

Abstract
Designing an effective vaccine against different subtypes of Influenza A virus is a critical issue in the field of medical biotechnology. At the current study, a novel potential multi-epitope vaccine candidate based on the neuraminidase proteins for seven subtypes of Influenza virus was designed, using the in silico approach. Potential linear B-cell and T-cell binding epitopes from each neuraminidase protein (N1, N2, N3, N4, N6, N7, N8) were predicted by in silico tools of epitope prediction. The selected epitopes were joined by three different linkers, and physicochemical properties, toxicity, and allergenecity were investigated. The final multi-epitope construct was modeled using GalaxyWEB server, and the molecular interactions with immune receptors were investigated and the immune response simulation assay was performed. A multi-epitope construct with GPGPGPG linker with the lowest allergenicity and highest stability was selected. The molecular docking assay indicated the interactions with immune system receptors, including HLA1, HLA2, and TLR-3. Immune response simulation detected both humoral and cellular response, including the elevated count of B-cells, T-cell, and Nk-cells.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40203-021-00095-w.
Keywords: In silico, Immunoinformatics, Multi-epitope vaccine, Influenza, Neuraminidase
Introduction
Influenza A viruses have a single-stranded RNA genome, and this virus has been isolated from different hosts (Chen and Holmes 2008). These viruses have been classified based on the structures of neuraminidase (NA) and hemagglutinin (HA) envelope glycoproteins. So far, eighteen HA (H1–H18) and eleven NA (N1–N11) subtypes have been discovered (Park et al. 2020). Although, in general the epitopes of HA are antigenically more significant than NA, both proteins have the potential for being used in the vaccine studies (Kosik and Yewdell 2019; Huang et al. 2013; Wohlbold and Krammer 2014). NA is an essential surface glycoprotein, which plays a substantial role in viral replication. Previously, it has been suggested that NA could also be considered for developing a highly efficient Influenza A vaccine (Eichelberger and Wan 2014).
It has been reported that the stimulation of a strong antibody response against NA in animal models, which is independent from the HA-based immunity, could provide protection against the Influenza virus (Vigil et al. 2018). Previous studies have claimed that the level of stability of NAs from different strains is extremely variable (Sultana et al. 2014; Wohlbold et al. 2017). It has been demonstrated that human CD8+ T-cell provides cross-reactivity across Influenza A, B and C viruses (Koutsakos et al. 2019). Also, it has been reported that Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies (Chen et al. 2018). Production of epitope-based vaccines by extremely conserved regions of Influenza virus proteins is considered as an important way to control the Influenza virus.
Several in silico tools are available to enable the growth of the epitope-based vaccines. The computational programs enable to use a large immunological data; including antigen presentation and processing them to acquire sensitive interpretations. Currently, epitope-based vaccine design studies are facilitated via application of the applied bioinformatics tools; such as protein modeling programs, epitope mapping and protein–protein interaction analysis tools (Kumar et al. 2008).
Bioinformatics methods have been used to design and produce vaccines for different subtypes of Influenza A; including those designed based on H1N1, H2N2, H3N2, and H5N1 (Bulimo et al. 2012). At the current investigation, in silico analysis was carried out to predict the exclusive B cell and T-cell epitope proteins of neuraminidase (N1, N2, N3, N4, N6, N7, N8) that are antigenically most significant for Influenza A virus subtypes.
In this study, some specific B cell and T-cell epitopes from seven subtypes of NA protein were chosen according to their antigenicity, stability, and length. The chosen epitopes were joined together by different linkers to construct the final potential multi-epitope constructs, and different properties of the structures were predicted via in silico approaches.
Methodology
Data collection
At the first step, the reference amino acid sequences for seven NA proteins (N1: YP_009118627.1, N2: BAN37214.1, N:3 AAO62039.1, N:4 Q6XV43.1, N6: AAO62071.1, N7: BAH69254.1, and N8: BAH69255.1), five HLA-1 (Human leukocyte antigen-1) sequences (NP_001229971.1, NP_001229687.1, NP_002118.1, NP_061823.2, and NP_005507.3) and six HLA-2 (Human leukocyte antigen-2) proteins (NP_001229454.1, NP_006111.2, NP_001230891.1, NP_002110.1, NP_061984.2, and NP_001020330.1) were fetched from NCBI data bank (https://www.ncbi.nlm.nih.gov) in FASTA format. SWISS-MODEL tool (https://swissmodel.expasy.org/) was used for template based homology modelling of HLA-1 and HLA-2. In the case of Toll Like Receptor (TLR)-3, which was selected due to the importance of TLR3 receptor in immunogenic response to viral sensing (Vercammen et al. 2008; Schneider-Ohrum et al. 2011; Sharma et al. 2021), the structure in PDB bank (ID: 2A0Z) was chosen. All of the structures were prepared for molecular docking by using Chimera 1.12 (Pettersen et al. 2004).
Multiple sequence alignment and antigen selection
In order to choose the particular conserved sequences of NA virus proteins, NCBI protein BLAST was carried out (https://blast.ncbi.nlm.nih.gov/Blast.cgi) by using Blosums 62 matrix, max target sequence 100, and expect threshold 0.05. Moreover, to define the conserved region (s) in the protein sequences, multiple sequence alignment was carried out using Multalin server (https://www.multalin.toulouse.inra.fr/multalin) by using default properties (Blosums 62-12-2, High consensus value of 90%, and Low consensus value of 50%). The antigenicity of NA subtypes were evaluated using VaxiJen 2.0 tool available at http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html (for more information about the average antigenicity, the readers are referred to references (Doytchinova and Flower 2007a,b,c,2008)). At the end of this stage, the conserved sequences for N1, N2, N3, N4, N6, N7, N8 proteins and the antigenic peptides were selected for further analysis.
Prediction and classification of T-cell epitopes and linear B-cell epitopes
The MHC-I epitopes were predicted using IEDB MHC-I prediction tool (http://tools.immuneepitope.org/analyze/html/mhc_binding.html) (Peters and Sette 2005; Lundegaard et al. 2008). Similarly, the MHC-II epitopes were predicted by IEDB MHC-II prediction tool (http://tools.immuneepitope.org/mhcii) (Jensen et al. 2018). The most particular conserved and antigenic peptides were chosen for further investigations. The linear B-cell epitopes in the peptide model were predicted by ElliPro (http://crdd.osdd.net/raghava/bcepred) (Ponomarenko et al. 2008) and IEDB population coverage tool (http://tools.iedb.org/population) (Bui et al. 2006) analysis programs.
Peptide construct design and physicochemical properties
One 10 amino acids sequence from each of N1, N2, N3, N4, N6, and N7and N8 were selected as the epitope and organized into the final multi-epitope structure. At the next step, all of the seven epitopes were joined by three different linker sequences (GPGPGPG, GPGP and PAPAPA).
The antigenicity of epitopes and multi-epitope peptide constructs were evaluated by Predicting Antigenic Peptides program available at http://imed.med.ucm.es/Tools/antigenic.pl (for more information please refer to the reference (Kolaskar and Tongaonkar 1990)). At this assay, various characteristics,such as molecular weight, theoretical Isoelectric pH, extinction coefficient, aliphatic index and grand average of hydropathicity were investigated. Multi-epitope peptide constructs were analyzed using ProtParam program available at http://web.expasy.org/protparam (for more information, about these properties and the server, readers are referred to reference (Gasteiger et al. 2005)). Composition Based Protein Identification (COPid) is a program that predicts the structure of various types of amino acids and is accessible at http://www.imtech.res.in/raghava/copid (Kumar et al. 2008). The amino acid sequences of the constructs were compared via this online application. The overall content of aliphatic amino acids (Val, Pro, Ala, Gly, Met, Iso, and Leu) was also investigated.
The antigenicity of three multi-epitope structures were analyzed using AlgPred (http://crdd.osdd.net/raghava/algpred/) (Saha and Raghava 2006), and toxicity was checked using Toxin pred (http://crdd.osdd.net/raghava/toxinpred/) (Gupta et al. 2013). The structure of selected construct was assessed with PepCalc tool (https://pepcalc.com/) (Lear and Cobb 2016) for hydrophobicity and pH.
The tertiary structure of the constructed multi-epitope peptide was modeled by GalaxyWEB server (http://galaxy.seoklab.org/tbm) (HeeShin 2014). For such a predicted tertiary structure modeling, investigation of different aspects, such as secondary structure, Ramachandran plot, hydrophobicity, and Z-score was required prior to studying molecular interactions (Haghighi et al. 2019; Haghighi and Moradi 2020). The secondary construct of the finally selected peptide was investigated in PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html) (Laskowski 2001). Verification of Vac2 modeling was performed using Ramachandran plot from PROCHECK tool (https://servicesn.mbi.ucla.edu/PROCHECK/) (Laskowski et al. 2012).
The quality of the model was verified using Qmean (https://swissmodel.expasy.org/qmean/) (Benkert et al. 2009) and ProSA (https://prosa.services.came.sbg.ac.at/prosa.php) (Wiederstein and Sippl 2007) tools. Verify 3D (https://servicesn.mbi.ucla.edu/Verify3D/) (Eisenberg et al. 1997) and ERRAT (https://servicesn.mbi.ucla.edu/ERRAT/) (Colovos and Yeates 1993) servers were used to confirm the three-dimensional structure of predicted model.
Molecular docking and immune response simulation
In order to investigate the molecular interactions between the multi-epitope peptide construct to HLA-I, II and TLR3, molecular docking was performed between the selected peptide and five HLA-1 structures, six HLA-2 proteins, and TLR-3. Molecular docking studies were carried out using HADDOCK2.4 server (https://wenmr.science.uu.nl/haddock2.4/) (Zundert et al. 2016) by using the default complex type. C-IMMSIM tool was used for simulating the immune response toward the selected multi-epitope peptide construct (Rapin et al. 2010).
Results
Epitopes selection
Initially, NA protein sequences from seven subtypes of Influenza A virus (N1, N2, N3, N4, N6, N7, N8) were investigated to determine the conserved regions of each protein between these serotypes. Outcomes of protein BLAST for NA proteins are provided in Table 1. It was demonstrated that seven subtypes of NA had conservancy between 94.04 and 100. Additionally, the average antigenicity VaxiJen score of these seven proteins showed high antigenicity. Due to having a predicted high antigenicity, and exposure to the immune system and conservancy, epitopes of NA protein were chosen for multi-epitope peptide design.
Table 1.
Results of the BLAST for different subtypes of NA proteins and antigenicity estimations
| Neuraminidase subtypes | Average antigenicity VaxiJen score |
Minimum identity (%) | Maximum identity (%) |
|---|---|---|---|
| N1 | 0.5086 | 99.36 | 100 |
| N2 | 0.5852 | 96.59 | 100 |
| N3 | 0.5120 | 95.74 | 100 |
| N4 | 0.4994 | 94.04 | 100 |
| N6 | 0.5388 | 96.60 | 100 |
| N7 | 0.5005 | 96.60 | 100 |
| N8 | 0.5648 | 97.66 | 100 |
Note: For all the structure, vaxijen program threshold was 0.4
Epitope prediction for T-cell and B-cells
The predicted MHC-I and MHC-II restricted epitopes were compared with B-cell epitopes. Finally, a 10 nucleotides epitope for each NA protein was selected as shown in Table 2.
Table 2.
The results of final T-cell and B-cell epitope prediction screening from seven neuraminidases
| Neuraminidase | Sequence | Length | Start | End |
|---|---|---|---|---|
| N1 | FWVELIRGRP | 10 | 422 | 431 |
| N2 | YVELIRGRKQ | 10 | 423 | 432 |
| N3 | TGYVCSKFHS | 10 | 313 | 322 |
| N4 | NDKHSNGTVK | 10 | 140 | 149 |
| N6 | VELIRGRPKE | 10 | 426 | 435 |
| N7 | IRNKHSNGTI | 10 | 139 | 148 |
| N8 | LLNDKHSNGT | 10 | 137 | 146 |
The epitopes with three different linkers were considered for designing the multi-epitope construct. Schematic diagrams of the designed constructs with their linkers are presented at Fig. 1.
Fig. 1.

Graphical diagram of the multi-epitope peptide constructs with a PAPAPA, b GPGPGPG, and c GPGP linkers constructs
Antigen selectivity of the constructed multi-epitope peptide sequences
The antigenicity scores of each epitope and seven joined epitopes with PAPAPA, GPGPGPG, and GPGP amino acid linkers (Vac1, Vac2, and Vac3, respectively) are presented in Table 3.
Table 3.
Average antigenicity of neuraminidase epitopes and the multi-epitope constructs predicted by IMED tool
| Neuraminidase | Sequences | Antigenicity predicted by IMED |
|---|---|---|
| N1 | FWVELIRGRP | 1.0304 |
| N2 | YVELIRGRKQ | 1.0362 |
| N3 | TGYVCSKFHS | 1.0889 |
| N4 | NDKHSNGTVK | 0.9561 |
| N6 | VELIRGRPKE | 1.0101 |
| N7 | IRNKHSNGTI | 0.9559 |
| N8 | LLNDKHSNGT | 0.9748 |
| Constructed multi-epitope peptide with PAPAPA (Vac1) | FWVELIRGRPPAPAPAYVELIRGRKQPAPAPATGYVCSKFHSPAPAPANDKHSNGTVKPAPAPAVELIRGRPKEPAPAPAIRNKHSNGTI PAPAPALLNDKHSNGT | 1.0267 |
| Constructed multi-epitope peptide with GPGPGPG (Vac2) | FWVELIRGRPGPGPGPGYVELIRGRKQGPGPGPGTGYVCSKFHSGPGPGPGNDKHSNGTVKGPGPGPGVELIRGRPKEGPGPGPGIRNKHSNGTIGPGPGPGLLNDKHSNGT | 0.9880 |
| Constructed multi-epitope peptide with GPGP (Vac3) | FWVELIRGRPGPGPYVELIRGRKQGPGPTGYVCSKFHSGPGPNDKHSNGTVKGPGPVELIRGRPKEGPGPIRNKHSNGTIGPGPLLNDKHSNGT | 0.9977 |
Structure analysis
Physicochemical properties of each epitope and the multi-epitope constructs were estimated using ProtParam. It was indicated that the construct with six GPGPGPG linkers has a high instability, and low aliphatic indices (Table 4). All of the three constructs demonstrated an equal isoelectric point of 10.28. The Vac2 and Vac3 constructs showed the lowest GRAVY indices of − 0.904, and − 0.92, respectively. From the instability point of view, the Vac1 showed the highest value of 68.96, which makes it an inappropriate candidate for further vaccine studies. The lowest instability score was that of Vac2 with value of 2.27, which indicates that it has a high stability compared to the other structures.
Table 4.
Physicochemical properties of different epitopes of neuraminidase and the proposed multi-epitope constructs
| Sequences | Extinction coefficient | Molecular weight | Isoelectric point | Aliphatic index | GRAVY | Instability |
|---|---|---|---|---|---|---|
| FWVELIRGRP | 5500 | 1272.52 | 9.60 | 107 | 0 | 11.28 |
| YVELIRGRKQ | 1490 | 1261.49 | 9.99 | 107 | − 0.91 | 24.19 |
| TGYVCSKFHS | 1490 | 1128.27 | 7.90 | 29 | − 0.160 | − 7.98 |
| NDKHSNGTVK | 0 | 1099.17 | 8.66 | 29 | − 1.92 | − 25.89 |
| VELIRGRPKE | 1490 | 1196.42 | 8.72 | 107 | − 0.94 | 19.77 |
| IRNKHSNGTI | 21.5 | 1139.28 | 11 | 78 | − 1.15 | 21.5 |
| LLNDKHSNGT | 0 | 1098.98 | 6.74 | 78 | − 1.19 | − 23 |
| Vac1 | 8480 | 11,114.74 | 10.28 | 67.45 | − 0.558 | 68.96 |
| Vac2 | 8480 | 11,204.57 | 10.28 | 47.77 | − 0.904 | 2.27 |
| Vac3 | 8480 | 9937.25 | 10.28 | 56.91 | − 0.92 | 6.45 |
The results of analyzing the amino acids composition of the designed constructs indicated that Vac2 has the highest overall content of aliphatic amino acids compared to the others. The results of amino acid composition are provided in Table S1. Allergenicity analysis indicated scores of 0.19 for Vac 2, 0.45 for Vac3, and 1.3 for Vac1. The lowest allergenicity was for Vac2 structure which made it a suitable candidate. Structure analysis by Toxin pred indicated that none of these peptide constructs were toxic. Vac 2 was chosen for further investigation due to having antigenicity properties, highest stability and lowest allergenicity.
Vac2 has 112 residues with molecular weight of 11,204.43 g/mol. The results of analysis with PepCalc showed this construct has a good water solubility, with isoelectric pH of 10.8, with a net charge of 8.3 at pH 7. Figure 2 presents the secondary structure of Vac2 construct using PDBsum.
Fig. 2.

Secondary structure of Vac2 construct predicted using PDBsum
Ramachandran plot analysis of Vac2 indicated that 94.9% of residues were in the most favored regions and 5.1% were in allowed regions. Figure 3. A represents the results of Ramachandran plot for VAC 2 construct. The 3D structure of Vac2 construct, the local quality estimate, along with the quality comparison graph are provided in parts B, C, and D of Fig. 3, respectively.
Fig. 3.

a The Ramachandran plot analysis graph of Vac2 construct b 3D predicted construct of Vac2 c Local quality estimate of Vac2 d Quality comparison of the predicted Vac2 construct
The results of ERRAT tool indicated an acceptable error for the construct, with all the residues having values below the warning level. Figure 4. A shows the output graph of the ERRAT server for Vac2 construct.
Fig. 4.

a The ERRAT analysis diagram of the Vac2 construct b The diagram of quality prediction provided by verify 3D for Vac2 construct
The results of Verify 3D software indicated that 88.39% of the residues had average 3D-1D score ≥ 0.2, and at least 80% of the amino acids are having this properties (Fig. 4b). Results of ProSA server indicated a Z. Score of − 0.46. The overall model quality predicted by ProSA tool is presented in Fig. 5.
Fig. 5.
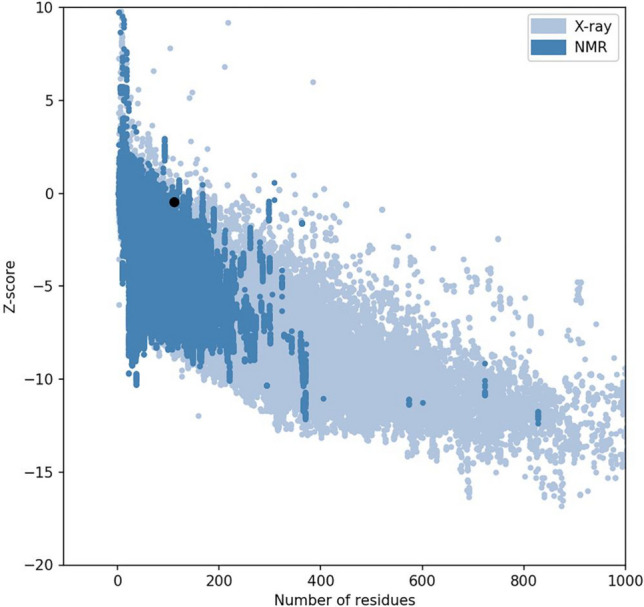
The overall quality of the predicted model for Vac2
Molecular docking and immune response simulation
The results of docking analysis for the proposed multi-epitope construct with HLA-1, HLA-2, and TLR3 are presented in Table 5. In the HLA-1 group, HLA-1-alpha-chainF showed that the highest Van der Waals energy, and the highest electrostatics energy was for interaction with HLA-1-alpha-chainE. In the HLA-2 group, HLA-2-DP showed the highest Van der Waals and electrostatics energy.
Table 5.
Molecular docking analysis of the predicted Vac2 construct with HLA, HLA2, and TLR
| Receptor | RMSD from the overall lowest-energy structure | Van der Waals energy | Electrostatic energy | Z-score |
|---|---|---|---|---|
| HLA-1-A1 | 0.7 ± 0.4 | − 96.9 ± 8.9 | − 395.2 ± 30.5 | − 1.7 |
| HLA-1-alpha-chainE | 13.4 ± 0.1 | − 87.8 ± 9.2 | − 455.2 ± 44.4 | − 1.5 |
| HLA-1-alpha-chainF | 18.6 ± 0.1 | − 109.3 ± 4.2 | − 413.7 ± 64.4 | − 1.6 |
| HLa-1-ChainG | 12.2 ± 0.4 | − 78.6 ± 10.5 | − 417.7 ± 63.7 | − 1.5 |
| HLA-1-cw-1 | 14.8 ± 0.2 | − 75.7 ± 6.7 | − 221.1 ± 15.3 | − 1.0 |
| HLA-2-DM | 7.7 ± 0.9 | − 88.1 ± 8.0 | − 298.8 ± 15.9 | − 1.0 |
| HLA-2-DO | 0.5 ± 0.3 | − 101.7 ± 8.0 | − 349.3 ± 7.5 | − 1.3 |
| HLA-2-DP | 10.5 ± 0.1 | − 109.6 ± 7.6 | − 567.2 ± 38.9 | − 1.5 |
| HLA-2-DQ | 1.9 ± 0.2 | − 93.3 ± 10.1 | − 305.5 ± 54.7 | − 1.3 |
| HLA-2-DR | 17.7 ± 0.3 | − 93.7 ± 7.4 | − 295.7 ± 16.9 | − 1.8 |
| HLA-2-gama chain | 4.4 ± 0.3 | − 72.3 ± 11.0 | − 405.2 ± 22.5 | − 1.7 |
| TLR-3 | 0.6 ± 0.4 | − 115.7 ± 16.2 | − 518.1 ± 19.8 | − 1.8 |
Molecular docking analysis showed that the predicted Vac2 construct has a high affinity toward TLR-3 receptor. Further analyzing with PDBsum showed that in the Vac2-TLR-3 complex, 47 residues from the TLR-3 and 37 residues of the Vac-2 construct were involved in the mutual interface, and provided an interface area of 1785 Ao2 for the TLR-3 and 1954 Ao2 for the Vac-2 construct. Among the interactions, 8 salt bridges, 17 hydrogen bonds, and 246 non-bounded contacts were detected and no disulphide bonds was found in the interaction area. Figure 6 shows the molecular interactions between TLR-3, and Vac2 construct. Figure 7 demonstrates molecular docking analysis of Vac2 construct with TLR-3 with regards to A. Hydrogen bonds, B. Interpolated charges C. Hydrophobicity, and D. Ionizability of the TLR-3 receptor.
Fig. 6.

The molecular interactions between TLR-3, and predicted Vac2 construct
Fig. 7.

Molecular docking analysis of Vac2 construct with TLR-3 regarding a hydrogen bonds, b interpolated charges c hydrophobicity, and d ionizability of the TLR-3 receptor surface
The results of immune response simulation by C-IMMSIM server indicated that upon the injection of the selected multi-epitope construct, the highest immunoglobulin response will be by IgM and IgG at days 15–20th (Fig. 8a). The B-cell response indicated that the total cell response will have a log phase of growth, starting around 5 days, which will continue to exponential phase until around day 20th, and after that remains at its high levels, close to 600 cells per mm3 (Fig. 8b). The number of NK cells will be increased and it will be 370 cells per mm3 at day 5th, and the lowest count will be less than 310 per mm3 at day 24th (Fig. 8c). In the case of T-cells, the cell counts start to increase immediately after injection and will drop at the day 25th (Fig. 8d). The TH-cells count start to increase at the day 5th, and will have an exponential phase to reach more than 10,000 cells per mm3 at day 15th, and after the day 20th, the cell count goes down (Fig. 8e). The TR (regulatory) cells also will have an immediate exponential growth which reaches its climax at the day 5th, and after that will start to reduce in the count (Fig. 8f).
Fig. 8.

The Immune simulation analysis of Vac2 construct influence of the a antigen and immunoglobulins loads b B-cell population c NK-cell count dT-cell population e TH-cell count f TR (regulatory) population per state at the following days upon injection
Discussion
Information regarding the three-dimensional (3D) structures of proteins and their complexes with their potential ligands are of critical importance for designing novel therapeutic agents; such as multi-epitope vaccines. Even though the laboratory methods; such as X-ray crystallography and NMR are considered as the efficient method for predicting 3D structure of proteins, they suffer from downsides; such as technically difficulties, and require a considerable time and financial resources. Application of the in silico methods for prediction of 3D structure of the protein has been proven an efficient method by using bioinformatics algorithms (Dev et al. 2016; Chou 2015).
In the post-genomic era, researchers have access to the vast resources of sequences and sequence-based knowledge; such as post translational modifications in proteins, which could be of critical importance for drug discovery. In fact, the accelerated development in sequential and structural bioinformatics have revolutionized the biology sciences. As a consequence of these substantial alterations, computational biology has been acting noticeably for stimulating the development of novel therapeutic agents (Chou 2011; Ju and Wang 2020). In this regard, in silico tools were utilized at the current research. Moreover, application of the graphic approaches for studying the biological and medical systems could present an intuitive knowledge to help analyzing the complicated molecular interactions (Chou et al. 1980; Chou and Forsén 1980).
Multi-epitope vaccines are a group of recombinant products with high specificity, safety, stability and low-cost of production (Nezafat et al. 2017); therefore, any study that could help to improve the quality of these agents is important. Since the laboratory-based works require application of a considerable amount of consumables, materials, human workforce, and sacrifice of animals; using computer-based methods could help to reduce the burden from laboratory, and decrease the costs of errors by using in silico methods.
Immunoinformatics or computational immunology is a branch of bioinformatics that includes investigations such as B- and T-cell epitope mapping, along with many other aspects including toxicity, and allergenicity. These methods represent high specificity, cost-effectiveness, potential and easy ways for vaccine development against infectious disorders (Nosrati et al. 2019). The potential of in silico designed vaccines for Influenza has also been confirmed by many in vivo and in vitro studies, for example, a study by Rodrigueza et al. reported a novel peptide-based vaccine (Vacc-FLU) candidate with protective efficacy against Influenza A in a mouse model, and the output confirmed efficacy of the designed peptide against the virus (Herrera-Rodriguez et al. 2018).
Generation of novel vaccines that can overcome antigenic diversity and inherent low immunogenicity of vaccines against Influenza A virus is critical for pandemic preparation. Despite the recent progresses in the growth and design of vaccines against epidemics threats, several issues still require the attention of the Influenza virus community (Vries et al. 2018). Currently, there is not enough reports on in silico design of multi-epitope vaccine based on neuraminidase protein. Several results have demonstrated that protection by vaccination with NA mostly relies on the induction of antibodies that could mediate inhibition of neuraminidase (NI) (Job et al. 2018).
Previously, many attempts have been devoted to develop a novel Influenza A vaccine that covers more subtypes of the virus. A 2015 study by Medina et al., reported an in silico identification of conserved epitopes of Influenza A. The valuable information provided by their study is useful for designing the future epitope based vaccine. In their study, they conducted an in silico investigation of the epitopes for four Influenza A proteins that are antigenically most significant (HA, NA, NP, and M2) in three strains with the highest world circulation in the last century (H1N1, H2N2, and H3N2), and one of the main aviary subtypes with a high importance at zoonosis (H5N1) (Muñoz-Medina et al. 2015).
At the current research, we aimed to design a potential multi-epitope vaccine candidate that could cover more subtypes of Influenza virus. Three peptide constructs were designed based on the conserved epitopes of neuraminidase protein, attached together using different linkers, and were subjected to different bioinformatics assays. Among the proposed constructs, Vac2 showed the highest stability, and lowest allergenicity, along with being none toxic. Structure of this construct was confirmed by different programs, considering different aspects, such as Ramachandran plot, the Z-score, and ERRAT values, along other features that certified the structure of proposed peptide construct.
The construct was ascertained to interact with immunologic receptors; including different HLA1, and HLA2 subtypes, along with TLR3. Finally, the immune responses including both humoral, and cellular responses (B-cell, T-cell, and NK-cells) were investigated through in silico immune response simulation.
Many of earlier studied have been focused on structural components of this virus, for example a study by Lohia et al. (2017) investigated immune responses to highly conserved Influenza A virus matrix 1 peptides. Another report by the same author was dedicated to the identification of conserved peptides comprising multiple T-cell epitopes of matrix 1 protein in H1N1 Influenza virus (Lohia and Baranwal 2015).
Several methods have been used to design epitope-based vaccines based on HA and NA proteins. Some of the candidates used in clinical trials show the possibilities in the decreasing Influenza infection (Sebastian and Lambe 2018; Sautto et al. 2018; Nachbagauer and Palese 2019). However, vaccine candidates which are currently in use at clinical studies are aimed to elicit an antibody response against more conserved Influenza proteins (Doorn et al. 2017a,2017b). Furthermore, the limitations of currently in market seasonal Influenza vaccines and the persistent threat of future pandemics have made it necessary for novel vaccine design.
In this study, a specific epitope from each subtype of seven NA proteins were chosen based on their antigenicity and stability. The results provided by the current in silico study showed that the multi-epitope construct with GPGPGPG linker and NA epitopes can provide promising outcomes against Influenza A virus and could be devoted for future in vitro, and in vivo studies.
Conclusion
This study provided a potential multi-epitope peptide vaccine candidate against Influenza A virus; based on the neuraminidase protein. This designed peptide could cover many subtypes of the virus and serve as wide rage protection against this seasonal disease. The multi-epitope construct presented by this study showed promising results through in silico step, which could be further investigated at in vitro and in vivo studies.
Supplementary Information
Below is the link to the electronic supplementary material.
Funding
No funding was received.
Availability of data and material
Not applicable.
Declarations
Conflict of interest
None to declare.
Research involving human participants and/or animals
No human or animal was involved in this study.
Informed consent
There was no human participant and consent was not required.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mandana Behbahani and Mohammad Moradi contributed equally to this work and hence are co-first authors.
References
- Benkert P, Künzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37(2):W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui H-H, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006;7(1):1–5. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulimo WD, Achilla RA, Majanja J, Mukunzi S, Wadegu M, Osunna F, et al. Molecular characterization and phylogenetic analysis of the hemagglutinin 1 protein of human influenza A virus subtype H1N1 circulating in Kenya during 2007–2008. J Infect Dis. 2012;206(1):S46–S52. doi: 10.1093/infdis/jis586. [DOI] [PubMed] [Google Scholar]
- Chen R, Holmes EC. The evolutionary dynamics of human influenza B virus. J Mol Evol. 2008;66(6):655. doi: 10.1007/s00239-008-9119-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-Q, Wohlbold TJ, Zheng N-Y, Huang M, Huang Y, Neu KE, et al. Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell. 2018;173(2):417–29.e10. doi: 10.1016/j.cell.2018.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou K-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol. 2011;273(1):236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou K-C. Impacts of bioinformatics to medicinal chemistry. Med Chem. 2015;11(3):218–234. doi: 10.2174/1573406411666141229162834. [DOI] [PubMed] [Google Scholar]
- Chou K-C, Forsén S. Diffusion-controlled effects in reversible enzymatic fast reaction systems-critical spherical shell and proximity rate constant. Biophys Chem. 1980;12(3–4):255–263. doi: 10.1016/0301-4622(80)80002-0. [DOI] [PubMed] [Google Scholar]
- Chou K-C, Li T-T, Forsén S. The critical spherical shell in enzymatic fast reaction systems. Biophys Chem. 1980;12(3–4):265–269. doi: 10.1016/0301-4622(80)80003-2. [DOI] [PubMed] [Google Scholar]
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries RD, Herfst S, Richard M. Avian influenza a virus pandemic preparedness and vaccine development. Vaccines. 2018;6(3):46. doi: 10.3390/vaccines6030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev J, Park D, Fu Q, Chen J, Ha HJ, Ghantous F, et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science. 2016;353(6295):172–175. doi: 10.1126/science.aaf7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8(1):4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8(1):1–7. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine. 2007;25(5):856–866. doi: 10.1016/j.vaccine.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. Bioinformatic approach for identifying parasite and fungal candidate subunit vaccines. Open Vaccine J. 2008;1(1):4. doi: 10.2174/1875035400801010022. [DOI] [Google Scholar]
- Eichelberger MC, Wan H. Influenza pathogenesis and control. Springer; 2014. Influenza neuraminidase as a vaccine antigen; pp. 275–299. [DOI] [PubMed] [Google Scholar]
- Eisenberg D, Lüthy R, Bowie JU. Methods in enzymology. Elsevier; 1997. [20] VERIFY3D: assessment of protein models with three-dimensional profiles; pp. 396–404. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. The proteomics protocols handbook. Springer; 2005. Protein identification and analysis tools on the ExPASy server; pp. 571–607. [Google Scholar]
- Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8(9):e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi O, Moradi M. In silico study of the structure and ligand interactions of alcohol dehydrogenase from Cyanobacterium Synechocystis sp. PCC 6803 as a key enzyme for biofuel production. Appl Biochem Biotechnol. 2020;192:1–22. doi: 10.1007/s12010-020-03400-z. [DOI] [PubMed] [Google Scholar]
- Haghighi O, Davaeifar S, Zahiri HS, Maleki H, Noghabi KA. Homology modeling and molecular docking studies of glutamate dehydrogenase (GDH) from Cyanobacterium Synechocystis sp PCC 6803. Int J Pept Res Ther. 2019;26:1–11. [Google Scholar]
- HeeShin W. Prediction of protein structure and interaction by GALAXY protein modeling programs. Biodesign. 2014;2:1–11. [Google Scholar]
- Herrera-Rodriguez J, Meijerhof T, Niesters HG, Stjernholm G, Hovden A-O, Sørensen B, et al. A novel peptide-based vaccine candidate with protective efficacy against Influenza A in a mouse model. Virology. 2018;515:21–28. doi: 10.1016/j.virol.2017.11.018. [DOI] [PubMed] [Google Scholar]
- Huang P, Yu S, Wu C, Liang L. Highly conserved antigenic epitope regions of hemagglutinin and neuraminidase genes between 2009 H1N1 and seasonal H1N1 influenza: vaccine considerations. J Transl Med. 2013;11(1):1–8. doi: 10.1186/1479-5876-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154(3):394–406. doi: 10.1111/imm.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job E, Ysenbaert T, Smet A, Christopoulou I, Strugnell T, Oloo E, et al. Broadened immunity against influenza by vaccination with computationally designed influenza virus N1 neuraminidase constructs. NPJ Vaccines. 2018;3(1):55. doi: 10.1038/s41541-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Z, Wang S-Y. Prediction of lysine formylation sites using the composition of k-spaced amino acid pairs via Chou's 5-steps rule and general pseudo components. Genomics. 2020;112(1):859–866. doi: 10.1016/j.ygeno.2019.05.027. [DOI] [PubMed] [Google Scholar]
- Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1–2):172–174. doi: 10.1016/0014-5793(90)80535-Q. [DOI] [PubMed] [Google Scholar]
- Kosik I, Yewdell JW. Influenza hemagglutinin and neuraminidase: Yin-Yang proteins coevolving to thwart immunity. Viruses. 2019;11(4):346. doi: 10.3390/v11040346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsakos M, Illing PT, Nguyen TH, Mifsud NA, Crawford JC, Rizzetto S, et al. Human CD8+ T cell cross-reactivity across influenza A, B and C viruses. Nat Immunol. 2019;20(5):613. doi: 10.1038/s41590-019-0320-6. [DOI] [PubMed] [Google Scholar]
- Kumar M, Thakur V, Raghava GP. COPid: composition based protein identification. Silico Biol. 2008;8(2):121–128. [PubMed] [Google Scholar]
- Laskowski RA. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 2001;29(1):221–222. doi: 10.1093/nar/29.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Thornton JM (2012) International tables for crystallography. vol F, chap 21.4. pp 684–687. 10.1107/97809553602060000882
- Lear S, Cobb SL. Pep-Calc. com: a set of web utilities for the calculation of peptide and peptoid properties and automatic mass spectral peak assignment. J Comput Aided Mol Des. 2016;30(3):271–277. doi: 10.1007/s10822-016-9902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohia N, Baranwal M. Identification of conserved peptides comprising multiple T cell epitopes of Matrix 1 protein in H1N1 influenza virus. Viral Immunol. 2015;28(10):570–579. doi: 10.1089/vim.2015.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohia N, Baranwal M. Immune responses to highly conserved Influenza A virus matrix 1 peptides. Microbiol Immunol. 2017;61(6):225–231. doi: 10.1111/1348-0421.12485. [DOI] [PubMed] [Google Scholar]
- Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24(11):1397–1398. doi: 10.1093/bioinformatics/btn128. [DOI] [PubMed] [Google Scholar]
- Muñoz-Medina JE, Sánchez-Vallejo CJ, Méndez-Tenorio A, Monroy-Muñoz IE, Angeles-Martínez J, Coy-Arechavaleta AS, et al. In silico identification of highly conserved epitopes of influenza A H1N1, H2N2, H3N2, and H5N1 with diagnostic and vaccination potential. Biomed Res Int. 2015;2015:813047. doi: 10.1155/2015/813047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachbagauer R, Palese P. Is a universal influenza virus vaccine possible? Ann Rev Med. 2019;71:315–327. doi: 10.1146/annurev-med-120617-041310. [DOI] [PubMed] [Google Scholar]
- Nezafat N, Eslami M, Negahdaripour M, Rahbar MR, Ghasemi Y. Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol Biosyst. 2017;13(4):699–713. doi: 10.1039/C6MB00772D. [DOI] [PubMed] [Google Scholar]
- Nosrati M, Hajizade A, Nazarian S, Amani J, Vansofla AN, Tarverdizadeh Y. Designing a multi-epitope vaccine for cross-protection against Shigella spp: an immunoinformatics and structural vaccinology study. Mol Immunol. 2019;116:106–116. doi: 10.1016/j.molimm.2019.09.018. [DOI] [PubMed] [Google Scholar]
- Park H-C, Shin J, Cho S-M, Kang S, Chung Y-J, Jung S-H. PAIVS: prediction of avian influenza virus subtype. Genom Inform. 2020;18(1):e5. doi: 10.5808/GI.2020.18.1.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005;6(1):132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008;9(1):514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE. 2010;5(4):e9862. doi: 10.1371/journal.pone.0009862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Raghava G. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34(suppl_2):W202–W209. doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautto GA, Kirchenbaum GA, Ross TM. Towards a universal influenza vaccine: different approaches for one goal. Virol J. 2018;15(1):17. doi: 10.1186/s12985-017-0918-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Ohrum K, Giles BM, Weirback HK, Williams BL, DeAlmeida DR, Ross TM. Adjuvants that stimulate TLR3 or NLPR3 pathways enhance the efficiency of influenza virus-like particle vaccines in aged mice. Vaccine. 2011;29(48):9081–9092. doi: 10.1016/j.vaccine.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian S, Lambe T. Clinical advances in viral-vectored influenza vaccines. Vaccines. 2018;6(2):29. doi: 10.3390/vaccines6020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kumari V, Kumbhar BV, Mukherjee A, Pandey R, Kondabagil K. Immunoinformatics approach for a novel multi-epitope subunit vaccine design against various subtypes of Influenza A virus. Immunobiology. 2021;226(2):152053. doi: 10.1016/j.imbio.2021.152053. [DOI] [PubMed] [Google Scholar]
- Sultana I, Yang K, Getie-Kebtie M, Couzens L, Markoff L, Alterman M, et al. Stability of neuraminidase in inactivated influenza vaccines. Vaccine. 2014;32(19):2225–2230. doi: 10.1016/j.vaccine.2014.01.078. [DOI] [PubMed] [Google Scholar]
- van Doorn E, Liu H, Ben-Yedidia T, Hassin S, Visontai I, Norley S, et al. Evaluating the immunogenicity and safety of a BiondVax-developed universal influenza vaccine (Multimeric-001) either as a standalone vaccine or as a primer to H5N1 influenza vaccine: phase IIb study protocol. Medicine. 2017;96(11):e6339. doi: 10.1097/MD.0000000000006339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doorn E, Pleguezuelos O, Liu H, Fernandez A, Bannister R, Stoloff G, et al. Evaluation of the immunogenicity and safety of different doses and formulations of a broad spectrum influenza vaccine (FLU-v) developed by SEEK: study protocol for a single-center, randomized, double-blind and placebo-controlled clinical phase IIb trial. BMC Infect Dis. 2017;17(1):241. doi: 10.1186/s12879-017-2341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zundert G, Rodrigues J, Trellet M, Schmitz C, Kastritis P, Karaca E, et al. The HADDOCK2. 2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428(4):720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev. 2008;21(1):13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil A, Estélles A, Kauvar LM, Johnson SK, Tripp RA, Wittekind M. Native human monoclonal antibodies with potent cross-lineage neutralization of influenza B viruses. Antimicrob Agents Chemother. 2018;62(5):e02269–e2317. doi: 10.1128/AAC.02269-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(suppl_2):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbold TJ, Krammer F. In the shadow of hemagglutinin: a growing interest in influenza viral neuraminidase and its role as a vaccine antigen. Viruses. 2014;6(6):2465–2494. doi: 10.3390/v6062465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbold TJ, Podolsky KA, Chromikova V, Kirkpatrick E, Falconieri V, Meade P, et al. Broadly protective murine monoclonal antibodies against influenza B virus target highly conserved neuraminidase epitopes. Nat Microbiol. 2017;2(10):1415. doi: 10.1038/s41564-017-0011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.