

Abstract
Clinical disorders that impair bile flow result in retention of bile acids and cholestatic liver injury, characterized by parenchymal cell death, bile duct proliferation, liver inflammation and fibrosis. However, the pathogenic role of bile acids in the development of cholestatic liver injury remains incompletely understood. In this review, we summarize the current understanding of this process focusing on the experimental and clinical evidence for direct effects of bile acids on each major cellular component of the liver: hepatocytes, cholangiocytes, stellate cells and immune cells. During cholestasis bile acids accumulated in the liver, causing oxidative stress and mitochondrial injury in hepatocytes. The stressed hepatocytes respond by releasing inflammatory cytokines through activation of specific signaling pathways and transcription factors. The recruited neutrophils and other immune cells then cause parenchymal cell death. In addition, bile acids also stimulate the proliferation of cholangiocytes and stellate cells that are responsible for bile duct proliferation and liver fibrosis. This review explores the evidence for bile acid involvement in these phenomena. The role of bile acid receptors, TGR5, FXR and the sphingosine-1-phosphate receptor 2 and the inflammasome are also examined. We hope that better understanding of these pathologic effects will facilitate new strategies for treating cholestatic liver injury.
Keywords: Bile acid, cholestatic liver injury, inflammation, hepatic cells
Introduction
Bile acids are metabolites of cholesterol and are the major constituents of bile. Primary bile acids are made in the liver and are mostly conjugated with glycine or taurine in humans and rodents, whereas secondary bile acids are formed by gut microbes (1). Thus, the bile acid pool consists of multiple species of bile acids which differ in the position and configuration of hydroxyl groups on their sterol rings, and side chain conjugates. Although all these species are considered amphipathic molecules, some are more hydrophobic than others, a physiochemical property that accounts for their functional differences (2). A primary function of bile acids is digestive. After a meal, bile is secreted into the gut where bile acids emulsify dietary lipids and facilitate their intestinal absorption. Enteric bile acids can also repress the growth of microbes and so are a component of innate immunity (3,4). In the ileum, bile acids are reabsorbed by enterocytes and return to the liver via the portal circulation. The intestinal and hepatic uptake of conjugated bile acids are mediated by specific membrane transporters that function to retain the majority of the bile acid within the enterohepatic system, with only ~5% daily loss in the stool. In contrast, unconjugated bile acids can diffuse across cell membranes in their protonated form (5).
In addition, bile acids are also signaling molecules. They regulate gene expression either by directly activating transcription factors via specific nuclear receptors (e.g., FXR/NR1H4, PXR/NR1I2, and VDR/NR1I1) or through signal transduction pathways by binding to membrane receptors, e.g., TGR5, and the sphingosine-1-phosphate receptor (S1PR2) (6-8). The bile acid pool size is highly regulated and the ability to maintain bile acid homeostasis is critical for health (9). In contrast, many diseases have been associated with dysregulation in bile acid homeostasis (10). In this review, we will focus on the effects of bile acids on specific cells in the liver that cause this dysregulation in cholestatic liver injury (Table 1).
Table 1. Proposed mechanisms of bile acid (BA) induced liver injury.
| Cell type | Gene: mechanistic actions | Identification/verification systems | References |
|---|---|---|---|
| Hepatocytes | NTCP (SLC10A1): Na+-driven BA entry into cells, deficiency or inhibition results in hypercholemia without cholestatic injury | Human and rodent cell cultures, animal models, and human subjects | (11-13) |
| BSEP (ABCB11): ATP-dependent BA excretion against concentration gradient, deficiency leads to parenchymal cell death | Animal models and human subjects | (14,15) | |
| FXR (NR1H4): BA nuclear receptor, activation may reduce NFκB mediated hepatic inflammation | Human and rodent cell cultures, animal models | (16-18) | |
| EGR1: a transcription factor stimulates the expression of inflammatory genes, KO in mice reduces cholestatic liver injury | Mouse cell cultures, BDL mouse model | (19,20) | |
| TLR9: cell signaling responds to mitochondrial damage and ER-stress, triggers inflammatory response by stimulating chemokine expression | Mouse cell cultures, BDL mouse model | (13,21) | |
| NFAT: cell signaling responds to Ca2+ dys-regulation, triggers inflammatory response by stimulating the expression of inflammatory genes | Mouse and human cell cultures, cholestatic mouse models, patients with PBC or PSC | (22) | |
| Acute bile duct obstruction causes bile lumen rupture, bile leak and results in cytolytic toxic injury | Animal models and human subjects | (23) | |
| RIPKs and MLKL: mediated via TNFa, activated by DAMPs release from necrotic and necroptotic cell death | Animal models and human subjects | (24,25) | |
| Cholangiocytes | OSTα-OSTβ: efflux of intracellular BA to blood, protect bile duct from BA overload, deficiency in humans developed bile duct injury and fibrosis | Human subjects. | (26,27) |
| TGR5: BA membrane receptor, activation of cAMP signaling, leading to cell proliferation, deficiency aggravate cholestatic injury | Animal models and patients with PBC or PSC | (28,29) | |
| S1PR2: activated by TCA, release of exosomal H19 to mediate inflammation, stimulates bile duct proliferation | Cell cultures, animal models, and patients with PBC or PSC | (30-32) | |
| Stellate cells | TGR5: BA membrane receptor, activation leads to cell proliferation | Rat stellate cell cultures and human LX-2 cells | (33,34) |
| Kupffer cells | TGR5: BA membrane receptor, activation represses LPS stimulated cytokine expression; inflammasomes: exacerbates cholestatic liver injury but bile acids do not directly activate the inflammasome | Mouse cell cultures, BDL mouse model, patient with PBC or PSC | (35,36) |
| NKT cells | FXR: activation represses the production of osteopontin, a proinflammatory mediator | Mouse cell cultures, and mouse hepatitis model | (37) |
Cholestasis is a syndrome caused by multiple disorders that share in common the impairment of bile formation that then results in the accumulation of bile acids in the liver and systemic circulation. Cholestasis can be caused by genetic and developmental defects (such as biliary atresia), as well as acquired diseases including the effects of drugs, viral hepatitis, alcoholic liver disease, primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), metabolic syndrome, and biliary blockage by gallstones or tumors (38-40). However, regardless of the etiology, elevated levels of bile acids in the blood and liver are a common characteristic of all forms of cholestasis. When bile acids accumulate in the liver, they cause oxidative stress and mitochondrial damage that results in the release of proinflammatory cytokines which triggers an inflammatory response. Recruited neutrophils and other immune cells then injure the hepatic parenchymal cells, leading to liver fibrosis, cirrhosis and eventually liver failure. Despite this sequence of pathological events, the mechanistic details as to the role that bile acids play in this injury remain elusive and still somewhat controversial which has limited the development of effective therapies. Here we review recent advances in our understanding of the role that bile acids play in cholestatic liver injury.
Cholestatic levels of bile acids injure hepatocytes
Liver parenchymal cells (hepatocytes) compose 90% of the tissue mass. They are the primary determinant of bile formation and the initial driver of bile flow (5). Hepatocytes synthesize bile acids from cholesterol and take up bile acids from blood and excrete them into the bile duct lumen (41). In humans, the major bile acids in the liver are glycine or taurine conjugated cholic acid and chenodeoxycholic acid. The more toxic unconjugated secondary bile acids, deoxycholic acid and lithocholic acid are formed in the intestine by bacterial dehydroxylases and are mostly excreted in the stool and do not rise to cholestatic levels in the blood. Thus hepatic uptake of bile acids from portal blood, are mainly conjugated and, are carried out by the high affinity sodium-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1, Km=5 µM for taurocholic acid), located on the basolateral sinusoidal membrane (42). Members of organic anion transporting polypeptides (OATPs/SLCOs) can also facilitate the entry of unconjugated bile acids. Because NTCP has a high affinity for conjugated bile acids and also because sodium concentrations in the blood are about 20 times higher than inside of the cell, NTCP can effectively transport bile acids from blood, maintaining low serum bile acid concentrations (≤10 µM) under normal physiologic conditions (43). Once in the hepatocytes, bile acids diffuse rapidly to the canalicular membrane where they are excreted into the bile duct lumen against a large concentration gradient by the bile salt export pump (BSEP/ABCB11). This is an energy-dependent process that requires the hydrolysis of ATP (44). Although estimates of hepatic bile acid concentrations are few, studies by Setchell et al. estimated that liver tissue concentrations of sham-operated rats were 21.3 nmol/g, which represented 2–4% of the bile acid pool; with cholic acid and D22-β-muricholic acids as the major bile acids identified. Concentrations of bile acids in bile duct ligated rats increased 7–8 fold and consisted mainly of chenodeoxycholic and cholic acids (45). It is estimated that the bile acid concentration gradient between bile and blood is ~1,000-fold. It is known that bile acids concentration in the bile duct lumen range from 10–200 mM depending on species. Because of their importance in hepatic bile acid transport, impairment of NTCP and BSEP are critically involved in the development of cholestatic liver injury. When NTCP is genetically absent in humans and mice, bile acids rise dramatically in serum and enter the hepatocyte only through facilitated transporters, including the OATPs and the Organic Solute Transporter alpha and beta, OSTα-OSTβ (SLC51A/SLC51B). In this case, bile acids are not effectively enriched in hepatocytes, even though their concentration in the blood reaches cholestatic levels as high as 1,500 µM yet there is no evidence of liver injury (11,12). These findings illustrate that bile acids must enter and accumulate in the cell for cholestatic hepatic injury to occur. In contrast, when BSEP is genetically deficient, bile acids accumulate in the cell since they can’t be excreted and hepatic injury occurs (14,15). Thus, it is clear that the induction of cholestatic liver injury starts from accumulation of bile acids in hepatocytes. How does this occur?
It was first proposed that bile acids kill hepatocytes through their direct cytolytic/detergent effects in cholestasis (46). Bile acid-induced hepatocyte apoptosis had also been speculated as a mechanism of cholestatic liver injury (47). Despite these “classic” concepts, recent studies favor the hypothesis that cholestatic hepatic injury is due in large part to an inflammatory response (48,49). This is due in part because prior studies often used non-pathophysiologic concentrations of bile acids or bile acids that were not appropriate for the species being studied. The current hypothesis was first proposed by Allen and colleagues (19), and is supported by evidence obtained from subsequent studies both in vitro and in vivo (13,50). First, at pathophysiological relevant concentrations, the major endogenous bile acids stimulated the expression of proinflammatory chemokines in primary hepatocyte cultures, including cells from humans and mice. Blocking the entry of bile acids with competitive inhibitors of NTCP reduces the production of these chemokines, indicating that it is the intracellular bile acids that specifically induce this production and lead to the liver injury (13). Second, mitigating the inflammatory response, either by knocking out genes (e.g., Icam1, Egr1, Ccl2) or blocking the signaling pathways [e.g., administration of antibody against IL-17A, or drug Cenicriviroc (a CCR2/CCR5 antagonist)] attenuates this response, and reduces hepatic injury in vivo in cholestatic rodent models (13,20,50-52).
The direct role of the bile acid nuclear receptor, FXR (NR1H4) in bile acid induction of inflammatory genes remains debatable. Qin et al. reported that activation of FXR in primary human hepatocytes and HepG2 cells using CDCA and GW4064 (a FXR agonist) increased ICAM-1 mRNA expression by 3-fold (53), suggesting that FXR plays a positive role in hepatic inflammation. However, when mouse hepatocytes were treated with GW4064, the expression of chemokines was not changed, including Cxcl2 (13). In contrast, a few studies indicate that activation of FXR mitigates hepatic inflammation by inhibiting NFkB signaling pathway (16-18). This is further supported by an abstract presented at the 2020 EASL meeting that showed that a potent FXR agonist EDP-297 (ENANTA Pharmaceuticals) decreased plasma ALT and the expression of tumor necrosis factor (TNF) α as well as Ccl2, Cxcl1 in a dose dependent manner when administered for 5 days to 2-day bile duct ligated rats (EASL 2020 poster SAT-042). Most importantly, several studies demonstrated that cholestatic liver injury was not significantly changed after BDL or cholic acid fed when comparing wild-type and FXR knockout mice (19,54,55), in which the hepatic neutrophil numbers and the expression of inflammatory genes were not significantly altered either. In contrast, the transcription factor Egr1 is involved because knockout of Egr1 significantly reduced bile acid induction of several inflammatory genes in mouse hepatocyte cultures, including Ccl2, Ccl20, Cxcl1, and Vcam-1 (19). Decreased liver injury was also found in Egr1 knockout mice after BDL (20).
Toll-like receptors may also be involved in the bile acid induced inflammatory response. However, Toll-like receptor (TLR) 4 which responds to lipopolysaccrides with release of IL-1β and other cytokines does not appear to be involved in cholestatic inflammation as C3H/HeJ mice, that contain an inactivating mutation in the TLR4 gene had plasma ALT activity and % areas of liver necrosis that were unaffected when subjected to bile duct ligation. Similarly, accumulation of neutrophils was also unaffected in these TLR4 mutant mice (49). However, TLR9 does seem to be involved (21). As mentioned previously, BA cause endoplasmic reticulum (ER) stress and mitochondrial damage in hepatocytes. These injured mitochondria release DNA that then activates the TLR9 signaling pathway that results in the up-regulation of inflammatory cytokine expression including Cxcl2 and Cxcl10. In Tlr9 deficient mouse hepatocyte cultures, induction of these chemokines were deceased after taurocholic acid (TCA) treatment (13). Also inflammation was reduced in Tlr9 liver-specific knockout mice albeit not eliminated, indicating that signaling pathways independent of Tlr9 must also play a role in the pathogenesis (13). While it is clear that damaged mitochondrial DNA can activate TLR9 (56), how intracellular TLR9 is activated by DNA remains to be elucidated.
Other studies (22) indicate that the transcription factor, Nuclear factor of activated T-cells (NFAT) also plays an important role in the induction of an inflammatory phenotype. NFAT is a family of transcription factors that consists of five members, i.e., NFATc1 through c4, and NFAT5. NFATc members are regulated by Ca2+ signaling (57,58). When activated, they translocate from cytosol to the nucleus to regulate gene expression. Their targets include cytokines. NFATc1 and NFATc3 are expressed in hepatocytes. Blocking their activation using inhibitors or knockdown of Nfatc3 in mouse hepatocytes greatly reduced bile acid induction of chemokine Ccl2, Cxcl1, Cxcl2 and Cxcl10. Since bile acids cause ER-stress and mitochondrial damage and both ER and mitochondria are the key players in Ca2+ signaling, it is very likely that dysregulated Ca2+ signaling activates NFAT in cholestatic hepatocytes and leads to the upregulation and synthesis of chemokines that are released into the systemic circulation. Altogether these studies provide a mechanistic framework for understanding how bile acids induce chemokines in hepatocytes. However, further studies are still needed to understand this phenomenon in greater detail.
Although it remains controversial if bile acids kill hepatocytes under cholestatic pathophysiologic conditions by their direct cytolytic effects, this may happen acutely after bile duct ligation (BDL) in mouse liver as recently described by Ghallab et al. (23). These authors used intravital two-photon microscopy to image live events in the liver by tracing the movement of fluorescent labeled bile acids and other cellular markers. During the first 3 days after BDL hepatic bile acid levels increased, resulting in loss of the mitochondrial membrane potential, followed by the rupture of the apical canalicular membrane in focal areas of the parenchyma. Thereafter, bile regurgitated initially into single cells and then spread to adjacent cells, resulting in more extensive sinusoidal membrane leakage and hepatocyte death and the formation of bile infarcts. While this bile leakage occurred in the early stage of obstructive cholestasis, these events were not seen in the livers 21 days after BDL in mice, nor in another cholestatic model, the Mdr2−/− mice. Because focal areas of hepatocyte necrosis were still detected in these 21-day BDL mouse livers, alternative mechanisms for necrotic hepatocyte death must also exist such as described earlier.
Necroptosis is a form of regulated necrotic cell death (59,60). However, it differs from apoptosis as the latter does not cause an inflammatory response. In contrast, necroptotic cell death can trigger inflammation by releasing Damage Associated Molecular Patterns (DAMPs) into the extracellular space. This process is mediated through TNFα signaling pathways where phosphorylation of receptor-interacting protein kinase (RIPK) 1, RIPK3 and mixed lineage kinase domain-like (MLKL) are the hallmarks. These hallmarks have been found in the livers of patients with PBC and BDL mice (24,25) and knockout of RIPK3 in mice reduced cholestatic liver injury after 3-day BDL. However, the liver injury was not altered in these mice after 14-day BDL, indicating that necroptosis is an early event in these BDL mice. To determine whether bile acids directly induce necroptotic cell death in hepatocytes, they treated primary hepatocytes from both wild-type and Ripk3−/− mice with 50 µM glycochenodeoxycholic acid (GCDCA). Of note, GCDCA is not a major bile acid in mouse so the results may not reflect the human condition. Although increased cell death was observed, no difference was found between the WT and Ripk3−/−, indicating that bile acids do not directly cause hepatic necroptosis. In support of this conclusion, Necrostatin-1 (a necroptosis inhibitor) did not reduce GCDCA-induced cell death in these cultures, and phosphorylation of Ripk3 and MLKL was not detected in these cells. We also did not see phosphorylation of these proteins when mouse hepatocyte cultures when treated with TCA (unpublished observation). Neither Necrostatin-1 nor Necrosulfonamide (another necroptosis inhibitor) blocked TCA induction of chemokines in our mouse hepatocyte cultures (13), consistent with the above findings.
Ferroptosis is a recently coined form of cell death that is defined by iron-dependence and accumulation of lipid peroxidation, resulting in plasma membrane rupture (61,62). This lethal process can be triggered by small molecules or conditions that inhibit glutathione biosynthesis or the glutathione-dependent antioxidant enzyme glutathione peroxidase 4 (GPX4). A second parallel protective pathway also exist, which involves the oxidoreductase FSP1/AIFM2 by generating a potent lipophilic antioxidant to suppresses the propagation of lipid peroxides (63,64). Hepatic ferroptosis has been found in the liver of animal models of alcoholic liver disease (ALD), nonalcoholic steatohepatitis, acetaminophen induced hepatotoxicity, and viral hepatitis (65-68). However, it remains to be determined whether bile acids cause hepatocyte ferroptosis during cholestasis. However, given that bile acids injure hepatocytes by causing ER-stress and mitochondrial damage, it would be reasonable to speculate that ferroptotic hepatocyte death occurs in cholestatic livers.
Bile acids induce cholangiocyte proliferation and inflammation
Cholangiocytes are the cells that form the bile duct epithelium which drains bile from hepatocytes and the gallbladder into the intestine. Cholangiocytes modify the bile by absorbing water and secreting bicarbonate via the anion exchange protein AE2 (5) driven by the chloride gradient via CFTR (5), and reabsorb bile acids via the apical sodium dependent bile acid transporter (ASBT) (69,70). Cholangiopathies are the direct pathologic cause of many cholestatic liver diseases, including biliary atresia, PBC and PSC (71-73). However, the role of bile acids in cholangiocyte injury in cholestasis remains debatable despite the presence of FXR, and bile acid transporters including ASBT and the heteromeric organic solute transporter alpha and beta (OSTα/OSTβ, SLC51A/SLC51B). While hepatocytes release inflammatory chemokines when exposed to cholestatic levels of bile acids, cholangiocytes do not do so under the same conditions (13). This difference is likely due to the relatively high expression of OSTα-OSTβ, at the basolateral membrane of cholangiocytes. OSTα-OSTβ is a facilitated bidirectional transporter system that functions to efflux intracellular bile acids when their concentration becomes higher than in blood. Thus, the intracellular level of bile acids in cholangiocytes may not reach levels that stress the cell as in cholestatic hepatocytes, despite both cells being exposed to millimolar levels of bile acids at their apical membranes. Several patients have been described with Ostα as well as OSTβ deficiencies that develop bile duct injury (26,27), supporting this hypothesis that OSTα-Ostβ protects the cholangiocyte from bile acid induced injury.
The expression of the Takeda G protein-coupled receptor 5 (TGR5, Gpbr-1, M-BAR) in cholangiocytes is another major difference compared to the hepatocyte (74,75). TGR5 is a plasma membrane receptor that is expressed on cholangiocytes and many other cells but not hepatocytes. It is localized in cilia of the apical membrane as well as basolateral membrane of cholangiocytes. TGR5 is characterized as a bile acid receptor because it can be activated by many bile acids, including conjugated and unconjugated forms of lithocholic acid (LCA), deoxycholic acid (DCA), chenodeoxycholic acid (CDCA) and cholic acid, with LCA being the most potent agonist (76,77). TGR5 expression is reduced in cholangiocytes from patients with PBC and PSC (28). Tgr5−/− mice are more susceptible to cholestatic liver injury with the bile duct epithelium showing increased cell damage and inflammation while cholangiocyte proliferation is attenuated (29). Together, these findings indicate that TGR5 also plays a protective role in cholangiocyte pathophysiology.
Recent reports indicate that TCA stimulates the expression of the long noncoding RNA (lncRNA) H19 in mouse cholangiocyte cultures. Release of the exosomal H19 from cholangiocytes promotes macrophage activation and hepatic inflammation that aggravates cholestatic liver injury in mouse, while elevated expression of H19 was also found in the liver of patients with PBC and PSC (30). This effect is mediated through the sphingosine 1-phosphate receptor 2 (S1PR2), also expressed on the apical membrane of cholangiocytes and activated by bile acids (31,32). Of note, TCA activation of S1PR2 also stimulates cholangiocyte proliferation in cholestatic mice (31). Interestingly, only TCA, but not GCA or DCA, stimulates H19 expression in these mouse cholangiocyte cultures. As TCA is the major endogenous bile acid in mouse, this pathway may play a role in the pathology of cholestatic murine models. Since GCDCA and GCA are the major bile acids in humans, it remains to be determined whether this mechanism can be extrapolated to human cholestatic liver injury.
Bile acids trigger stellate cell activation and proliferation
Liver fibrosis is mainly due to the activation and proliferation of hepatic stellate cells that produce excessive deposition of extracellular matrix components in response to the injury of liver parenchyma (78,79). In cholestasis, elevated levels of bile acid cause liver parenchymal injury. The injured cells release cytokines and DAMPs that can activate stellate cells, in addition to other signals from responding immune cells (80). However, the role of bile acids in stellate cell activation and proliferation has not been fully explored. Svegliati-Baroni et al. demonstrated that both conjugated and unconjugated bile acids at cholestatic levels (≥25 µM) stimulated the proliferation of rat hepatic stellate cells in culture, where the expression of collagen I and TGFβ was not altered (33). This proliferation is mediated via activation of epidermal growth factor receptor (EGFR) in a ligand-independent fashion where protein kinase C and extracellular signal-regulated kinase are involved. They also showed that bile acids did not enter these cells. However, the details of this signaling remain to be determined. Similarly, two recent reports indicate that bile acids (CA, CDCA, LCA and TCA) stimulated activation and proliferation of LX-2 cells, a human hepatic stellate cell line, where the expression of α-smooth muscle actin (SMA) and procollagen-1 are also increased (34). However mechanistic details for these regulatory pathways are also lacking. Hepatic stellate cells express TGR5 (28), yet it is not known if TGR5 plays the same role in cholestatic hepatic stellate cell activation and proliferation as it does for cholangiocyte proliferation. If so, it will be interesting to know whether this signaling pathway cross-talks with the EGFR pathway mentioned above. Most importantly, in vivo animal studies are needed to verify the role of these signal pathways in cholestatic liver fibrosis.
Role of immune cells and inflammasomes in cholestatic liver injury- the interaction of bile acids and innate immunity
It is well known that bile acids play a major role in the regulation of the immune system in the intestine and that these interactions predominately involve the bile acid receptors TGR5 and FXR. These interactions are well described in a recent review (3). However, much less is known about bile acid interactions with immune cells within the liver although resident macrophages express both TGR5 and FXR. Immune cells do play a very important role in cholestatic liver injury by responding to cytokines and DAMPs released from liver parenchymal cells (48,49,80,81). Although hepatocytes are protected from injury in cholestatic rodent models by various maneuvers that mitigate this inflammatory response, there is no evidence that bile acids at pathophysiologic concentrations directly activate immune cells. This is likely because immune cells lack bile acid transporters for entry to these cells while unconjugated bile acids that can diffuse across cell membranes are very low in concentration (43). However, Kupffer cells do express TGR5 and activation of this receptor by bile acids may play a yet to be defined role in reducing cholestatic liver injury. Keitel et al. localized TGR5 to the plasma membrane of isolated Kupffer cells and noted that bile acids inhibited cytokine expression but only if pretreated with LPS (35). Further, TGR5-immunoreactivity increased in Kupffer cells in rat liver after BDL (34). Similar findings are described for bile acid effects on human macrophages (36). Further studies from this group recently performed a pathway analysis of TLCA’s effect on human macrophage phenotype that revealed that TLCA suppressed the expression of genes that mediated pro-inflammatory effects, including phagocytosis, interactions with pathogens and autophagy, in addition to the recruitment of immune cells, such as NK cells, neutrophils and T cells (82). FXR is also expressed in liver NKT cells and when activated in-vivo inhibits their ability to produce osteopontin, a potent pro-inflammatory mediator, in an acute liver injury model induced by concanavalin A (37). These studies emphasize that FXR and TGR5 activation by bile acids may be a protective mechanism. However the role of Kupffer cells in cholestatic liver injury remains controversial and in need of further study before it can be concluded whether Kupffer cells are responsible for promotion or protection of liver damage and if depletion of Kupffer cells can be a therapeutic approach or only lead to exacerbation of the liver injury (81).
The inflammasome is a cytosolic protein complex that senses intracellular danger stimuli, e.g., endogenous and exogenous pathogen-associated or danger-associated molecular patterns (PAMPs and DAMPs). When it is activated, a nod-like receptor (e.g., NLRP3) activates caspase-1 by cleaving pro-caspase-1 protein. Activated caspase-1 then is able to cleave pro-IL-1β and pro-IL-18, which releases these cytokines from the cell thereby amplifying the inflammatory response. Proteins that comprise the inflammasome are prominently expressed in macrophages or Kupffer cells but are less abundant in liver parenchymal cells (83). NLRP3 is the most prominent family member and is expressed in rodent and human liver and can be activated by both FXR and TGR5, where it plays a role in the inflammatory response in alcohol and metabolic liver disease (84). Recent studies have also examined the role of the inflammasome and bile acids in cholestatic liver diseases (85). Inflammasome genes NLRP1, NLPR3, caspase-1, and IL-1β (mRNA and or protein) were found to be increased in cholestatic patients with PBC or PSC and cholestatic mouse livers after BDL. Yet bile acids did not activate the inflammasome in mouse hepatocytes or macrophages. Furthermore, deficiency of caspase-1 in mice promoted an increase of the M2 macrophage’s anti-inflammatory phenotype in the liver after BDL, thus presumably partially converting a proinflammatory phenotype to an anti-inflammatory state. Therefore, in the liver, the inflammasome exacerbates cholestatic liver injury but bile acids do not directly activate the inflammasome (85).
Conclusions
Decades of research have provided insights into the role of bile acids in cholestatic liver injury. It is clear that different cells in the liver respond differently to bile acids due either to accumulation in the cell (hepatocytes) or through activation of bile acid membrane receptors on cholangiocytes, stellate cells, Kupffer cells, although many of the details are still not understood. As illustrated in Figure 1, during cholestasis bile acids cause hepatocyte stress and release of inflammatory signals and can even cause direct toxic damage in the acute phase of bile duct obstruction, while they also stimulate the proliferation of cholangiocyte and stellate cells that are responsible for bile duct proliferation and liver fibrosis respectively. Although responding immune cells attack the injured parenchyma, bile acid activation of TGR5 in these cells may also mitigate this response and attenuate liver injury. These issues and whether bile acids could have direct effects on other immune cells remains to be studied.
Figure 1.
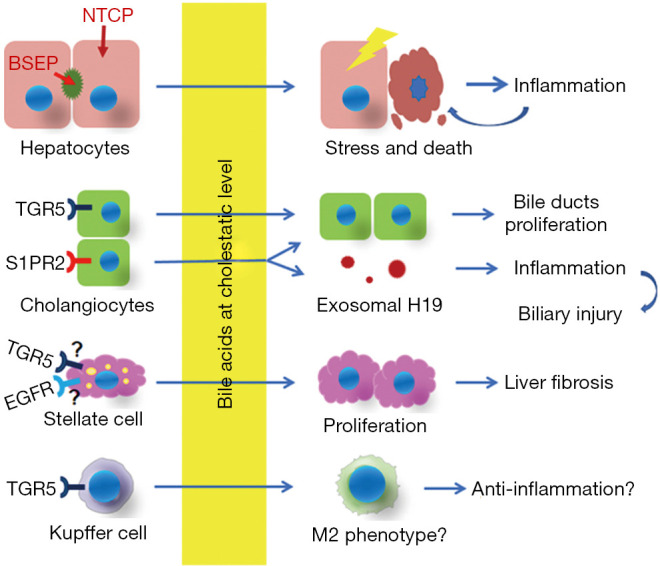
A schematic view of hepatic cells responding to bile acids in cholestatic liver.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This study was supported by National Institutes of Health Grants DK34989 (Yale Liver Center), DK25636 (to J.L.B.).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editors (Ralf Weiskirchen and Wolfgang Stremmel) for the series “Unresolved Basic Issues in Hepatology” published in Annals of Translational Medicine. The article has undergone external peer review.
Peer Review File: Available at http://dx.doi.org/10.21037/atm-20-5110
Conflicts of Interest: Both authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-5110). The series “Unresolved Basic Issues in Hepatology” was commissioned by the editorial office without any funding or sponsorship. The authors have no other conflicts of interest to declare.
References
- 1.Hofmann AF. The enterohepatic circulation of bile acids in mammals: form and functions. Front Biosci (Landmark Ed) 2009;14:2584-98. 10.2741/3399 [DOI] [PubMed] [Google Scholar]
- 2.Hofmann AF. Bile acids: trying to understand their chemistry and biology with the hope of helping patients. Hepatology 2009;49:1403-18. 10.1002/hep.22789 [DOI] [PubMed] [Google Scholar]
- 3.Fiorucci S, Biagioli M, Zampella A, et al. Bile Acids Activated Receptors Regulate Innate Immunity. Front Immunol 2018;9:1853. 10.3389/fimmu.2018.01853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen ML, Takeda K, Sundrud MS. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol 2019;12:851-61. 10.1038/s41385-019-0162-4 [DOI] [PubMed] [Google Scholar]
- 5.Boyer JL. Bile formation and secretion. Compr Physiol 2013;3:1035-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science 1999;284:1362-5. 10.1126/science.284.5418.1362 [DOI] [PubMed] [Google Scholar]
- 7.Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999;284:1365-8. 10.1126/science.284.5418.1365 [DOI] [PubMed] [Google Scholar]
- 8.Liu R, Zhao R, Zhou X, et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology 2014;60:908-18. 10.1002/hep.27085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmann AF, Hagey LR. Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades. J Lipid Res 2014;55:1553-95. 10.1194/jlr.R049437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang JYL, Ferrell JM. Bile Acid Metabolism in Liver Pathobiology. Gene Expr 2018;18:71-87. 10.3727/105221618X15156018385515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaz FM, Paulusma CC, Huidekoper H, et al. Sodium taurocholate cotransporting poly-peptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2015;61:260-7. 10.1002/hep.27240 [DOI] [PubMed] [Google Scholar]
- 12.Slijepcevic D, Kaufman C, Wichers CG, et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+) -taurocholate cotransporting polypeptide knockout mice. Hepatology 2015;62:207-19. 10.1002/hep.27694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai SY, Ouyang X, Chen Y, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2017;2:e90780. 10.1172/jci.insight.90780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998;20:233-8. 10.1038/3034 [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Lam P, Liu L, et al. Severe cholestasis induced by cholic acid feeding in knockout mice of sister of P-glycoprotein. Hepatology 2003;38:1489-99. 10.1016/j.hep.2003.09.037 [DOI] [PubMed] [Google Scholar]
- 16.Gai Z, Visentin M, Gui T, et al. Effects of Farnesoid X Receptor Activation on Arachidonic Acid Metabolism, NF-kB Signaling, and Hepatic Inflammation. Mol Pharmacol 2018;94:802-11. 10.1124/mol.117.111047 [DOI] [PubMed] [Google Scholar]
- 17.Verbeke L, Mannaerts I, Schierwagen R, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep 2016;6:33453. 10.1038/srep33453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang YD, Chen WD, Wang M, et al. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008;48:1632-43. 10.1002/hep.22519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol 2011;178:175-86. 10.1016/j.ajpath.2010.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim ND, Moon JO, Slitt AL, et al. Early growth response factor-1 is critical for cholestatic liver injury. Toxicol Sci 2006;90:586-95. 10.1093/toxsci/kfj111 [DOI] [PubMed] [Google Scholar]
- 21.Gäbele E, Muhlbauer M, Dorn C, et al. Role of TLR9 in hepatic stellate cells and experi-mental liver fibrosis. Biochem Biophys Res Commun 2008;376:271-6. 10.1016/j.bbrc.2008.08.096 [DOI] [PubMed] [Google Scholar]
- 22.Cai SY, Yu D, Soroka CJ, et al. Hepatic NFAT signaling regulates the expression of in-flammatory cytokines in cholestasis. J Hepatol 2021;74:550-9. 10.1016/j.jhep.2020.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghallab A, Hofmann U, Sezgin S, et al. Bile Microinfarcts in Cholestasis Are Initiated by Rupture of the Apical Hepatocyte Membrane and Cause Shunting of Bile to Sinusoidal Blood. Hepatology 2019;69:666-83. 10.1002/hep.30213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Afonso MB, Rodrigues PM, Simao AL, et al. miRNA-21 ablation protects against liver injury and necroptosis in cholestasis. Cell Death Differ 2018;25:857-72. 10.1038/s41418-017-0019-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Afonso MB, Rodrigues PM, Simao AL, et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis 2016;7:e2390. 10.1038/cddis.2016.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sultan M, Rao A, Elpeleg O, et al. Organic solute transporter-beta (SLC51B) deficiency in two brothers with congenital diarrhea and features of cholestasis. Hepatology 2018;68:590-8. 10.1002/hep.29516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao E, Cheema H, Waheed N, et al. Organic Solute Transporter Alpha Deficiency: A Disorder With Cholestasis, Liver Fibrosis, and Congenital Diarrhea. Hepatology 2020;71:1879-82. 10.1002/hep.31087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deutschmann K, Reich M, Klindt C, et al. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim Biophys Acta Mol Basis Dis 2018;1864:1319-25. 10.1016/j.bbadis.2017.08.021 [DOI] [PubMed] [Google Scholar]
- 29.Guo C, Qi H, Yu Y, et al. The G-Protein-Coupled Bile Acid Receptor Gpbar1 (TGR5) In-hibits Gastric Inflammation Through Antagonizing NF-kappaB Signaling Pathway. Front Pharmacol 2015;6:287. 10.3389/fphar.2015.00287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Liu R, Wang Y, et al. Cholangiocyte-Derived Exosomal lncRNA H19 Promotes Macrophage Activation and Hepatic Inflammation under Cholestatic Conditions. Cells 2020;9:190. 10.3390/cells9010190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Aoki H, Yang J, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017;65:2005-18. 10.1002/hep.29076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao Y, Liu R, Li X, et al. Long Noncoding RNA H19 Contributes to Cholangiocyte Pro-liferation and Cholestatic Liver Fibrosis in Biliary Atresia. Hepatology 2019;70:1658-73. 10.1002/hep.30698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svegliati-Baroni G, Ridolfi F, Hannivoort R, et al. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology 2005;128:1042-55. 10.1053/j.gastro.2005.01.007 [DOI] [PubMed] [Google Scholar]
- 34.Liu Z, Zhang Z, Huang M, et al. Taurocholic acid is an active promoting factor, not just a biomarker of progression of liver cirrhosis: evidence from a human metabolomic study and in vitro experiments. BMC Gastroenterol 2018;18:112. 10.1186/s12876-018-0842-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keitel V, Donner M, Winandy S, et al. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun 2008;372:78-84. 10.1016/j.bbrc.2008.04.171 [DOI] [PubMed] [Google Scholar]
- 36.Haselow K, Bode JG, Wammers M, et al. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J Leukoc Biol 2013;94:1253-64. 10.1189/jlb.0812396 [DOI] [PubMed] [Google Scholar]
- 37.Mencarelli A, Renga B, Migliorati M, et al. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J Immunol 2009;183:6657-66. 10.4049/jimmunol.0901347 [DOI] [PubMed] [Google Scholar]
- 38.Hirschfield GM. Genetic determinants of cholestasis. Clin Liver Dis 2013;17:147-59. 10.1016/j.cld.2012.12.002 [DOI] [PubMed] [Google Scholar]
- 39.Keitel V, Droge C, Stepanow S, et al. Intrahepatic cholestasis of pregnancy (ICP): case report and review of the literature. Z Gastroenterol 2016;54:1327-33. 10.1055/s-0042-118388 [DOI] [PubMed] [Google Scholar]
- 40.Pollheimer MJ, Fickert P, Stieger B. Chronic cholestatic liver diseases: clues from histo-pathology for pathogenesis. Mol Aspects Med 2014;37:35-56. 10.1016/j.mam.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 41.Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci 2008;65:2461-83. 10.1007/s00018-008-7568-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004;126:322-42. 10.1053/j.gastro.2003.06.005 [DOI] [PubMed] [Google Scholar]
- 43.Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury. World J Gastroenterol 2012;18:4985-93. 10.3748/wjg.v18.i36.4985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerloff T, Stieger B, Hagenbuch B, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem 1998;273:10046-50. 10.1074/jbc.273.16.10046 [DOI] [PubMed] [Google Scholar]
- 45.Setchell KD, Rodrigues CM, Clerici C, et al. Bile acid concentrations in human and rat liver tissue and in hepatocyte nuclei. Gastroenterology 1997;112:226-35. 10.1016/S0016-5085(97)70239-7 [DOI] [PubMed] [Google Scholar]
- 46.Schölmerich J, Becher MS, Schmidt K, et al. Influence of hydroxylation and conjugation of bile salts on their membrane-damaging properties--studies on isolated hepatocytes and lipid membrane vesicles. Hepatology 1984;4:661-6. 10.1002/hep.1840040416 [DOI] [PubMed] [Google Scholar]
- 47.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev 2010;90:1165-94. 10.1152/physrev.00061.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woolbright BL. Inflammation: Cause or consequence of chronic cholestatic liver injury. Food Chem Toxicol 2020;137:111133. 10.1016/j.fct.2020.111133 [DOI] [PubMed] [Google Scholar]
- 49.Li M, Cai SY, Boyer JL. Mechanisms of bile acid mediated inflammation in the liver. Mol Aspects Med 2017;56:45-53. 10.1016/j.mam.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Brien KM, Allen KM, Rockwell CE, et al. IL-17A synergistically enhances bile ac-id-induced inflammation during obstructive cholestasis. Am J Pathol 2013;183:1498-507. 10.1016/j.ajpath.2013.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allen K, Kim ND, Moon JO, et al. Upregulation of early growth response factor-1 by bile acids requires mitogen-activated protein kinase signaling. Toxicol Appl Pharmacol 2010;243:63-7. 10.1016/j.taap.2009.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu D, Cai SY, Mennone A, et al. Cenicriviroc, a cytokine receptor antagonist, potentiates all-trans retinoic acid in reducing liver injury in cholestatic rodents. Liver Int 2018;38:1128-38. 10.1111/liv.13698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qin P, Borges-Marcucci LA, Evans MJ, et al. Bile acid signaling through FXR induces intracellular adhesion molecule-1 expression in mouse liver and human hepatocytes. Am J Physiol Gastrointest Liver Physiol 2005;289:G267-73. 10.1152/ajpgi.00043.2005 [DOI] [PubMed] [Google Scholar]
- 54.Wagner M, Fickert P, Zollner G, et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology 2003;125:825-38. 10.1016/S0016-5085(03)01068-0 [DOI] [PubMed] [Google Scholar]
- 55.Zollner G, Wagner M, Fickert P, et al. Role of nuclear receptors and hepatocyte-enriched transcription factors for Ntcp repression in biliary obstruction in mouse liver. Am J Physiol Gastrointest Liver Physiol 2005;289:G798-805. 10.1152/ajpgi.00319.2004 [DOI] [PubMed] [Google Scholar]
- 56.Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep 2020;21:e49799. 10.15252/embr.201949799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 2005;5:472-84. 10.1038/nri1632 [DOI] [PubMed] [Google Scholar]
- 58.Oh-hora M, Rao A. The calcium/NFAT pathway: role in development and function of regulatory T cells. Microbes Infect 2009;11:612-9. 10.1016/j.micinf.2009.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gautheron J, Gores GJ, Rodrigues CMP. Lytic cell death in metabolic liver disease. J Hepatol 2020;73:394-408. 10.1016/j.jhep.2020.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738-52. 10.1038/s41575-018-0065-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci 2016;73:2195-209. 10.1007/s00018-016-2194-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol 2016;26:165-76. 10.1016/j.tcb.2015.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019;575:688-692. 10.1038/s41586-019-1705-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019;575:693-8. 10.1038/s41586-019-1707-0 [DOI] [PubMed] [Google Scholar]
- 65.Li X, Wang TX, Huang X, et al. Targeting ferroptosis alleviates methionine-choline defi-cient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int 2020;40:1378-94. 10.1111/liv.14428 [DOI] [PubMed] [Google Scholar]
- 66.Liu CY, Wang M, Yu HM, et al. Ferroptosis is involved in alcohol-induced cell death in vivo and in vitro. Biosci Biotechnol Biochem 2020;84:1621-8. 10.1080/09168451.2020.1763155 [DOI] [PubMed] [Google Scholar]
- 67.Macías-Rodríguez RU, Inzaugarat ME, Ruiz-Margain A, et al. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis-A Novel Form of Non-Apoptotic Cell Death? Int J Mol Sci 2020;21:1651. 10.3390/ijms21051651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsurusaki S, Tsuchiya Y, Koumura T, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis 2019;10:449. 10.1038/s41419-019-1678-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lazaridis KN, Pham L, Tietz P, et al. Rat cholangiocytes absorb bile acids at their apical domain via the ileal sodium-dependent bile acid transporter. J Clin Invest 1997;100:2714-21. 10.1172/JCI119816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dawson PA, Hubbert ML, Rao A. Getting the mOST from OST: Role of organic solute transporter, OSTalpha-OSTbeta, in bile acid and steroid metabolism. Biochim Biophys Acta 2010;1801:994-1004. 10.1016/j.bbalip.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fabris L, Fiorotto R, Spirli C, et al. Pathobiology of inherited biliary diseases: a roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol 2019;16:497-511. 10.1038/s41575-019-0156-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yokoda RT, Carey EJ. Primary Biliary Cholangitis and Primary Sclerosing Cholangitis. Am J Gastroenterol 2019;114:1593-605. 10.14309/ajg.0000000000000268 [DOI] [PubMed] [Google Scholar]
- 73.Sato K, Glaser S, Kennedy L, et al. Preclinical insights into cholangiopathies: disease modeling and emerging therapeutic targets. Expert Opin Ther Targets 2019;23:461-72. 10.1080/14728222.2019.1608950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keitel V, Ullmer C, Haussinger D. The membrane-bound bile acid receptor TGR5 (Gpbar-1) is localized in the primary cilium of cholangiocytes. Biol Chem 2010;391:785-9. 10.1515/bc.2010.077 [DOI] [PubMed] [Google Scholar]
- 75.Keitel V, Haussinger D. TGR5 in the biliary tree. Dig Dis 2011;29:45-7. 10.1159/000324127 [DOI] [PubMed] [Google Scholar]
- 76.Maruyama T, Miyamoto Y, Nakamura T, et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 2002;298:714-9. 10.1016/S0006-291X(02)02550-0 [DOI] [PubMed] [Google Scholar]
- 77.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem 2003;278:9435-40. 10.1074/jbc.M209706200 [DOI] [PubMed] [Google Scholar]
- 78.Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev 2017;121:27-42. 10.1016/j.addr.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397-411. 10.1038/nrgastro.2017.38 [DOI] [PubMed] [Google Scholar]
- 80.Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol 2017;66:1300-12. 10.1016/j.jhep.2017.02.026 [DOI] [PubMed] [Google Scholar]
- 81.Sato K, Hall C, Glaser S, et al. Pathogenesis of Kupffer Cells in Cholestatic Liver Injury. Am J Pathol 2016;186:2238-47. 10.1016/j.ajpath.2016.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wammers M, Schupp AK, Bode JG, et al. Reprogramming of pro-inflammatory human macrophages to an anti-inflammatory phenotype by bile acids. Sci Rep 2018;8:255. 10.1038/s41598-017-18305-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014;157:1013-22. 10.1016/j.cell.2014.04.007 [DOI] [PubMed] [Google Scholar]
- 84.Guo C, Xie S, Chi Z, et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016;45:802-16. 10.1016/j.immuni.2016.09.008 [DOI] [PubMed] [Google Scholar]
- 85.Cai SY, Ge M, Mennone A, et al. Inflammasome Is Activated in the Liver of Cholestatic Patients and Aggravates Hepatic Injury in Bile Duct-Ligated Mouse. Cell Mol Gastroenterol Hepatol 2020;9:679-88. 10.1016/j.jcmgh.2019.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as