

Abstract
The combination of direct sampling ionization and miniature mass spectrometer presents a promising technical pathway of point-of-care analysis in clinical applications. In this work, a miniature mass spectrometry system was used for analysis of tissue samples. Direct tissue sampling coupled with extraction spray ionization was used with a home-built miniature mass spectrometer, Mini 12. Lipid species in tissue samples were well profiled in rat brain, kidney, and liver in a couple of minutes. By incorporating a photochemical (Paternò–Büchi) reaction, fast identification of lipid C=C location was realized. Relative quantitation of the lipid C=C isomer was performed by calculating the intensity ratio C=C diagnostic product ions, by which FA 18:1 (Δ9)/FA 18:1 (Δ11) was found to change significantly in mouse cancerous breast tissue samples. Accumulation of 2-hydroxylglutarate in human glioma samples, not in normal brains, can also be easily identified for rapid diagnosis.
Graphical Abstract
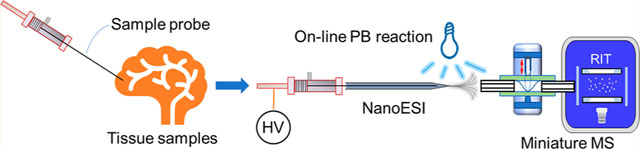
Mass spectrometry (MS) has played an important role in biological analysis.1,2 MS measures the molecular weights of the analytes, in the form of mass-to-charge ratio (m/z), while tandem mass spectrometry (MS/MS) can provide fragmentation information useful for structure identification and confirmation. MS is widely applicable and has great quantitative performance even at low concentrations in complicated matrices. With significant improvement of performance during the past decades, MS has become a powerful tool for clinical analysis.3 Currently, there is a strong push for transferring analytical techniques toward point-of-care (POC) analysis.4,5 New development of ionization and miniature mass spectrometers made it promising to realize POC MS analysis.6
Implementation of ambient ionization methods with miniature mass spectrometers in clinical analysis outside the laboratory presents a huge opportunity for POC testing.6–8 The development of miniature mass spectrometers started from early 1990s.9 A series of miniature mass spectrometers have been developed in the past decade, such as Mini 10 of 10 kg,10 Mini 11 of 4 kg,11 a backpack miniature MS,12,13 as well as Mini 12,14,15 which maintains an adequate performance for MS analysis. Other types of miniature mass spectrometers were also reported recently, such as linear wire ion trap,16 brick size miniature mass spectrometer with sinusoidal frequency scanning,17 planar-electrode linear ion trap mass spectrometer,18 miniature magnetic sector mass spectrometer,19 and miniature laser ablation time-of-flight mass spectrometer.20 To fulfill the demand of analyzing different kinds of molecules including nonvolatile chemical and biological compounds, we also developed an atmospheric interface for miniature MS systems. The discontinuous atmospheric pressure introduction (DAPI)21 enabled a pulsed ion produced by ion sources being introduced into mass analyzer, it can lower the demand for pumping capacity significantly while ensuring adequate number of ions being transferred. Miniature MS has been applied to therapeutic drug monitoring, determination of drug of abuse and pharmacokinetic analysis.14,15,22 Although good capability of structure identification and quantitation of lipids and other compounds were obtained, efficient direct analysis of tissue samples by miniature MS is still hindered by sampling methods.
Direct sampling ionization such as ambient ionization, produces gas-phase ions from samples in their ambient status.23 They are simple, fast and free of sample preparation or only require minimum sample pretreatment.24 Desorption electrospray ionization (DESI)25 is a representative ambient ionization method that is suitable for tissue analysis. Tissue samples can be smeared onto a glass slide, on which the chemical information can be profiled directly by DESI-MS.26 DESI-based MS imaging has also been used for biomarker discovery in breast,27 bladder28 and kidney29 tissues. Other ambient ionization methods such as touch spray30 and probe electrospray ionization (PESI)31 were also developed to simplify the sampling procedures. Extraction methods such as solid phase microextraction (SPME) were also introduced for direct MS analysis. SPME enables solventless microextraction and can selectively extract compounds from tissue or blood into a thin layer of solid adsorbent.32,33 Some direct sampling or ambient ionization methods developed on benchtop mass spectrometers, however, may not be transferrable for miniature MS systems. For example, DESI and DART usually utilize gases at high flow rates to produce ions or facilitate ion transferring, which is hard to be coupled with miniature MS systems directly. Paper spray has been proved to be efficient for analysis of drugs in blood samples on both large mass spectrometers34,35 and Mini 12.23 Although tissue analysis was also demonstrated by paper spray with a commercial triple quadrupole mass spectrometer, tissue analysis by paper spray miniature MS is limited by dilution effect coming from relative large volumes of elution solvents and possible adsorption of some lipids onto hydrophilic paper substrates. Therefore, it is necessary to develop an efficient direct tissue sampling method that is compatible with miniature MS for sensitive and accurate lipid analysis in tissue samples, which is important to understand the physiological status of a biological system and some cellular processes,36,37 as well as biomarker determination.38–43
In this work, we used a simple yet effective direct sampling method for MS-based POC analysis of tissue samples. A thin wire probe was used for fast sampling from tissue samples, following with extraction spray and lipid profiling by Mini 12. By incorporating online photochemical (Paternò–Büchi, PB) reaction during the extraction spray ionization, determination of C=C location of unsaturated lipids was realized. More importantly, the intensity ratio of lipid C=C location isomers was calculated conveniently after direct sampling-based PB-MS/MS analysis, giving an opportunity of targeted quantification of possible C=C location isomer biomarkers in intact or trace tissue samples. The capability of this analytical system was also demonstrated for analysis of 2-hydroxylglutarate (2-HG), associated with the mutation of the isocitrate dehydrogenase (IDH), in human glioma tissues.
EXPERIMENTAL SECTION
Sample Preparation.
All animals involved in this study were handled with the protocol approved by the Purdue Animal Care and Use Committee (PACUC). Male Sprague–Dawley rats ranging from 200 to 400 g were used. Rat brain, kidney, and liver tissue samples were carefully collected for the method development and characterization. All of the tissue samples were freshly frozen and stored at −80 °C.
Fatty acid (FA 18:1 (9Z), FA 18:1 (11Z)) standards and phospholipid standard (PC 16:0/18:1 (9Z)) (Avanti Polar Lipids, Inc., Alabaster, AL) were used to establish PB-MS/MS method for identification of lipid C=C location isomers. Standards were dissolved in 50/50 (v/v) acetone/water and stored at −20 °C. Polar lipid extracts from normal or cancerous mouse breast tissues were dissolved in 50/50 (v/v) acetone/water for MS analysis and stored at −20 °C.
Direct Sampling of Lipids in Animal Tissue Samples.
Animal tissue samples stored under −80 °C were thawed to room temperature for approximately 10 min before analysis. A straight stainless-steel wire (diameter of 0.5 mm, length about 50 mm) was gently inserted into a tissue sample (~2 mm in depth) for sampling. The wire was then inserted into a borosilicate glass capillary (1.5 mm o.d. and 0.86 mm i.d., Sutter Instrument, Novato, CA) with a pull tip for nanoESI. The capillary was filled with 5–10 μL polar solvents (acetonitrile/acetone/water) for fast extraction of sampled lipids under room temperature within 1 min. The capillary was then placed in the front of inlet of Mini 12, a home-built miniature mass spectrometer, a discontinuous atmospheric interface mini 12 for analysis. A DC of 1.5 kV was applied to induce nanoESI. Full MS scan and ion trap collision-induced dissociation (CID) were performed for lipid profiling and fragmentation in both positive and negative mode.
PB Reaction and Lipid C=C Isomer Analysis.
Lipids from tissue samples were extracted with a mixture solvent of acetone/acetonitrile/water in a pulled borosilicate capillary. A low-pressure mercury UV lamp (LP-Hg, BHK Inc., Ontario, CA) of 6 W with an emission at 254 nm was placed parallel to capillary for induction of PB reaction. Ionization and ion introduction by DAPI were performed after reaction for about 2 min. Ion trap CID was performed for determination of lipid C=C location in standard lipids and tissue samples. “Δ” (as for FA 18:1 (Δ9)) is used when C=C configurations (E/Z) fatty acyls are not specified; “_” (as for PC 16:0_18:1 (Δ9)) is used when sn positions of the glycerophospholipid fatty acyl chains are not specified.
Analysis of the Human Glioma Tissue Samples.
The study was conducted in conformity with institutional protocols of Tsinghua University (IRB No. 20180030). The glioma tissue sample (stage IV, IDH1 mutant) and the control (discarded normal brain tissues in some special patients) were obtained from Huashan Hospital, Fudan University. Consent forms were all obtained from the patients. Tissue samples stored under −80 °C were thawed under room temperature for approximately 10 min before analysis. A piece of glioma or control tissue sample (about 5 mm × 5 mm × 5 mm) was placed on a glass slide for sampling by the stainless-steel wire, followed by nanoESI MS analysis on the miniature mass spectrometer. Full scan MS was performed in negative ion mode for comparing the metabolite profiles in the range of m/z 100–200. Tandem MS was also performed to further confirm the presence of the metabolite biomarker.
RESULTS AND DISCUSSION
In this work, a direct sampling method was developed for fast lipid and metabolite analysis. A straight stainless-steel wire, typically used as electrode for nanoESI, was used as the sample probe. It was then combined with lipid extraction and desorption by solvents inside a pulled capillary, as well as nanoESI induced by high voltages. Conductive wires made of other materials (e.g., copper and platinum) could also be applicable. The diameter and surface properties of the wire can affect the sampling efficiency. Wires with larger diameters or rougher surfaces are expected to extract more tissue samples for each sampling. However, for ultimate use for in vivo tissue sampling, thinner wire would certainly cause less damages to the organ. Besides serving as the eluting reagent, the solvent is also facilitating the ionization of targeted compounds. Polar solvents, such as methanol, acetonitrile and acetone, are demonstrated to be suitable for the extraction and ESI of fatty acids and phospholipids. Other solvents such as chloroform, dichloromethane and isooctane can be used for nonpolar lipid analysis, although the ESI efficiencies are relatively low.
As a demonstration shown in Figure 1, the stainless-steel wire was inserted into the rat organ for fast tissue sampling. Lipids in the small amount of collected tissue samples on the wire were extracted by merging the wire into small volume of polar solvents inside a pulled capillary. A DC voltage of 1.5 kV, either in positive or negative mode, was then applied to induce the nanoESI. The extracted lipids from the organ tissue were ionized and injected by DAPI of Mini 12 for MS analysis. The direct sampling-based nanoESI MS method was first evaluated by profiling lipids in tissue samples. Three types of rat organs were collected from same rats and stored under the same condition. Fatty acids and phospholipids could be observed in negative or positive ionization modes.
Figure 1.
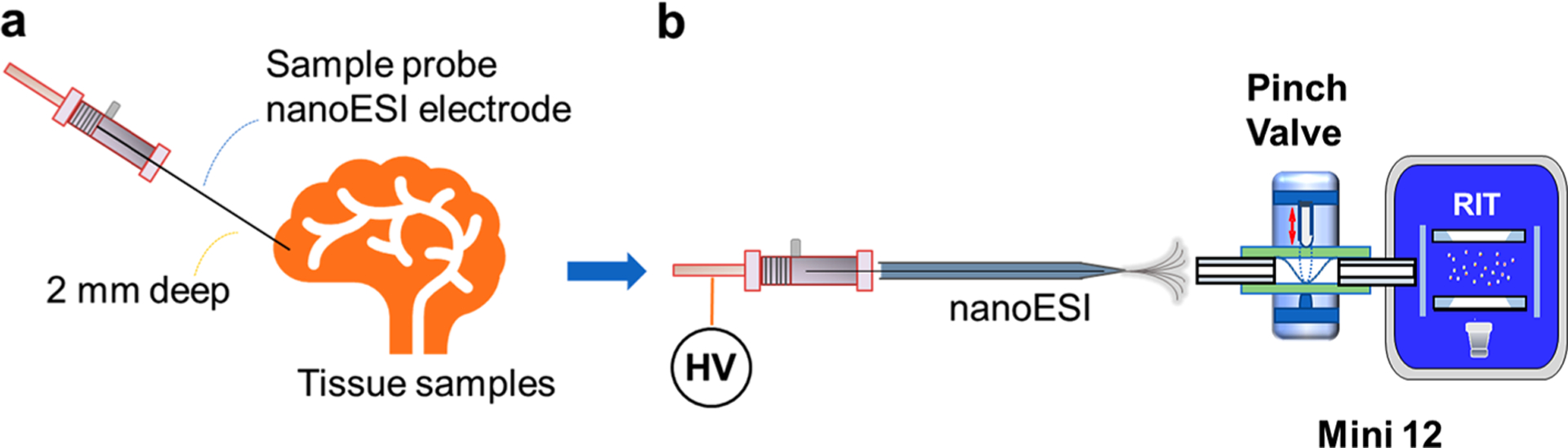
(a) Sampling of organ tissue using a stainless-steel wire, which also served as the noanESI electrode. (b) Extraction of the compounds adsorbed onto the probe and analysis by nanoESI Mini 12 with DAPI.
As shown in Figure 2a–f, fatty acids and phospholipids in tissues of rat brain, kidney, and liver were observed. For analysis of lipids in the lower mass range m/z 150–450 under negative MS mode, the rectilinear ion trap (RIT) of Mini 12 was operated at RF frequency at 1000 kHz, with resonance ejection at 350 kHz (q = 0.80). Some fatty acids were identified in all of the tissue samples, including palmitic acid (FA 16:0 m/z 255), oleic/vis-vaccenic acid (FA 18:1 m/z 281) (FA 18:0 m/z 283), arachidonic acid (FA 20:4 m/z 303), and docosahexaenoic acid (FA 22:6, m/z 327). Rat brain (Figure 2a) and liver (Figure 2c) showed similar FA profiles, with FA 20:1 as the highest peak in the mass spectra. However, FA 16:0 was found to be the highest peak in the FA mass spectrum of rat kidney (Figure 2b). We also observed that the abundance of FA 18:1 was lower than that of FA 18:0 in both rat brain and kidney, while the abundance of FA 18:1 was higher than that of FA 18:0 in the rat liver sample. For analysis of lipids in the higher mass range m/z 650–1000 in negative MS mode, the RIT was operated at a decreased RF frequency of 760 kHz with resonance ejection at 250 kHz to extend the m/z range. Some typical phospholipids, including PE-P 34:1 (m/z 700), PE 36:0 (m/z 746), PS 36:1 (m/z 788), PS 38:4 (m/z 810), PS 22:6 (m/z 834), PI 36:2 (m/z 861), and PI 38:4 (m/z 885) were observed. PI 38:4 at m/z 885 was detected at high abundance in all the rat tissue samples (Figure 2d–f), which is consistent with the results in a previous report.44
Figure 2.
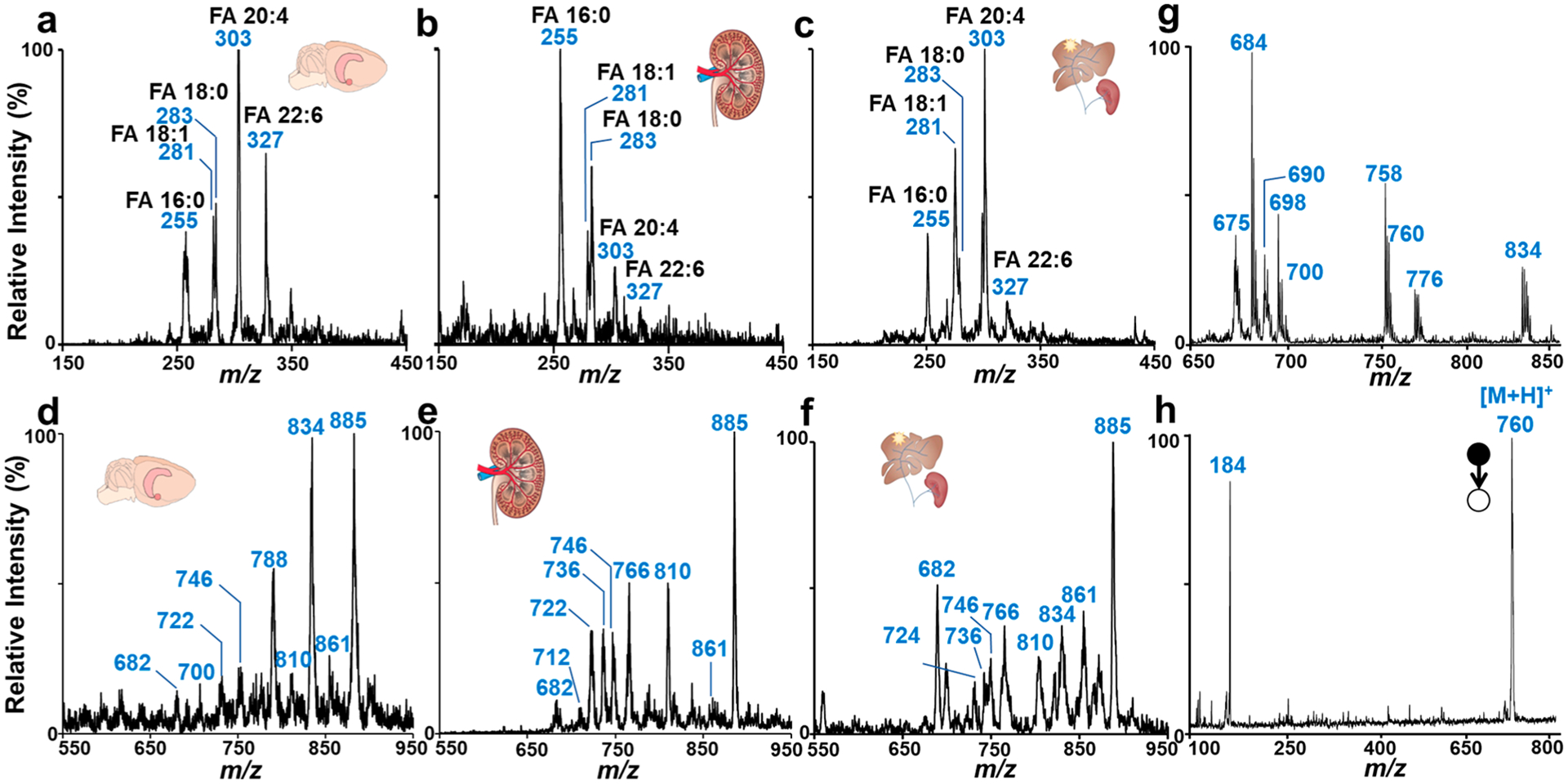
Mass spectra of fatty acids and phospholipids of rat brain (a, d), kidney (b, e), and liver (c, f) in negative mode. Mass spectrum of phospholipids of porcine brain in positive ion mode (g) and MS/MS spectrum of PC 34:1 (m/z 760 in positive ion mode) detected the porcine brain (h).
MS/MS analysis could also be performed by Mini 12 for lipid identity confirmation. As a well-developed protocol in MS-based lipidomics, lipid subclass species can be identified by their characteristic head groups. Figure 2h shows the MS/MS spectrum of PC 34:1 (m/z 760) in the porcine brain sample (full scan mass spectrum is shown in Figure 2g). An isolation window of 5 Da was used, and normalized collision energy Vpp of 2.2 V was applied. The characteristic fragment ion of the PC headgroup was observed at m/z 184 in positive mode.
Isomeric structures with different locations of C=C bonds are commonly found in lipids.45 MS/MS by CID is usually not effective for locating C=C bonds, since higher energy deposition is required for dissociation of C=C bonds. In a previous study, we developed an online photochemical (Paternò–Büchi, PB) reaction to locate the C=C bonds in lipids and showed that the abundance ratios of the lipid C=C location isomers could be used for differentiating the normal and diseased status of tissues.45,46 In this work, we incorporated the PB reaction with the extraction-spray process to allow the identification of the lipid C=C location isomers. A mercury UV lamp of 6 W with an emission at 254 nm was placed parallel to nanoESI emitter (Figure 1. Acetone was added in the extraction/spray solvent as the reagent. Under UV irradiation of 254 nm, [2 + 2] cycloaddition occurred at the C=C bond to form two product isomers of the oxetane ring for each lipid C=C location isomer, both with a + 58 Da mass shift. With CID, both product ions produced two characteristic fragment ions which can be used for the determination of the location of the C=C bonds.
We first evaluated this method on Mini 12 using lipid standard solutions containing C18:1 fatty acids and phospholipids with C18:1 acyl chains, since their C=C isomers are widely distributed in biological systems and some isomers have been identified as potential biomarkers.45 The standard solutions including FA 18:1 (9Z) (m/z 281, negative ion mode), FA 18:1 (11Z) (m/z 281, negative ion mode), and PC 16:0/18:1 (9Z) (m/z 760, positive ion mode) were prepared separately. Solvent of 50/50 acetone/water with 1% (v/v) ammonium hydroxide was used for FAs, and 70/20/10 acetone/acetonitrile/water with 1% (v/v) acetic acid was used for the phospholipid. A UV exposure time of 2 min was applied. As shown Figure 3a, PB product with adduct of 58 Da (oxetane ring isomers) at m/z 339 was produced for FA 18:1 (11Z), which fragmented at the octane ring through CID to produce a pair of diagnostic ions at m/z 199 and m/z 225, corresponding to the C=C bond at the 11th position. Similarly, fragments at m/z 171 and m/z 197 were observed by performing PB-MS/MS on FA 18:1 (9Z), this pair of diagnostic ions separated by 26 Da indicated the C=C located on ninth position (Figure 3b). Apart from fatty acids, phospholipids were also analyzed by PB-MS/MS on the Mini 12. PC 16:0/18:1 (9Z) was analyzed in positive mode and a solvent of 70/20/10 acetone/acetonitrile/water with 1% (v/v) acetic acid was used. After online PB reaction, PB product with adduct of 58 Da at m/z 818 were produced. By performing ion trap CID, beside the PC headgroup ion at m/z 184, diagnostic ions at m/z 650 and m/z 676 were observed in the mass spectrum, indicating the C=C located on ninth position of the acyl chain C18:1 (Figure 3c).
Figure 3.
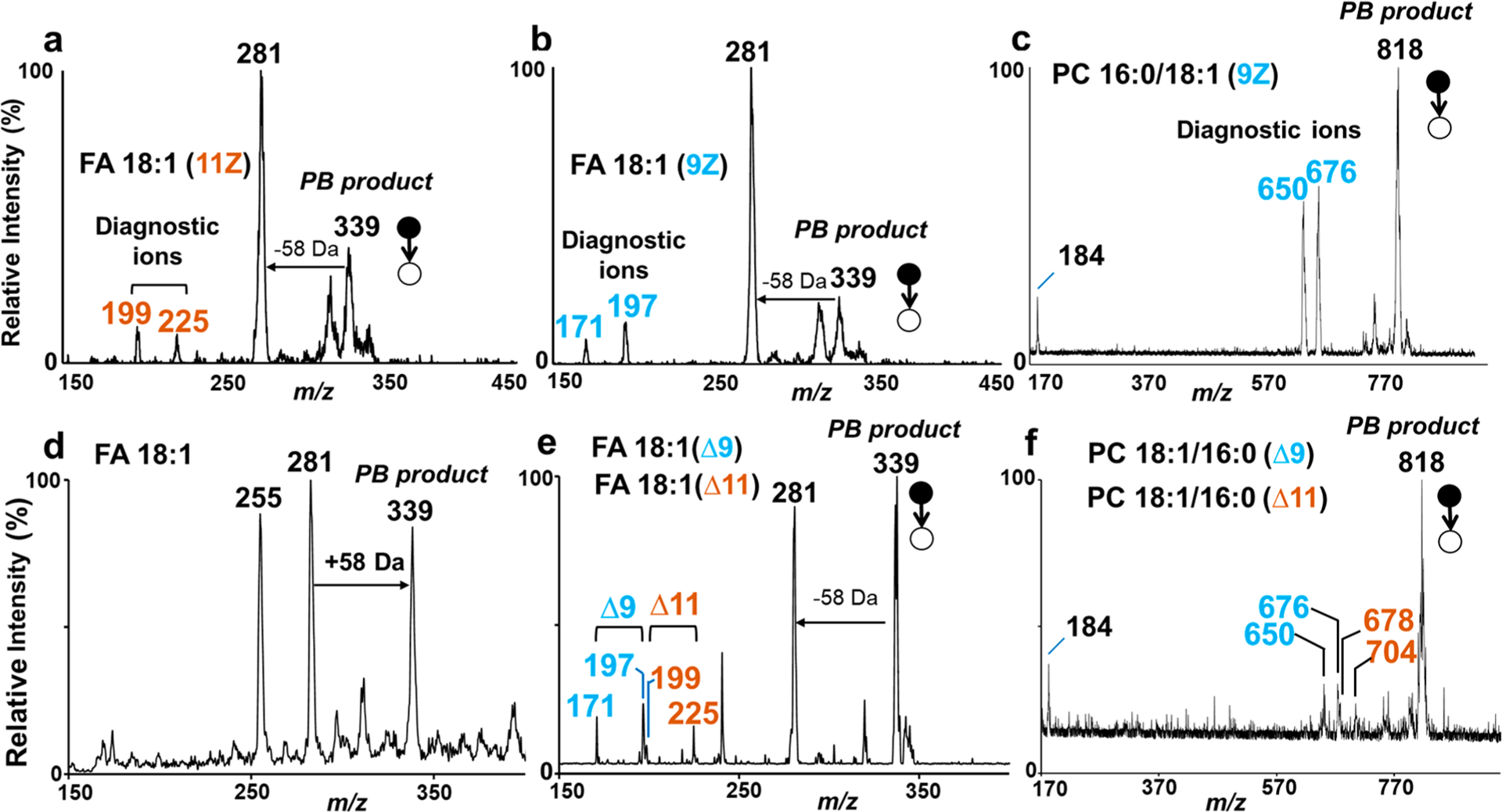
(a) MS/MS spectrum of PB reaction product ion m/z 339 for analysis of 10 ppm cis-vaccenic acid (FA 18:1 (11Z)) in 50/50 acetone/water with 1% (v/v) ammonium hydroxide, negative ion mode. (b) MS/MS spectrum of PB product ion m/z 339 for analysis of 10 ppm oleic acid (FA 18:1 (9Z)) in 50/50 acetone/water with 1% (v/v) ammonium hydroxide, negative ion mode. (c) MS/MS spectrum of PB reaction product ion m/z 818 for 10 ppm PC 16:0/18:1 (9Z) in 70/20/10 acetone/acetonitrile/water with 1% (v/v) acetic acid, positive ion mode. (d) MS spectrum of fatty acids sampled from rat brain, direct extraction spray with PB reaction, negative ion mode. (e) MS/MS of PB product ion m/z 339 from FA 18:1 sampled from rat brain. (f) MS/MS spectrum of PB product ion m/z 818 for PC 16:0_18:1 sampled from rat brain, extracted using solvent 70/20/10 acetone/acetonitrile/water with 1% (v/v) acetic acid, positive ion mode.
The results described above showed that it is possible to transfer PB-MS/MS from benchtop mass spectrometers to miniature mass spectrometers. Here we developed a direct sampling-based PB-MS/MS method for analysis of C=C locations of lipids in tissue sample on the Mini 12. Rat brain tissue was used for evaluation of the performance of the method. After direct sampling, the probe was insert carefully into a capillary tip filled with acetone-contained solution. Figure 3d shows the MS spectrum recorded by Mini 12 in negative mode after extraction of the fatty acids from the probe and performing PB reaction. Both FA 18:1 at m/z 281 and its PB product at m/z 339 were observed. By applying CID to the PB product ion at m/z 339, two pairs of C=C diagnostic ions at m/z 171/197 and m/z 199/225 were obtained (Figure 3e). According to these diagnostic ions, FA 18:1 in rat brain was found to have two C=C location isomers, namely, FA 18:1 (Δ9) and FA 18:1 (Δ11). Figure 3f shows the PB-MS/MS spectrum for PC 34:1 (m/z 760) sampled from the rat brain. By applying CID on the PB product ion m/z 818, two pairs of diagnostic ions at m/z 650/676 and m/z 678/704 were obtained. According to these diagnostic ions, PC 34:1 in rat brain was found to have two C=C location isomers, PC 16:0_18:1 (Δ9) and PC 16:0_18:1 (Δ11).
This analytical method, with steps including direct sampling of tissue, extraction with lipids, online PB reaction, extraction spray and mass analysis by Mini 12 system, was also applied to differentiate mouse normal breast tissue and cancerous tissue. Figure 4 shows the MS/MS spectra of FA 18:1 in normal breast tissue (Figure 4a) and cancerous tissue (Figure 4b). Both FA 18:1 (Δ9) and FA 18:1 (Δ11) were identified in the two tissue samples. In order to quantitatively compare the abundance difference of the C=C location isomers, the isomer ratio was calculated from the summed diagnostic ion intensities of one pair of C=C location isomers versus by the other summed diagnostic ion intensities:
| (1) |
where I171, I197, I199, and I225 are the intensity of diagnostic ions at m/z 171, 197, 199, and 225, respectively. The ratio refers to the intensity ratio of FA 18:1 (Δ9) and FA 18:1 (Δ11). The intensity ratio of FA 18:1 Δ9/Δ11 was calculated to be 2.881 for the normal breast tissue, but 0.667 for cancerous tissue, which are consistent with results obtained on a large MS.45 The significant difference in the relative abundances for lipid C=C location isomers could potentially serve as biomarkers for cancer diagnosis.45,47
Figure 4.
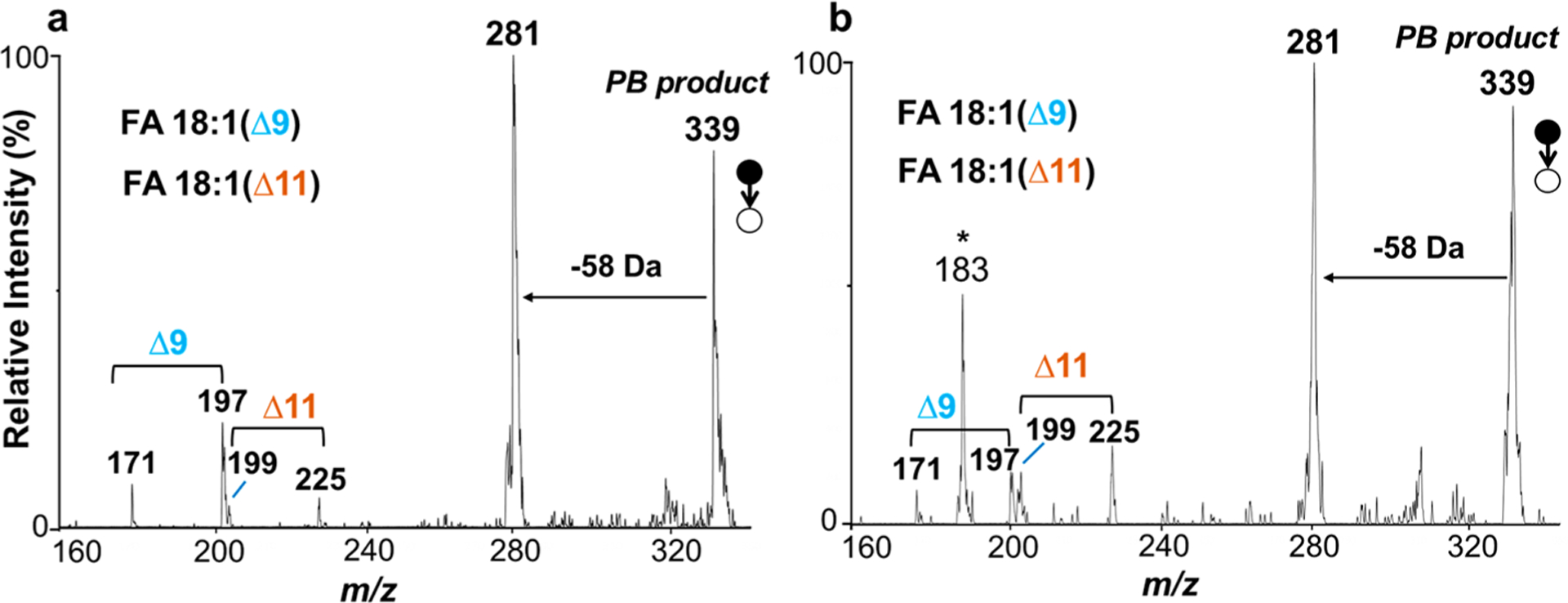
MS/MS spectra of PB reaction products m/z 339 for FA 18:1 sampled from (a) normal and (b) cancerous mouse breast tissues, solvent of 50/50 acetone/water with 1% (v/v) ammonium hydroxide used for extraction, reaction and analyzed in negative ion mode. * Product ion from m/z 339 existed in the tissue sample.
Besides lipids, this miniature direct sampling MS system can also be used for fast analysis of other metabolite biomarkers in tissue samples for clinical diagnosis of some diseases, such as glioma. Mutation of the isocitrate dehydrogenase (IDH) have been revealed in some cancer processes, resulting in a significant increase of 2-hydroxylglutarate (2-HG) in gliomas, which has been identified as a biomarker via comprehensive metabolism analysis.48–50 Due to the limitation in brain tissue biopsy for presurgery analysis, it is highly desirable to develop a method capable of fast analyzing 2-HG in glioma tissue for real-time surgery decision making. Previously, desorption electrospray ionization was used for direct analysis of 2-HG in tissue samples,43,44 which, however, required fast-speed gas for sampling ionization and represents a difficulty for its implementation with miniature systems. The direct sampling analysis with miniature MS system reported here was explored for rapid intraoperative of 2-HG in human glioma samples. A significant peak of 2-HG was detected at m/z 147 (negative ion mode) in the MS1 spectrum of the glioma (IDH1 mutant, grade IV) sample (Figure 5a), while the peak at m/z 147 (negative ion mode) was negligible in the normal brain (Figure 5b). MS2 of m/z 147 and MS3 of m/z 147/129 (Figure 5c,d) further confirmed the presence of 2-HG in the glioma tissue sample.49,50 Although large scale analysis may be required for further evaluation of this method for intrasurgical analysis, the results showed that the direct sampling miniature MS system is applicable to fast and POC analysis of 2-HG in small amount of glioma samples, which can be potentially used in an operating room for fast molecular profiling of gliomas.
Figure 5.
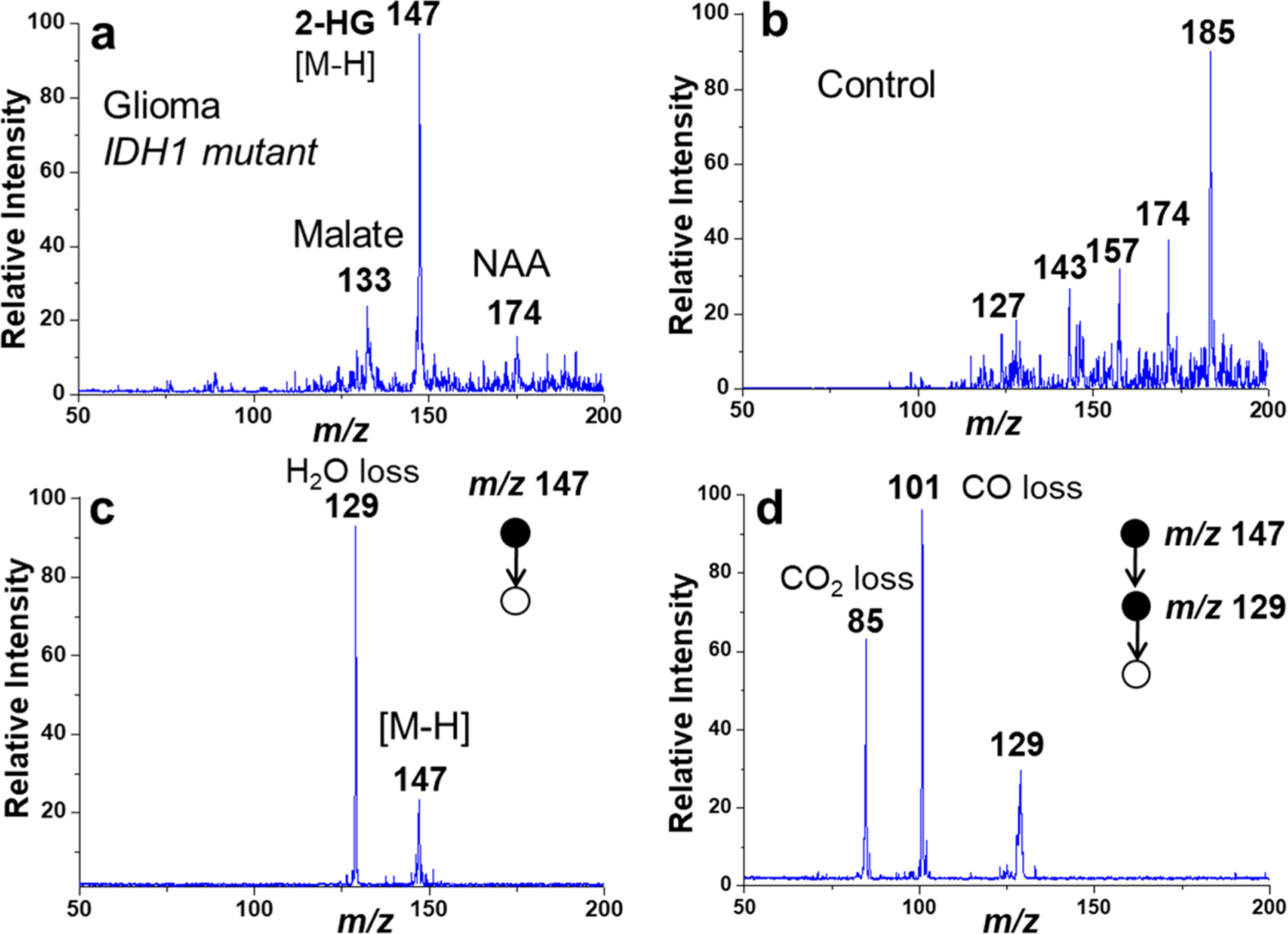
Mass spectra of metabolites sampled from (a) glioma and (b) control (normal brain) tissues. (c) MS2 spectrum of 2-HG (m/z 147) in the glioma tissue sample, and (d) MS3 spectra of 2-HG (m/z 147/129) in the glioma tissue sample. Methanol/water (7/3, v/v) was used as the extraction solvent, and ionization was performed by nanoESI in the negative ion mode. NAA, N-acetylaspartate.
Although the results demonstrated a great potential of the miniature MS system for POC analysis of tissue samples, some issues still need to be further considered to transfer this methodology into clinical analysis. Since a single linear ion trap was used here as the mass analyzer with MS/MS capability, comprehensive structure confirmation through versatile tandem MS modes might be desirable for analysis of lipids and other metabolites.51 While the direct sampling MS analysis eliminates extensive sample preparation, including purification and chromatographic separation, its tolerance to matrix effect and impacts on the reproducibility of quantitation for biological samples remain to be further validated. This can be addressed by development of specialized cartridges and sample-dependent calibration methods. While the total lipid intensities can be measured with incorporation of internal standards, relative quantitation through lipid isomeric ratios could be powerful and suitable for POC analysis of tissue samples.52,53
CONCLUSIONS
In this work, we developed a POC system for lipid and metabolite analysis by coupling direct sampling and miniature MS. The direct sampling method was found to be simple, fast, and effective by evaluation in different types of tissue samples. Lipids and metabolites were extracted by a thin stainless-steel wire, following fast elution by polar solvents inside a nanoESI emitter. Through tuning the Mini 12 system, direct sampling-based nanoESI MS showed good performance for lipid profiling in different tissue samples. By incorporating with online photochemical PB reaction, this system can also be used to determine the C=C location of unsaturated fatty acids and phospholipids in tissue samples. The significant change of C=C isomer intensity ratios in rat cancerous breast tissue samples was also testified by direct sampling nanoESI mini MS system, showing its capability of relative quantitation of potential lipid biomarkers. Successful analysis of 2-HG accumulation in glioma samples showed that this system has potential to be a useful POC tool for rapid intraoperative molecular subtyping of gliomas. Development of disposable cartridges for direct sampling and simple calibration methods is ongoing to improve the performance as well as the applicability of the system for clinical analysis.
ACKNOWLEDGMENTS
Financial support from National Natural Science Foundation of China (Project 21627807) and National Institutes of Health (Project R44GM119584, 1R01AI122298 and R01AI122932) is greatly appreciated.
Footnotes
The authors declare the following competing financial interest(s): Z.O. is the founder of the PURSPEC Technologies Inc.
REFERENCES
- (1).Eberlin LS; Ferreira CR; Dill AL; Ifa DR; Cooks RG Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2011, 1811, 946–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Aksenov AA; da Silva R; Knight R; Lopes NP; Dorrestein PC Nat. Rev. Chem 2017, 1, 0054. [Google Scholar]
- (3).Harris GA; Galhena AS; Fernandez FM Anal. Chem 2011, 83, 4508–4538. [DOI] [PubMed] [Google Scholar]
- (4).NIH. Point-of-Care Diagnostic Testing. https://report.nih.gov/nihfactsheets/ViewFactSheet.aspx?csid=112 (accessed Dec. 10, 2017).
- (5).Chen S; Wan Q; Badu-Tawiah AK J. Am. Chem. Soc 2016, 138, 6356–6359. [DOI] [PubMed] [Google Scholar]
- (6).Ferreira CR; Yannell KE; Jarmusch AK; Pirro V; Ouyang Z; Cooks RG Clin. Chem 2016, 62, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Corso G; D’Apolito O; Gelzo M; Paglia G; Russo AD Bioanalysis 2010, 2, 1883–1891. [DOI] [PubMed] [Google Scholar]
- (8).Ma X; Ouyang Z TrAC, Trends Anal. Chem 2016, 85, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sinha M; Tomassian A Rev. Sci. Instrum 1991, 62, 2618–2620. [Google Scholar]
- (10).Gao L; Song Q; Patterson GE; Cooks RG; Ouyang Z Anal. Chem 2006, 78, 5994–6002. [DOI] [PubMed] [Google Scholar]
- (11).Gao L; Sugiarto A; Harper JD; Cooks RG; Ouyang Z Anal. Chem 2008, 80, 7198–7205. [DOI] [PubMed] [Google Scholar]
- (12).Hou K; Xu W; Xu J; Cooks RG; Ouyang Z Anal. Chem 2011, 83, 1857–1861. [DOI] [PubMed] [Google Scholar]
- (13).Hendricks PI; Dalgleish JK; Shelley JT; Kirleis MA; McNicholas MT; Li L; Chen T-C; Chen C-H; Duncan JS; Boudreau F; et al. Anal. Chem 2014, 86, 2900–2908. [DOI] [PubMed] [Google Scholar]
- (14).Ma Q; Bai H; Li W; Wang C; Cooks RG; Ouyang Z Talanta 2015, 142, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li L; Chen T-C; Ren Y; Hendricks PI; Cooks RG; Ouyang Z Anal. Chem 2014, 86, 2909–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wu Q; Li A; Tian Y; Zare RN; Austin DE Anal. Chem 2016, 88, 7800–7806. [DOI] [PubMed] [Google Scholar]
- (17).Jiang T; Zhang H; Tang Y; Zhai Y; Xu W; Xu H; Zhao X; Li D; Xu W Anal. Chem 2017, 89, 5578–5584. [DOI] [PubMed] [Google Scholar]
- (18).Li A; Hansen BJ; Powell AT; Hawkins AR; Austin DE Rapid Commun. Mass Spectrom 2014, 28, 1338–1344. [DOI] [PubMed] [Google Scholar]
- (19).Guo M; Li D; Cheng Y; Wang Y; Sun W; Pei X; Dong M; Sheng X; Zhao L; Li Y Eur. J. Mass Spectrom 2018, 24, 206–213. [DOI] [PubMed] [Google Scholar]
- (20).Riedo A; Meyer S; Heredia B; Neuland MB; Bieler A; Tulej M; Leya I; Iakovleva M; Mezger K; Wurz P Planet. Space Sci 2013, 87, 1–13. [Google Scholar]
- (21).Gao L; Cooks RG; Ouyang Z Anal. Chem 2008, 80, 4026–4032. [DOI] [PubMed] [Google Scholar]
- (22).Pu F; Zhang W; Bateman KP; Liu Y; Helmy R; Ouyang Z Bioanalysis 2017, 9, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Venter A; Nefliu M; Cooks RG TrAC, Trends Anal. Chem 2008, 27, 284–290. [Google Scholar]
- (24).Ren Y; McLuckey MN; Liu J; Ouyang Z Angew. Chem., Int. Ed 2014, 53, 14124–14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Takáts Z; Wiseman JM; Gologan B; Cooks RG Science 2004, 306, 471–473. [DOI] [PubMed] [Google Scholar]
- (26).Jarmusch AK; Pirro V; Baird Z; Hattab EM; Cohen-Gadol AA; Cooks RG Proc. Natl. Acad. Sci. U. S. A 2016, 113, 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Calligaris D; Caragacianu D; Liu X; Norton I; Thompson CJ; Richardson AL; Golshan M; Easterling ML; Santagata S; Dillon DA; Jolesz FA; Agar NYR Proc. Natl. Acad. Sci. U. S. A 2014, 111, 15184–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dill AL; Eberlin LS; Costa AB; Zheng C; Ifa DR; Cheng L; Masterson TA; Koch MO; Vitek O; Cooks RG Chem. - Eur. J 2011, 17, 2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wiseman JM; Ifa DR; Zhu Y; Kissinger CB; Manicke NE; Kissinger PT; Cooks RG Proc. Natl. Acad. Sci. U. S. A 2008, 105, 18120–18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kerian K; Jarmusch A; Pirro V; Koch M; Masterson T; Cheng L; Cooks R Analyst 2015, 140, 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mandal MK; Saha S; Yoshimura K; Shida Y; Takeda S; Nonami H; Hiraoka K Analyst 2013, 138, 1682–1688. [DOI] [PubMed] [Google Scholar]
- (32).Piri-Moghadam H; Ahmadi F; Gómez-Ríos GA; Boyacı E; Reyes-Garcés N; Aghakhani A; Bojko B; Pawliszyn J Angew. Chem., Int. Ed 2016, 55, 7510–7514. [DOI] [PubMed] [Google Scholar]
- (33).Goryński K; Goryńska P; Górska A; Harężlak T; Jaroch A; Jaroch K; Lendor S; Skobowiat C; Bojko BJ Pharm. Biomed. Anal 2016, 130, 55–67. [DOI] [PubMed] [Google Scholar]
- (34).Wang H; Liu J; Cooks RG; Ouyang Z Angew. Chem., Int. Ed 2010, 122, 889–892. [Google Scholar]
- (35).Espy RD; Manicke NE; Ouyang Z; Cooks RG Analyst 2012, 137, 2344–2349. [DOI] [PubMed] [Google Scholar]
- (36).Griffin JL; Shockcor JP Nat. Rev. Cancer 2004, 4, 551–561. [DOI] [PubMed] [Google Scholar]
- (37).Piomelli D; Astarita G; Rapaka R Nat. Rev. Neurosci 2007, 8, 743–754. [DOI] [PubMed] [Google Scholar]
- (38).Blanksby SJ; Mitchell TW Annu. Rev. Anal. Chem 2010, 3, 433–465. [DOI] [PubMed] [Google Scholar]
- (39).Pirro V; Guffey S; Sepúlveda M; Mahapatra C; Ferreira C; Jarmusch A; Cooks R Mol. BioSyst 2016, 12, 2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Anderson JL; Carten JD; Farber SA Methods Cell Biol. 2011, 101, 111–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Brunelle A; Laprévote O Anal. Bioanal. Chem 2009, 393, 31–35. [DOI] [PubMed] [Google Scholar]
- (42).Murphy RC; Hankin JA; Barkley RM J. Lipid Res 2009, 50, S317–S322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Dill AL; Ifa DR; Manicke NE; Ouyang Z; Cooks RGJ Chromatogr. B: Anal. Technol. Biomed. Life Sci 2009, 877, 2883–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Liu J; Cooks RG; Ouyang Z Anal. Chem 2011, 83, 9221–9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ma X; Chong L; Tian R; Shi R; Hu TY; Ouyang Z; Xia Y Proc. Natl. Acad. Sci. U. S. A 2016, 113, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ma X; Xia Y Angew. Chem., Int. Ed 2014, 126, 2630–2634. [Google Scholar]
- (47).Paine MRL; Poad BLJ; Eijkel GB; Marshall DL; Blanksby SJ; Heeren RMA; Ellis SR Angew. Chem., Int. Ed 2018, 57, 10530–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dang L; White DW; Gross S; Bennett BD; Bittinger MA; Driggers EM; Fantin VR; Jang HG; Jin S; Keenan MC; Marks KM; Prins RM; Ward PS; Yen KE; Liau LM; Rabinowitz JD; Cantley LC; Thompson CB; Vander Heiden MG; Su SM Nature 2009, 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Santagata S; Eberlin LS; Norton I; Calligaris D; Feldman DR; Ide JL; Liu X; Wiley JS; Vestal ML; Ramkissoon SH; Orringer DA; Gill KK; Dunn IF; Dias-Santagata D; Ligon KL; Jolesz FA; Golby AJ; Cooks RG; Agar NYR Proc. Natl. Acad. Sci. U. S. A 2014, 111, 11121–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Pirro V; Alfaro CM; Jarmusch AK; Hattab EM; Cohen-Gadol AA; Cooks RG Proc. Natl. Acad. Sci. U. S. A 2017, 114, 6700–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Liu X; Bu J; Zhou X; Ouyang Z Anal. Chem 2018, DOI: 10.1021/acs.analchem.8b03958. [DOI] [PubMed] [Google Scholar]
- (52).Zhang W; Wang X; Xia Y; Ouyang Z Theranostics 2017, 7, 2968–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Banerjee S; Zare RN; Tibshirani RJ; Kunder CA; Nolley R; Fan R; Brooks JD; Sonn GA Proc. Natl. Acad. Sci. U. S. A 2017, 114, 3334–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]