
Figure 2.
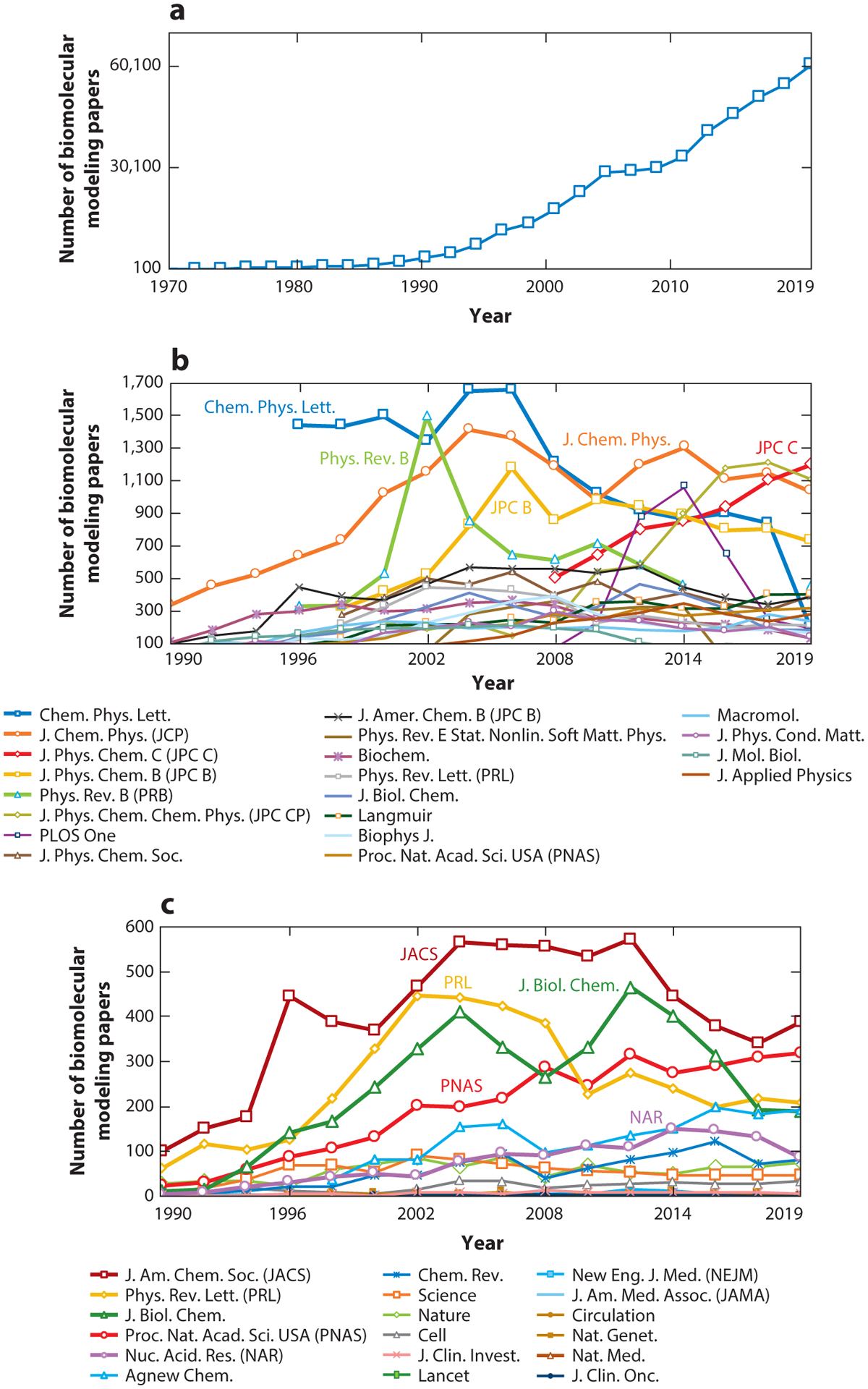
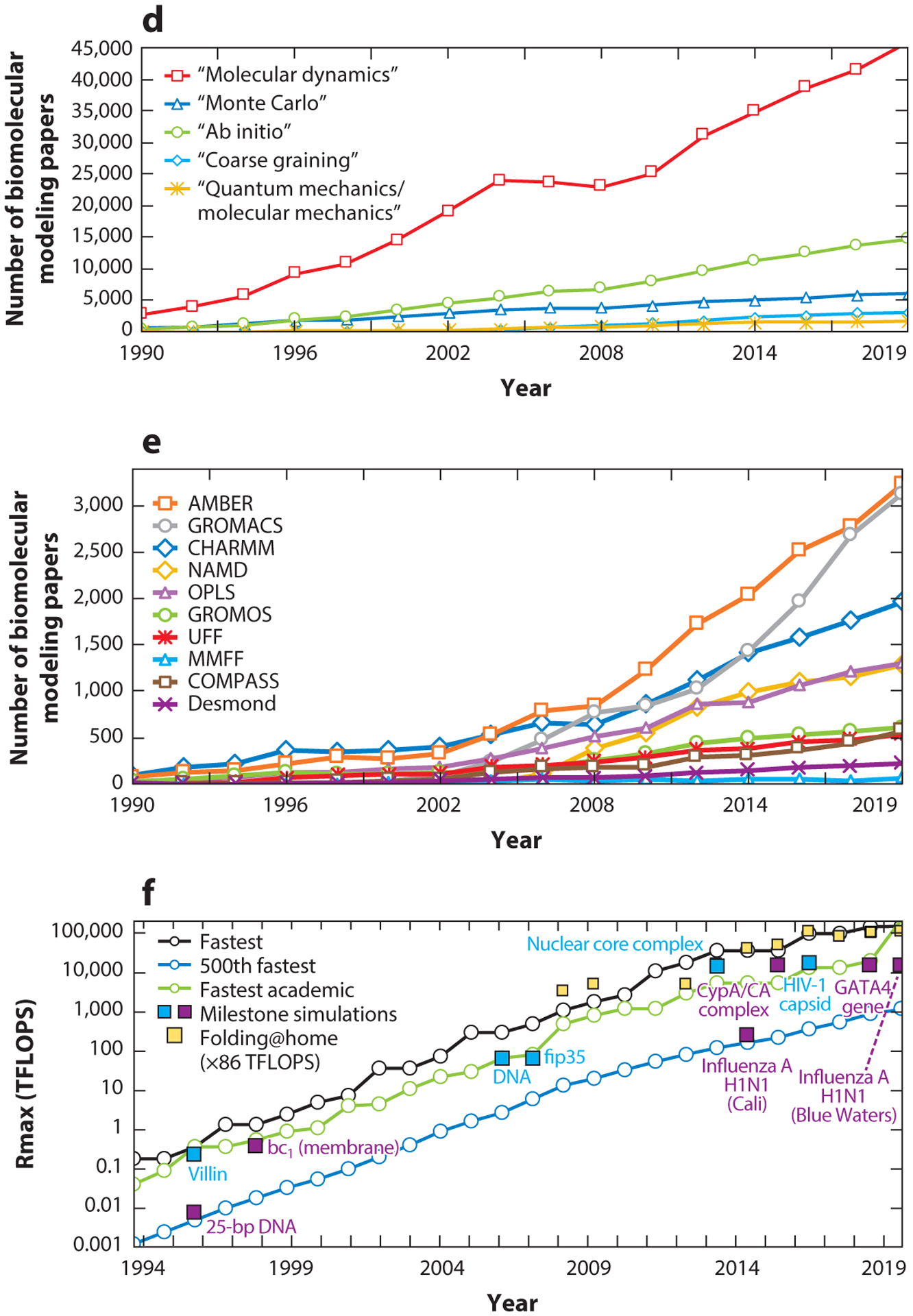
Metrics of the field’s rise in popularity and the evolution of computational performance. (a) Biomolecular modeling papers per year in peer-reviewed journals, as found in Scopus using the query search: “molecular dynamics” OR “biomolecular simulation” OR “molecular modeling” OR “molecular simulation” OR “biomolecular modeling”. (b) Biomolecular modeling papers from panel a in the 20 journals with the most numbers of modeling papers, rank-ordered according to the average number of modeling papers across the years sampled. (c) Biomolecular modeling papers from panel a appearing in high-impact-factor journals, rank-ordered by the SCImago Journal Rank (SJR) h-index. (d) Biomolecular modeling papers from panel a decomposed by method using the query search: ((“molecular modeling” OR “molecular simulation” OR “biomolecular modeling” OR ‘biomolecular simulation”) AND (“method name”)), where method name is “molecular dynamics”, “Monte Carlo”, “ab initio,” “coarse graining”, or “quantum mechanics/molecular mechanics”. (e) Biomolecular modeling papers from panel a decomposed by use of seven popular molecular dynamics packages and force fields using the query search: ((“molecular modeling” OR “molecular simulation” OR “biomolecular modeling” OR ‘biomolecular simulation”) AND (“package/force field”)), where package/force field is Amber (140), CHARMM (24), GROMACS (16), NAMD (145), OPLS (81), GROMOS (57), UFF (157), COMPASS (191), MMFF (60), and Desmond (22). (f) Ranked overall and academic computational systems as reported according to the LINPACK benchmark, as assembled in the Top500 supercomputer lists (www.top500.org). The estimated total speed for the Folding@home distributed computing project is shown in x86 TFLOPS for direct comparison with LINPACK speeds. Biomolecular modeling milestones are dated assuming the computations were performed about a year prior to publication, except for the two 1998 publications, which we associate with computations started in 1996. These include the 25-base-pair DNA system using NCSA SGI machines (204); villin using the Cray T3E900 (43); the bc1 membrane complex using the Cray T3E900 (70); the B-DNA dodecamer using MareNostrum/Barcelona (141); the fip35 protein run on NCSA Abe clusters (49); influenza A H1N1 using the Jade supercomputer (158); the HIV capsid using the Titan Cray XK7 (143); the GATA4 gene using the Trinity Phase 2 (83); and three simulations on Blue Waters, the nuclear core complex (50), the CypA/CA complex (111), and influenza A H1N1 (44). For the simulations in Blue Waters, which has opted out of the Top500 benchmark since 2012, we use estimates of sustained system performance/sustained petascale performance from 2012 and 2020 (14).