

Abstract
Alzheimer's disease (AD) is the most common neurodegenerative disease, affecting millions of people worldwide. Extracellular beta‐amyloid (Aβ) plaques and neurofibrillary tau tangles are classical hallmarks of AD pathology and thus are the prime targets for AD therapeutics. However, approaches to slow or stop AD progression and dementia by reducing Aβ production, neutralizing toxic Aβ aggregates, or inhibiting tau aggregation have been largely unsuccessful in clinical trials. The contribution of dysregulated vascular components and inflammation is evident in AD pathology. Vascular changes are detectable early in AD progression, so treatment of vascular defects along with anti‐Aβ/tau therapy could be a successful combination therapeutic strategy for this disease. Here, we explain how vascular dysfunction mechanistically contributes to thrombosis as well as inflammation and neurodegeneration in AD pathogenesis. This review provides evidence that addressing vascular dysfunction in people with AD could be a promising therapeutic strategy.
Keywords: Alzheimer's disease, beta‐amyloid, blood‐brain barrier, coagulation factors, contact system, dementia, fibrinogen
Essentials.
Crosstalk between beta‐amyloid (Aβ) and blood proteins contributes to Alzheimer's disease (AD).
Contact system activation correlates with memory impairment in AD and mild cognitive impairment.
Blood clots can block blood flow and cause inflammation and cell death, which contribute to AD.
Preventing contact system activation or resistant blood clots are promising therapies for AD.
1. INTRODUCTION
Alzheimer's disease (AD) is the most prevalent form of neurodegenerative dementia affecting nearly 30 million people worldwide. 1 , 2 Though beta‐amyloid (Aβ) plaques and oligomers as well as aggregated phosphorylated tau tangles are the most studied therapeutic targets for AD, 3 it remains unclear how these proteinaceous inclusions lead to neuronal death and cognitive impairment. Aβ plaques are composed of aggregated Aβ peptides that are generated from the amyloid precursor protein (APP) upon a series of enzymatic cleavages. Aβ peptides can be of various lengths, ranging from 38 to 49 amino acids in length (ie, Aβ38‐Aβ49), depending on the type of APP cleavage. 4 , 5 , 6 Among these different lengths, Aβ42 and Aβ43 aggregates are the most neurotoxic and pathogenic. 5 , 6 However, approaches to prevent or slow neurodegeneration and dementia by reducing Aβ production, neutralizing toxic Aβ aggregates, or inhibiting tau aggregation have been largely unsuccessful. 7 , 8 , 9 , 10 There are several other factors that may play a role in neurodegeneration and cognitive decline in AD, 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 however, which should also be considered when defining new targets and developing effective AD therapeutics.
AD is recognized as a multifactorial disease, where numerous components, including cerebrovascular dysfunction and inflammation, drive disease pathology. 16 , 20 , 21 Cerebrovascular abnormalities, such as decreased blood flow, hemorrhage, microinfarcts, small‐vessel disease, dysregulated plasma contact system, and whitematter hyperintensities, are observed in >50% of people with AD. 13 , 31 In a large data‐driven study of people with late‐onset AD (LOAD), imaging techniques were used to analyze brain Aβ aggregates, cerebral glucose metabolism, cerebral blood flow, and functional activity/brain structural patterns in relation to disease progression. Temporal ordering of these abnormalities was assigned using trajectory models of aging. This analysis revealed that dysregulated cerebral blood flow occurs before Aβ deposition in people with LOAD. 16 Similarly, white matter hyperintensities are an early pathological feature observed in people with early‐onset AD (EOAD). 30
The most clear‐cut and classic example of crosstalk between neurodegeneration and vascular damage in AD is cerebral amyloid angiopathy (CAA). Between 80% and 95% of AD patients develop CAA, a condition characterized by deposition of Aβ aggregates in and around cerebral blood vessels. 3 , 32 It results in damage to endothelial cells and the blood vessel wall, which can lead to a loss in blood‐brain barrier (BBB) integrity 33 , 34 and inflammatory activity. In animal models of CAA, Aβ can trigger structural as well as functional damage in smooth muscle cells, pericytes, and endothelial cells. 17 , 35 , 36 , 37 CAA also leads to production of superoxide radicals, 36 which impairs the perivascular drainage system, an Aβ clearance pathway, in both AD mouse models and human patients, 32 , 38 , 39 , 40 thereby augmenting Aβ‐driven damage within the brain. Thus, CAA is a major contributor to cerebrovascular dysfunction, which can lead to microhemorrhage, vessel occlusion, loss of smooth muscle cells, and disruption of vasoactivity in AD. 32 , 41 , 42
Impaired BBB integrity can lead to abnormal levels of Aβ in the plasma as well as extravasation of blood proteins, such as fibrin(ogen), into the brain. In the circulation, Aβ can activate the plasma contact system, which induces coagulation and inflammation. 13 , 43 , 44 , 45 , 46 Extravasation of fibrin(ogen) into the brain can lead to its persistent accumulation as well as neuroinflammation. Below, we provide detailed evidence of the role of vascular dysfunction in AD pathogenesis. Table 1 summarizes the human and animal model evidence for vascular dysfunction and contact system activation in AD pathophysiology discussed herein. This review emphasizes the importance of better understanding the mechanistic link between Aβ and components of the contact system and the vasculature in AD, as it could provide alternative therapeutic strategies for many people with AD.
TABLE 1.
Human and animal model evidence of vascular dysfunction and contact system activation in AD
| Symptom/Phenotype | Reference | |
|---|---|---|
| Patients with AD | ||
| Vascular Pathology | Vascular dysfunction is an early pathology. | [16, 30] |
| CAA is observed in 80%‐95% of patients. | [3, 32] | |
| Impaired BBB integrity and extravasation of fibrin(ogen) into the brain parenchyma is observed. | [17, 33, 34, 79] | |
| Dutch and Iowa APP mutations increase cerebral fibrin deposits. | [55] | |
| Patient plasma exhibits increased aPTT. | [57] | |
| Contact System Activation | Aβ42 activates FXII to trigger the contact system. | [43, 44, 45, 46] |
| FXII is found in Aβ plaques in postmortem brain tissue. | [50] | |
| FXIIa, cHK, and bradykinin levels are increased in patient plasma. | [13, 64, 65] | |
| Kallikrein activity is increased in patient plasma. | [13] | |
| Intact HK levels are decreased in patient plasma. | [13] | |
| FXI levels are decreased in patient plasma. | [43] | |
| cHK levels are increased in patient CSF. | [70] | |
| Memory impairment correlates with vascular deficits and cHK levels. | [64, 65, 71] | |
| Animal models | ||
|
Vascular Pathology |
Anticoagulation treatment delays AD pathogenesis. | [54] |
| Fibrin(ogen) is associated with abnormal thrombosis, fibrinolysis, inflammation, neuronal damage, and cognitive impairment in AD. | [15, 33, 52, 79] | |
| BBB integrity is impaired in AD. | [17, 54] | |
| Cerebrovascular lesions induce Aβ deposition in AD. | [82, 83] | |
| Contact System Activation | Aβ42 activates FXII to trigger the contact system. | [13] |
| Contact system activation is associated with BBB damage. | [62, 63, 66] | |
| Inhibition of contact system activation ameliorates AD pathology and cognitive decline. | [69] | |
Abbreviations: Aβ, beta‐amyloid; AD, Alzheimer's disease; APP, amyloid precursor protein; aPTT, activated partial thromboplastin time; BBB, blood‐brain barrier; CAA, cerebral amyloid angiopathy; cHK, cleaved high molecular weight kininogen; CSF, cerebrospinal fluid; FXI, factor XI; FXII, factor XII; FXIIa, activated factor XII; HK, high molecular weight kininogen.
2. Aβ ACTIVATES FACTOR XII‐DRIVEN INTRINSIC COAGULATION
The plasma contact system is comprised of a group of plasma proteins, including factor XII (FXII), factor XI (FXI), prekallikrein (PK), and high molecular weight kininogen (HK). The role of the plasma contact system is to (i) promote thrombin generation and fibrin clotting via activated FXI (FXIa) and (ii) induce inflammation upon cleavage of HK and release of bradykinin (Figure 1). When FXII is activated (FXIIa) by its binding to a negatively charged surface, both of these pathways can be triggered. 47 Though it is primarily produced in the liver and found in the blood, 48 FXII has been found in the brain and even localized with Aβ plaques in the postmortem brain tissue of people with AD. 49 , 50
FIGURE 1.
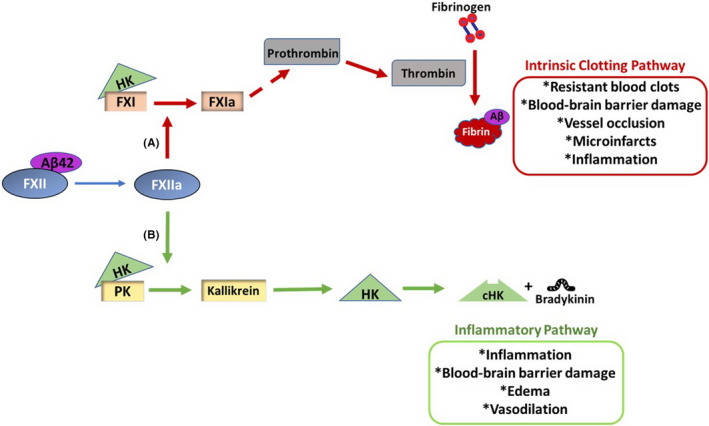
Activation of the contact system induces thrombotic and inflammatory pathways in Alzheimer's disease (AD). Activation of coagulation factor XII (FXII) by Aβ42 can trigger the intrinsic clotting pathway as well as an inflammatory pathway. A, Intrinsic clotting occurs when activated FXII (FXIIa) activates factor XI (FXI) to FXIa. Eventually, prothrombin is cleaved to thrombin, which cleaves fibrinogen into fibrin. Aβ42 can interact with fibrinogen, and fibrin clots formed in the presence of Aβ42 are resistant to degradation. These resistant blood clots can increase the incidence of vessel occlusion, leading to microinfarcts, blood‐brain barrier (BBB) damage, and inflammation. Extravasated fibrin(ogen) can also induce cerebral inflammation. B, FXIIa cleaves prekallikrein (PK) to generate kallikrein. The proinflammatory peptide, bradykinin, is released upon high molecular weight kininogen (HK) cleavage by kallikrein. Bradykinin can induce vasodilation, edema, inflammation, and BBB damage. HK is essential for normal operation of both thrombotic and inflammatory pathways of the contact system, as PK and FXI need to bind HK to be activated by FXIIa
Aβ42 can bind to and activate FXII and thus trigger the contact system. 43 , 44 , 45 , 46 Addition of Aβ42 to normal human plasma accelerates the generation of FXIIa. 45 To trigger intrinsic clotting, FXIIa cleaves FXI. This activated coagulation factor, FXIa, sets off a series of activation events that eventually result in fibrin clot formation. 51 The level of FXI is significantly lower in the plasma of people with AD compared to age‐matched nondemented individuals, 43 suggesting increased FXIa levels and increased activation of the intrinsic clotting system in AD. Furthermore, using both human plasma and purified protein systems, it has been shown that Aβ can promote thrombin generation required for fibrin clot formation. 43 Additionally, the procoagulant effect of Aβ42 is specifically via FXII activation since Aβ42 has no effect on thrombin generation in FXII‐deficient human plasma. 43 Moreover, antibody‐mediated blocking of FXII activation abolishes Aβ42‐induced thrombin generation in plasma. 43 Increased FXIIa levels are also reported in the plasma of people with AD. 13 Together, these findings support the hypothesis that Aβ42‐induced FXIIa generation promotes fibrin production. These fibrin deposits, which aggregate with Aβ and are resistant to degradation (discussed in detail below), may not only increase the number of occlusive thrombi in vessels but could also drive inflammation associated with AD pathology. 52 , 53 , 54 , 55 , 56 Therefore, blocking Aβ42‐induced FXII activation could be a therapeutic strategy to reduce vascular pathologies in AD.
AD patient plasma also often shows prolonged activated partial thromboplastin time (aPTT), a test that measures intrinsic clotting, compared to age‐matched healthy control plasma. 57 Prolonged aPTT correlates with cognitive impairment in people with AD, 57 suggesting that a coagulation defect could affect memory. Extrinsic clotting is not significantly altered in these patients with AD, 57 indicating that the issue lies within the intrinsic clotting pathway. Moreover, analysis of plasma from an AD mouse model (5XFAD) that overexpresses human APP also shows significantly prolonged intrinsic clotting time compared to that of their wild‐type littermates. 57 It should be noted that although Aβ42 is a procoagulant, 58 its precursor APP possesses an anticoagulant property. 59 , 60 Furthermore, AD is a heterogeneous disease, and it is possible that depletion of coagulation factors over time due to a continuously activated contact system or the presence of some other protease inhibitors could also affect intrinsic clotting in AD.
3. Aβ TRIGGERS INFLAMMATION THROUGH FXII‐DRIVEN PK ACTIVATION AND BRADYKININ GENERATION
FXII‐driven contact system activation not only produces fibrin clots but also generates proinflammatory bradykinin. 61 FXIIa cleaves PK to generate kallikrein, which in turn cleaves HK. 51 , 61 The proinflammatory molecule, bradykinin, is liberated upon HK cleavage (Figure 1). In addition to having increased FXIIa, 13 AD patient plasma also shows increased kallikrein‐kinin activity 13 , 43 that leads to inflammation. It has been suggested that kallikrein could drive hemorrhagic conditions, 62 , 63 and therefore AD patients with increased kallikrein activity could be at greater risk of cerebral hemorrhage. Furthermore, levels of full‐length/intact HK are decreased, while levels of cleaved HK (cHK) 13 , 64 and bradykinin are increased in the plasma of people with AD compared to that of controls. 65 This increased bradykinin not only induces inflammation but also can lead to edema, vasodilation, and increased BBB permeability. 61 , 66 , 67 , 68 Consistently, an impaired BBB is often observed in people with AD. 17 Furthermore, the addition of Aβ42 to human plasma from nondemented individuals shows significantly higher levels of bradykinin than untreated plasma. 12 , 65 It has also been shown that AD mice that overexpress Aβ have increased plasma cHK levels. 69 Additionally, it has been reported that intravenous injection of Aβ42 increases HK cleavage and kallikrein activity in wild‐type mouse plasma. 13 The connection between a dysregulated contact system and AD is further supported by findings that people with AD have increased cHK in their cerebrospinal fluid (CSF). 70 These findings suggest that both the thrombotic (via FXI) and inflammatory (via HK cleavage and bradykinin) arms of the contact system are activated in AD.
Vascular abnormalities alone can lead to memory impairment. 71 However, the coexistence of AD and cerebrovascular pathology could exert synergistic effects on brain and memory function. If the peripheral contact activation system contributes to AD progression and pathology, there should be a correlation between these changes and memory impairment and/or AD pathologies.
The level of CSF Aβ42 is a well‐established marker of AD as the amount of CSF Aβ42 decreases with AD progression. 72 , 73 , 74 The concentration of plasma HK is positively correlated with the level of CSF Aβ42; as HK levels decrease in plasma, levels of Aβ42 decrease in the CSF. 13 Additionally, the level of plasma cHK is significantly correlated with memory test scores in AD patients. 64 For example, as cHK levels increase, indicating contact system activation, cognitive status decreases as defined by the Mini‐Mental State Examination (MMSE) or clinical dementia rating. 64 Moreover, increased plasma bradykinin level is associated with memory impairment in people with AD. 65 Plasma cHK levels also positively correlate with Consortium to Establish a Registry for Alzheimer’s Disease scores, which define the extent of Aβ plaque pathology at postmortem analysis. 64 , 75 As these associations were obtained in a cross‐sectional analysis, a longitudinal analysis of contact activation in AD and its association with memory decline needs to be performed.
These findings suggest that a dysregulated plasma contact system could be a reliable predictor of cerebral pathology and memory impairment in AD. However, contact activation is not specific to AD, and therefore it should be used along with established fluid biomarkers (CSF Aβ42, phosphorylated tau), imaging modalities (positron emission tomography, magnetic resonance imaging), and/or memory tests 76 for diagnosing AD patients with vascular dysfunction.
4. DYSREGULATED CONTACT SYSTEM: AN EARLY PREDICTOR OF COGNITIVE DECLINE
It has been suggested that subtle losses in cognitive function may be an early sign of AD development. 76 , 77 Mild cognitive impairment (MCI) refers to the intermediary stage between the cognitive decline associated with normal aging and mild dementia. 72 , 76 People with MCI show only slight cognitive defects, which do not significantly affect their daily functioning. 72 , 76 , 77 However, people with MCI are at a higher risk of developing AD compared to cognitively normal individuals. Specifically, each year 10% to 15% of people with MCI will develop AD, while only 1% to 2% of cognitively normal people will develop AD. 72 , 76 It was recently reported that the contact system is also dysregulated in people with MCI. For example, kallikrein activity and levels of cHK and bradykinin are significantly higher in the plasma of people with MCI than age‐matched cognitively normal individuals. 77 Moreover, these changes are more pronounced in MCI patients enorr with impaired short‐term recall memory. 77 A significant inverse correlation is found between short‐term recall scores and kallikrein activity or level of cHK in plasma. 77 Because short‐term recall memory is one of the earliest cognitive changes observed in people with AD, its correlation with increased contact system activation suggests that evidence of contact activation could predict cognitive changes and AD progression.
5. FIBRINOGEN: A SIGNIFICANT CONTRIBUTOR TO AD PATHOGENESIS
In the circulatory system, conversion of the blood protein fibrinogen into a fibrin clot by thrombin is essential to stop or prevent bleeding. 78 However, a damaged BBB allows for extravasation of fibrin(ogen) and other blood proteins into the brain parenchyma, where it can induce inflammation and neuronal damage. 15 , 33 , 54 Brains of AD mouse models and people with AD often exhibit impaired BBB integrity, 17 which can be exacerbated by coexistence of vascular disease or cerebrovascular dysfunction. 17 Extravasation of fibrinogen into the brain parenchyma also leads to the formation of fibrin deposits. 79 The extent of fibrin deposition positively correlates with neurodegeneration, suggesting that the accumulation of fibrin(ogen) directly affects cognitive function. 79
In cerebral blood vessels, fibrin(ogen) can interact directly with Aβ, which leads to degradation‐resistant blood clots, vessel occlusion, and subsequently ischemic conditions and neuronal death. 52 , 53 , 54 , 55 , 56 The inflammatory response caused by vessel occlusion can also increase the production of Aβ and lead to greater Aβ plaque deposition, which has been shown in animal models. 80 , 81 , 82 , 83 Accordingly, it was recently shown that long‐term treatment with dabigatran, a direct thrombin inhibitor and anticoagulant, 84 prevents occlusive thrombi formation in an AD mouse model. 54 This reduction of occlusive thrombi/fibrin deposits also prevents neuroinflammation, BBB damage, and cognitive impairment in an AD mouse model. 54 Similar correlations are observed when fibrinogen itself is pharmacologically or genetically reduced in AD mouse models, as these strategies result in less neuronal death, synaptic dysfunction, and amyloid pathology as well as improved cognition compared to AD control mice. 54 , 79 Anticoagulant treatment has been found to slow cognitive dysfunction in people with dementia. 85 , 86 Oral anticoagulant treatment was also associated with a lower risk of cognitive decline in people with atrial fibrillation. 87 Despite these findings, anticoagulant therapy is controversial since elderly patients are at higher risk of bleeding. 88
Fibrinogen can directly drive disease pathology via activating microglial inflammatory responses and increasing the generation of reactive oxygen species. 15 Fibrinogen‐mediated microglial activation via CD11b receptor binding leads to cognitive deficits in an AD mouse model through elimination of dendritic spines, tiny extensions on neurons that harbor synaptic receptors of excitatory connections in the brain and are critical for memory and cognitive function. 89 Genetic disruption of the fibrinogen domain that binds to and activates the CD11b receptor on microglia results in reduced neurodegeneration and memory impairment in AD mice. 15 Cerebral injection of fibrinogen itself can induce neuronal injury such as spine elimination and dendritic loss, 15 and in the presence of Aβ, the deleterious effect of fibrinogen is much more pronounced. 15 Aβ42 can directly interact with fibrinogen and reduce its clearance. 52 , 53 , 56 Microscopic analysis of fibrin clots formed in the presence of Aβ42 demonstrates significant structural abnormalities. 52 Radiographic crystallography analysis revealed that the central region of Aβ binds to the outer D domain of fibrinogen, which alters its structure and blocks its cleavage by plasmin. 56 This Aβ42‐induced structural change could also be responsible for increased levels of a plasmin‐resistant fibrin degradation fragment. 56 These results suggest that both vascular and extravasated fibrin(ogen) could synergize the Aβ‐induced pathology in AD. In line with these results, a prospective population‐based study that measured plasma fibrinogen levels in 2835 individuals longitudinally found that high fibrinogen levels were associated with increased risk of AD and dementia. 90 Furthermore, a meta‐analysis involving 3649 patients with dementia had significantly higher plasma fibrinogen levels than nondemented individuals. 91
It is important to note that some Aβ mutations that are associated with EOAD, particularly the Dutch (E22Q) and Iowa (D23 N) mutations, increase Aβ’s toxicity as well as its vascular deposition. 55 , 92 , 93 , 94 , 95 , 96 , 97 More specifically, the Dutch and Iowa Aβ mutations have a 50‐fold higher binding affinity for fibrinogen. 55 It has been suggested that this stronger affinity leads to greater structural clot abnormalities and further delayed fibrinolysis compared to wild‐type Aβ. 55 Furthermore, AD patients who harbor the Dutch or Iowa mutations have significantly more fibrin deposits and Aβ‐fibrin(ogen) codeposits in postmortem brain tissue compared to patients without these mutations. 55 Taken together, these findings reveal that the Aβ‐fibrin(ogen) interaction is instrumental in driving cerebrovascular pathologies and cognitive decline in AD. Thus, inhibiting the interaction between fibrinogen and Aβ could be a promising therapeutic strategy in AD.
6. NOVEL THERAPEUTIC STRATEGIES FOR AD
6.1. Inhibiting the plasma contact system
If a dysregulated contact system is contributing to AD pathology, blocking or inhibiting this system should show beneficial effects in AD. It has been shown that peripheral reduction of FXII (by FXII antisense oligonucleotide, FXII‐ASO) in an AD mouse model not only prevents contact activation in plasma, but also significantly reduces microglial and astrocytic activation in the brain parenchyma. 69 Furthermore, AD mice treated with FXII‐ASO show reduced neuronal loss and less extravasated fibrin(ogen) compared to control AD mice. More importantly, FXII‐ASO treated AD mice perform significantly better in cognitive tests compared to vehicle‐treated AD mice. 69 Collectively, these results suggest that dysregulation of the contact system contributes to AD pathology and memory impairment and that inhibiting this system could prove beneficial in AD. Further studies are needed to establish whether AD pathology initially triggers the contact system or an activated contact system exacerbates AD progression.
Contact system activation could also be a significant contributor to the inflammation observed in AD, both via fibrin(ogen) and bradykinin. Directly blocking HK cleavage to prevent bradykinin generation is another promising strategy to reduce the contact system‐driven inflammatory response in AD. We have recently shown that a monoclonal anti‐HK antibody, 3E8, can block Aβ42‐induced HK cleavage and bradykinin generation in human plasma. 98 Also, an oral PK‐targeting therapy, another promising approach for blocking contact system activation, is undergoing clinical trial testing. 99 , 100 It should be noted that people who lack contact system components (FXII‐, HK‐, or PK‐deficient individuals) are not prone to bleeding, 101 , 102 , 103 , 104 , 105 , 106 , 107 , 108 since intrinsic clotting is not the main coagulation pathway in the body. 61 , 109 , 110 Therefore, blocking the contact system in patients would not increase the risk of hemorrhage. The 3E8 anti‐HK antibody or other contact system inhibitors could be effective at ameliorating not only vascular and inflammatory pathologies in AD but also other diseases and vascular pathologies in which the contact system is also dysregulated, such as hereditary angioedema, 111 sickle cell anemia, 112 lupus, 113 rheumatoid arthritis, 114 multiple sclerosis–associated neuroinflammation, 66 , 115 infection (sepsis/endotoxemia), 116 , 117 and colitis. 118
6.2. Blocking the Aβ‐fibrinogen interaction
Treatment of AD mice with RU‐505, a small molecule that inhibits the Aβ42‐fibrinogen interaction, leads to reduced cerebral inflammation, less vascular Aβ deposition, and improved cognitive function compared to untreated AD mice. 14 The inhibition of the Aβ‐fibrinogen interaction also prevents the Aβ‐induced structural abnormalities of fibrin clots and the altered thrombosis and fibrinolysis observed in AD mice. 14 These findings suggest that blocking the Aβ‐fibrinogen interaction not only reduces thrombotic abnormalities but also lessens neuroinflammation, cognitive impairment, and other AD pathologies. Moreover, some of the Aβ aggregation inhibitors simultaneously inhibit the Aβ‐fibrinogen interaction, and their inhibitory activity could be further increased by making chemical modifications. 119 Collectively, these data suggest that the interaction between vascular fibrinogen and pathogenic Aβ could be extremely detrimental and that minimizing this crosstalk could be beneficial in AD.
7. CONCLUSION AND FUTURE DIRECTIONS
AD is a multifactorial condition in which various pathways may synergize to produce the deleterious effects on brain function. Therefore, targeting various pathways in a combination therapy could maximize the beneficial outcome in AD treatment. Treating vascular dysregulation in AD could also improve brain function and memory along with minimizing vascular defects. We have provided evidence to support a mechanistic link between vascular components and AD pathologies. The crosstalk between pathogenic Aβ and circulatory fibrinogen as well as between Aβ and components of contact or intrinsic pathway (FXII, HK, PK) could dramatically affect the numerous pathologies found in people with AD. Targeting the contact system and amyloid pathology together or targeting contact system activation alone could improve cognitive function in people with AD with vascular deficits. Moreover, a monoclonal anti‐HK antibody could be a promising drug candidate to treat thrombotic and inflammatory conditions in AD. Markers of contact activation—cHK, kallikrein activity, FXIIa, delayed intrinsic clotting (aPTT)—could be used to quickly diagnose people with MCI and AD with a vascular component to their pathology, and preventive or therapeutic care could be provided before severe AD onset.
RELATIONSHIP DISCLOSURE
The 3E8 anti‐HK antibody has been licensed to Millipore Sigma. The authors have no other conflicts of interest or disclosures.
AUTHOR CONTRIBUTIONS
PKS reviewed the literature and wrote the first draft of the review; AB, ZLC, SS, and EHN edited the review and designed the figure and table; EHN supervised the process.
Acknowledgements
The authors thank members of the Laboratory of Neurobiology & Genetics as well as their funders for their support. The laboratory is funded by National Institutes of Health (NIH) Grants NS102721, NS106668, and AG069987; National Center for Advancing Translational Sciences and NIH Clinical and Translational Science Award UL1 TR001866; Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation (ADDF) Diagnostics Accelerator; May and Samuel Rudin Family Foundation; Samuel Newhouse Foundation; Robertson Therapeutic Development Fund; and Mr. John A. Herrmann, Jr.
Handling Editor: Dr Mary Cushman.
Funding information
Our laboratory is supported by National Institutes of Health (NIH) Grants NS102721, NS106668, and AG069987; National Center for Advancing Translational Sciences and NIH Clinical and Translational Science Award UL1 TR001866; Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation Diagnostics Accelerator; May and Samuel Rudin Family Foundation; Samuel Newhouse Foundation; Robertson Therapeutic Development Fund; and Mr. John A. Herrmann Jr.
Contributor Information
Ana Badimon, @ana_badimon.
Erin H. Norris, Email: enorris@rockefeller.edu, @ErinNor22331025.
REFERENCES
- 1. Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11(2):111‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120(4):545‐555. [DOI] [PubMed] [Google Scholar]
- 3. Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saito T, Suemoto T, Brouwers N et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci. 2011;14(8):1023‐1032. [DOI] [PubMed] [Google Scholar]
- 6. Szaruga M, Munteanu B, Lismont S et al. Alzheimer’s‐causing mutations shift Aβ length by destabilizing γ‐secretase‐Aβn interactions. Cell. 2017;170(3):443‐56.e14. [DOI] [PubMed] [Google Scholar]
- 7. Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug‐development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2020;1‐6. 10.1002/alz.12213 [DOI] [PubMed] [Google Scholar]
- 9. Honig LS, Vellas B, Woodward M et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med. 2018;378(4):321‐330. [DOI] [PubMed] [Google Scholar]
- 10. Gauthier S, Feldman HH, Schneider LS et al. Efficacy and safety of tau‐aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double‐blind, parallel‐arm, phase 3 trial. Lancet. 2016;388(10062):2873‐2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen ZL, Singh P, Wong J, Horn K, Strickland S, Norris EH. An antibody against HK blocks Alzheimer’s disease peptide beta‐amyloid‐induced bradykinin release in human plasma. Proc Natl Acad Sci U S A. 2019;116(46):22921‐22923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zamolodchikov D, Chen ZL, Conti BA, Renne T, Strickland S. Activation of the factor XII‐driven contact system in Alzheimer's disease patient and mouse model plasma. Proc Natl Acad Sci U S A. 2015;112(13):4068‐4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahn HJ, Glickman JF, Poon KL et al. A novel Abeta‐fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer’s disease mice. J Exp Med. 2014;211(6):1049‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Merlini M, Rafalski VA, Rios Coronado PE et al. Fibrinogen induces microglia‐mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron. 2019;101(6):1099‐108 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iturria‐Medina Y, Sotero RC, Toussaint PJ, Mateos‐Perez JM, Evans AC. Early role of vascular dysregulation on late‐onset Alzheimer’s disease based on multifactorial data‐driven analysis. Nat Commun. 2016;7:11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: a matter of blood‐brain barrier dysfunction? J Exp Med. 2017;214(11):3151‐3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120(Pt 23):4081‐4091. [DOI] [PubMed] [Google Scholar]
- 19. Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66(3):300‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157‐172. [DOI] [PubMed] [Google Scholar]
- 21. Grammas P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J Neuroinflammation. 2011;8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gouw AA, Seewann A, Vrenken H et al. Heterogeneity of white matter hyperintensities in Alzheimer’s disease: post‐mortem quantitative MRI and neuropathology. Brain. 2008;131(Pt 12):3286‐3298. [DOI] [PubMed] [Google Scholar]
- 23. Brickman AM, Provenzano FA, Muraskin J et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch Neurol. 2012;69(12):1621‐1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cruz Hernandez JC, Bracko O, Kersbergen CJ et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat Neurosci. 2019;22(3):413‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer’s disease–lessons from pathology. BMC Med. 2014;12:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kalaria RN. Small vessel disease and Alzheimer’s dementia: pathological considerations. Cerebrovasc Dis. 2002;13(suppl 2):48‐52. [DOI] [PubMed] [Google Scholar]
- 27. Mazza M, Marano G, Traversi G, Bria P, Mazza S. Primary cerebral blood flow deficiency and Alzheimer’s disease: shadows and lights. J Alzheimers Dis. 2011;23(3):375‐389. [DOI] [PubMed] [Google Scholar]
- 28. Leeuwis AE, Benedictus MR, Kuijer JPA et al. Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer’s disease. Alzheimers Dement. 2017;13(5):531‐540. [DOI] [PubMed] [Google Scholar]
- 29. van Rooden S, Goos JD, van Opstal AM et al. Increased number of microinfarcts in Alzheimer disease at 7‐T MR imaging. Radiology. 2014;270(1):205‐211. [DOI] [PubMed] [Google Scholar]
- 30. Lee S, Viqar F, Zimmerman ME et al. White matter hyperintensities are a core feature of Alzheimer's disease: evidence from the dominantly inherited Alzheimer network. Ann Neurol. 2016;79(6):929‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Markus HS. Cerebrovascular abnormalities in Alzheimer’s dementia: a more tractable treatment target? Brain. 2017;140(7):1822‐1825. [DOI] [PubMed] [Google Scholar]
- 32. Greenberg SM, Bacskai BJ, Hernandez‐Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease ‐ one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ryu JK, McLarnon JG. A leaky blood‐brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med. 2009;13(9A):2911‐2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sweeney MD, Sagare AP, Zlokovic BV. Blood‐brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iadecola C, Zhang F, Niwa K et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2(2):157‐161. [DOI] [PubMed] [Google Scholar]
- 36. Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. beta‐Amyloid‐mediated vasoactivity and vascular endothelial damage. Nature. 1996;380(6570):168‐171. [DOI] [PubMed] [Google Scholar]
- 37. Christie R, Yamada M, Moskowitz M, Hyman B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol. 2001;158(3):1065‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arbel‐Ornath M, Hudry E, Eikermann‐Haerter K et al. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 2013;126(3):353‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Charidimou A, Boulouis G, Pasi M et al. MRI‐visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology. 2017;88(12):1157‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Veluw SJ, Biessels GJ, Bouvy WH et al. Cerebral amyloid angiopathy severity is linked to dilation of juxtacortical perivascular spaces. J Cereb Blood Flow Metab. 2016;36(3):576‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Soontornniyomkij V, Lynch MD, Mermash S et al. Cerebral microinfarcts associated with severe cerebral beta‐amyloid angiopathy. Brain Pathol. 2010;20(2):459‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Magaki S, Tang Z, Tung S et al. The effects of cerebral amyloid angiopathy on integrity of the blood‐brain barrier. Neurobiol Aging. 2018;70:70‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zamolodchikov D, Renne T, Strickland S. The Alzheimer’s disease peptide beta‐amyloid promotes thrombin generation through activation of coagulation factor XII. J Thromb Haemost. 2016;14(5):995‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shibayama Y, Joseph K, Nakazawa Y, Ghebreihiwet B, Peerschke EI, Kaplan AP. Zinc‐dependent activation of the plasma kinin‐forming cascade by aggregated beta amyloid protein. Clin Immunol. 1999;90(1):89‐99. [DOI] [PubMed] [Google Scholar]
- 45. Maas C, Govers‐Riemslag JW, Bouma B et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118(9):3208‐3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bergamaschini L, Donarini C, Foddi C, Gobbo G, Parnetti L, Agostoni A. The region 1–11 of Alzheimer amyloid‐beta is critical for activation of contact‐kinin system. Neurobiol Aging. 2001;22(1):63‐69. [DOI] [PubMed] [Google Scholar]
- 47. Renne T. The procoagulant and proinflammatory plasma contact system. Semin Immunopathol. 2012;34(1):31‐41. [DOI] [PubMed] [Google Scholar]
- 48. Stavrou E, Schmaier AH. Factor XII: what does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis. Thromb Res. 2010;125(3):210‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zamolodchikov D, Bai Y, Tang Y, McWhirter JR, Macdonald LE, Alessandri‐Haber N. A short isoform of coagulation factor XII mRNA is expressed by neurons in the human brain. Neuroscience. 2019;413:294‐307. [DOI] [PubMed] [Google Scholar]
- 50. Yasuhara O, Walker DG, McGeer PL. Hageman factor and its binding sites are present in senile plaques of Alzheimer’s disease. Brain Res. 1994;654(2):234‐240. [DOI] [PubMed] [Google Scholar]
- 51. Gailani D, Renné T. The intrinsic pathway of coagulation: a target for treating thromboembolic disease? J Thromb Haemost. 2007;5(6):1106‐1112. [DOI] [PubMed] [Google Scholar]
- 52. Cortes‐Canteli M, Paul J, Norris EH et al. Fibrinogen and beta‐amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron. 2010;66(5):695‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zamolodchikov D, Berk‐Rauch HE, Oren DA et al. Biochemical and structural analysis of the interaction between beta‐amyloid and fibrinogen. Blood. 2016;128(8):1144‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cortes‐Canteli M, Kruyer A, Fernandez‐Nueda I et al. Long‐term dabigatran treatment delays Alzheimer’s disease pathogenesis in the TgCRND8 mouse model. J Am Coll Cardiol. 2019;74(15):1910‐1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cajamarca SA, Norris EH, van der Weerd L, Strickland S, Ahn HJ. Cerebral amyloid angiopathy‐linked β‐amyloid mutations promote cerebral fibrin deposits via increased binding affinity for fibrinogen. Proc Natl Acad Sci U S A. 2020;117(25):14482‐14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zamolodchikov D, Strickland S. Abeta delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood. 2012;119(14):3342‐3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Suidan GL, Singh PK, Patel‐Hett S et al. Abnormal clotting of the intrinsic/contact pathway in Alzheimer disease patients is related to cognitive ability. Blood Adv. 2018;2(9):954‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zamolodchikov D, Strickland S. A possible new role for Abeta in vascular and inflammatory dysfunction in Alzheimer’s disease. Thromb Res. 2016;141(suppl 2):S59‐61. [DOI] [PubMed] [Google Scholar]
- 59. Smith RP, Higuchi DA, Broze GJ Jr. Platelet coagulation factor XIa‐inhibitor, a form of Alzheimer amyloid precursor protein. Science. 1990;248(4959):1126‐1128. [DOI] [PubMed] [Google Scholar]
- 60. Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin‐II (amyloid beta‐protein precursor): a platelet alpha‐granule protein. Science. 1990;248(4956):745‐748. [DOI] [PubMed] [Google Scholar]
- 61. Maas C, Renné T. Coagulation factor XII in thrombosis and inflammation. Blood. 2018;131(17):1903‐1909. [DOI] [PubMed] [Google Scholar]
- 62. Simão F, Ustunkaya T, Clermont AC, Feener EP. Plasma kallikrein mediates brain hemorrhage and edema caused by tissue plasminogen activator therapy in mice after stroke. Blood. 2017;129(16):2280‐2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu J, Gao BB, Clermont AC et al. Hyperglycemia‐induced cerebral hematoma expansion is mediated by plasma kallikrein. Nat Med. 2011;17(2):206‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamamoto‐Imoto H, Zamolodchikov D, Chen ZL et al. A novel detection method of cleaved plasma high‐molecular‐weight kininogen reveals its correlation with Alzheimer’s pathology and cognitive impairment. Alzheimers Dement (Amst). 2018;10:480‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Singh PK, Chen ZL, Ghosh D, Strickland S, Norris EH. Increased plasma bradykinin level is associated with cognitive impairment in Alzheimer’s patients. Neurobiol Dis. 2020;139:104833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Göbel K, Asaridou CM, Merker M et al. Plasma kallikrein modulates immune cell trafficking during neuroinflammation via PAR2 and bradykinin release. Proc Natl Acad Sci U S A. 2019;116(1):271‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Abbott NJ. Inflammatory mediators and modulation of blood‐brain barrier permeability. Cell Mol Neurobiol. 2000;20(2):131‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Golias C, Charalabopoulos A, Stagikas D, Charalabopoulos K, Batistatou A. The kinin system–bradykinin: biological effects and clinical implications. Multiple role of the kinin system–bradykinin. Hippokratia. 2007;11(3):124‐128. [PMC free article] [PubMed] [Google Scholar]
- 69. Chen ZL, Revenko AS, Singh P, MacLeod AR, Norris EH, Strickland S. Depletion of coagulation factor XII ameliorates brain pathology and cognitive impairment in Alzheimer disease mice. Blood. 2017;129(18):2547‐2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bergamaschini L, Parnetti L, Pareyson D, Canziani S, Cugno M, Agostoni A. Activation of the contact system in cerebrospinal fluid of patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12(2):102‐108. [DOI] [PubMed] [Google Scholar]
- 71. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. DeCarli C. Mild cognitive impairment: prevalence, prognosis, aetiology, and treatment. Lancet Neurol. 2003;2(1):15‐21. [DOI] [PubMed] [Google Scholar]
- 73. Seppälä TT, Koivisto AM, Hartikainen P, Helisalmi S, Soininen H, Herukka SK. Longitudinal changes of CSF biomarkers in Alzheimer’s disease. J Alzheimers Dis. 2011;25(4):583‐594. [DOI] [PubMed] [Google Scholar]
- 74. Andreasen N, Hesse C, Davidsson P et al. Cerebrospinal fluid beta‐amyloid(1–42) in Alzheimer disease: differences between early‐ and late‐onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56(6):673‐680. [DOI] [PubMed] [Google Scholar]
- 75. Hyman BT, Phelps CH, Beach TG et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nestor PJ, Scheltens P, Hodges JR. Advances in the early detection of Alzheimer’'s disease. Nat Med. 2004;10(S7):S34‐S41. [DOI] [PubMed] [Google Scholar]
- 77. Singh PK, Chen ZL, Strickland S, Norris EH. Increased contact system activation in mild cognitive impairment patients with impaired short‐term memory. J Alzheimers Dis. 2020;77(1):59‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3(8):1894‐1904. [DOI] [PubMed] [Google Scholar]
- 79. Cortes‐Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol Aging. 2015;36(2):608‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hur JY, Frost GR, Wu X et al. The innate immunity protein IFITM3 modulates γ‐secretase in Alzheimer’s disease. Nature. 2020;586(7831):735‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nihashi T, Inao S, Kajita Y et al. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir (Wien). 2001;143(3):287‐295. [DOI] [PubMed] [Google Scholar]
- 82. Garcia‐Alloza M, Gregory J, Kuchibhotla KV et al. Cerebrovascular lesions induce transient β‐amyloid deposition. Brain. 2011;134(Pt 12):3697‐3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. van Groen T, Puurunen K, Mäki HM, Sivenius J, Jolkkonen J. Transformation of diffuse beta‐amyloid precursor protein and beta‐amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke. 2005;36(7):1551‐1556. [DOI] [PubMed] [Google Scholar]
- 84. Kopec AK, Joshi N, Towery KL et al. Thrombin inhibition with dabigatran protects against high‐fat diet‐induced fatty liver disease in mice. J Pharmacol Exp Ther. 2014;351(2):288‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ratner J, Rosenberg G, Kral VA, Engelsmann F. Anticoagulant therapy for senile dementia. J Am Geriatr Soc. 1972;20(11):556‐559. [DOI] [PubMed] [Google Scholar]
- 86. Walsh AC. Anticoagulant therapy for senile dementia. JAMA. 1983;249(24):3305. [PubMed] [Google Scholar]
- 87. Zeng D, Jiang C, Su C, Tan Y, Wu J. Anticoagulation in atrial fibrillation and cognitive decline: a systematic review and meta‐analysis. Medicine (Baltimore). 2019;98(7):e14499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Torn M, Bollen WL, van der Meer FJ, van der Wall EE, Rosendaal FR. Risks of oral anticoagulant therapy with increasing age. Arch Intern Med. 2005;165(13):1527‐1532. [DOI] [PubMed] [Google Scholar]
- 89. Sanders J, Cowansage K, Baumgärtel K, Mayford M. Elimination of dendritic spines with long‐term memory is specific to active circuits. J Neurosci. 2012;32(36):12570‐12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. van Oijen M, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke. 2005;36(12):2637‐2641. [DOI] [PubMed] [Google Scholar]
- 91. Zhou Z, Liang Y, Zhang X et al. Fibrinogen and risk of dementia: a systematic review and meta‐analysis. Neurosci Biobehav Rev. 2020;112:353‐360. [DOI] [PubMed] [Google Scholar]
- 92. Levy E, Carman MD, Fernandez‐Madrid IJ et al. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248(4959):1124‐1126. [DOI] [PubMed] [Google Scholar]
- 93. Kamp JA, Moursel LG, Haan J et al. Amyloid β in hereditary cerebral hemorrhage with amyloidosis‐Dutch type. Rev Neurosci. 2014;25(5):641‐651. [DOI] [PubMed] [Google Scholar]
- 94. Wang Z, Natté R, Berliner JA, van Duinen SG, Vinters HV. Toxicity of Dutch (E22Q) and Flemish (A21G) mutant amyloid beta proteins to human cerebral microvessel and aortic smooth muscle cells. Stroke. 2000;31(2):534‐538. [DOI] [PubMed] [Google Scholar]
- 95. Whalen BM, Selkoe DJ, Hartley DM. Small non‐fibrillar assemblies of amyloid beta‐protein bearing the Arctic mutation induce rapid neuritic degeneration. Neurobiol Dis. 2005;20(2):254‐266. [DOI] [PubMed] [Google Scholar]
- 96. Nilsberth C, Westlind‐Danielsson A, Eckman CB et al. The “Arctic” APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4(9):887‐893. [DOI] [PubMed] [Google Scholar]
- 97. Basun H, Bogdanovic N, Ingelsson M et al. Clinical and neuropathological features of the arctic APP gene mutation causing early‐onset Alzheimer disease. Arch Neurol. 2008;65(4):499‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen TB, Lai YH, Ke TL et al. Changes in plasma amyloid and tau in a longitudinal study of normal aging, mild cognitive impairment, and Alzheimer’s disease. Dement Geriatr Cogn Disord. 2020;48(3‐4):180‐195. [DOI] [PubMed] [Google Scholar]
- 99. Hofman ZLM, Clark CHZ, de Hack CE, Maat S, Maas C. Detecting oral kallikrein‐targeting therapy through triggered contact activation: a phase I study. J Allergy Clin Immunol. 2020;146(6):1446‐1449. [DOI] [PubMed] [Google Scholar]
- 100. Hofman ZLM, Clark CC, Hack CE, de Maat S, Maas C. Detecting oral kallikrein‐targeting therapy through triggered contact activation: a phase I study. J Allergy Clin Immunol. 2020;146(6):1446‐9.e5. [DOI] [PubMed] [Google Scholar]
- 101. Ratnoff OD, Colopy JE. A familial hemorrhagic trait associated with a deficiency of a clot‐promoting fraction of plasma. J Clin Invest. 1955;34(4):602‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lämmle B, Wuillemin WA, Huber I et al. Thromboembolism and bleeding tendency in congenital factor XII deficiency–a study on 74 subjects from 14 Swiss families. Thromb Haemost. 1991;65(2):117‐121. [PubMed] [Google Scholar]
- 103. Wuepper KD. Prekallikrein deficiency in man. J Exp Med. 1973;138(6):1345‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Saito H, Goodnough LT, Soria J, Soria C, Aznar J, España F. Heterogeneity of human prekallikrein deficiency (Fletcher trait): evidence that five of 18 cases are positive for cross‐reacting material. N Engl J Med. 1981;305(16):910‐914. [DOI] [PubMed] [Google Scholar]
- 105. Hathaway WE, Belhasen LP, Hathaway HS. Evidence for a new plasma thromboplastin factor. I. Case report, coagulation studies and physicochemical properties. Blood. 1965;26(5):521‐532. [PubMed] [Google Scholar]
- 106. Lacombe MJ, Varet B, Levy JP. A hitherto undescribed plasma factor acting at the contact phase of blood coagulation (Flaujeac factor): case report and coagulation studies. Blood. 1975;46(5):761‐768. [PubMed] [Google Scholar]
- 107. Saito H, Ratnoff OD, Waldmann R, Abraham JP. Fitzgerald trait: deficiency of a hitherto unrecognized agent, Fitzgerald factor, participating in surface‐mediated reactions of clotting, fibrinolysis, generation of kinins, and the property of diluted plasma enhancing vascular permeability (PF/Dil). J Clin Invest. 1975;55(5):1082‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yang J, Fan L, Qiao Y, Zhao Y, Zhu T. Severe high‐molecular‐weight kininogen deficiency due to a homozygous c.1456C > T nonsense variant in a large Chinese family. J Thromb Thrombolysis. 2020;50(4):989‐994. [DOI] [PubMed] [Google Scholar]
- 109. Weidmann H, Heikaus L, Long AT, Naudin C, Schlüter H, Renné T. The plasma contact system, a protease cascade at the nexus of inflammation, coagulation and immunity. Biochim Biophys Acta Mol Cell Res. 2017;1864(11):2118‐2127. [DOI] [PubMed] [Google Scholar]
- 110. Scott CF, Colman RW. Fibrinogen blocks the autoactivation and thrombin‐mediated activation of factor XI on dextran sulfate. Proc Natl Acad Sci U S A. 1992;89(23):11189‐11193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. De Maat S, Hofman ZLM, Maas C. Hereditary angioedema: the plasma contact system out of control. J Thromb Haemost. 2018;16(9):1674‐1685. [DOI] [PubMed] [Google Scholar]
- 112. Sparkenbaugh EM, Kasztan M, Henderson MW et al. High molecular weight kininogen contributes to early mortality and kidney dysfunction in a mouse model of sickle cell disease. J Thromb Haemost. 2020;18(9):2329‐2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vanarsa K, Henderson J, Soomro S et al. Upregulation of proinflammatory bradykinin peptides in systemic lupus erythematosus and rheumatoid arthritis. J Immunol. 2020;205(2):369‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hargreaves KM, Troullos ES, Dionne RA, Schmidt EA, Schafer SC, Joris JL. Bradykinin is increased during acute and chronic inflammation: therapeutic implications. Clin Pharmacol Ther. 1988;44(6):613‐621. [DOI] [PubMed] [Google Scholar]
- 115. Göbel K, Eichler S, Wiendl H, Chavakis T, Kleinschnitz C, Meuth SG. The coagulation factors fibrinogen, thrombin, and factor XII in inflammatory disorders‐a systematic review. Front Immunol. 2018;9:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yang A, Xie Z, Wang B, Colman RW, Dai J, Wu Y. An essential role of high‐molecular‐weight kininogen in endotoxemia. J Exp Med. 2017;214(9):2649‐2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Raghunathan V, Zilberman‐Rudenko J, Olson SR, Lupu F, McCarty OJT, Shatzel JJ. The contact pathway and sepsis. Res Pract Thromb Haemost. 2019;3(3):331‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang B, Yang A, Zhao Z et al. The plasma kallikrein‐kininogen pathway is critical in the pathogenesis of colitis in mice. Front Immunol. 2018;9:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Singh PK, Kawasaki M, Berk‐Rauch HE et al. Aminopyrimidine class aggregation inhibitor effectively blocks abeta‐fibrinogen interaction and abeta‐induced contact system activation. Biochemistry. 2018;57(8):1399‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]