadmium chloride together with 10× OP50-1. After 24 hours, parent populations were bleached to obtain naïve, infection-primed, or cadmium-primed embryos. Immunity to N. parisii was tested as described in the “Basic infection and priming assays” section. Immunity to P. aeruginosa was tested as described in the “P. aeruginosa infection assays” section. To test for protection against cadmium stress, 1000 F1 larvae were maintained on standard NGM with 10× OP50-1 until L4 stage and then plated on NGM containing 50 mM cadmium chloride. After 24 hours, animals were fixed and stained with DY96 for embryo counting.
Auxin-inducible depletion experiments
Auxin plates were prepared by adding auxin stock solution [400 mM auxin (Alfa Aesar) in ethanol] to NGM, for a final concentration of 200 μM auxin, immediately before pouring plates. Control plates were prepared by adding ethanol to NGM, for a final concentration of 0.15%. Auxin plates were stored in the dark at 4°C and used within 1 month.
Embryos obtained from bleaching gravid adults were plated on auxin or ethanol control plates following M9 washes. 10× OP50-1 was added to plates 18 to 24 hours after plating to allow embryos to hatch and synchronize. Worms were bleached 72 hours after L1 arrest and F1 immunity to N. parisii tested as described in the “Basic infection and priming assays” section.
Cross-progeny generation
For testing maternal effects, 50 L4 ERT054 males and 30 L4 hermaphrodites were plated on a 3.5-cm NGM plate for mating. To test paternal effects, L4 mutant males carrying the jyIs8 transgene were crossed with L4 N2 hermaphrodites (Fig. 6C). After 22 to 24 hours, animals were bleached to obtain embryos and F1 immunity to N. parisii tested as described in the “Basic infection and priming assays” section. During quantification, the presence of myo-2p::mCherry was used to distinguish cross-progeny from self-progeny.
Fixation
For visualization of germ lines of JMC101 (GFP::3xFLAG::CSR-1) animals, bead-feeding assays, and staining of N. parisii or Orsay virus with FISH probes, worms were fixed in 1 ml of 4% paraformaldehyde (PFA) in PBS containing 0.1% Tween 20 (PBST), for 20 min at RT or overnight at −20°C. For P. aeruginosa burden assays and DY96 staining, worms were fixed in 1-ml acetone for 10 min at RT or overnight at 4°C. For pelleting of worms during fixing protocols, animals were centrifuged in microcentrifuge tubes for 30 s at 10,000g.
Staining with DY96
To assay parasite burden and worm gravidity, microsporidia spores and embryos were visualized with the chitin-binding dye DY96. Acetone-fixed animals were washed twice in 1 ml of PBST, resuspended in 500-μl staining solution [PBST, 0.1% SDS, and DY96 (20 μg/ml)], and rotated at 21°C for 30 min in the dark. Stained worms were resuspended in 20-μl EverBrite Mounting Medium (Biotium) and mounted on slides for imaging. For pelleting of worms during staining protocols, animals were centrifuged in microcentrifuge tubes for 30 s at 10,000g.
FISH assays
For FISH staining of N. parisii 18S ribosomal RNA or Orsay virus RNA, worms were fixed in PFA as above and washed twice in 1 ml of PBST. Worms were then washed once in 1 ml hybridization buffer [900 mM NaCl, 20 mM tris (pH 8.0), and 0.01% SDS] and incubated overnight at 46°C in 100-μl hybridization buffer containing FISH probe (5 to 10 ng/μl) conjugated to a Cal Fluor 610 dye (LGC Biosearch Technologies). MicroB (5 ng/μl; ctctcggcactccttcctg) (26) was used to detect N. parisii 18S RNA. A solution of Orsay 1 (gacatatgtgatgccgagac) and Orsay 2 (gtagtgtcattgtaggcagc) mixed 50:50 (10 ng/μl) was used to detect Orsay virus. Stained animals were washed once in 1-ml wash buffer (hybridization buffer containing 5 mM EDTA) and incubated in 500-μl fresh wash buffer for a further 30 min at 46°C. Worms were resuspended in 20-μl EverBrite Mounting Medium (Biotium) and mounted on slides for imaging. To pellet worms during staining protocols, animals were centrifuged in microcentrifuge tubes for 30 s at 10,000g. For costaining of N. parisii RNA with DY96, wash buffer was supplemented with DY96 (20 μg/ml) for the final 30-min incubation.
Microscopy and image analysis
For measurement of worm body size and quantification of embryos and gravid or infected animals, as well as N. parisii or P. aeruginosa burden, worms were imaged using an Axio Imager 2 (Zeiss). Worms carrying one or more embryos were considered gravid. Worms having any quantity of DY96-stained microsporidia spores were considered infected. Worms with any number of cells stained with FISH probe to detect Orsay virus RNA were considered infected.
Bead ingestion and precise pathogen burdens were determined using ImageJ/FIJI (67); here, each worm was defined as an individual “region of interest,”, and fluorescence from GFP (DY96-stained microsporidia, or GFP beads) or dsRed (dsRed-expressing PA14) was subjected to “threshold” and “measure area percentage” functions on ImageJ. For DY96-stained samples, images were thresholded to capture the brighter signal from microsporidia spores while eliminating the dimmer GFP signal from worm embryos. Final values are given as percentage of fluorescence for single animals.
Transcriptional analyses
WormExp (40) was used to search for published expression datasets that have a significant overlap with the set of genes enriched in N. parisii–infected animals. FPKM (fragments per kilobase of transcript per million mapped reads) values from RNA sequencing data were obtained for pals-22 (31) mutants and five N. parisii infection time points (28), and fold changes were calculated. Fold change values were obtained for lin-35 mutant microarray data (68), and replicates were averaged. Log2 fold changes greater than 2 or less than −2 were used to determine differentially expressed genes. Genes that were either up- or down-regulated in lin-35, pals-22, and at least one infection time point were plotted as a heatmap with dendrograms using heatmap.2 function from gplots package in R with all arguments (70) set to default except for trace that was set to none.
Statistical analyses
Unless otherwise stated, P values were determined by two-tailed unpaired Student’s t test. All P values not meeting significance requirements are displayed in figures for clarity. These P values were calculated using Prism software (GraphPad Software Inc.). Statistical significance is defined as P < 0.05, unless otherwise stated (i.e., when using Bonferroni correction for multiple testing).
Acknowledgments
We thank E. R. Troemel for mentorship and support in allowing A.W.R. to start this project as a postdoctoral fellow in the Troemel laboratory. We thank M. A. Bakowski and L. B. Cohen for sharing initial results on the shared transcriptional similarity between lin-35 mutants and microsporidia infection. We thank A. P. Ryan and K. C. Reddy for sharing initial findings on the shared transcriptional similarity between cadmium exposure and microsporidia infection. We thank N. O. Burton for advice on the intergenerational osmotic stress assay. We are grateful to C. Mok and N. O. Burton for providing helpful comments on the manuscript. Additional C. elegans strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs Grant P40 OD010440. Schematics were created using BioRender.com. Funding: This work was supported by the Natural Sciences and Engineering Research Council of Canada (grant no. 522691522691 to A.W.R.), the Canadian Institutes of Health Research (grant no. PJT-156083 to J.M.C.), an Alfred P. Sloan Research Fellowship FG2019-12040 (to A.W.R.), and a CRC Tier II in Small RNA Biology (J.M.C.). Author contributions: A.R.W., W.Z., and A.W.R. designed and analyzed the experiments and cowrote the paper, with consolation from all the authors. A.R.W. and W.Z. conducted all the experiments, except where noted. R.S. conducted heat-killed, osmotic stress, and cadmium assays. L.W. conducted bioinformatic analysis of similarity of transcriptional response between N. parisii infection and pals-22 and lin-35 mutants. H.T.E.J. conducted the pals-22 clearance assay. J.M.C. designed the small-RNA inheritance factors experiment. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabf3114/DC1
REFERENCES AND NOTES
- 1.Tetreau G., Dhinaut J., Gourbal B., Moret Y., Trans-generational immune priming in invertebrates: Current knowledge and future prospects. Front. Immunol. 10, 1938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barribeau S. M., Schmid-Hempel P., Sadd B. M., Royal Decree: Gene expression in trans-generationally immune primed bumblebee workers mimics a primary immune response. PLOS ONE 11, e0159635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salmela H., Amdam G. V., Freitak D., Transfer of immunity from mother to offspring is mediated via egg-yolk protein vitellogenin. PLOS Pathog. 11, e1005015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Little T. J., O’Connor B., Colegrave N., Watt K., Read A. F., Maternal transfer of strain-specific immunity in an invertebrate. Curr. Biol. 13, 489–492 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Norouzitallab P., Baruah K., Vandegehuchte M., Van Stappen G., Catania F., Bussche J. V., Vanhaecke L., Sorgeloos P., Bossier P., Environmental heat stress induces epigenetic transgenerational inheritance of robustness in parthenogenetic Artemia model. FASEB J. 28, 3552–3563 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Dubuffet A., Zanchi C., Boutet G., Moreau J., Teixeira M., Moret Y., Trans-generational immune priming protects the eggs only against gram-positive bacteria in the mealworm beetle. PLOS Pathog. 11, e1005178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D. H., Feinbaum R., Alloing G., Emerson F. E., Garsin D. A., Inoue H., Tanaka-Hino M., Hisamoto N., Matsumoto K., Tan M.-W., Ausubel F. M., A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297, 623–626 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Weinhouse C., Truong L., Meyer J. N., Allard P., Caenorhabditis elegans as an emerging model system in environmental epigenetics: C. elegans as an environmental epigenetics model. Environ. Mol. Mutagen. 59, 560–575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fire A., Xu S., Montgomery M. K., Kostas S. A., Driver S. E., Mello C. C., Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Willis A. R., Sukhdeo R., Reinke A. W., Remembering your enemies: mechanisms of within-generation and multigenerational immune priming in Caenorhabditis elegans. FEBS J. 288, 1759–1770 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Rechavi O., Minevich G., Hobert O., Transgenerational inheritance of an acquired small RNA-based antiviral response in C. elegans. Cell 147, 1248–1256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashe A., Sarkies P., Le Pen J., Tanguy M., Miska E. A., Antiviral RNA interference against Orsay virus is neither systemic nor transgenerational in Caenorhabditis elegans. J. Virol. 89, 12035–12046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gammon D. B., Ishidate T., Li L., Gu W., Silverman N., Mello C. C., The antiviral RNA interference response provides resistance to lethal arbovirus infection and vertical transmission in Caenorhabditis elegans. Curr. Biol. 27, 795–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sterken M. G., Snoek L. B., Bosman K. J., Daamen J., Riksen J. A. G., Bakker J., Pijlman G. P., Kammenga J. E., A heritable antiviral RNAi response limits Orsay virus infection in Caenorhabditis elegans N2. PLoS One. 9, e89760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaletsky R., Moore R. S., Vrla G. D., Parsons L. R., Gitai Z., Murphy C. T., C. elegans interprets bacterial non-coding RNAs to learn pathogenic avoidance. Nature 586, 445–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore R. S., Kaletsky R., Murphy C. T., Piwi/PRG-1 argonaute and TGF-β mediate transgenerational learned pathogenic avoidance. Cell 177, 1827–1841.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pereira A. G., Gracida X., Kagias K., Zhang Y., C. elegans aversive olfactory learning generates diverse intergenerational effects. J. Neurogenet. 34, 378–388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palominos M. F., Verdugo L., Gabaldon C., Pollak B., Ortíz-Severín J., Varas M. A., Chávez F. P., Calixto A., Transgenerational diapause as an avoidance strategy against bacterial pathogens in Caenorhabditis elegans. MBio 8, e01234-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burton N. O., Riccio C., Dallaire A., Price J., Jenkins B., Koulman A., Miska E. A., Cysteine synthases CYSL-1 and CYSL-2 mediate C. elegans heritable adaptation to P. vranovensis infection. Nat. Commun. 11, 1741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wadi L., Reinke A. W., Evolution of microsporidia: An extremely successful group of eukaryotic intracellular parasites. PLOS Pathog. 16, e1008276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stentiford G. D., Becnel J. J., Weiss L. M., Keeling P. J., Didier E. S., Williams B. A. P., Bjornson S., Kent M. L., Freeman M. A., Brown M. J. F., Troemel E. R., Roesel K., Sokolova Y., Snowden K. F., Solter L., Microsporidia–Emergent pathogens in the global food chain. Trends Parasitol. 32, 336–348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luallen R. J., Reinke A. W., Tong L., Botts M. R., Félix M.-A., Troemel E. R., Discovery of a natural microsporidian pathogen with a broad tissue tropism in Caenorhabditis elegans. PLOS Pathog. 12, e1005724 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balla K. M., Andersen E. C., Kruglyak L., Troemel E. R., A wild C. elegans strain has enhanced epithelial immunity to a natural microsporidian parasite. PLOS Pathog. 11, e1004583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balla K. M., Luallen R. J., Bakowski M. A., Troemel E. R., Cell-to-cell spread of microsporidia causes Caenorhabditis elegans organs to form syncytia. Nat. Microbiol. 1, 16144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Estes K. A., Szumowski S. C., Troemel E. R., Non-lytic, actin-based exit of intracellular parasites from C. elegans intestinal cells. PLOS Pathog. 7, e1002227 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Troemel E. R., Félix M.-A., Whiteman N. K., Barrière A., Ausubel F. M., Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLOS Biol. 6, e309 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jarkass H. T. E., Reinke A. W., The ins and outs of host-microsporidia interactions during invasion, proliferation and exit. Cell. Microbiol. 22, e13247 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Bakowski M. A., Desjardins C. A., Smelkinson M. G., Dunbar T. A., Lopez-Moyado I. F., Rifkin S. A., Cuomo C. A., Troemel E. R., Ubiquitin-mediated response to microsporidia and virus infection in C. elegans. PLOS Pathog. 10, e1004200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dierking K., Yang W., Schulenburg H., Antimicrobial effectors in the nematode Caenorhabditis elegans: An outgroup to the Arthropoda. Philos. Trans. R. Soc. B Biol. Sci. 371, 20150299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy K. C., Dror T., Sowa J. N., Panek J., Chen K., Lim E. S., Wang D., Troemel E. R., An intracellular pathogen response pathway promotes proteostasis in C. elegans. Curr. Biol. 27, 3544–3553.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy K. C., Dror T., Underwood R. S., Osman G. A., Elder C. R., Desjardins C. A., Cuomo C. A., Barkoulas M., Troemel E. R., Antagonistic paralogs control a switch between growth and pathogen resistance in C. elegans. PLOS Pathog. 15, e1007528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balla K. M., Lažetić V., Troemel E. R., Natural variation in the roles of C. elegans autophagy components during microsporidia infection. PLOS ONE 14, e0216011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luallen R. J., Bakowski M. A., Troemel E. R., Characterization of microsporidia-induced developmental arrest and a transmembrane leucine-rich repeat protein in Caenorhabditis elegans. PLOS ONE 10, e0124065 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan M.-W., Rahme L. G., Sternberg J. A., Tompkins R. G., Ausubel F. M., Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc. Natl. Acad. Sci. U.S.A. 96, 2408–2413 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lev I., Bril R., Liu Y., Ceré L. I., Rechavi O., Inter-generational consequences for growing Caenorhabditis elegans in liquid. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374, 20180125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Félix M.-A., Ashe A., Piffaretti J., Wu G., Nuez I., Bélicard T., Jiang Y., Zhao G., Franz C. J., Goldstein L. D., Sanroman M., Miska E. A., Wang D., Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLOS Biol. 9, e1000586 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burton N. O., Furuta T., Webster A. K., Kaplan R. E. W., Baugh L. R., Arur S., Horvitz H. R., Insulin-like signalling to the maternal germline controls progeny response to osmotic stress. Nat. Cell Biol. 19, 252–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu X., Horvitz H. R., lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell 95, 981–991 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Yang W., Dierking K., Schulenburg H., WormExp: A web-based application for a Caenorhabditis elegans-specific gene expression enrichment analysis. Bioinformatics 32, 943–945 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Zhang L., Ward J. D., Cheng Z., Dernburg A. F., The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development 142, 4374–4384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma C., Niu R., Huang T., Shao L.-W., Peng Y., Ding W., Wang Y., Jia G., He C., Li C.-Y., He A., Liu Y., N6-methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat. Cell Biol. 21, 319–327 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Kishimoto S., Uno M., Okabe E., Nono M., Nishida E., Environmental stresses induce transgenerationally inheritable survival advantages via germline-to-soma communication in Caenorhabditis elegans. Nat. Commun. 8, 14031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eggert H., Buhr M. F. D., Kurtz J., A temperature shock can lead to trans-generational immune priming in the Red Flour Beetle, Tribolium castaneum. Ecol. Evol. 5, 1318–1326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klosin A., Casas E., Hidalgo-Carcedo C., Vavouri T., Lehner B., Transgenerational transmission of environmental information in C. elegans. Science 356, 320–323 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Sadd B. M., Schmid-Hempel P., A distinct infection cost associated with trans-generational priming of antibacterial immunity in bumble-bees. Biol. Lett. 5, 798–801 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Félix M.-A., Duveau F., Population dynamics and habitat sharing of natural populations of Caenorhabditis elegans and C. briggsae. BMC Biol. 10, 59 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kudron M., Niu W., Lu Z., Wang G., Gerstein M., Snyder M., Reinke V., Tissue-specific direct targets of Caenorhabditis elegans Rb/E2F dictate distinct somatic and germline programs. Genome Biol. 14, R5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui M., Cohen M. L., Teng C., Han M., The tumor suppressor Rb critically regulates starvation-induced stress response in C. elegans. Curr. Biol. 23, 975–980 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.González-Rangel A. A., Navarro R. E., LIN-35 beyond its classical roles: Its function in the stress response. Int. J. Dev. Biol. 65, 377–382 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Hutcheson J., Witkiewicz A. K., Knudsen E. S., The RB tumor suppressor at the intersection of proliferation and immunity: Relevance to disease immune evasion and immunotherapy. Cell Cycle 14, 3812–3819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kirienko N. V., Fay D. S., Transcriptome profiling of the C. elegans Rb ortholog reveals diverse developmental roles. Dev. Biol. 305, 674–684 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rechavi O., Houri-Ze’evi L., Anava S., Goh W. S. S., Kerk S. Y., Hannon G. J., Hobert O., Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 158, 277–287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Norouzitallab P., Baruah K., Vanrompay D., Bossier P., Teaching shrimps self-defense to fight infections. Trends Biotechnol. 37, 16–19 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Huang C.-C., Song Y.-L., Maternal transmission of immunity to white spot syndrome associated virus (WSSV) in shrimp (Penaeus monodon). Dev. Comp. Immunol. 23, 545–552 (1999). [DOI] [PubMed] [Google Scholar]
- 55.Moret Y., Schmid-Hempel P., Immune defence in bumble-bee offspring. Nature 414, 506–506 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Roy S., Kumar V., Bossier P., Norouzitallab P., Vanrompay D., Phloroglucinol treatment induces transgenerational epigenetic inherited resistance against vibrio infections and thermal stress in a brine shrimp (Artemia franciscana) model. Front. Immunol. 10, 2745 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.S. Bjørnson, D. Oi, Microsporidia (John Wiley & Sons, Ltd., 2014), pp. 635–670. [Google Scholar]
- 58.Lorenz L. M., Koella J. C., Maternal environment shapes the life history and susceptibility to malaria of Anopheles gambiae mosquitoes. Malar. J. 10, 382 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herren J. K., Mbaisi L., Mararo E., Makhulu E. E., Mobegi V. A., Butungi H., Mancini M. V., Oundo J. W., Teal E. T., Pinaud S., Lawniczak M. K. N., Jabara J., Nattoh G., Sinkins S. P., A microsporidian impairs Plasmodium falciparum transmission in Anopheles arabiensis mosquitoes. Nat. Commun. 11, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brenner S., The genetics of Caenorhabtidis elegans. Genetics 77, 71–94 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwartz M. L., Jorgensen E. M., SapTrap, a toolkit for high-throughput CRISPR/Cas9 gene modification in Caenorhabditis elegans. Genetics 202, 1277–1288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dickinson D. J., Slabodnick M. M., Chen A. H., Goldstein B., SapTrap assembly of repair templates for Cas9-triggered homologous recombination with a self-excising cassette. MicroPubl. Biol. 2018, 10.17912/W2KT0N (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dickinson D. J., Pani A. M., Heppert J. K., Higgins C. D., Goldstein B., Streamlined genome engineering with a self-excising drug selection cassette. Genetics 200, 1035–1049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frokjaer-Jensen C., Davis M. W., Hopkins C. E., Newman B. J., Thummel J. M., Olesen S.-P., Grunnet M., Jorgensen E. M., Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375–1383 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y., Lu H., Bargmann C. I., Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature 438, 179–184 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Yu Y., Zhi L., Wu Q., Jing L., Wang D., NPR-9 regulates the innate immune response in Caenorhabditis elegans by antagonizing the activity of AIB interneurons. Cell. Mol. Immunol. 15, 27–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petrella L. N., Wang W., Spike C. A., Rechtsteiner A., Reinke V., Strome S., synMuv B proteins antagonize germline fate in the intestine and ensure C. elegans survival. Development 138, 1069–1079 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.R Development Core Team, R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2019); www.R-project.org.
- 70.Ouyang J. P. T., Folkmann A., Bernard L., Lee C.-Y., Seroussi U., Charlesworth A. G., Claycomb J. M., Seydoux G., P granules protect RNA interference genes from silencing by piRNAs. Dev. Cell 50, 716–728.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raizen D. M., Lee R. Y., Avery L., Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans. Genetics 141, 1365–1382 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu F., Feng X., Chen X., Weng C., Yan Q., Xu T., Hong M., Guang S., A cytoplasmic Argonaute protein promotes the inheritance of RNAi. Cell Rep. 23, 2482–2494 (2018). [DOI] [PubMed] [Google Scholar]
- 73.C. elegans Deletion Mutant Consortium , Large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 2, 1415–1425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabf3114/DC1