

Abstract
Neuroinflammation has become an important underlying factor in many cardiovascular disorders, including hypertension. Previously we showed that elevated angiotensin II (Ang II) and angiotensin II type I receptor (AT1R) expression levels can increase neuroinflammation leading to hypertension. We also found that kinin B1 receptor (B1R) expression increased in the hypothalamic paraventricular neurons resulting in neuroinflammation and oxidative stress in neurogenic hypertension. However, whether there are any potential interactions between AT1R and B1R in neuroinflammation is not clear. In the present study, we aimed to determine whether Ang II-mediated effects on inflammation and oxidative stress are mediated by the activation of B1R in mouse neonatal primary hypothalamic neuronal cultures. Gene expression and immunostaining revealed that both B1R and AT1R are expressed on primary hypothalamic neurons. Ang II stimulation significantly increased the expression of the B1R, decreased mitochondrial respiration, increased the expression of two NADPH oxidase subunits (Nox2 and Nox4), increased the oxidative potential, upregulated several proinflammatory genes (IL-1β, IL-6 and TNFα) and increased NF-kB p65 DNA binding activity. These changes were prevented by pretreatment with the B1R-specific peptide antagonist, R715. In summary, our study demonstrates a causal relationship between B1R expression after Ang II stimulation, suggesting a possible cross talk between AT1R and B1R in neuroinflammation and oxidative stress.
Keywords: Angiotensin II, AT1R, Kinin B1R, Mitochondrial respiration, Neuroinflammation, Oxidative stress
Introduction
Hypertension is a major risk factor for the development of many cardiovascular diseases and considered as one of the leading causes of morbidity and mortality worldwide (Bartoloni et al. 2017). Despite the availability of various anti-hypertensive therapies, the treatment of hypertension is often unsatisfactory (Winklewski et al. 2015). Increasing evidence suggest that uncontrolled, resistant hypertension predominantly involves elevated sympathetic drive and neuroinflammation within the brain, hence referred as neurogenic hypertension (Chobanian 2009; Mowry and Biancardi 2019; Winklewski et al. 2015). Inflammation within the brain cardiovascular regulatory centers that control the sympathetic outflow from the brain resulting in elevated sympathetic drive to target organs, ultimately leading to increase blood pressure, represents an evolving concept of the pathogenesis of neurogenic hypertension (Mowry and Biancardi 2019; Sriramula and Lazartigues 2017; Winklewski et al. 2015). Multiple studies have shown evidence that dysregulated neuroinflammation is involved in angiotensin II (Ang II)-mediated sympathoexcitation and development of hypertension (Haspula and Clark 2018; Xue et al. 2016; Zubcevic et al. 2017). The peripheral inflammatory mediators can gain access to the brain through leaky vasculature in regions where the blood-brain barrier has been disturbed (Abbott 2000; Erickson et al. 2012). On the other hand, proinflammatory cytokines are produced by glia and neurons within the brain and can induce sympathoexcitation and regulate the peripheral immune responses (Alvarez et al. 2013; Willis 2011). This bidirectional interaction between the nervous and immune systems may eventually lead to the development of neurogenic hypertension.
Previous work with experimental and transgenic animal models has shown that angiotensin type 1 receptors (AT1R) are expressed in different cell types within the brain and play a role in the development of neuroinflammation and neurogenic hypertension (Saavedra 2017; Xu et al. 2017). In contrast to previous immunohistochemistry and in vitro studies that have localized AT1R and AT2R to non-neuronal cells in the brain (Li et al. 2009; Miyoshi et al. 2008; Wu et al. 2013), recent studies with genetic mouse models and advances in in situ hybridization provide evidence that, at least under normal conditions, these receptor subtypes are predominantly expressed in neurons as opposed to glia (Chen et al. 2012; de Kloet et al. 2016; Gonzalez et al. 2012). Neuron-specific knockdown of prorenin receptor was shown to reduce Ang II formation, expression of inflammatory mediators, and blood pressure in the DOCA-salt mouse model of hypertension (Li et al. 2012; Li et al. 2014). On the other hand, we previously showed that neuron specific overexpression of ACE2 decreased inflammation, oxidative stress, and reduced hypertension (Sriramula et al. 2015; Xia et al. 2013). Despite these observations, molecular mechanisms that lead to enhanced neuroinflammation remain unknown.
The kallikrein-kinin system (KKS) is a hormonal system that plays an important role in inflammation and blood pressure regulation (Leeb-Lundberg et al. 2005). The KKS system is composed of vasoactive proinflammatory peptides known as kinins, mainly bradykinin and kallidin. Bradykinin, the well-known kinin, and its active metabolite, des-arg9-bradykinin, mediate their physiological effects via G-protein-coupled kinin receptor subtypes B1 (B1R) and B2 (B2R) (Leeb-Lundberg et al. 2005). The B2R is constitutively expressed whereas the B1R is inducible by inflammation or oxidative stress (Leeb-Lundberg et al. 2005). However, under normal physiological conditions, the B1R is expressed in lower levels in certain organs such as brain and heart (McLean 2000). Direct stimulation of the B1 receptor is shown to be responsible for an increase in the overall number of B1R due to positive feedback of the receptor-mediated signaling, forming a vicious cycle of B1R activation (Campos et al. 1996; Campos et al. 1998). This unique aspect of the receptor distinguishes it from other receptors involved in inflammation and neurological disorders. Interestingly, components of the KKS have been shown to mediate various pathophysiological conditions within the nervous system. Both B1R and B2R were expressed in neurons, more specifically the B1R is expressed in human thalamus, hypothalamus and spinal cord neurons (Raidoo and Bhoola 1997). It was also shown that the B1R was detected on dopaminergic neurons of the ventral tegmental area in SHR (De Brito Gariepy et al. 2013). We and others have shown that elevated levels of angiotensin II in the brain or upregulated neuronal AT1R expression, results in increased neuroinflammation and the development of neurogenic hypertension (Li et al. 2014; Xia et al. 2013; Xu et al. 2017). In our previous study, we first associated neurogenic hypertension with increased expression of B1R in the hypothalamic paraventricular neurons in a model of DOCA-salt hypertension (Sriramula and Lazartigues 2017). In the present study, we investigated the direct interaction between Ang II signaling and B1R activation using mouse neonatal primary hypothalamic neuronal cultures. We hypothesized that B1R activation in primary hypothalamic neurons plays a role in Ang II-mediated inflammation and oxidative stress. Our data demonstrate a causal relationship between B1R expression after Ang II stimulation. Overall, this report supports a mechanistic role for B1R in inflammation and oxidative stress in Ang II-stimulated primary hypothalamic neurons.
Materials and Methods
Primary Neuronal Cell Culture
Primary hypothalamic neuronal cultures were prepared and maintained according to a previously described method with minor modifications (Xu et al. 2018). The experimental protocols used for breeding mice were approved by East Carolina University Animal Care and Use Committee (AUP #W254) and were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Briefly, neonatal or 1-day-old mouse pups were anesthetized with isoflurane (4%) in an oxygen flow (1 L/min) before decapitation and brains were collected in ice-cold Hank’s balanced salt solution (HBSS) (Gibco 14175-079). Hypothalamic tissue was dissected under sterile conditions, rinsed in HBSS, and minced into small pieces. The minced tissue was transferred into a 15 ml conical tube, washed once with HBSS, then treated with HBSS containing 1% trypsin (Sigma-Aldrich T1426) and 1.5 kU/mL DNaseI (Sigma-Aldrich D5025), and digested for 10 min at 37 °C. Then the tissue was washed with HBSS with 10% FBS twice followed by washing twice with HBSS. The tissue was further triturated in HBSS containing DNase I, using pipette with 1ml pipette tip (six times) and then with 200 μl pipette tip (six times) attached to 10 ml serological pipette. Following the disassociation, the cell pellet was collected by centrifugation and resuspended in complete Neurobasal culture medium supplemented with 2% B27, 0.5 mM GlutaMax and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively) (Gibco). Then, the resuspended cells were plated into poly-L-lysine-coated 6 well plates at a density of 50,000 cells per ml. The neurons were grown in a humidified atmosphere of 5% CO2–95% air at 37°C. After 24 hours, additional fresh medium was added to the cells. On the fourth day, cytosine arabinofuranoside (Ara-C, 2 μM, Sigma-Aldrich C1768) was supplemented to the cultures to suppress glial cell proliferation. Hypothalamic primary neurons were cultured at least 10 days and then used for experiments.
Immunofluorescence staining
Cultured neurons were validated using immunofluorescence labeling with a neuron-specific cytoskeletal marker, MAP2 (microtubule-associated protein 2) and a glial cell-specific marker, GFAP (glial fibrillary acidic protein). Primary neurons were grown on poly-L-lysine-coated glass cover slips in 12-well plates for 10 days and fixed with 4% paraformaldehyde for 15 min. The cells were washed with 100 mM Glycine in 1× PBS for 5 min each for 3 times followed by incubating with 0.1% Triton X-100 in 1× PBS for 15 min to permeabilize cells. After blocking with 5% donkey serum in 1× PBS containing 0.1% Tween-20 for 1 h, cells were incubated with MAP2 antibody (#13–1500, lot T5275359, ThermoFisher, 1:200 dilution) overnight at 4 °C. Cells were washed with 0.1% Triton X-100 in 1× PBS for 10 min each for 3 times and then incubated with specific secondary antibody (Donkey anti-Mouse Alexa Fluor Plus 488, A32766 or Donkey anti-Mouse Alexa Fluor Plus 594 A32744, Invitrogen, 1:1000 dilution) for 1 h at room temperature in a humidifying chamber in the dark. For GFAP staining, an Alexa Fluor 488 labeled antibody (#53-9892-82, lot 1,914,350, ThermoFisher, 1:50 dilution) was used. Triple immunostaining was performed with specific validated antibodies for detection of AT1R (#AAR-011, lot AN2002, Alomone labs, 1:200 dilution) and B1R (#ABR-011, lot An-01, Alomone labs, 1:200 dilution) coupled with MAP2 and DAPI staining. After 3 washes, coverslips were counterstained with DAPI, then mounted with ProLong Diamond (ThermoFisher) antifade medium and examined under a confocal microscope (Zeiss LSM 700).
Measurement of Reactive Oxygen Species Generation
Reactive oxygen species (ROS) levels were determined using dihydroethidium (DHE), which is relatively specific for superoxide anion measurement, oxidized by superoxide to form DNA binding fluorophore ethidium bromide. Primary neuronal cultures grown on poly-L-lysine-coated glass coverslips in 12-well plates were treated with 10 μM DHE and incubated in a light protected humidified chamber at 37°C for 15 minutes. Fluorescent images of ethidium-stained cells were obtained with a florescent microscope (Leica). Ethidium bromide was excited at 488 nm, and fluorescence was detected at 560 nm filter. Generation of superoxide was determined by red florescent labeling. Mean fluorescence intensity of the digitized image was measured with ImageJ software (version 1.52p, National Institutes of Health) for quantification. The data was expressed as corrected total cell fluorescence (CTCF) and calculated by the formula CTCF = Integrated Density – (Area of selected cell X Mean fluorescence of background readings). In addition, the production of ROS was measured spectrofluorometrically by using the probe DHE. Briefly, the primary neurons were cultured in 48-well plates and treated with 10 μM DHE (Invitrogen) for 30 minutes. Cells were exposed to Ang II or vehicle for 30 minutes with or without a pretreatment of R715 for 30 minutes. Then the DHE medium was removed and cells were washed twice with PBS and fluorescence was detected on a microplate spectrofluorometer (Tecan infinite m200) at an excitation wavelength of 488 nm and emission wavelength of 610 nm. Treatment with antimycin A, an inhibitor of complex III of the mitochondrial electron transport chain, is used as a positive control for ROS generation. Treatment with N-acetyl Cysteine is used as an antioxidant control. Data are expressed as total florescence (relative florescent units, RFU).
Gene Expression Analysis by Real Time qRT-PCR
Gene expression was measured using real time RT-PCR as described previously (Xu et al. 2018). Total RNA from the primary hypothalamic neurons was extracted using the Direct-Zol RNA miniprep plus kit (Zymo Research) according to manufacturer’s protocol. RNA concentration was measured using the spectrophotometer (NanoDrop One). Real Time PCR amplification reactions were performed with Power SYBR Green RNA-to-CT one-step Kit (Applied Biosystems) using a QuantStudio 6 Flex real time PCR machine (Applied Biosystems). Data were normalized to β-actin expression by the 2−(ΔΔCT) comparative method and expressed as a fold change compared to control.
Seahorse XF-24 Metabolic Flux Analysis
The Seahorse analyzer XF24 (Agilent) was used to continuously monitor OCR. Primary hypothalamic mouse neurons were cultured on Seahorse XF-24 plates at a density of 80,000 cells per well. The cells were treated with vehicle or Ang II with or without R715 for 24 hours. On the day of metabolic flux analysis, unbuffered DMEM (DMEM base medium supplemented with 25 mM glucose, 1 mM sodium pyruvate, 31 mM NaCl, 2 mM GlutaMax, pH 7.4) was added to cells and incubated at 37 °C in a non-CO2 incubator for 1 h. All medium and injection reagents were adjusted to pH 7.4 on the day of assay. After preparation and application of components of the XF Cell Mito Stress Test Kit (Agilent Technologies, Seahorse Bioscience) into cartridge ports, the cartridge and subsequently the cell culture plate were loaded into the Seahorse analyzer. Four baseline measurements of OCR and ECAR were taken before sequential injection of mitochondrial inhibitors. Three readings were taken after each addition of mitochondrial inhibitor before injection of the subsequent inhibitors. The mitochondrial inhibitors used were oligomycin (1 μM), FCCP (1 μM), and antimycin A and rotenone (1 μM). OCR and ECAR were automatically calculated and recorded by the Seahorse XF-24 software. At the end of the assay protocol, the cells in plates were lysed with NP-40 lysis buffer, and protein concentration was determined using Pierce BCA protein assay. The data were expressed as percentage of control.
NF-κB Binding Activity Assay
NF-κB activation was measured using a DNA binding assay. Primary hypothalamic neurons cultured in 6-well plates for 14 days were treated with vehicle and angiotensin II for 24 hours with or without R715 pretreatment. The neurons were harvested, and nuclear extracts were prepared using a nuclear extraction kit (Active Motif). Protein concentrations were then quantified using a BCA protein assay kit (Thermo Fisher). Equal amounts of protein were used for the determination of the activity of NF-κB with a colorimetric NF-κB assay specific for the activated form of the p65 subunit of NF-κB using a commercially available kit (TransAm NF-κB p65, Active Motif) according to the manufacturer’s instructions.
Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism 7 (GraphPad Software). Multiple comparisons were made using 1-way analysis of variance followed by Tukey’s multiple comparisons test. Differences were considered statistically significant at P < 0.05.
Results
Ang II Stimulation Leads to an Increase in the Expression of the B1R in Primary Hypothalamic Neurons
In this study, we used mouse neonatal primary hypothalamic neuronal cultures (Fig. 1A) to test the hypothesis that the B1R mediates the effects of Ang II in hypothalamic neurons. We first validated the primary hypothalamic neurons via immunofluorescence labeling with MAP2, a neuron-specific cytoskeletal marker, and GFAP, a glial cell-specific marker. Ara-C treatment was used for glial cell suppression. As expected, in the absence of Ara-C both glial cells and neurons were observed (Fig. 1B) whereas treatment with Ara-C suppressed the proliferation of glial cells (Fig. 1C). The neurons cultured for 14 days with Ara-C treatment showed predominantly neuronal population, demonstrating numerous processes and discrete cellular morphology of neurons with cell-cell interaction, as shown in Fig. 1D. Primary hypothalamic neurons showed immunopositivity for the presence of the B1R (Fig. 1E) and the AT1R (Fig.1F) confirming the expression of these receptors in our neuron preparation. Next, we wanted to know whether Ang II effects on the expression of the B1R is recapitulated in our system. As shown in Fig.1G, treatment of the neurons with 300 nM of Ang II increased the expression of the B1R at 3 and 6 hours. Interestingly, this effect of Ang II at 6 hours was largely prevented by 1-hour pretreatments with either 10 μM of the AT1R antagonist, losartan or 10 μM of the B1R antagonist, R715 (Fig.1H). These results confirm that Ang II increases the expression the B1R in hypothalamic neurons via activation of AT1R.
Fig. 1. Kinin B1 receptor gene expression is induced by angiotensin II in primary hypothalamic neurons.
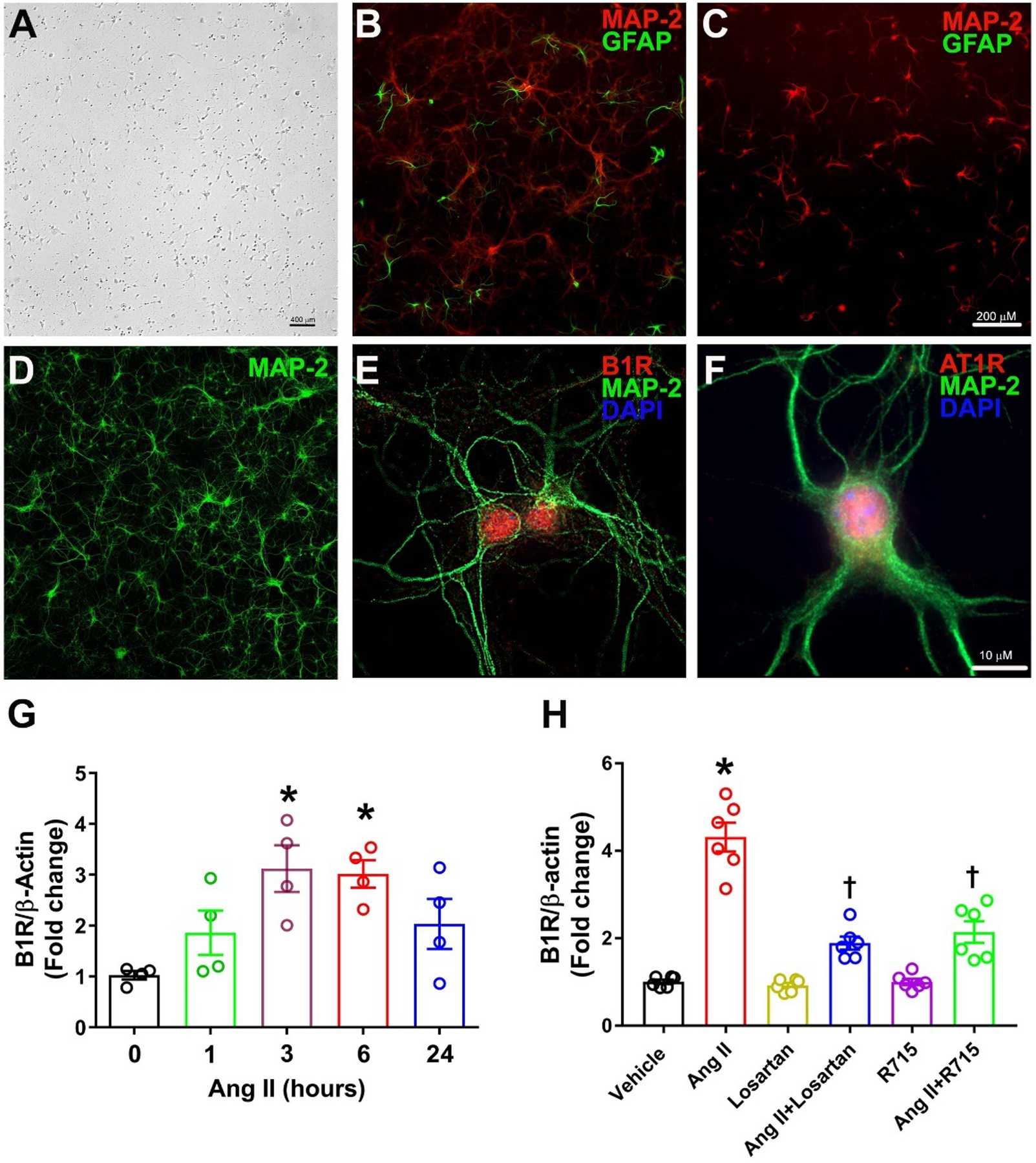
Brightfield photomicrograph showing primary mouse hypothalamic neuron cultures grown for 5 days (A). Representative photomicrographs showing immunofluorescence staining for neuron specific marker microtubule associated protein 2, MAP-2 (Red) and glial cell specific marker glial fibrillary acidic protein, GFAP (Green) in primary neurons cultured for 10 days without Ara-C (B) and with Ara-C (C) treatment. Treatment with Ara-C for 14 days resulted in primarily predominant neuronal population as stained for neuronal marker MAP-2 (D). Triple immunostaining revealed that Kinin B1R (E) and AT1R (F) are expressed in primary hypothalamic neurons. Treatment with angiotensin II (300 nM) induced increase in B1R mRNA levels in cultured neurons, measured by real time PCR (G). (n=4 independent cultures/group). Statistical significance: One-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05 compared 0-hour group. Angiotensin II-induced increase in B1R mRNA was prevented by pre-treatment with losartan or R715 (H). The cultured primary neurons are pre-treated with a specific AT1R antagonist (losartan, 10 μM) or a specific B1R antagonist (R715, 10 μM) for 1 hour, followed by treatment with angiotensin II (Ang II, 300 nM) for 6 hours. (n=6 independent cultures/group). Statistical significance: One-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05 compared to vehicle, †p<0.05 compared to Ang II.
The B1R Mediates the Increase in the Expression of Proinflammatory Mediators Induced by Ang II in Primary Hypothalamic Neurons
Both the AT1R and the B1R have been shown to be independently involved in neuroinflammation and neurogenic hypertension. To determine whether a blockade of the B1R prevents Ang II-induced expression of proinflammatory mediators we incubated the cells with 300 nM Ang II for 6 hours with or without a preincubation of 1 hour with 10 μM R715 and quantified the steady state levels of the mRNA of cyclooxygenase 2 (Cox2), interleukin-1β (IL-1β), IL-6 and tumor necrosis factor α (TNFα). The results presented in Fig 2 illustrates that preincubation with R715 prevents the increase in expression of the four proinflammatory mediators tested (Fig 2A, 2B, 2C and 2D).
Fig. 2. Angiotensin II-induced inflammatory gene expression is reduced by treatment with B1R antagonist in primary hypothalamic neurons.
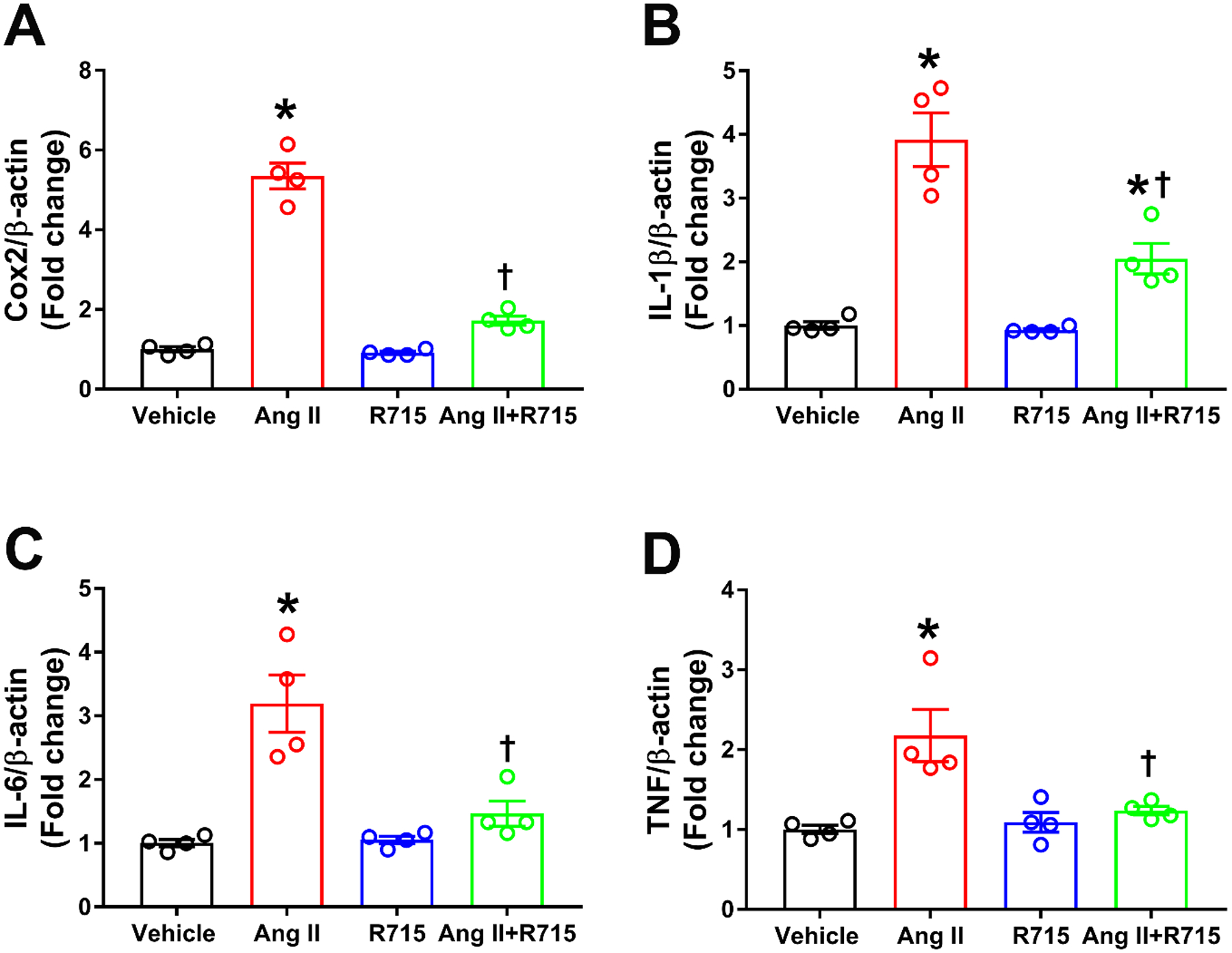
Angiotensin II treatment induced neuroinflammation as indicated by increase in pro-inflammatory genes (A) cyclooxygenase (Cox2), (B) Interleukin (IL)-1β, (C) IL-6, and (D) tumor necrosis factor (TNF). This increase in neuroinflammation was prevented by pre-treatment with R715. The cultured primary neurons are pre-treated with a specific B1R antagonist (R715, 10 μM) for 1 hour, followed by treatment with angiotensin II (Ang II, 300 nM) for 6 hours. The gene expression was measured using real time RT-PCR in triplicates. (n=4 independent cultures/group). Statistical significance: One-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05 compared to vehicle, †p<0.05 compared to Ang II.
The B1R Mediates the Increase in the Oxidative Potential as Well as the Expression of NADPH oxidase 2 (Nox2) and Nox4 Induced by Ang II in Primary Hypothalamic Neurons
The importance of reactive oxygen species (ROS) or more generally oxidants in the pathophysiology of neurogenic hypertension has been suggested (Chan et al. 2009; Zimmerman et al. 2004). To determine whether the B1R are also involved in oxidant production in cultured primary hypothalamic neurons, dihydroethidium (DHE) a molecule that can readily be oxidized by various cellular oxidants was used to measure the oxidative potential of the cells (Kalyanaraman et al. 2012). As shown in Fig 3a, Ang II exposure led to increased DHE red fluorescence (2-OH-ethidium and ethidium) indicating increased oxidative potential. The quantification of DHE florescence showed significant increase in CTCF by Ang II stimulation which was attenuated by pretreatment with R715 (Fig 3b). We further confirmed our DHE staining results with a microplate DHE assay. Using this method, we showed that stimulation with Ang II produced a significant 3-fold increase in ROS production (Fig 3c). This Ang II-induced ROS production was greatly attenuated by pretreatment with B1R antagonist. In the DHE plate assay, antimycin A, an inhibitor of complex III of the mitochondrial electron transport chain was used as a positive control as blocking complex III should lead to a progressive increase in ROS and reactive nitrogen species levels, which should be largely prevented by N-Acetyl-L-Cysteine (NAC).
Fig. 3. B1R antagonist treatment reduced angiotensin II induced reactive oxygen species (ROS) production in primary hypothalamic neurons.
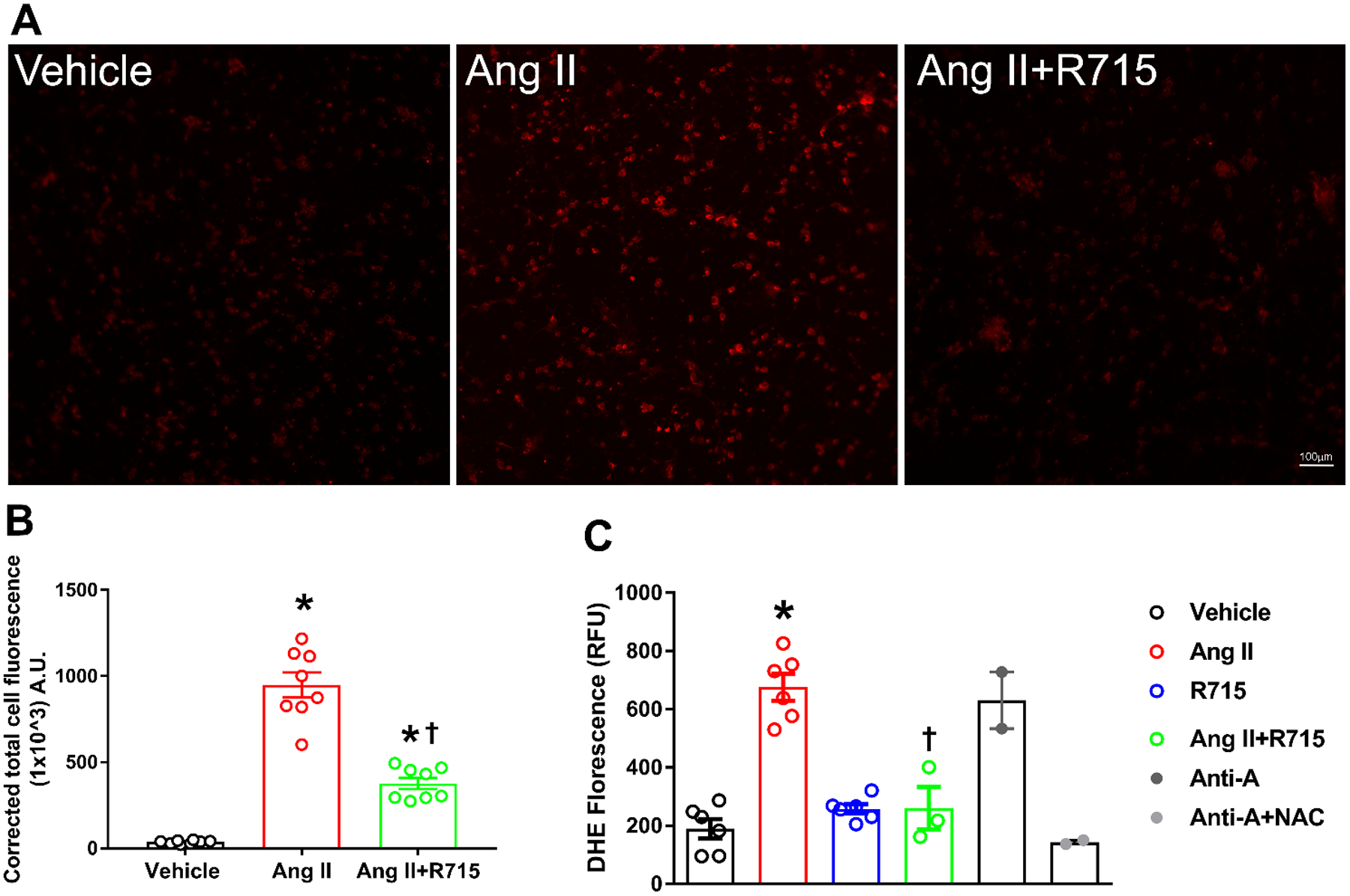
(A). Representative photomicrographs showing dihydroethidium (DHE) stained primary hypothalamic neurons. (B). DHE staining quantified as corrected total cell fluorescence shows increased superoxide generation by treatment with angiotensin II (Ang II), which was attenuated by R715 treatment. (n=8/group). Statistical significance: One-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05 compared to vehicle. †p<0.05 compared to Ang II (C). ROS production measured by microplate DHE assay indicates that Ang II-induced a significant increase in total ROS production indicating increased oxidative stress, and treatment with R715 prevented this Ang II-induced ROS production. (n=6 independent culture wells/group). Treatment with antimycin A (Ani-A), an inhibitor of complex III of the mitochondrial electron transport chain, is used as a positive control for ROS generation and treatment with N-Acetyl-L-Cysteine (NAC) is used as an antioxidant control. Statistical significance: One-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05 compared to vehicle, †p<0.05 compared to Ang II.
NADPH oxidases have been implicated in the ROS production caused by Ang II in neurons (Case et al. 2013; Peterson et al. 2009). However, the role of the B1R in Ang-II regulated Nox2 and Nox4 expression have not been investigated. As shown in Fig 4, R715 largely prevented Ang II induction of the expression of Nox2 (Fig 4a) and Nox4 (Fig 4b). Overall, our results provide evidence that in primary cultured hypothalamic neurons Ang II-induced ROS production is mediated at least in part through activation of the B1R and suggest that it may involve an increase in the expression of Nox2 and Nox4.
Fig. 4. Angiotensin II-induced Nox2 and Nox4 gene expression was reduced by treatment with B1R antagonist in primary hypothalamic neurons.
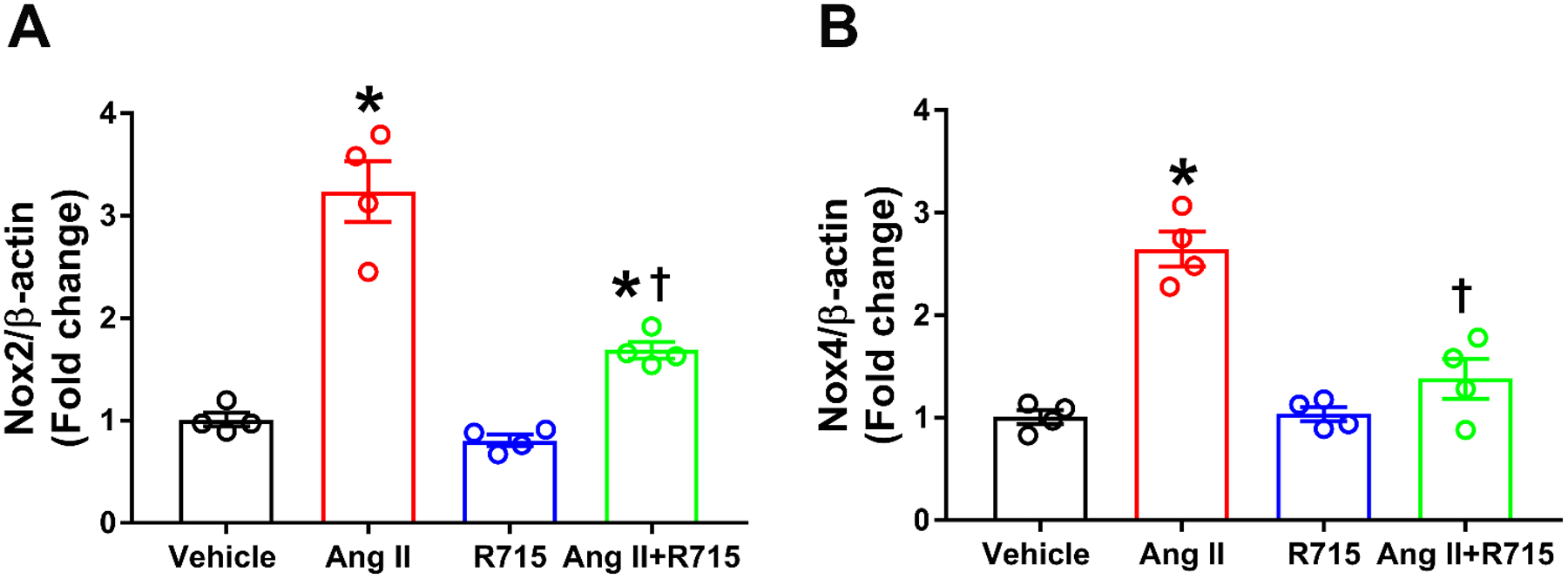
Angiotensin II treatment induced oxidative stress by increased gene expression of (A) Nox2 and (B) Nox4. This increase was attenuated or prevented by pre-treatment with R715. The cultured primary neurons are pre-treated with a specific B1R antagonist (R715, 10 μM) for 1 hour, followed by treatment with angiotensin II (Ang II, 300 nM) for 6 hours. The gene expression was measured using real time RT-PCR in triplicates. (n=4 independent cultures/group). Statistical significance: One-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05 compared to vehicle, †p<0.05 compared to Ang II.
The B1R is Largely Responsible for the Decrease Mitochondrial Respiration Elicited by Ang II in Primary Hypothalamic Neurons
A decrease in mitochondrial bioenergetic efficiency has been suggested in Ang II induced neurogenic hypertension (Zimmerman et al. 2004). To investigate if Ang II impacts mitochondrial respiration, a reasonable assumption in cells experiencing such an increase in oxidative potential, we measured the oxygen consumption rate (Fig 5). Primary hypothalamic neurons were treated for 24 h with 300 nM Ang II with or without a 1-h pretreatment with 10 μM R715. As illustrated in Fig. 5a, Ang II decreased basal and FCCP-driven maximal respiration. Although not as clean as in isolated mitochondria where the substrates and ADP can be provided in a nearly unlimited amount, the observed decrease in oxygen consumption in the presence of FCCP is highly suggestive of a defect at the level of the electron transport chain. This is also consistent with a generalized decrease in all calculated respiratory components including basal respiration (Fig. 5b), the respiration driven by the ATP regeneration need (Fig. 5c), maximal respiration (Fig. 5d), and spare respiratory capacity (Fig. 5E). These effects of Ang II were all partly (~50–60%) prevented by the pretreatment with R715 suggesting a role the B1R in Ang II-induced mitochondrial dysfunction.
Fig. 5. B1R antagonist treatment prevented angiotensin II-induced decrease in mitochondrial respiration in primary hypothalamic neurons.
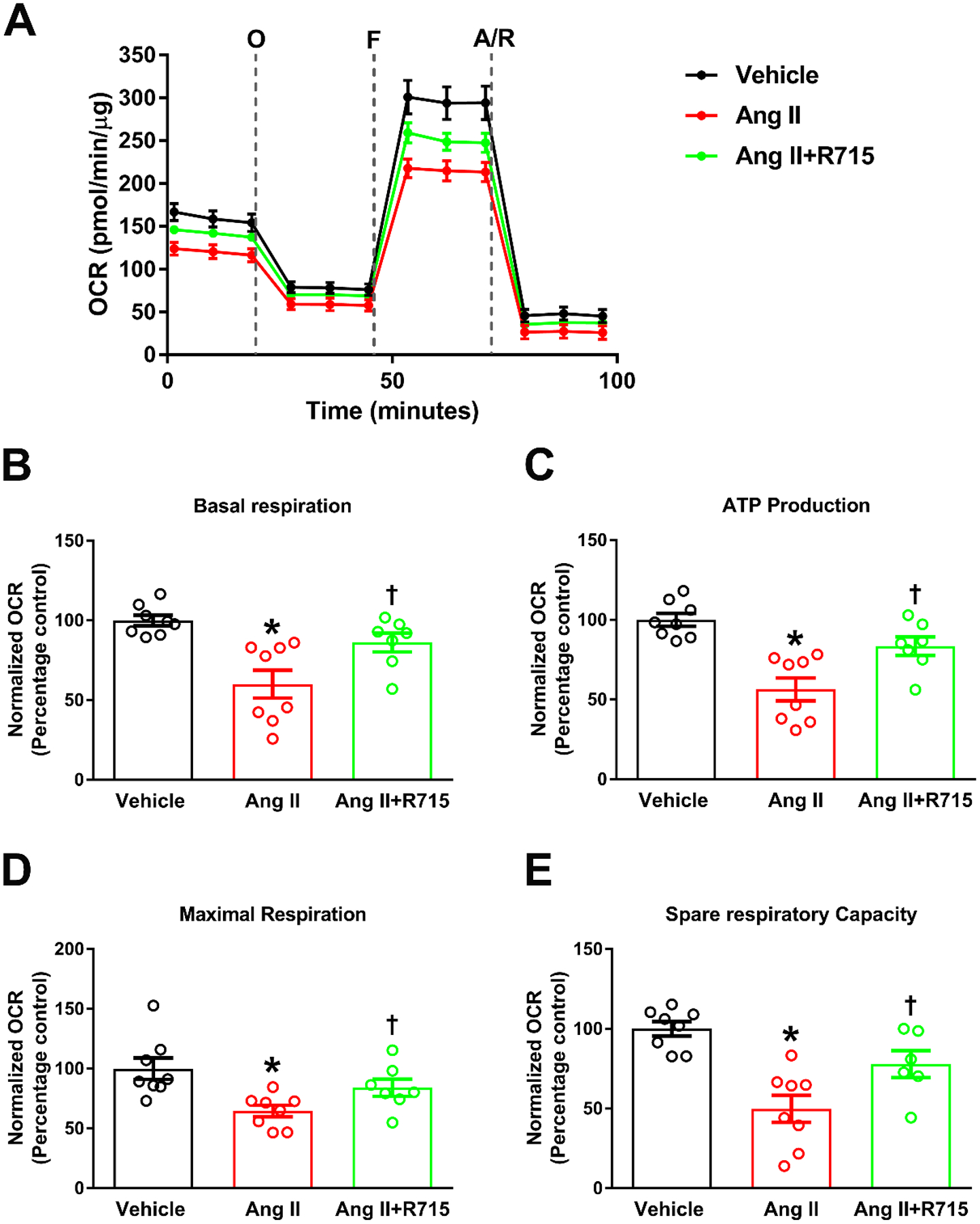
Primary hypothalamic mouse neurons were cultured on Seahorse XF-24 plates and treated with vehicle or Ang II with or without R715 for 24 hours. Seahorse mito stress assay was performed to measure oxygen consumption rate (OCR). (A). Bioenergetic profile following sequential injection of oligomycin (O), FCCP (F) and antimycin A/rotenone (A/R) indicating the key parameters of mitochondrial respiration in neurons. OCR measurements showed that Ang II stimulation resulted in significant decrease in basal respiration (B), ATP production (C), maximal respiration (D), and spare respiratory capacity (E) indicating mitochondrial dysfunction, and treatment with R715 prevented this Ang II-induced mitochondrial dysfunction. (n=8 independent culture wells/group). Statistical significance: One-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05 compared to vehicle, †p<0.05 compared to Ang II.
The B1R mediates angiotensin II-induced NF-κB binding activity in primary hypothalamic neurons
The AT1R has been shown to be able to promote NF-κB activation in vivo and in vitro (Kalra et al. 2002; Kang et al. 2009; Mariappan et al. 2010). Because we hypothesized that NF-κB in the most likely common denominator with respect to the induction Cox2, IL-1β, IL-6 and TNF in our cells, we tested the hypothesis that the B1R is involved in these effects of Ang II. The cultured primary hypothalamic neurons were pretreated or not with R715 for 1 h, followed by treatment with vehicle or Ang II for 24 h. NF-κB DNA binding activity was analyzed by the NF-kB p65 transcription factor assay kit. The results showed that treatment with Ang II significantly increased NF-kB p65 binding activity compared to the vehicle treated control group (Fig. 6). R715 treatment alone did not have any effect on NF-κB activity. Ang II-induced NF-κB activity was significantly attenuated by the treatment with R715 indicating that B1R activation is at least in part is involved in Ang II-mediated NF-κB activation in primary neurons.
Fig. 6. B1R antagonist treatment reduced angiotensin II induced NF-κB binding activity in primary hypothalamic neurons.
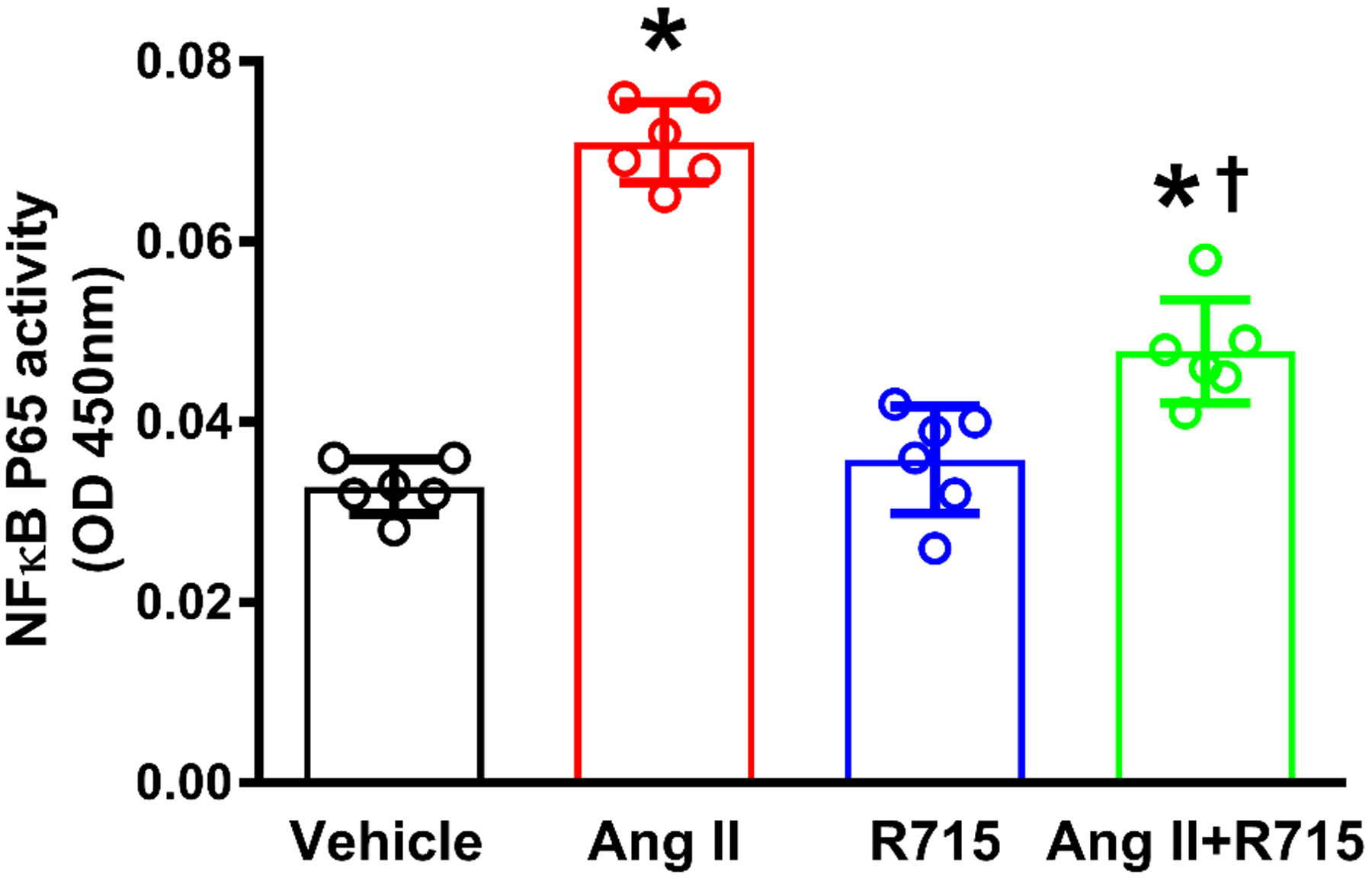
The cultured primary neurons are pre-treated with B1R specific antagonist (R715, 10 μM) for 1 hour, followed by treatment with vehicle or angiotensin II (Ang II, 300 nM) for 24 hours. NF-kB DNA binding activity was analyzed by NF-kB p65 transcription factor assay kit. The result of NF-kB DNA binding showed that Ang II treatment significant increased NF-kB p65 DNA binding activity in primary neurons, and treatment with R715 significantly attenuated this effect. (n=6 independent culture wells/group). Statistical significance: One-way ANOVA followed by Tukey’s multiple comparisons test. *p<0.05 compared to vehicle, †p<0.05 compared to Ang II.
Discussion
The primary neuronal cells provide a standard system for the investigation of neuronal structure and function at a high resolution compared to immortalized cells. Previous studies have been shown that immortalized neuronal cell lines derived from tumors differ biologically from normal, differentiated primary neurons (Edwards et al. 2007; LePage et al. 2005). The hypothalamus contains pivotal cardiovascular regulatory nuclei including subfornical organ and paraventricular nucleus (PVN) that have been shown to be play a role in blood pressure homeostasis. Therefore, in this study we have used primary hypothalamic neuronal cultures to examine the interaction between B1R and AT1R.
We and others have previously shown that B1R expression is significantly upregulated in brain cardiovascular regulatory nuclei during DOCA-salt hypertension (Sriramula and Lazartigues 2017) or in spontaneously hypertensive rats (Cloutier et al. 2004; Qadri et al. 2002). Using a double immunolabeling of B1R and a neuronal marker - microtubule associated protein 2, we demonstrated that B1R expression was upregulated specifically in neurons of hypothalamic PVN during neurogenic hypertension (Sriramula and Lazartigues 2017). It is well established that elevated brain Ang II levels and upregulated AT1R can increase neuroinflammation and lead to the development of neurogenic hypertension (Xia et al. 2013; Xu et al. 2017). However, there has been no study investigating the direct interaction of AT1R, the main receptor of Ang II, and B1R, in hypothalamic neurons in inducing neuroinflammation. In the present study, we demonstrate that Ang II stimulation induces B1R gene expression in mouse primary hypothalamic neurons and treatment with pharmacological B1R antagonist R715 blunts this B1R expression suggesting a direct effect of Ang II on B1R gene expression. In addition, pretreatment with AT1R antagonist losartan attenuated Ang II-mediated increase in gene expression of B1R suggesting the involvement of AT1R in this increased B1R expression. Our data is similar to a previous study which showed that in vascular smooth muscle cells enhanced expression of B1R by Ang II is mediated by AT1R possibly via activation of phosphatidylinositol 3-kinase and NF-κB (Morand-Contant et al. 2010). A growing body of evidence suggest that cross talk exists between RAS and kinins in multiple physiological and pathological conditions (Ceravolo et al. 2007; Ceravolo et al. 2014; Fernandes et al. 2006). It has been shown that chronic infusion of Ang II for 14 days can increase B1R expression in aorta of Wistar rats (Ceravolo et al. 2007). In 2 kidneys-1 clip hypertensive rats with elevated plasma Ang II levels, B1R mRNA levels were upregulated in cardiac but not in renal and aortic tissues, suggesting a tissue-specific induction of B1R expression (Fernandes et al. 2006). Thus, expression of B1R and AT1R in primary hypothalamic neurons and induction of B1R by Ang II treatment support the evidence that B1R within the neurons play an important role in Ang II-mediated hypertension.
A previous study has shown that treatment with LPS upregulated B1R and Cox2 mRNA, and this B1R upregulation via Cox2 resulted in endothelium-independent contractions in endotoxin treated pig coronary arteries (More et al. 2014). In cultured vascular smooth muscle cells, Ang II induces Cox2 expression in a time-and concentration- dependent manner (Morinelli et al. 2008). In addition, treatment with Ang II resulted in increased inflammatory cytokines TNF, IL-1β, and IL-6 production in CATH.a neuronal cell line (Agarwal et al. 2013). However, whether Ang II-induced inflammatory cytokine expression is mediated by B1R is not known. Our data show that blocking B1R prevents the Ang II-induced inflammatory gene expression in primary neurons and support the hypothesis that Ang-II induced neuroinflammation is indeed mediated by B1R expression.
Increasing evidence implicates oxidative stress and mitochondrial dysfunction within the brain in neurogenic hypertension. Previous evidence suggest that mitochondria are a major subcellular cite for superoxide generation in neurons stimulated with Ang II (Chan et al. 2009; Yin et al. 2010). In this study, we found that elevated oxidative stress and reduced mitochondrial respiration by Ang II treatment was abrogated by B1R blockade. The increased ROS production in primary neurons might be from mitochondrial electron transport complexes or from mitochondrial Nox4, or both. Indeed, a study by Case et al., proposed that Nox4 is a major source of superoxide production in neuron mitochondria that contributes to Ang II intraneuronal signaling (Case et al. 2013). In addition, contribution from extramitochondrial sites, possibly via Nox2 upregulation can also be implicated in Ang II-induced oxidative stress (Peterson et al. 2009). Since Ang II-induced oxidative stress was mediated by B1R, blocking B1R signaling might be a target to prevent ROS production, reduce oxidative stress, and improve mitochondrial bioenergetics. Indeed, our recent report that B1R blockade prevents hypertension by reducing oxidative stress in the brain hypothalamic PVN supports this hypothesis (Sriramula and Lazartigues 2017).
Ang II activates multiple signaling pathways involving p38 mitogen activated protein kinase leading to activation of NF-κB and cAMP response element binding protein, which feedback to upregulate the AT1R gene expression in neurons (Haack et al. 2013). B1R expression shown to be involved in the activation of NF-κB pathway in human (Moreau et al. 2007) and rat (Morand-Contant et al. 2010) vascular smooth muscle cells. In our study, treatment with R715 attenuated Ang II-induced NF-κB activity indicating that B1R is at least in part is involved in Ang II-mediated NF-κB activation in neurons. This result is in agreement with our previous in vivo study where mice DOCA-salt hypertension induced increase in nuclear NF-κB expression and decrease in cytosolic NF-κB expression in the hypothalamic PVN while mice with B1R gene deletion prevented these changes (Sriramula and Lazartigues 2017). These data further support the evidence that blocking B1R activation prevents the NF-κB activation and expression of inflammatory genes mediated by AT1R, thereby decreasing neuroinflammation.
In summary, to our knowledge, these data provide first direct evidence for involvement of B1R signaling in neuroinflammation induced by Ang II in mouse neonatal primary hypothalamic neurons (Fig. 7). We first demonstrated that the cultured primary hypothermic neurons express both B1R and AT1R. Then, we have demonstrated that Ang II stimulation of neurons increased expression of B1R and resulted in elevated inflammatory cytokine production, which was blunted by R715 treatment. Furthermore, we have shown that AngII is associated with impaired mitochondrial respiratory function and increased oxidative stress, and B1R antagonism preserved the mitochondrial respiratory function and prevented oxidative stress. Although the current study identifies causal relationship and the involvement of B1R in Ang-II mediated neuroinflammation and oxidative stress in neurons, whether there is a direct interaction between AT1R and B1R in mediating these effects is still not clear. Further studies are needed to clarify this interaction. Nevertheless, our results present evidence to support the beneficial effects of B1R blockade in elevated neuronal inflammation and oxidative stress by Ang II treatment in primary hypothalamic neurons.
Fig 7. Angiotensin II-induced neuroinflammation and oxidative stress are mediated by kinin B1R.
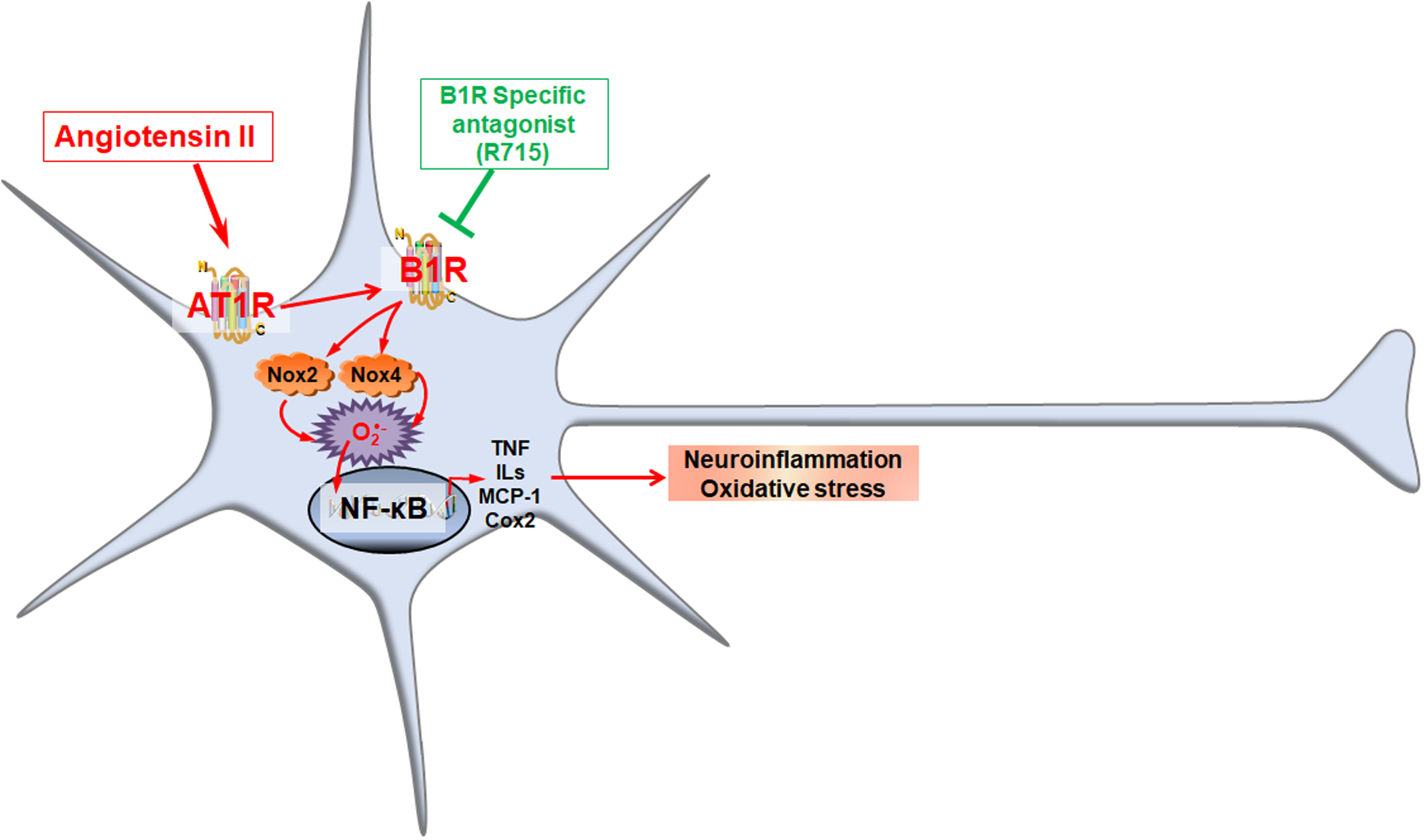
Mouse neonatal primary hypothermic neurons express both B1R and AT1R. Ang II stimulation of neurons increased expression of B1R, which in turn increased resulted in upregulation of Nox2 and Nox4 expression, and NF-κB activation, and ultimately leading to expression of pro-inflammatory cytokine production. Treatment with R715, the specific B1R antagonist blunted angiotensin II-induced neuroinflammation and oxidative stress by reducing Nox gene expression and attenuating NF-κB activation.
Funding
This work was supported, in part, by The American Heart Association Scientist Development Grant (15SDG25720021), and startup funds from the East Carolina University Division of Research Economic Development and Engagement and the Department of Pharmacology and Toxicology to S. Sriramula.
Footnotes
Conflicts of interest: The authors report no conflicts of interest related to the study.
Ethical Approval: This article does not contain any studies with human participants. The experimental protocols used for breeding mice were approved by East Carolina University Animal Care and Use Committee (AUP #W254) and were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
References
- Abbott NJ (2000) Inflammatory mediators and modulation of blood-brain barrier permeability Cell Mol Neurobiol 20:131–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal D, Dange RB, Raizada MK, Francis J (2013) Angiotensin II causes imbalance between pro- and anti-inflammatory cytokines by modulating GSK-3beta in neuronal culture Br J Pharmacol 169:860–874 doi: 10.1111/bph.12177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A (2013) Glial influence on the blood brain barrier Glia 61:1939–1958 doi: 10.1002/glia.22575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoloni E et al. (2017) Role of Inflammatory Diseases in Hypertension High Blood Press Cardiovasc Prev 24:353–361 doi: 10.1007/s40292-017-0214-3 [DOI] [PubMed] [Google Scholar]
- Campos MM, Souza GE, Calixto JB (1996) Upregulation of B1 receptor mediating des-Arg9-BK-induced rat paw oedema by systemic treatment with bacterial endotoxin Br J Pharmacol 117:793–798 doi: 10.1111/j.1476-5381.1996.tb15262.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos MM, Souza GE, Calixto JB (1998) Modulation of kinin B1 but not B2 receptors-mediated rat paw edema by IL-1beta and TNFalpha Peptides 19:1269–1276 doi: 10.1016/s0196-9781(98)00087-4 [DOI] [PubMed] [Google Scholar]
- Case AJ, Li S, Basu U, Tian J, Zimmerman MC (2013) Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons Am J Physiol Heart Circ Physiol 305:H19–28 doi: 10.1152/ajpheart.00974.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceravolo GS et al. (2007) Angiotensin II chronic infusion induces B1 receptor expression in aorta of rats Hypertension 50:756–761 doi: 10.1161/HYPERTENSIONAHA.107.094706 [DOI] [PubMed] [Google Scholar]
- Ceravolo GS et al. (2014) An interaction of renin-angiotensin and kallikrein-kinin systems contributes to vascular hypertrophy in angiotensin II-induced hypertension: in vivo and in vitro studies PLoS One 9:e111117 doi: 10.1371/journal.pone.0111117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Wu KL, Chang AY, Tai MH, Chan JY (2009) Oxidative impairment of mitochondrial electron transport chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertension Hypertension 53:217–227 doi: 10.1161/HYPERTENSIONAHA.108.116905 [DOI] [PubMed] [Google Scholar]
- Chen D et al. (2012) Angiotensin type 1A receptors in C1 neurons of the rostral ventrolateral medulla modulate the pressor response to aversive stress J Neurosci 32:2051–2061 doi: 10.1523/JNEUROSCI.5360-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chobanian AV (2009) Shattuck Lecture. The hypertension paradox--more uncontrolled disease despite improved therapy N Engl J Med 361:878–887 doi: 10.1056/NEJMsa0903829 [DOI] [PubMed] [Google Scholar]
- Cloutier F, Ongali B, Campos MM, Thibault G, Neugebauer W, Couture R (2004) Correlation between brain bradykinin receptor binding sites and cardiovascular function in young and adult spontaneously hypertensive rats Br J Pharmacol 142:285–296 doi: 10.1038/sj.bjp.0705759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Brito Gariepy H, Talbot S, Senecal J, Couture R (2013) Brain kinin B(1) receptor contributes to the onset of stereotypic nocifensive behavior in rat Behav Brain Res 241:17–26 doi: 10.1016/j.bbr.2012.11.032 [DOI] [PubMed] [Google Scholar]
- de Kloet AD et al. (2016) Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system Brain Struct Funct 221:891–912 doi: 10.1007/s00429-014-0943-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MA, Loxley RA, Williams AJ, Connor M, Phillips JK (2007) Lack of functional expression of NMDA receptors in PC12 cells Neurotoxicology 28:876–885 doi: 10.1016/j.neuro.2007.04.006 [DOI] [PubMed] [Google Scholar]
- Erickson MA, Dohi K, Banks WA (2012) Neuroinflammation: a common pathway in CNS diseases as mediated at the blood-brain barrier Neuroimmunomodulation 19:121–130 doi: 10.1159/000330247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes L et al. (2006) Modulation of kinin B1 receptor expression by endogenous angiotensin II in hypertensive rats Regul Pept 136:92–97 doi: 10.1016/j.regpep.2006.04.018 [DOI] [PubMed] [Google Scholar]
- Gonzalez AD et al. (2012) Distribution of angiotensin type 1a receptor-containing cells in the brains of bacterial artificial chromosome transgenic mice Neuroscience 226:489–509 doi: 10.1016/j.neuroscience.2012.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack KK, Mitra AK, Zucker IH (2013) NF-kappaB and CREB are required for angiotensin II type 1 receptor upregulation in neurons PLoS One 8:e78695 doi: 10.1371/journal.pone.0078695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspula D, Clark MA (2018) Neuroinflammation and sympathetic overactivity: Mechanisms and implications in hypertension Auton Neurosci 210:10–17 doi: 10.1016/j.autneu.2018.01.002 [DOI] [PubMed] [Google Scholar]
- Kalra D, Sivasubramanian N, Mann DL (2002) Angiotensin II induces tumor necrosis factor biosynthesis in the adult mammalian heart through a protein kinase C-dependent pathway Circulation 105:2198–2205 doi: 10.1161/01.cir.0000015603.84788.47 [DOI] [PubMed] [Google Scholar]
- Kalyanaraman B et al. (2012) Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations Free Radic Biol Med 52:1–6 doi: 10.1016/j.freeradbiomed.2011.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM, Francis J (2009) Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension Cardiovasc Res 82:503–512 doi: 10.1093/cvr/cvp073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL (2005) International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences Pharmacol Rev 57:27–77 doi: 10.1124/pr.57.1.2 [DOI] [PubMed] [Google Scholar]
- LePage KT, Dickey RW, Gerwick WH, Jester EL, Murray TF (2005) On the use of neuro-2a neuroblastoma cells versus intact neurons in primary culture for neurotoxicity studies Crit Rev Neurobiol 17:27–50 [DOI] [PubMed] [Google Scholar]
- Li JJ, Lu J, Kaur C, Sivakumar V, Wu CY, Ling EA (2009) Expression of angiotensin II and its receptors in the normal and hypoxic amoeboid microglial cells and murine BV-2 cells Neuroscience 158:1488–1499 doi: 10.1016/j.neuroscience.2008.11.046 [DOI] [PubMed] [Google Scholar]
- Li W et al. (2012) Brain-targeted (pro)renin receptor knockdown attenuates angiotensin II-dependent hypertension Hypertension 59:1188–1194 doi: 10.1161/HYPERTENSIONAHA.111.190108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W et al. (2014) Neuron-specific (pro)renin receptor knockout prevents the development of salt-sensitive hypertension Hypertension 63:316–323 doi: 10.1161/HYPERTENSIONAHA.113.02041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan N, Elks CM, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, Francis J (2010) NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes Cardiovasc Res 85:473–483 doi: 10.1093/cvr/cvp305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean AD (2000) Extraordinary case of shoulder trauma caused by a foreign bone J Trauma 49:793 doi: 10.1097/00005373-200010000-00045 [DOI] [PubMed] [Google Scholar]
- Miyoshi M, Miyano K, Moriyama N, Taniguchi M, Watanabe T (2008) Angiotensin type 1 receptor antagonist inhibits lipopolysaccharide-induced stimulation of rat microglial cells by suppressing nuclear factor kappaB and activator protein-1 activation Eur J Neurosci 27:343–351 doi: 10.1111/j.1460-9568.2007.06014.x [DOI] [PubMed] [Google Scholar]
- Morand-Contant M, Anand-Srivastava MB, Couture R (2010) Kinin B1 receptor upregulation by angiotensin II and endothelin-1 in rat vascular smooth muscle cells: receptors and mechanisms Am J Physiol Heart Circ Physiol 299:H1625–1632 doi: 10.1152/ajpheart.00735.2009 [DOI] [PubMed] [Google Scholar]
- More AS et al. (2014) Des-Arg9-bradykinin causes kinin B1 receptor mediated endothelium-independent contractions in endotoxin-treated porcine coronary arteries Pharmacol Res 90:18–24 doi: 10.1016/j.phrs.2014.09.001 [DOI] [PubMed] [Google Scholar]
- Moreau ME, Bawolak MT, Morissette G, Adam A, Marceau F (2007) Role of nuclear factor-kappaB and protein kinase C signaling in the expression of the kinin B1 receptor in human vascular smooth muscle cells Mol Pharmacol 71:949–956 doi: 10.1124/mol.106.030684 [DOI] [PubMed] [Google Scholar]
- Morinelli TA, Walker LP, Ullian ME (2008) COX-2 expression stimulated by Angiotensin II depends upon AT1 receptor internalization in vascular smooth muscle cells Biochim Biophys Acta 1783:1048–1054 doi: 10.1016/j.bbamcr.2008.01.012 [DOI] [PubMed] [Google Scholar]
- Mowry FE, Biancardi VC (2019) Neuroinflammation in hypertension: the renin-angiotensin system versus pro-resolution pathways Pharmacol Res 144:279–291 doi: 10.1016/j.phrs.2019.04.029 [DOI] [PubMed] [Google Scholar]
- Peterson JR et al. (2009) Genetic silencing of Nox2 and Nox4 reveals differential roles of these NADPH oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II Hypertension 54:1106–1114 doi: 10.1161/HYPERTENSIONAHA.109.140087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadri F, Hauser W, Johren O, Dominiak P (2002) Kinin B1 and B2 receptor mRNA expression in the hypothalamus of spontaneously hypertensive rats Can J Physiol Pharmacol 80:258–263 doi: 10.1139/y02-051 [DOI] [PubMed] [Google Scholar]
- Raidoo DM, Bhoola KD (1997) Kinin receptors on human neurones J Neuroimmunol 77:39–44 doi: 10.1016/s0165-5728(97)00048-9 [DOI] [PubMed] [Google Scholar]
- Saavedra JM (2017) Beneficial effects of Angiotensin II receptor blockers in brain disorders Pharmacol Res 125:91–103 doi: 10.1016/j.phrs.2017.06.017 [DOI] [PubMed] [Google Scholar]
- Sriramula S, Lazartigues E (2017) Kinin B1 Receptor Promotes Neurogenic Hypertension Through Activation of Centrally Mediated Mechanisms Hypertension 70:1122–1131 doi: 10.1161/HYPERTENSIONAHA.117.09744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramula S, Xia H, Xu P, Lazartigues E (2015) Brain-targeted angiotensin-converting enzyme 2 overexpression attenuates neurogenic hypertension by inhibiting cyclooxygenase-mediated inflammation Hypertension 65:577–586 doi: 10.1161/HYPERTENSIONAHA.114.04691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis CL (2011) Glia-induced reversible disruption of blood-brain barrier integrity and neuropathological response of the neurovascular unit Toxicol Pathol 39:172–185 doi: 10.1177/0192623310385830 [DOI] [PubMed] [Google Scholar]
- Winklewski PJ, Radkowski M, Wszedybyl-Winklewska M, Demkow U (2015) Brain inflammation and hypertension: the chicken or the egg? J Neuroinflammation 12:85 doi: 10.1186/s12974-015-0306-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY et al. (2013) Expression of angiotensin II and its receptors in activated microglia in experimentally induced cerebral ischemia in the adult rats Mol Cell Biochem 382:47–58 doi: 10.1007/s11010-013-1717-4 [DOI] [PubMed] [Google Scholar]
- Xia H, Sriramula S, Chhabra KH, Lazartigues E (2013) Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension Circ Res 113:1087–1096 doi: 10.1161/CIRCRESAHA.113.301811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Sriramula S, Lazartigues E (2018) Excessive Glutamate Stimulation Impairs ACE2 Activity Through ADAM17-Mediated Shedding in Cultured Cortical Neurons Cell Mol Neurobiol 38:1235–1243 doi: 10.1007/s10571-018-0591-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J et al. (2017) Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension Circ Res 121:43–55 doi: 10.1161/CIRCRESAHA.116.310509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Thunhorst RL, Yu Y, Guo F, Beltz TG, Felder RB, Johnson AK (2016) Central Renin-Angiotensin System Activation and Inflammation Induced by High-Fat Diet Sensitize Angiotensin II-Elicited Hypertension Hypertension 67:163–170 doi: 10.1161/HYPERTENSIONAHA.115.06263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JX, Yang RF, Li S, Renshaw AO, Li YL, Schultz HD, Zimmerman MC (2010) Mitochondria-produced superoxide mediates angiotensin II-induced inhibition of neuronal potassium current Am J Physiol Cell Physiol 298:C857–865 doi: 10.1152/ajpcell.00313.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL (2004) Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system Circ Res 95:210–216 doi: 10.1161/01.RES.0000135483.12297.e4 [DOI] [PubMed] [Google Scholar]
- Zubcevic J et al. (2017) A Single Angiotensin II Hypertensive Stimulus Is Associated with Prolonged Neuronal and Immune System Activation in Wistar-Kyoto Rats Front Physiol 8:592 doi: 10.3389/fphys.2017.00592 [DOI] [PMC free article] [PubMed] [Google Scholar]