

SAMD1 is a newly identified repressive CpG island regulator.
Abstract
CpG islands (CGIs) are key regulatory DNA elements at most promoters, but how they influence the chromatin status and transcription remains elusive. Here, we identify and characterize SAMD1 (SAM domain-containing protein 1) as an unmethylated CGI-binding protein. SAMD1 has an atypical winged-helix domain that directly recognizes unmethylated CpG-containing DNA via simultaneous interactions with both the major and the minor groove. The SAM domain interacts with L3MBTL3, but it can also homopolymerize into a closed pentameric ring. At a genome-wide level, SAMD1 localizes to H3K4me3-decorated CGIs, where it acts as a repressor. SAMD1 tethers L3MBTL3 to chromatin and interacts with the KDM1A histone demethylase complex to modulate H3K4me2 and H3K4me3 levels at CGIs, thereby providing a mechanism for SAMD1-mediated transcriptional repression. The absence of SAMD1 impairs ES cell differentiation processes, leading to misregulation of key biological pathways. Together, our work establishes SAMD1 as a newly identified chromatin regulator acting at unmethylated CGIs.
INTRODUCTION
Vertebrate CpG islands (CGIs) are specific genomic regions characterized by the accumulation of CpG dinucleotides. They are commonly found at gene promoters and play important roles in gene regulation (1). Most of the CGIs are in an unmethylated state, and the associated genes are typically actively transcribed. The CXXC domain, which contains two zinc fingers, was first identified to specifically recognize unmethylated but not methylated CpG motifs (2). Proteins having CXXC domains are often subunits of larger protein complexes involved in modifying the chromatin state. For example, CXXC1 (CFP1) is part of an H3K4me3 methyltransferase complex (3) that is important to establish an active chromatin state. In contrast, KDM2B is associated with the Polycomb repressive complex 1 (PRC1) and is involved in establishing a repressive state (4). Recently, we identified the winged-helix (WH) domain of the Polycomb-like (PCL) proteins as a second type of an unmethylated CpG motif–binding domain. The PCL proteins are responsible for the recruitment of PRC2 to Polycomb-targeted CGIs (5). Because of the action of different CpG-binding proteins, distinct chromatin states can be established at CGIs. Most CGIs are enriched in active H3K4me3 marks and are typically associated with highly expressed genes. In contrast, unmethylated CGIs at the Polycomb target genes are decorated by the repressive H3K27me3 and H2Aub marks, deposited by the PRCs. A third group of CGIs are bivalent in that they have both H3K4me3 and H3K27me3 marks. This bivalent state is proposed to allow a rapid activation during differentiation processes (6). Despite major progresses in understanding the molecular mechanisms that govern the activity of CGIs, many aspects regarding the regulation of these abundant and fundamental genomic elements remain poorly understood (1).
Here, we identify and characterize the essentially uncharacterized protein sterile alpha motif (SAM) domain-containing protein 1 (SAMD1) as a novel unmethylated CGI-binding protein and show that SAMD1 directly binds to unmethylated CpG motifs through an atypical WH domain. SAMD1 acts in concert with chromatin regulators, such as KDM1A and L3MBTL3, to modulate the function of active CGIs in mouse embryonic stem (ES) cells, a process required for proper ES cell differentiation.
RESULTS
SAMD1 is a nuclear protein that binds to unmethylated CGIs
We recently found that the WH domains of the PCL proteins functions as a new type of unmethylated CGI-binding domains (5). Thus, we speculated that there could be other unknown domains or proteins that regulate CGI function through direct interaction with the CpG motifs. To identify potential unmethylated CpG-binding proteins, we surveyed available literature and data. Investigation of mass spectrometry datasets revealed that a protein called SAMD1 (also named Atherin) behaves like known CGI-binding proteins. Specifically, SAMD1 was pulled down with CpG-rich DNA (7, 8) and repelled by methylated DNA (9, 10). SAMD1 is also associated with chromatin and chromatin-related protein complexes (11–13), suggesting a function at chromatin. Together, we concluded that SAMD1 may be a potential new CGI regulator.
SAMD1 is a vertebrate-specific protein that has a C-terminal SAM domain, a central unstructured region, and an unannotated globular N-terminal domain, computationally predicted (14, 15) to be a WH domain (Fig. 1A and fig. S1, A and B). Related sequences to this domain were identified in the transcriptional regulators KAT6A, KAT6B, and ZMYND11 (fig. S1, B and C), which we grouped together as a novel class of WH domains (fig. S1D). Since SAMD1 is pulled down by CpG-rich sequences and is repelled from hydroxy-methylated counterparts, similar to the PCL protein MTF2 (7, 9), we hypothesized that the WH domain of SAMD1 might facilitate binding to CpG-rich sequences. Through EMSA (electrophoretic mobility shift assay) experiments, we observed that it is the WH, but not the SAM domain, that is responsible for binding to CpG-containing DNA (Fig. 1B). No binding was observed for AT-rich DNA for both domains. To investigate the DNA binding of the WH domain in an unbiased fashion, we made use of universal protein binding microarrays (PBMs) that represent all possible 10–base pair (bp) sequences (16). This assay identified the GCGC sequence as a preferred motif recognized by the SAMD1-WH domain in vitro (Fig. 1C and fig. S1E), consistent with our hypothesis.
Fig. 1. Characterization of SAMD1 CpG-binding in vitro and in vivo.
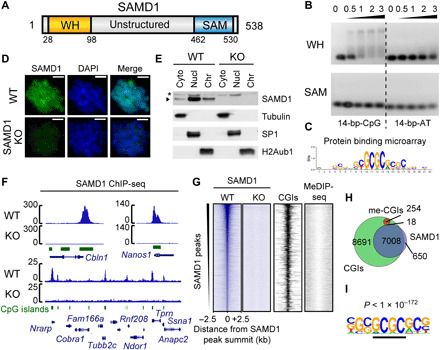
(A) Domain structure of SAMD1. (B) EMSA of the SAMD1-WH and SAMD1-SAM domain using CpG-rich and AT-rich DNA. The numbers indicate the molar ratio. (C) Representative DNA binding motif of the SAMD1-WH domain, derived from PBM experiment (16). The experiment was performed in two replicates with two distinct WH domain constructs (fig. S1E). (D) Immunofluorescence of the endogenous SAMD1 in wild-type (WT) and SAMD1 knockout KO cells. Scale bars, 20 μM. (E) Cellular fractionation of mouse ES cells followed by Western blotting. Asterisks indicate a nonspecific band (Cyto, cytoplasm; Nucl, nucleoplasm; Chr, chromatin). (F) Example ChIP-seq peaks of the endogenous SAMD1 in mouse ES cells. (G) Heatmap showing SAMD1 enrichment at SAMD1 peaks (n = 8733). The heatmaps of the KO, CGI position, and DNA methylation (MeDIP-seq) are shown in comparison. (H) Venn diagram showing the overlap of SAMD1 peaks (blue) with all CGIs (green) and methylated CGIs (red). (I) Top enriched motif at SAMD1-bound versus unbound CGIs is obtained by HOMER. DAPI, 4′,6-diamidino-2-phenylindole.
To verify our in vitro findings, we determined the cellular localization and genomic binding loci of SAMD1 in vivo. SAMD1 is expressed to similar levels in different mouse organs, with the strongest expression shown in mouse ES cells (fig. S2A). Thus, we used mouse ES cells as the model system for further investigations. We generated SAMD1 knockout (KO) cells (fig. S2B), which proliferated normally without any obvious phenotype (fig. S2, C and D). Using a custom-made antibody, we found that endogenous SAMD1 is predominantly nuclear localized, with a substantial proportion associated with chromatin (Fig. 1, D and E), supporting a potential chromatin-related function. Subsequently, using chromatin immunoprecipitation sequencing (ChIP-seq), we identified 8733 significant peaks and discovered that they strongly (>90%) overlap with nonmethylated CGIs but not with methylated CGIs (Fig. 1, F to H). The ChIP-seq signal was absent in SAMD1 KO cells, demonstrating the specificity of the antibody (Fig. 1, F and G). SAMD1 is highly enriched at some CGIs such as those of the Cbln1, Nanos1, and Pth2 genes, while it shows only a subtle or no binding to other CGIs, suggesting preferential binding to certain CGIs (Fig. 1F). Comparing the sequences of the SAMD1-bound versus the unbound CGIs, a GCGC-containing motif is enriched (Fig. 1I), consistent with the motif identified by the in vitro PBM (Fig. 1C).
SAMD1’s WH domain interacts with the minor and major groove of DNA
To address the molecular details of the interaction of SAMD1 with DNA, we solved the crystal structure of the SAMD1-WH domain in complex with 5′-GCGC-3′–containing double-stranded DNA (dsDNA) at a resolution of 1.78 Å (Table 1). Unlike a typical WH domain that contains three β strands and two wing-like loops (named W1 and W2) (17), SAMD1-WH contains only two β strands (named β1 and β2) and the W1 loop connecting both strands in addition to three conserved α helices (Fig. 2A). The C-terminal end of helix α1 and its following loop are inserted into the CpG-containing major groove and make sequence-specific contacts with the CpG-containing region, while the W1 loop reaches deep into the neighboring minor groove to recognize bases flanking the CpG motif (Fig. 2A). In detail, Arg45 and Lys46 at the C-terminal end of α1 are mainly responsible for CpG recognition in the major groove. The main chain carbonyl oxygen atoms of both residues form a hydrogen bond each with bases C6 and its symmetric related C7′, respectively (Fig. 2B). In addition, the side chain of Lys46 forms hydrogen bonds with G7 from the CpG motif and its flanking base C8. The side chain of Arg45 also forms a hydrogen bond with the phosphate backbone of T4. The major groove recognition is further strengthened by a hydrogen bond between Arg56 and the phosphate backbone of C2 (Fig. 2, B and C). The minor groove recognition is mainly mediated by two long side-chain residues, Tyr87 and Lys88 from the W1 loop. The side chain of Tyr87 hydrogen bonds with the G10′ base, while the side chain of Lys88 forms hydrogen bonds with the bases C10 and T9′, respectively (Fig. 2D). The phosphate backbone connecting both the major and the minor grooves is also extensively recognized. Arg43 from α1 and Arg48 from the loop following α1 each form a pair of hydrogen bonds with the phosphate backbone of T9′ and C7′, respectively (Fig. 2E). The side chains of Ser92 and Arg94 from β2 form one and a pair of hydrogen bonds with the phosphate backbone of G8′, respectively. Overall, SAMD1-WH recognizes the CpG-containing target DNA over a 9-bp footprint, with the major groove, the minor groove, and the phosphate backbone in-between both grooves extensively recognized (Fig. 2C).
Table 1. Data collection and refinement statistics (molecular replacement).
Each dataset is collected from one crystal. RMS, root mean square.
|
SAMD1-WH (27–105)/DNA Se-Met–labeled |
SAMD1-SAM (459–523) |
SAMD1-SAM (459–526) |
SAMD1-SAM (459–530) Se-Met–labeled |
|
| PDB code | 6LUI | 6LUJ | 6LUK | – |
| Data collection | ||||
| Space group | P3221 | P3121 | P21 | P32 |
| Cell dimensions | ||||
| a, b, c (Å) | 45.93, 45.93, 132.64 | 69.34, 69.34, 181.89 | 66.43, 182.84, 66.97 | 67.75, 67.75, 293.39 |
| α, β, γ (°) | 90.00, 90.00, 120.00 | 90.00, 90.00, 120.00 | 90.00, 93.32, 90.00 | 90.00, 90.00, 120.00 |
| Resolution (Å) | 50.00–1.78 (1.81–1.78)* |
50.00–1.12 (1.14–1.12) |
50.00–2.06 (2.12–2.06) |
50.00–2.90 (2.95–2.90) |
| Rmerge | 0.091 (0.797) | 0.071 (0.560) | 0.099 (0.520) | 0.191 (1.262) |
| I / σI | 60.0 (3.1) | 49.7 (2.3) | 12.1 (1.9) | 16.3 (1.7) |
| Completeness (%) | 100.0 (100.0) | 97.9 (81.1) | 99.9 (100.0) | 100.0 (100.0) |
| Redundancy | 18.5 (16.8) | 12.5 (5.9) | 3.4 (3.4) | 10.2 (10.4) |
| Refinement | ||||
| Resolution (Å) | 25.47–1.78 | 23.85–1.12 | 45.71–2.06 | – |
| No. of reflections | 16,241 | 190,644 | 98,371 | – |
| Rwork/Rfree | 0.202/0.242 | 0.175/0.178 | 0.185/0.226 | – |
| No. atoms | ||||
| Protein | 598 | 2640 | 11,000 | – |
| DNA | 527 | – | – | – |
| Ligand/ion | – | 80 | 90 | – |
| Water | 103 | 715 | 833 | – |
| B-factors (Å2) | ||||
| Protein | 37.1 | 11.4 | 26.5 | – |
| DNA | 42.0 | – | – | – |
| Ligand/ion | – | 23.3 | 43.4 | – |
| Water | 40.3 | 22.3 | 33.6 | – |
| RMS deviations | ||||
| Bond lengths (Å) | 0.007 | 0.004 | 0.002 | – |
| Bond angles (°) | 0.963 | 0.913 | 0.515 | – |
*Values in parentheses are for highest-resolution shell.
Fig. 2. Structural and biochemical analysis of recognition between the SAMD1-WH domain and various DNA substrates.
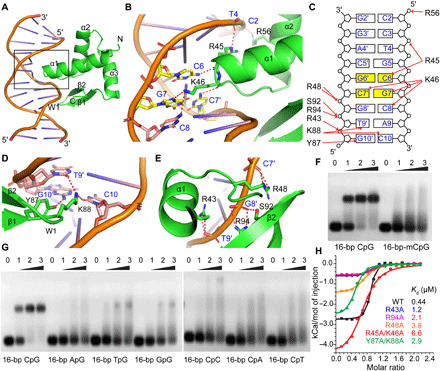
(A) An overall view of the SAMD1-WH domain/DNA complex [Protein Data Bank (PDB): 6LUI]. SAMD1-WH is colored in green with the secondary structural elements labeled. (B) A zoom-in view of the bases in the DNA major groove recognized by SAMD1-WH. (C) A schematic representation of the interaction network between SAMD1-WH and the target DNA. (D and E) Zoom-in views of the recognition in the minor groove (D) and the phosphate backbone (E) by SAMD1-WH. (F and G) EMSA analysis of the binding affinity of SAMD1-WH for a 16-bp CpG-containing DNA and its CpG-methylated (F) or substituted (G) counterpart. Numbers indicate molar ratio. (H) ITC measurements of the binding affinities for a 16-bp CpG-containing DNA by the WT SAMD1-WH domain and its mutants. Dissociation constants (Kd) are shown as inserts.
The DNA binding mode is different from that shown for PCL proteins (5), as well as those exhibited by the other WH domains (17). Since three bases from the CpG motif are specifically recognized (Fig. 2, B and C), methylation of either cytosine, or replacement of either base from the CpG duplex would disrupt the interaction or cause a steric clash with the main-chain backbone of the SAMD1-WH domain. Consistently, through EMSA analysis, we found that SAMD1-WH showed a marked loss of binding affinity toward the DNA substrates when the cytosines of the CpG motif are fully methylated (Fig. 2F). Furthermore, replacement of either the cytosine or the guanine of the CpG motif in the substrate DNA resulted in a marked loss of binding affinity by SAMD1-WH (Fig. 2G). In contrast, replacement of single bases flanking either side of the CpG motifs did not substantially lower the binding affinity of the SAMD1-WH domain (fig. S3), further confirming the importance of an unmethylated CpG motif for recognition. Through isothermal titration calorimetry (ITC) measurements, we found that wild-type (WT) SAMD1-WH binds a 16-bp CpG-containing DNA with a dissociation constant (Kd) of 0.44 μM (Fig. 2H and Table 2). The double mutant R45A/K46A that disrupts the recognition in the DNA major groove displayed a 15-fold weaker binding affinity (Kd = 6.6 μM). A double mutation Y87A/K88A that disrupts recognition at the minor groove decreased the binding for the target DNA by 6.6-fold. R43A, R94A, and R48A mutations that disrupt recognition of the phosphate backbone between the major and minor grooves reduce the binding affinity of SAMD1-WH to the target DNA by 2.7-, 4.8-, and 8.6-fold, respectively (Fig. 2H and Table 2). This indicates that in addition to the CpG motif, recognition of the minor groove and the phosphate backbone between the major and minor grooves also play important roles for high binding affinity of SAMD1-WH domain to CpG-containing DNA.
Table 2. ITC-based measurements of Kd between the SAMD1-WH domain or its mutants with 16-bp CpG DNA.
| DNA | Protein sample | Kd (μM) | ΔH (kcal/mol) |
| 16-bp CpG | SAMD1-WH(16–110) | 0.44 ± 0.11 | −2.75 ± 0.04 |
| 16-bp CpG | SAMD1-WH(16–110)- R45A/K46A |
6.6 ± 0.9 | −4.60 ± 0.14 |
| 16-bp CpG | SAMD1-WH(16–110)- Y87A/K88A |
2.9 ± 0.5 | −2.60 ± 0.08 |
| 16-bp CpG | SAMD1-WH(16–110)- R43A |
1.2 ± 0.2 | −0.62 ± 0.01 |
| 16-bp CpG | SAMD1-WH(16–110)- R48A |
3.8 ± 0.4 | −1.48 ± 0.10 |
| 16-bp CpG | SAMD1-WH(16–110)- R94A |
2.1 ± 0.4 | −0.58 ± 0.02 |
SAMD1 acts as a repressor at active CGIs
To gain insights into the potential function of SAMD1 at CGIs, we compared the genomic binding pattern of SAMD1 with those of other CpG-binding proteins, such as MTF2 (PCL2) (5), KMT2B (MLL2) (18), KDM2A (FBXL11), KDM2B (FBXL10) (4), and CXXC1 (CFP1) (3) and related histone modifications. Using correlation analysis, we found that SAMD1 clusters together with CXXC1, KMT2B, KDM2A, KDM2B, and H3K4me3 but is distant from MTF2 and H3K27me3 (Fig. 3A). To investigate this further, we categorized all CGIs into active (H3K4me3 only), repressed (H3K27me3 only), bivalent (H3K4me3 and H3K27me3), and undecorated (neither) CGIs. Comparison of the respective heatmaps revealed that the binding pattern of SAMD1 is most similar to CXXC1 and KMT2B, which selectively bind to CGIs that have the active histone mark H3K4me3, while KDM2A and KDM2B are more broadly distributed across all CGI categories (Fig. 3B and fig. S4A). SAMD1 overlaps with the PRC2-associated factor MTF2 only at bivalent CGIs but not at repressed CGIs. Overall, SAMD1-bound genes are predominantly highly expressed (fig. S4B) and belong to many distinct biological processes, such as transcription, translation, cell cycle, intracellular transport, and development (fig. S4C).
Fig. 3. Investigation of SAMD1’s role at CGIs.
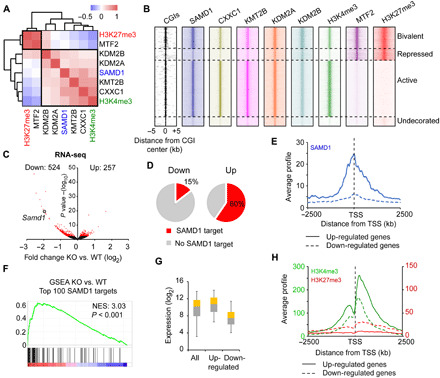
(A) Correlation analysis of SAMD1, KMT2B, KDM2A, KDM2B, CXXC1, MTF2, H3K4me3, and H3K27me3 at CGIs. (B) Heatmap showing the distribution of the factors from (A) at distinct CGIs. Based on H3K4me3 and H3K27me3 levels, the CGIs were divided into bivalent (H3K4me3 and H3K27me3), repressed (only H3K27me3), active (only H3K4me3), or undecorated (neither) CGIs. (C) Volcano plot of RNA sequencing (RNA-seq) data of SAMD1 KO versus WT cells. Cutoff: P < 0.01. Four biological replicates were performed. (D) Occupancy of up- and down-regulated genes with SAMD1. (E) Promoter profile of SAMD1 at up- and down-regulated genes. (F) Gene set enrichment analysis (GSEA) of the top 100 SAMD1-bound genes, using the RNA-seq data. (G) Expression level of SAMD1-bound up- and down-regulated genes in comparison to all SAMD1-bound genes. The whisker-box plots represent the lower quartile, median, and upper quartile of the data with 5 and 95% whiskers. (H) Promoter profiles of H3K4me3 and H2K27me3 at up- and down-regulated genes. NES, Normalized Enrichment Score.
To gather information about the functional role of SAMD1 at those genes, we performed RNA sequencing (RNA-seq) in SAMD1 KO versus WT ES cells. This experiment identified 524 significantly (P < 0.01) down-regulated and 257 up-regulated genes (Fig. 3C). Further investigation showed that the up-regulated but not the down-regulated genes are strongly occupied by SAMD1 (Fig. 3, D and E), suggesting that direct targets of SAMD1 become derepressed upon SAMD1 deletion. We confirmed via gene set enrichment analysis (GSEA) that the 100 genes with the highest levels of SAMD1 are, on average, significantly up-regulated upon SAMD1 KO (Fig. 3F), further supporting a repressive role for SAMD1. Gene Ontology analysis of the genes that are up-regulated and bound by SAMD1 showed that they are related to transcription, cell division, chromatin remodeling, and developmental processes (fig. S4D). These genes are generally highly expressed and rich in H3K4me3 but lack H3K27me3 modifications (Fig. 3, G and H), suggesting that SAMD1 restricts the expression of a subset of highly active genes.
SAMD1 interacts with L3MBTL3 and the KDM1A complex
To determine the molecular mechanism of the repressive function of SAMD1, we purified SAMD1 from HeLa-S cells and identified SAMD1-associated proteins by mass spectrometry (Fig. 4A). Consistent with previous reports, we found that SAMD1 associates with L3MBTL3, SFMBT1, and SFMBT2 (11, 19), as well as the KDM1A complex (12), which demethylates the active H3K4me2 mark (20). Using coimmunoprecipitation experiments after ectopic overexpression, we validated the interaction of SAMD1 with L3MBTL3 and KDM1A (Fig. 4B). The endogenous interaction between SAMD1 and L3MBTL3 could also be confirmed in mouse ES cells (fig. S5A).
Fig. 4. Identification of SAMD1 interaction partners.
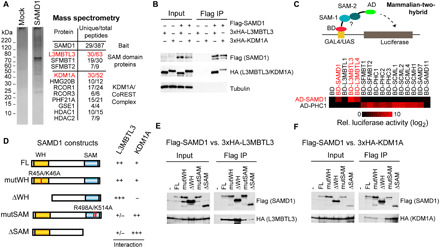
(A) Tandem affinity purification of Flag-HA-SAMD1 from Hela-S cells followed by mass spectrometry analysis. Shown are unique and total peptide numbers. (B) Coimmunoprecipitation experiment in human embryonic kidney–293 cells, demonstrating the interaction of SAMD1 with L3MBTL3 and KDM1A. (C) Schematic representation and results of mammalian-two hybrid using various SAM domains (see also fig. S5B). (D) Overview of constructs used and the results from mapping experiments in (E) and (F). (E) Coimmunoprecipitation of HA-L3MBTL3 with distinct versions of Flag-SAMD1. (F) Coimmunoprecipitation of HA-KDM1A with distinct versions of Flag-SAMD1. HDAC, histone deacetylase.
SAMD1, L3MBTL3, SFMBT1, and SFMBT2 all have a SAM domain at their C termini. This domain is also present in several Polycomb-related proteins, such as the PHC proteins (fig. S5B), and is important for protein-protein interactions by forming polymers, which contribute to the formation of Polycomb bodies (21, 22). To address the interplay of SAMD1 with other SAM domain–containing proteins in more detail, we performed mammalian-two-hybrid experiments and examined the association of distinct SAM domains (Fig. 4C, top, and fig. S5B). We used the SAM domain of the Polycomb protein PHC1 as a positive control, which is known to interact with several other SAM domain proteins (21, 22). We found that the SAM domain of SAMD1 specifically interacts with the SAM domain of L3MBTL3, L3MBTL4, and itself but not with the other investigated SAM domains (Fig. 4C, bottom). These SAM-SAM interactions can be disrupted by mutating critical residues of the SAMD1-SAM domain (fig. S5, C and D). Via coimmunoprecipitation, we confirmed the self-association feature of SAMD1 and validated that this interaction requires an intact SAM domain (fig. S5, E and F). The SAM domain of PHC1 interacts with the SAM domains of several proteins involved in Polycomb repression but not with SAMD1 or L3MBTL3/4 (Fig. 4C, bottom), suggesting a restricted selection of interacting partners among SAM domains. Together, these results support that L3MBTL3 associates with SAMD1 through direct SAM-SAM domain interaction.
As KDM1A appeared to be a strong SAMD1 interactor (Fig. 4, A and B), we also studied the association between SAMD1 and KDM1A. Previous work suggests that L3MBTL3 interacts with KDM1A (19). Thus, SAMD1 may associate with KDM1A either directly or indirectly through L3MBTL3. To test these possibilities, we created different SAMD1 deletion and point mutants (Fig. 4D). Deletion or mutation of the SAM domain strongly reduced the interaction with L3MBTL3 (Fig. 4E) but not the association with KDM1A (Fig. 4F), supporting that the SAMD1-L3MBTL3 association is not required for the interaction between SAMD1 and KDM1A. By contrast, deletion of the WH domain reduced the association with KDM1A but not with L3MBTL3 (Fig. 4, E and F), suggesting that SAMD1 interacts with KDM1A and L3MBTL3 through distinct binding sites.
SAMD1 modulates the chromatin landscape at CGIs
We next addressed how SAMD1, KDM1A, and L3MBTL3 may cooperate at CGIs. First, we investigated the binding pattern of L3MBTL3 and KDM1A relative to that of SAMD1. For L3MBTL3, we generated two polyclonal antibodies directed against the N- or C terminus of L3MBTL3 and determined its genome-wide binding pattern via ChIP-seq. The N-terminal antibody led to more significant peaks (n = 576) and was used for this initial analysis. For KDM1A, we used publicly available ChIP-seq data (23). Analysis of the data revealed that in mouse ES cells, L3MBTL3 mainly localizes to CGIs (Fig. 5A), with about 57% of its binding sites overlapping with SAMD1. In contrast, KDM1A binds to many non-CGI locations, such as enhancers, that are hardly targeted by SAMD1. However, most of the SAMD1 binding sites (>95%) are also bound by KDM1A (Fig. 5B). At the four CGI categories established above (Fig. 3B), SAMD1, KDM1A, and L3MBTL3 are similarly distributed (fig. S6A), demonstrating that all three proteins preferentially bind to actively transcribed genes. Together, these results suggest that most SAMD1-targeted CGIs are also targeted by KDM1A, while around half of the L3MBTL3 binding sites are cobound by SAMD1.
Fig. 5. Chromatin regulation by SAMD1.
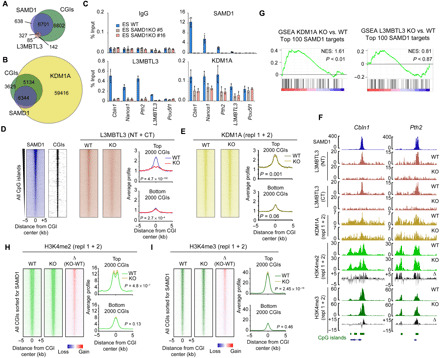
(A) Venn diagram showing the genome-wide overlap of SAMD1 peaks with L3MBTL3 peaks and CGIs (B) Overlap of SAMD1 with KDM1A and CGIs. (C) ChIP–quantitative polymerase chain reaction (qPCR) experiment for L3MBTL3 and KDM1A in WT and SAMD1 KO cells. (D) Heatmap of ChIP-seq experiments for L3MBTL3 in WT and SAMD1 KO cells at all CGIs, sorted after SAMD1 levels. The ChIP-seq results using two distinct L3MBTL3 antibodies were merged. The profiles at the top 2000 and bottom 2000 SAMD1-bound CGIs are shown. (E) Heatmap and profiles of ChIP-seq experiments using KDM1A antibody in WT versus SAMD1 KO cells, sorted as in (D). Two replicates for KDM1A were merged. (F) Genome browser view of ChIP-seq experiments at two SAMD1 targets. (G) GSEA analysis of the top 100 SAMD1 genes in KDM1A (left) and L3MBTL3 (right) KO cells, compared to WT cells. (H) Heatmap and profiles of H3K4me2 at CGI, sorted as in (D), in WT and SAMD1 KO cells. The difference is shown in the right heatmap. Two biological replicates were merged. See also fig. S6C. (I) Heatmaps and profiles as described in (H) but for H3K4me3. See also fig. S6E. P values in (D), (E), (H), and (I) were calculated by were calculated by two-sided Student’s t tests. NT, N terminus. CT, C terminus.
To determine whether SAMD1 influences the binding of L3MBTL3 and KDM1A to chromatin, we performed ChIP–quantitative polymerase chain reaction (qPCR) experiments at selected SAMD1 target genes in SAMD1 KO cells. SAMD1 deletion strongly reduced the chromatin binding of L3MBTL3 (Fig. 5C), suggesting that L3MBTL3 binding to chromatin might substantially depend on SAMD1 at those locations. Given that L3MBTL3 is related to the Drosophila Polycomb group protein L(3)mbt (24), we also determined the influence on the classical Polycomb group proteins, but we did not observe any obvious consequences on the chromatin binding of those proteins in the absence of SAMD1 (fig. S6B), supporting that SAMD1 is not affecting the canonical Polycomb system. In contrast to the strong reduction of L3MBTL3, the KDM1A levels were only partially reduced upon SAMD1 deletion (Fig. 5C). At a genome-wide level, we confirmed a strong reduction of the L3MBTL3 levels, particularly at CGIs with strong SAMD1 binding (Fig. 5, D and F), while at CGIs where SAMD1 is not present the L3MBTL3 level is not affected (Fig. 5D). This effect can be observed with both L3MBTL3 antibodies (Fig. 5F). For KDM1A, only a subtle reduction can be detected at a genome-wide level (Fig. 5, E and F). These results suggest that SAMD1 is involved in tethering L3MBTL3 to chromatin at SAMD1 binding sites, while SAMD1 may contribute only marginally to KDM1A recruitment. Given that KDM1A interacts with many proteins (12), the recruitment of KDM1A is likely facilitated also by other chromatin-binding factors.
To assess the contribution of L3MBTL3 and KDM1A to gene repression by SAMD1, we created L3MBTL3 and KDM1A KO cells using CRISPR-Cas9 and performed RNA-seq experiments. Via GSEA, we found that the top SAMD1 targets become up-regulated upon KDM1A but not L3MBTL3 KO (Fig. 5G), suggesting that KDM1A may cooperate with SAMD1 for regulating gene expression, while L3MBTL3 may be less relevant for this aspect. Given that KDM1A demethylates the active H3K4me2 histone mark (20), we analyzed the consequence of SAMD1 KO on H3K4me2. We discovered a subtle but significant increase of H3K4me2 at CGIs (Fig. 5H and fig. S6C), which is similar to the consequences observed upon KDM1A knockdown or its chemical inhibition (25, 26). The increase is particularly evident at CGIs with robust SAMD1 binding, suggesting that SAMD1 deletion impairs the function of the KDM1A histone demethylase complex at those CGIs. The H3K4me2 level is significantly reduced at enhancer sites (fig. S6D), where SAMD1 is barely present. This reduction is likely an indirect effect and may explain why more genes become down-regulated than up-regulated upon SAMD1 deletion (Fig. 3C). H3K4me2 serves also as substrate for histone methyltransferases that deposit H3K4me3. Consistently, we also observed a subtle increase of H3K4me3 at CGIs (Fig. 5I and fig. S6E). Given that the KDM1A complex is also associated with histone deacetylases (HDACs), and HDACs were identified as interacting partners by IP-MS (Immunoprecipitation followed by mass-spectrometry) (Fig. 4A), we asked whether SAMD1 deletion would affect histone acetylation. Via ChIP-seq, we observed no major changes for the histone acetylation mark H3K27ac (fig. S6F), suggesting that SAMD1 does not alter the function or recruitment of those enzymes. Similarly, we found only minimal consequences on H3K27me3 (fig. S6G), supporting the view that SAMD1 is not directly influencing the chromatin regulation by the canonical Polycomb proteins. Together, our results suggest that SAMD1 directly binds to unmethylated CGIs and modulates gene transcription by influencing the function of KDM1A and possibly other chromatin regulators.
SAMD1 requires both the WH and the SAM domain for efficient chromatin binding
To determine the recruitment mechanism of SAMD1 to CGIs in vivo, we performed ChIP-qPCR experiment in SAMD1 KO cells in which SAMD1 expression was restored by ectopically expressed WT or mutated SAMD1 (Fig. 6A). We found that mutation of either the WH domain or the SAM domain reduced the chromatin association of SAMD1 (Fig. 6B). The fact that mutations of the WH domain affect chromatin binding is consistent with our in vitro data showing that this domain is directly involved in DNA binding. This is also in line with what has been shown for the WH domain of MTF2 (5). Regarding the SAM domain, we speculated that the SAM domain could mediate the interaction with other SAM domain-containing proteins, such as L3MBTL3, which in turn might contribute to the chromatin association of SAMD1. Given that L3MBTL3 can bind to chromatin via its MBT domains (27), we hypothesized that L3MBTL3 may be relevant for SAMD1 chromatin binding. Unlike the strong effect of an almost complete loss of chromatin association of L3MBTL3 upon SAMD1 KO (Fig. 5C), we found that L3MBTL3 deletion only moderately reduced the association of SAMD1 with chromatin (fig. S7A). This reduction of SAMD1 chromatin binding may also be due to a slightly reduced protein level of SAMD1 in the L3MBTL3 KO cells (fig. S7B). Thus, the association with L3MBTL3 cannot explain the importance of the SAM domain integrity for SAMD1 chromatin association. We speculated therefore that SAMD1 may increase its chromatin-binding affinity via SAM homopolymerization, similar to other SAM domain proteins (21, 22, 28).
Fig. 6. Structural details of the SAMD1-SAM pentamer.
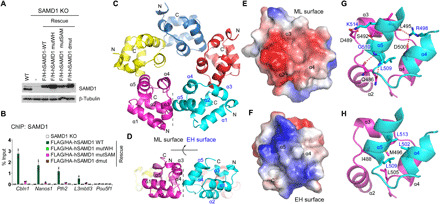
(A) Western blot of ectopically expressed SAMD1 in SAMD1 KO ES cells. (B) ChIP-qPCR of SAMD1 in rescued cells at SAMD1 target genes. Two biological replicates were performed. Error bars indicate ± SD. (C and D) Top (C) and side (D) views of the SAMD1-SAM pentamer (PDB: 6LUJ). Five molecules are colored in different colors, with the secondary elements of the SAM domain labeled. The interface between mid-loop (ML) and end-helix (EH) surfaces is indicated by a dotted line. (E and F) Electrostatic potentials of the ML (E) and EH (F) surfaces. Negative electrostatic potential is colored in red, while the positive electrostatic potential is colored in blue. (G and H) Details of hydrogen bonding interactions (G) and hydrophobic interactions (H) between ML surface colored in magenta and EH surface colored in cyan.
The SAMD1-SAM domain homopolymerizes into a pentameric ring
To explore the self-association mechanism of the SAMD1-SAM domain, we crystallized two SAMD1-SAM–containing constructs and solved their structures at the resolution of 1.12 and 2.06 Å (Table 1), respectively. The shorter construct of the SAM domain self-associates into a pentamer (Fig. 6C), while in the structure of the longer construct, two pentamers stack together into a decamer (fig. S7C). Gel filtration and mass spectrometry analysis confirmed that WT SAMD1-SAM, but not its mutated version, forms a pentamer in solution (fig. S7, D and E), indicating that the pentameric state is a stable form of the SAMD1-SAM polymer. In the structure of the pentameric SAMD1-SAM polymer, five molecules associate with one another to form a donut-shaped closed ring (Fig. 6C), which is different from the spirally associated SAM domain polymers, such as those of the Drosophila Ph-SAM and human Translocated ETS leukemia (TEL)–SAM domains (29, 30). SAMD1-SAM adopts the fold of a typical SAM domain in which five α helices fold into a compact globular structure (Fig. 6, C and D). Similar to the canonical SAM domain polymers (31), the SAMD1-SAM pentamer is stabilized through close contacts between the mid-loop (ML) surface of one molecule and the end-helix (EH) surface of a neighboring molecule (Fig. 6, C and D). The ML surface—formed by α helices 2, 3, and 4—exhibits a negative electrostatic potential (Fig. 6E), which is complemented by the EH surface, that is composed of helices 4 and 5 and displays a positive electrostatic potential (Fig. 6F). The interactions between ML and EH surfaces are mediated by both polar and nonpolar interactions. Arg498 and Lys514 from EH surface each form a salt bridge with Asp500 and Asp489 from the ML surface (Fig. 6G), respectively. Arg498 also forms a hydrogen bond with Leu495 from ML surface. Gly510 from EH surface forms a hydrogen bond each with Ser492 and Gln486 from ML surface. The side chain of Gln486 also hydrogen bonds with the main chain of Leu509 (Fig. 6G). Intermolecular contacts between two surfaces are further stabilized by hydrophobic interactions, with the long hydrophobic side chains of Leu502, Leu509, and Leu513 from the EH surface pointing toward the hydrophobic patch composed of Ile488, Met496, and Leu505 from the ML surface (Fig. 6H). Together, these findings demonstrate an unusual self-association of the SAMD1-SAM domain and supports that SAMD1 possibly can interact with CGIs in a multivalent manner. Given that the SAMD1-SAM pentamer is distinct to other nuclear SAM domain polymers (29, 30), it may facilitate unknown chromatin-related functions, which warrants further investigation in the future.
SAMD1 is required for normal ES cell differentiation
Currently, not much is known about the biological role of SAMD1. To address the potential biological function of SAMD1, we performed undirected ES cell differentiation, via leukemia inhibitory factor (LIF) removal, in the presence and absence of SAMD1 and investigated the consequence on the differentiation process. The expression of SAMD1 itself remains largely constant during the differentiation (Fig. 7A). Observation of the cells showed no obvious differences between WT and SAMD1 KO cells, suggesting that the absence of SAMD1 does not impair the general differentiation process. Consistently, in reverse transcription (RT)–qPCR experiments, we observed only minor differences of classical pluripotency and differentiation markers (Fig. 7B). However, we observed that SAMD1-targeted genes, which are slightly up-regulated in SAMD1 KO cells in the undifferentiated state, remain higher expressed in the KO cells throughout the differentiation process (Fig. 7B). To gain deeper insights into the role of SAMD1, we performed RNA-seq after 7 days of differentiation. Principal components analysis (PCA) showed that the difference in the expression pattern between WT and SAMD1 KO cells becomes larger upon differentiation (Fig. 7C), demonstrating that the absence of SAMD1 impairs the differentiation. We identified more than 5000 genes of which the expression changes significantly (P < 0.01), including more than 1500 genes that are more than twofold misregulated (Fig. 7D). Consistent with the RT-qPCR experiment, the top 100 SAMD1 targets remain significantly up-regulated in SAMD1 KO cells (Fig. 7E). Also, the up-regulated, but not the down-regulated, genes are typically occupied by SAMD1 (Fig. 7F and fig. S8A), suggesting that SAMD1 restricts the expression of its target genes throughout the differentiation process. To gain deeper insights into the pathways regulated by SAMD1, we performed unbiased GSEA followed by network analysis. This analysis demonstrates that SAMD1 deletion leads to an up- and down-regulation of numerous biological processes (Fig. 7G). Specifically, pathways related to immune system and metabolism become down-regulated in SAMD1 KO cells (fig. S8B), while pathways related to neuronal function and heart muscle cells become up-regulated (fig. S8C). Key regulators such as Sall3, Nanos1, Gata4, and Pparg are strongly differentially expressed in SAMD1 KO versus WT cells (Fig. 7F). Together, these analyses demonstrate that SAMD1 is required for proper ES cell differentiation, and it suggest that SAMD1 plays a pleiotropic biological role, similar to the role of KDM1A (32). Future investigations of the role of SAMD1 during specific differentiation processes and embryogenesis will shed further light on its biological importance.
Fig. 7. Investigation of the role of SAMD1 during ES cell differentiation.
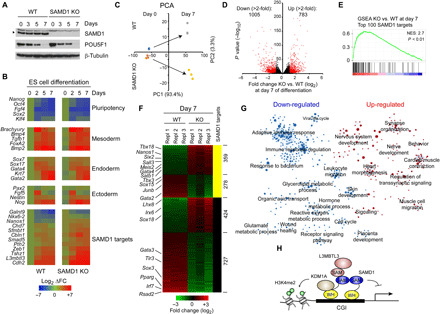
(A) Western blot of SAMD1 and POU5F1 during undirected ES cell differentiation by LIF removal. (B) RT-qPCR experiments of stem cell and differentiation markers, as well as SAMD1 target genes during the differentiation. Three biological replicates were performed. (C) PCA analysis of RNA-seq data at days 0 and 7 of ES cell differentiation. Three biological replicates were performed. (D) Volcano plot of RNA-seq data after 7 days of differentiation in WT and SAMD1 KO cells. In red are genes that are significant (P < 0.01) and at least twofold differentially expressed. (E) GSEA of top 100 SAMD1 target genes, comparing SAMD1 KO versus WT cells at day 7 of the differentiation. (F) Genes from (D) shown has heatmap and sorted for SAMD1-bound and unbound genes. (G) GSEA followed by network analysis, demonstrating up- and down-regulated pathways in SAMD1 KO cells after 7 days of differentiation. The circle radius indicates the size of the gene sets. (H) Model of putative SAMD1 function at CGIs.
DISCUSSION
In this study, we uncovered a regulatory mechanism that links unmethylated CGIs to chromatin regulation, mediated by SAMD1. Our in vivo and in vitro data robustly demonstrate that SAMD1 directly interacts with unmethylated CpG motifs and links key chromatin modifiers, such as KDM1A and L3MBTL3, to CGIs. Our findings highlight a previously unknown regulatory pathway at CGIs and open avenues of investigation in chromatin regulation and translational fields.
In mammals, a large proportion of RNA polymerase II promoters have CGIs (1) but lack other classical promoter elements such as the TATA box. The chromatin environments of the CGIs are largely modulated by CpG motif–binding proteins that recruit or are themselves part of the histone modifying complexes. Our study establishes SAMD1 as a new player in CGI regulation that directly binds to unmethylated CGIs. Several features distinguish SAMD1 from those previously identified unmethylated CpG-binding proteins. First, the SAMD1 WH domain is structurally different from the CXXC domains (Fig. 2), which are well-documented unmethylated CpG-binding domains that adopt a special fold through coordinating two zinc ions (33). Second, although the SAMD1 WH domain also exhibits a WH fold, its DNA recognition mode is completely different from those shown by the WH domains of PCL proteins (5). The PCL WH domain recognizes only the major groove of the unmethylated CpG-containing DNA through the W1 loop whose sequence is conserved only among different PCL proteins. By contrast, the SAMD1 WH domain recognizes the major groove of the CpG-containing DNA through its first α helix, with its W1 loop recognizing the minor groove of the DNA. Last, the SAMD1 WH and PCL WH domains have different DNA sequence preferences (GCGC versus TCGG) (5). Through sequence analysis, we found that the SAMD1 WH domain and the WH domains from several other proteins represent a new subgroup of WH domains that have the potential to recognize unmethylated CpG motifs (fig. S1, C and D). This substantially expands the repertoire of the unmethylated CpG-binding proteins. Given the diversity of the unmethylated CpG-containing motifs and the various binding modes that the CpG-binding domains exhibited, it is quite possible that there are other uncharacterized unmethylated CpG motif–binding domains, which await identification.
Our further work found that SAMD1 requires not only the DNA-recognizing WH domain but also the self-polymerizing SAM domain for efficient chromatin binding. SAM domains are one of the most abundant protein-protein interaction motifs in eukaryotes that can mediate complex formation through homo- or heterodimerization/polymerization (21, 31). In Drosophila, the canonical PRC1 is tethered to the DNA binding Pho-repressive complex through SAM-SAM interactions (28). In this study, SAMD1 was also found to interact with several SAM domain-containing proteins through SAM-SAM interactions. Although the physiological function of this interaction remains to be explored, it does provide an explanation for the diverse regulatory function of SAMD1. Unexpectedly, interactions with proteins, such as the interaction with L3MBTL3, contribute only marginally to the recruitment of SAMD1, which prompted us to assess the self-oligomerization feature of the SAMD1-SAM domain. The SAMD1-SAM domain self-associates into a pentameric circle in solution (Fig. 6). This pentamer structure is homogeneous and stable as verified by mass spectrometry. The SAMD1-SAM pentamer is the second ring-shaped SAM domain structure formed by self-association identified in higher eukaryotes and the other being the recently described octameric ring of the SARM1 SAM domain (34); previously described SAM domain structures in eukaryotes tend to form an open-ringed polymer. Oligomerization of the SAMD1 through its SAM domain would most likely enhance the avidity of its WH binding to the target DNAs, thus enhancing its recruitment to the chromatin. Protein oligomerization, including SAM polymerization, has also been shown to play an important role in phase separation, a process that has been observed for many chromatin regulators, and which contributes to chromatin organization (21, 22). A similar mechanism may also apply to SAMD1. Future work is required to determine whether SAMD1 regulates chromatin structure in vivo.
In the genome of mouse ES cells, SAMD1 is preferentially associated with active CGIs where it cooperates with chromatin regulators, such as KDM1A, to modulate the chromatin landscape and down-regulate its target genes (Figs. 3 to 5). The interplay of SAMD1 with KDM1A at CGIs may contribute to the role of KDM1A at CpG-rich regions in sperm cells (35). The repressive function of SAMD1 places SAMD1 between the CGI-binding proteins that activate transcription, such as CXXC1 (3) and the PCL proteins (5), which represses transcription at Polycomb target genes. SAMD1 binds to over 7000 genes in mouse ES cells, involved in numerous biological pathways (fig. S4C), demonstrating that SAMD1 has a global gene regulatory function. The consequence of loss of function of SAMD1 in undifferentiated ES cells on gene expression is rather minor (Fig. 3C), similar to what is observed upon loss of function of many other DNA binding factors (36). However, upon ES cell differentiation, SAMD1 KO cells exhibited a substantial alteration of their differentiation program, leading to the up- and down-regulation of numerous genes (Fig. 7D). Those genes are involved in many biological pathways (Fig. 7G), supporting a versatile role of SAMD1, and a potential pleiotropic biological function. Notably, SAMD1 is commonly up-regulated in many cancer types (fig. S8D) (37) and its high expression is often associated with a poorer prognosis in patients with cancer, such as in liver cancer and acute myeloid leukemia (fig. S8E), suggesting that SAMD1 could play a role in multiple cancer types. Moreover, SAMD1 has recently been found to be essential for the growth of the K562 erythroleukemic cells (38), supporting a putative protumorigenic function of SAMD1 in this context.
In summary, we have identified SAMD1 as a CGI-binding protein that links H3K4me3-decorated CGIs in mouse ES cells to gene repression. We have provided structural insight into SAMD1’s CGI recognition and homopolymerization, which revealed a previously undiscovered mode of binding to CGIs. We provide evidence that SAMD1 acts in concert with the KDM1A demethylase complex, L3MBTL3, and most likely other factors to modulate the chromatin landscape at its target CGIs (Fig. 7H). Future investigations of SAMD1 in a physiological and pathophysiological context will provide further details about the mechanistic and biological function of SAMD1.
MATERIALS AND METHODS
Experimental design
The present study aimed to characterize the molecular mechanisms of the protein SAMD1 at CpG-containing DNA in vitro and in vivo. All experiments performed in vitro and in vivo were carefully controlled. In vitro experiments performed using recombinant proteins or cell lysates were performed independently in at least two replicates. Experiments performed in cells, including chromatin immunoprecipitation and differentiation experiments, were performed in at least two biological replicates.
SAMD1 constructs
Because of high CG content, the annotation of the SAMD1 gene is conflicting. We used constructs corresponding to human SAMD1 as published previously (39) and annotated in UniProt (Q6SPF0), National Center for Biotechnology Information (NM_138352) and Ensembl (ENSG00000288488). Note that the standard annotation for human SAMD1 in Ensembl (ENSG00000141858) is likely incorrect. To reduce the CG content in our constructs, the sequence of the open reading frame was synonymously mutated. Point mutations were introduced by PCR or by DNA synthesis.
Cell culture
E14 mouse ES cells (E14TG2a) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and GlutaMAX (Gibco, catalog no. 61965-026), 15% fetal calf serum (FCS) (Biochrom, S0115, Lot: 1247B), 1× nonessential amino acids (Gibco, 11140-035), 1× sodium pyruvate (Gibco, 11160-039), 1× penicillin/streptomycin (Gibco, 15140-122), 0.15% β-mercaptoethanol, and LIF (1000 U/ml; Millipore, ESG1107, lot: 3060038) on gelatin-coated plates. Human embryonic kidney–293 (HEK293) cells were culture with DMEM/F-12 (Gibco, 31331-028), 1× penicillin/streptomycin, and 10% FCS.
SAMD1, L3MBTL3, and KDM1A mES KO cells were created by transient transfection using the jetPRIME transfection reagent (Polyplus) with LentiCRISPRv2 (Addgene no. 52961) (40) constructs with following guide RNA sequences: mSAMD1 (#1: AGCGCATCTGCCGGATGGTG; #2: GAGCATCTCGTACCGCAACG), mL3MBTL3 (#1: AGCAGTTGGGACCATCCATG; #2: GCGAAGATCTAAGCAGCGGT), and mKDM1A (#1: GGAATAGCCGAGACCCCGGA; #2: GTTCGATCACGGCCTCACCT). After puromycin selection (3 μg/ml) for 3 days, single-cell clones were obtained and further validated. The KO of SAMD1 was confirmed by Western blot, ChIP, and immunofluorescence. Because of the high CG content of the targeted sequence, a validation of potential indels by sequencing was not possible. The KO of KDM1A was confirmed by Western blot, ChIP, and sequencing. The KO of L3MBTL3 was confirmed by sequencing, ChIP, and immunofluorescence. Detection of the endogenous L3MBTL3 by Western blotting was not successful.
Mouse ES cells were undirected differentiated by removal of LIF as described previously (41). In short, 3 × 105 WT and SAMD1 KO ES cells were plated in the absence of LIF on ungelatinized six-well plates and grown to confluency. The cells were then transferred to ungelatinized bacteria petri dishes and grown for 3 days, during which they were unable to adhere and generated spherical aggregates. These aggregates were then replated on gelatin-coated six-well tissue culture plates where cells adhered and formed differentiating outgrowths. At different time points after replating, the total RNA was isolated to analyze gene expression changes. RT-qPCR primers are presented in table S1.
Antibodies
Polyclonal antibodies for SAMD1 and L3MBTL3 were made using purified glutathione S-transferase (GST)–fusion proteins as antigen. For SAMD1, the antibody is directed against the SAM domain of human SAMD1 (amino acids 452 to 538). For L3MBTL3, the antibodies are directed against the N terminus (amino acids 3 to 233) or the C terminus (amino acids 778 to 883) of mouse L3MBTL3. Antibodies were made with Eurogentec using the 28-day speedy protocol. Obtained antibodies were affinity-purified. The following commercial antibodies were used: H3K4me3 (Diagenode, C15410003), H3K4me2 (Diagenode, C15410035), H3K27ac (Active Motif, 39133), H3K27me3 (Diagenode, C15410195), KDM1A (Abcam, 17721), tubulin (Millipore, MAB3408), Pou5f1/Oct4 (Santa Cruz Biotechnology, SC-5279), FLAG (Sigma-Aldrich, F3165), hemagglutinin (Roche, 11867423001), H2AUb1 (Cell Signaling Technology, 8240), Lamin B (Santa Cruz Biotechnology, sc-6217), EZH2 (Diagenode, C15410039), L3MBTL2 (Active Motif, 39569), PCGF6 (ProteinTech, 24103-1-AP), RING2 (Abcam, ab101273), RYBP (Sigma-Aldrich, PRS2227), SFMBT1 (Bethyl Laboratories, A303-221A), rabbit immunoglobulin G (IgG) control (Diagenode, C15410206), and H2Av (Drosophila) (Active Motif, 39716). The Sp1 antibody was described previously (42).
Mammalian-two-hybrid
Mammalian-two-hybrid was performed using the Mammalian Two-Hybrid Assay Kit from Stratagene/Agilent [catalog no. 211344 (discontinued)]. SAMD1-SAM and PHC1-SAM was cloned into the pCMV-activation domain (AD) vectors. All other SAM domains were cloned into the pCMV-DNA binding domain (BD) vectors. SAM domains from following human proteins were used: SAMD1 (452-538), SAMD7 (307-410), SAMD11 (533-628), L3MBTL1 (743-840), L3MBTL3 (705-780), L3MBTL4 (537-623), SFMBT1 (751-866), SFMBT2 (804-894), SCML1 (240-329), SCML2 (623-700), SCML4 (338-414), and SCMH1 (579-660). For the experiment, 30,000 HEK293 cells were plated into one 24-well plate. Two technical replicates were performed for each experiment. The next day, the cells were transfected with 200 ng of pFR-Luc, 0.5 ng of SV-40-RLuc, and 50 ng of the pCMV-AD and pCMV-BD constructs using FuGeneHD (Promega). Two days after transfection, cells were washed one time with phosphate-buffered saline (PBS) and lysed for 20 min with 1× passive lysis buffer (reagents from the Dual-Luciferase Reporter Assay System Kit, Promega). Firefly luciferase and renilla luciferase activity was determined by using the Dual-Luciferase Reporter Assay System Kit (Promega). Firefly values were normalized to renilla activity.
ChIP, ChIP-seq, and RNA-seq
ChIP experiments were performed in accordance to the One Day ChIP Kit protocol (Diagenode) using antibodies described above. ChIP-qPCRs with gene-specific primers (table S1) were performed using the ImmoMix PCR reagent (Bioline) in the presence of 0.1× SYBR Green (Molecular Probes). ChIP-qPCR experiments have been repeated at least twice. For ChIP-seq, three individual ChIPs were pooled and purified on QIAquick columns (Qiagen). For ChIP-seq of histone marks, 1 μg of Drosophila S2 chromatin (1:200 relative to mouse ES cell chromatin) was added to each reaction as a spike-in control along with 1 μg of a spike-in antibody directed against the Drosophila-specific H2Av variant (Active Motif). Five nanograms of precipitated DNA was used for indexed sequencing library preparation using the Microplex Library Preparation Kit v2 (Diagenode). Libraries were purified on AMPure magnetic beads (Beckman). For RNA-seq, total RNA was extracted from WT mouse ES cells and three different SAMD1 KO clones by using the RNeasy Mini system (QIAGEN) including an on-column deoxyribonuclease I digestion. RNA integrity was assessed on an Experion StdSens RNA Chip (Bio-Rad). RNA-seq libraries were prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina). RNA-seq and ChIP-seq libraries were quantified on a Bioanalyzer (Agilent Technologies). Next-generation sequencing was performed on Illumina HiSeq1500 or NextSeq550.
Bioinformatical analysis
ChIP-seq data were aligned to mouse genome mm9 using Bowtie (43), allowing one mismatch. SAM files were converted to BAM using SAMtools (44). Bigwig files were obtained using deepTools/bamCoverage (45) and normalized based on reads per kilo base per million mapped reads (RPKM). ChIP-seq for Histone marks were normalized according to the spike-in controls (46). Replicates were merged using SAMtools. Downstream data analysis was performed using Galaxy (47), Cistrome (48), and Bioconductor/R (49). SAMD1-bound peaks were identified by MACS2 (50) using standard settings. Heatmaps and profiles were created using deepTools (45). The P values were calculated by performing at two-tailed Student’s t test at RPKM-normalized and log2-transformed read counts at top or bottom 2000 SAMD1-bound CGIs, comparing the data from WT and SAMD1 KO cells. Methylated CGIs were identified using Methylated DNA immunoprecipitation (MeDIP)–seq data (GSM881346) (51). CGI and Promoter definitions were downloaded from the University of California, Santa Cruz (UCSC) Table Browser. Motif enrichment was performed using HOMER (52) by using SAMD1-bound CGIs as input and SAMD1-unbound CGIs as background control. Correlation analysis was performed using the “Multiple wiggle files correlation in given regions” tool within the Galaxy/Cistrome platform (48) using all CGIs as given region. Gene ontology analysis was performed using GREAT (53) using all significant SAMD1 peaks as input. Subsequent network analysis was performed using the EnrichmentMap app in Cytoscape (54). Gene Ontology analysis of SAMD1-bound and up-regulated genes was performed using DAVID (55). The phylogenetic tree of the WH domains was made using interactive Tree Of Life (56). Gene expression of mouse SAMD1 was investigated using BioGPS (57). The analysis of the expression of SAMD1 in cancer and survival analysis was performed using gene expression profiling interactive analysis (GEPIA) (37).
RNA-seq data were aligned to mouse transcriptome GenCode.M23 using RNA Star (2.7.2b) (58). Counts per gene were determined using FeatureCounts (1.6.4). Differentially expressed genes and normalized reads were determined using DeSeq2 (2.11.40.6) (59). GSEA (60) was performed with standard setting. For top SAMD1 target genes, the 100 genes with the highest SAMD1 promoter occupancy, excluding Samd1 itself, were used as gene set. Network analysis was performed using the EnrichmentMap app in Cytoscape (54).
The following publicly available datasets were used: CXXC1 [GSM2454338 and GSM2454339 (61)], MTF2 [GSM2472747 and GSM2472748 (7)], KMT2B [GSM2073033 (18)], KDM2A (GSM1003593) and KDM2B [GSM1003594 (4)], KDM1A [GSM2630507 (23)], H3K27me3 [GSM2472743 and GSM2472744 (7)], H3K4me3 [GSM2472745 and GSM2472746 (7)], and MeDIP-seq [GSM881346 (51)].
Coimmunoprecipitation
All ectopic coimmunoprecipitation (Co-IP) experiments were performed in HEK293 cells. Cells were seeded in 10-cm dishes with 2 × 106 cells per dish. One day later, the expression constructs for 3xHA or N-FLAG–tagged proteins were transfected using FuGENE HD Transfection Reagent (Promega, E2311). Two days after transfection, cell lysis was done using Co-IP lysis buffer [50 mM tris-Cl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% Glycerol, 1× protease inhibitor cocktail, and 0.5 mM phenylmethylsulfonyl fluoride]. Cells were shaken for 30 min at 4°C followed by centrifugation for 10 min at 13,000 rpm at 4°C. Protein concentration was determined with the DC Protein Assay (Bio-Rad, 5000116). For each IP, 1 mg of protein was applied, and extract was filled up to a total volume of 500 μl using Co-IP lysis buffer. To remove unspecific binding proteins, a preclearing was performed for 1 hour using mouse IgG−Agarose (Merck, A0919). Beads were equilibrated by washing two times with 1× tris-buffered saline and one time with Co-IP lysis buffer. To bind FLAG-tagged proteins, precleared extracts were added to 50 μl of ANTI-FLAG M2 Affinity Gel (Merck, A2220) and incubated for approximately 3 hours at 4°C. After incubation, three washing steps with Co-IP lysis buffer were performed. The FLAG beads were boiled 3 min in 30 μl of 2× Laemmli buffer without β-mercaptoethanol. Afterward, 1 μl of β-mercaptoethanol was added and the supernatants were cooked again for 5 min. Detection of proteins in the input, supernatant, and IP fractions was conducted via Western blotting using an 8% gel. Co-IP experiments were repeated at least two times. Endogenous Co-IP was performed in mouse ES cells, comparing WT and SAMD1 KO cells.
Complex purification and mass spectrometry
Flag-HA–tagged human SAMD1 was expressed after retroviral infection of HeLa-S. Nuclear extract was prepared from the established stable cell lines, and the SAMD1 complex was purified using anti-Flag (M2)–conjugated agarose beads (Sigma-Aldrich, A2220) by incubation in Tandem Affinity Purification (TAP) buffer [50 mM tris-HCl (pH 7.9), 100 mM KCl, 5 mM MgCl2, 10% glycerol, 0.1% NP-40, 1 mM dithiothreitol (DTT), and protease inhibitors] for 4 hours and three times washing with TAP buffer. Proteins were eluted with Flag peptides. A second purification was performed using anti-HA–conjugated agarose beads (Santa Cruz Biotechnology, sc-7392), followed by elution with HA peptides. For mass spectrometry, the sample was TCA-precipitated and peptides were identified via liquid chromatography–tandem mass spectrometry at the Taplin Core facility/Harvard Medical School.
Immunofluorescence
WT or SAMD1 KO mouse ES cells were seeded (5 × 105 cells) on coverslips coated with gelatine in six-well plates; after 1 day, cells were washed three times with PBS and incubated for 25 min in 4% paraformaldehyde (in PBS). Cells were washed one time with wash buffer (0.5% Triton in PBS), permeabilized with wash buffer for 25 min, and blocked (wash buffer and 10% FCS) for 1 hour. One-hundred fifty microliters of primary antibody (1:1000 in wash buffer and 10% FCS) was added and incubated for 1 hour. Cells were washed with wash buffer three times for 10 min each, and 150 μl of secondary antibody [1:2000, goat anti-rabbit IgG H&L (Alexa Fluor 488, Thermo Fisher Scientific, A-11008) in wash buffer and 10 % FCS] was added and incubated for 1 hour (dark). Cells were washed with wash buffer three times for 10 min each and subsequently one time with PBS for 10 min. Coverslips were mounted with VECTASHIELD antifade mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories, H1200), transferred to microscope slides, and sealed. Microscopy was performed using a Leica DM5500 microscope, and data were analyzed using ImageJ (Fiji).
Cellular fractionation
Cellular fractionations were performed using “Subcellular Protein Fractionation Kit for Cultured Cells” (Thermo Fisher Scientific, 78840) according to the manufacturer’s instructions, followed by Western blotting.
PBM experiments and analysis
Sequences of two distinct hSAMD1-WH regions [amino acids 1 to 110 (WH1) and 28 to 110 (WH2)] were cloned into the pT7CFE1-NHis-GST-CHA plasmid (Thermo Fisher Scientific, 88871). GST-fusion proteins were expressed using the 1-Step Human Coupled IVT Kit (Thermo Fisher Scientific). Expressed protein concentrations were estimated from anti-GST Western blots. Subsequently, custom-designed “all-10mer” universal oligonucleotide arrays in 8 × 60 K GSE array format (Agilent Technologies, AMADID 030236) were double-stranded, and PBM experiments were performed essentially as described previously (16) with Alexa 488–conjugated anti-GST antibody (Invitrogen, A-11131). Each of the two WH domain constructs (hSAMD1-WH1 and hSAMD1-WH2) was assayed in duplicate at a final concentration of 600 nM in PBS-based binding and wash buffers on fresh slides. Scans were acquired using a GenePix 4400A (Molecular Devices) microarray scanner. Microarray data quantification, normalization, and motif derivation were performed essentially as described previously using the Universal PBM Analysis Suite and the Seed-and-Wobble motif-derivation algorithm (16).
Protein expression and purification
Open reading frame of human SAMD1-WH is chemically synthesized with codon optimized for efficient bacterial expression. SAMD1-WH–containing fragments (residues 27 to 105, 16 to 110, and its mutants) and SAMD1-SAM–containing fragments (residues 459 to 523, 459 to 526 and 459 to 530) were inserted into a hexahistidine-SUMO–tagged pRSFDuet-1 vector. The target proteins were expressed in Escherichia coli strain BL21(DE3) cells, which were shaken at 37°C until the OD600 (optical density at 600 nm) reached around 1.0, and then cooled at 20°C for around 1 hour before 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) were added to induce expression overnight. Cells were collected by centrifugation at 5000g for 10 min. Cell pellets were resuspended with the initial buffer containing 20 mM tris at pH 7.0, 500 mM NaCl, and 20 mM imidazole and then sonicated for around 5 min. The supernatant was collected by centrifugation of the cell lysate at 25,000g for 1 hour. Histidine-SUMO–tagged target protein was isolated through a nickel-charged HiTrap Chelating FF column (GE Healthcare). The histidine-SUMO tag was cleaved by incubating with a histidine-tagged ubiquitin-like-specific protease 1 (ULP1) protease and then dialyzed with the initial buffer at 4°C overnight.
For SAMD1-WH, the dialyzed solution was reloaded onto a nickel-charged chelating column to remove both the histidine-tagged SUMO and ULP1 protease. The flow-through was diluted twofold with 20 mM tris at pH 7.0 and 2 mM DTT to yield a solution at half the initial salt concentration (250 mM NaCl), which was then loaded directly onto a heparin column (GE Healthcare) to remove bound DNA. Target protein was separated by increasing the salt concentration of the low-salt buffer (20 mM tris at pH 7.0, 250 mM NaCl, and 2 mM DTT) from 250 mM to 1 M NaCl through a linear gradient. The target protein was further purified by a HiLoad 200 16/600 gel filtration column (GE Healthcare) equilibrated with the low-salt buffer through which the resulting product was pooled. Purified proteins were concentrated to around 20 mg/ml and stored in a −80°C freezer.
For SAMD1-SAM, after elution of the histidine-SUMO–tagged protein, SAMD1-SAM was incubated with histidine-tagged ULP1 protease and dialyzed with the low-salt buffer containing 20 mM tris at pH 8.0, 100 mM NaCl, and 2 mM DTT at 4°C overnight. The dialyzed sample was loaded onto a HiTrap Q FF column (GE Healthcare) and then eluted by increasing the salt concentration from 100 mM NaCl to 1 M NaCl to remove histidine-SUMO tag. The eluted target protein was then purified through a HiLoad 200 16/600 gel filtration column before loading onto a Mono Q 5/50 column (GE Healthcare) for further purification. After these steps, the target protein was concentrated to around 16 mg/ml and stored in a freezer.
The selenomethionine-labeled SAMD1-WH and SAMD1-SAM were expressed in the methionine auxotrophic B834 (DE3) strain. Cells (1 liter) grown in LB media at the OD600 of 1.2 were harvested by centrifugation at 4000 rpm for 10 min. The cells were washed twice with M9 media and then were used to inoculate 2 liter of methionine-depleted medium supplemented with l-selenomethionine (50 mg/liter) and nutrient mix (SelenoMet, Molecular Dimensions). After shaking for an additional 30 min at 37°C, the cells were induced with 0.2 mM IPTG and shaken overnight at 20°C. Selenomethionine-labeled proteins were purified similarly as those of the native proteins, except that 2 mM β-mercaptoethanol was added in the buffer in the initial stage of protein purification. All mutations of SAMD1-WH and SAMD1-SAM were generated by PCR-based method.
Crystallization and structure resolution
Crystallization was carried out using the hanging-drop, vapor-diffusion method through mixing equal volume of protein and well solution. The complex of SAMD1-WH (27 to 105), and DNA was prepared by mixing the target protein with a 13-bp CpG-containing dsDNA (5′-ACCTGCGCACCAT-3′ as the sequence of one strand) at the molar ratio of 2:1.1.
The crystals of both native and selenomethionine-labeled SAMD1-WH/DNA complex were grown in the solution containing 0.2 M calcium acetate hydrate, 20% (w/v) polyethylene glycol 3350 at 4°C. Crystals were flash-frozen in the cryoprotectant composed of crystallization buffer containing 12% 2,3-butanediol.
The crystals of SAMD1-SAM (459 to 523) were grown in the solution containing 2.1 M ammonium sulfate and 0.2 M magnesium chloride hexahydrate at 20°C. Crystals of SAMD1-SAM (459 to 530) were grown in the solution of 0.1 M Hepes at pH 7.0, 23% (w/v) polyethylene glycol 3350 at 20°C. Crystallization buffer with addition of 10% 2,3-butanediol was used as the cryoprotectant for both crystals. The crystals of SAMD1-SAM (459 to 526) were grown in the solution containing 0.1 M bis-tris at pH 7.5 and 2.1 M ammonium sulfate at 20°C. Crystallization buffer containing 20% glycerol was used as the cryoprotectant.
All the datasets were collected at the Shanghai Synchrotron Radiation Facility beamlines in China at the temperature of −196°C. Datasets for selenomethionine-labeled SAMD1-WH/DNA complex crystals were collected at the beamline BL19U1 at the wavelength of 0.97855 Å. Datasets for SAMD1-SAM (459 to 523) crystals were collected at the beamline BL17U1 at the wavelength of 0.97922 Å. Datasets for SAMD1-SAM (459 to 526) crystals were collected at the beamline BL19U1 at the wavelength of 0.97891 Å. Datasets for selenomethionine-labeled SAMD1-SAM (459 to 530) crystals were collected at the beamline BL19U1 at the wavelength of 0.97917. The datasets were processed using the program HKL2000 (62). Structures of SAMD1-WH/DNA complex and SAMD1-SAM (459 to 530) were solved by PHENIX (63) using the SAD method with the anomalous signals from selenomethionine-labeled crystals. The initial partial model was manually built in Coot (64) and further refined by PHENIX. High-resolution structures of SAMD1-SAM (459 to 523) and SAMD1-SAM (459 to 526) were solved by molecular replacement method using the model of SAMD1-SAM (459 to 530). There is one SAMD1-WH/DNA complex molecule in one crystallographic asymmetric unit. In the final model, 98.55 and 1.45% residues are refined in the favored and allowed regions in the Ramachandran plot, respectively. There is one SAMD1-SAM pentamer in one asymmetric unit of the SAMD1-SAM (459 to 523) crystals. In the final model, 99.38 and 0.62% residues are refined in the favored and allowed regions in the Ramachandran plot, respectively. There are two SAMD1-SAM decamers in one asymmetric unit of the SAMD1-SAM (459 to 530) and SAMD1-SAM (459 to 526) crystals. In both crystals, each decamer is formed by two head-to-head stacked SAMD1-SAM pentamers. In the final model of the SAMD1-SAM (459 to 526) structure, 98.28 and 1.72% residues are refined in the favored and allowed regions in the Ramachandran plot, respectively. X-ray statistics are listed in Table 1.
ITC measurement
Calorimetric experiments were carried out at 20°C with a MicroCal iTC200 instrument. Purified WT or mutant proteins and dsDNA molecules were dialyzed overnight at 4°C in the titration buffer containing 20 mM tris at pH 7.5, 150 mM NaCl, and 2 mM β-mercaptoethanol. Titration was performed by injecting DNA molecules into the protein samples. Calorimetric titration data were fitted with the Origin software under the algorithm of one binding site model. All ITC measurements have been repeated at least twice. ITC binding parameters are listed in Table 2.
Electrophoretic mobility shift assay
dsDNA (50 pmol) was mixed with increasing amounts of recombinant SAMD1-WH proteins in the reaction buffer containing 20 mM tris at pH 7.0, 200 mM NaCl, and 2 mM DTT and incubated at 4°C for 10 min. The mixture was then loaded onto a 1.2% agarose gel in the tris-acetate-EDTA buffer for electrophoresis and detected by ethidium bromide staining. SAMD1-WH (16 to 110), and its mutants were used for the analysis. All EMSA experiments were repeated at least three times. DNA names and sequences are listed in table S2.
Statistical analysis
Statistical analyses were performed as indicated in the figure legends or Materials and Methods.
Acknowledgments
Funding: R.L. is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, 109546710 - TRR81/3, 416910386 - GRK 2573/1) and the Fritz-Thyssen Foundation (Az. 10.20.1.005). Z.W. is supported by the National Natural Science Foundation of China (32071204 and 31870725). M.L.B. was supported by NIH grant R01 HG010501. Y.S. is an American Cancer Society Research Professor. Author contributions: R.L. initiated the project, performed complex purification of SAMD1, performed bioinformatical analyses, and supervised experiments. B.S. performed most biochemical, in vivo, and genome-wide experiments. Y.Z. and Y.C. performed structural and biochemical studies on the SAMD1-WH and SAMD1-SAM domains, respectively. J.J. and Y.R. helped for the structural work. C.S. performed Co-IP and immunofluorescence experiments. I.R. performed Mammalian-Two-Hybrid experiments. A.N. performed next generation sequencing. S.K.P. performed PBM experiments and analysis. H.-M.P. performed biological experiments. Z.W., M.H, M.L.B, T.S., and Y.S. supervised experiments. R.L. and Z.W. wrote the manuscript with input from all authors. Competing interests: M.L.B. is a coinventor on a U.S. patent related to this work filed by The Brigham and Women’s Hospital Inc. (no. 8,530,638, published 10 September 2013). Universal PBM array designs used in this study are available via a materials transfer agreement with The Brigham and Women’s Hospital. Y.S. is a scientific cofounder and holds equity of Constellation Pharmaceuticals Inc. and Athelas Therapeutics Inc. Y.S. is also a scientific cofounder and a member of the scientific advisory board of K36 Therapeutics Inc. Y.S. also holds equity of Imago Biosciences and is a consultant for Active Motif Inc. The other authors declare that they have no competing interests. Data and materials availability: ChIP-seq and RNA-seq data are available at the GEO repository under the accession number GSE144396. Coordinates for the crystal structures of the SAMD1-WH/DNA complex and the SAMD1-SAM domain in two crystal forms have been deposited into the Protein Data Bank (PDB) under the accession codes of 6LUI, 6LUJ, and 6LUK, respectively. Mass spectrometry data are available at the PRIDE Repository under the accession number PXD016781. PBM data are available in the UniPROBE database under the accession number STI20A. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf2229/DC1
REFERENCES AND NOTES
- 1.Deaton A. M., Bird A., CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Voo K. S., Carlone D. L., Jacobsen B. M., Flodin A., Skalnik D. G., Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol. Cell. Biol. 20, 2108–2121 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomson J. P., Skene P. J., Selfridge J., Clouaire T., Guy J., Webb S., Kerr A. R., Deaton A., Andrews R., James K. D., Turner D. J., Illingworth R., Bird A., CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464, 1082–1086 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farcas A. M., Blackledge N. P., Sudbery I., Long H. K., McGouran J. F., Rose N. R., Lee S., Sims D., Cerase A., Sheahan T. W., Koseki H., Brockdorff N., Ponting C. P., Kessler B. M., Klose R. J., KDM2B links the Polycomb repressive complex 1 (PRC1) to recognition of CpG islands. eLife 1, e00205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H., Liefke R., Jiang J., Kurland J. V., Tian W., Deng P., Zhang W., He Q., Patel D. J., Bulyk M. L., Shi Y., Wang Z., Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549, 287–291 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voigt P., Tee W. W., Reinberg D., A double take on bivalent promoters. Genes Dev. 27, 1318–1338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perino M., van Mierlo G., Karemaker I. D., van Genesen S., Vermeulen M., Marks H., van Heeringen S. J., Veenstra G. J. C., MTF2 recruits Polycomb repressive complex 2 by helical-shape-selective DNA binding. Nat. Genet. 50, 1002–1010 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Viturawong T., Meissner F., Butter F., Mann M., A DNA-centric protein interaction map of ultraconserved elements reveals contribution of transcription factor binding hubs to conservation. Cell Rep. 5, 531–545 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Xiong J., Zhang Z., Chen J., Huang H., Xu Y., Ding X., Zheng Y., Nishinakamura R., Xu G. L., Wang H., Chen S., Gao S., Zhu B., Cooperative action between SALL4A and TET proteins in stepwise oxidation of 5-methylcytosine. Mol. Cell 64, 913–925 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Bartke T., Vermeulen M., Xhemalce B., Robson S. C., Mann M., Kouzarides T., Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143, 470–484 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Bonasio R., Strino F., Kluger Y., Holloway J. K., Modzelewski A. J., Cohen P. E., Reinberg D., SFMBT1 functions with LSD1 to regulate expression of canonical histone genes and chromatin-related factors. Genes Dev. 27, 749–766 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malovannaya A., Li Y., Bulynko Y., Jung S. Y., Wang Y., Lanz R. B., O’Malley B. W., Qin J., Streamlined analysis schema for high-throughput identification of endogenous protein complexes. Proc. Natl. Acad. Sci. U.S.A. 107, 2431–2436 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelen E., Brandsma J. H., Moen M. J., Signorile L., Dekkers D. H., Demmers J., Kockx C. E., Ozgür Z., van IJcken W. F., van den Berg D. L., Poot R. A., Proteins that bind regulatory regions identified by histone modification chromatin immunoprecipitations and mass spectrometry. Nat. Commun. 6, 7155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., Schwede T., SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berger M. F., Philippakis A. A., Qureshi A. M., He F. S., Estep P. W. III, Bulyk M. L., Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 24, 1429–1435 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harami G. M., Gyimesi M., Kovacs M., From keys to bulldozers: Expanding roles for winged helix domains in nucleic-acid-binding proteins. Trends Biochem. Sci. 38, 364–371 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Hu D., Gao X., Cao K., Morgan M. A., Mas G., Smith E. R., Volk A. G., Bartom E. T., Crispino J. D., Di Croce L., Shilatifard A., Not All H3K4 methylations are created equal: Mll2/COMPASS dependency in primordial germ cell specification. Mol. Cell 65, 460–475.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu T., Park S. S., Giaimo B. D., Hall D., Ferrante F., Ho D. M., Hori K., Anhezini L., Ertl I., Bartkuhn M., Zhang H., Milon E., Ha K., Conlon K. P., Kuick R., Govindarajoo B., Zhang Y., Sun Y., Dou Y., Basrur V., Elenitoba-Johnson K. S., Nesvizhskii A. I., Ceron J., Lee C. Y., Borggrefe T., Kovall R. A., Rual J. F., RBPJ/CBF1 interacts with L3MBTL3/MBT1 to promote repression of Notch signaling via histone demethylase KDM1A/LSD1. EMBO J. 36, 3232–3249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi Y., Lan F., Matson C., Mulligan P., Whetstine J. R., Cole P. A., Casero R. A., Shi Y., Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Isono K., Endo T. A., Ku M., Yamada D., Suzuki R., Sharif J., Ishikura T., Toyoda T., Bernstein B. E., Koseki H., SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell 26, 565–577 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Wani A. H., Boettiger A. N., Schorderet P., Ergun A., Munger C., Sadreyev R. I., Zhuang X., Kingston R. E., Francis N. J., Chromatin topology is coupled to Polycomb group protein subnuclear organization. Nat. Commun. 7, 10291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao K., Collings C. K., Morgan M. A., Marshall S. A., Rendleman E. J., Ozark P. A., Smith E. R., Shilatifard A., An Mll4/COMPASS-Lsd1 epigenetic axis governs enhancer function and pluripotency transition in embryonic stem cells. Sci. Adv. 4, eaap8747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonasio R., Lecona E., Reinberg D., MBT domain proteins in development and disease. Semin. Cell Dev. Biol. 21, 221–230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egolf S., Aubert Y., Doepner M., Anderson A., Maldonado-Lopez A., Pacella G., Lee J., Ko E. K., Zou J., Lan Y., Simpson C. L., Ridky T., Capell B. C., LSD1 inhibition promotes epithelial differentiation through derepression of fate-determining transcription factors. Cell Rep. 28, 1981–1992.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris W. J., Huang X., Lynch J. T., Spencer G. J., Hitchin J. R., Li Y., Ciceri F., Blaser J. G., Greystoke B. F., Jordan A. M., Miller C. J., Ogilvie D. J., Somervaille T. C., The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Nady N., Krichevsky L., Zhong N., Duan S., Tempel W., Amaya M. F., Ravichandran M., Arrowsmith C. H., Histone recognition by human malignant brain tumor domains. J. Mol. Biol. 423, 702–718 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Frey F., Sheahan T., Finkl K., Stoehr G., Mann M., Benda C., Muller J., Molecular basis of PRC1 targeting to Polycomb response elements by PhoRC. Genes Dev. 30, 1116–1127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim C. A., Gingery M., Pilpa R. M., Bowie J. U., The SAM domain of polyhomeotic forms a helical polymer. Nat. Struct. Biol. 9, 453–457 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Kim C. A., Phillips M. L., Kim W., Gingery M., Tran H. H., Robinson M. A., Faham S., Bowie J. U., Polymerization of the SAM domain of TEL in leukemogenesis and transcriptional repression. EMBO J. 20, 4173–4182 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiao F., Bowie J. U., The many faces of SAM. Sci. STKE 2005, re7 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Wang J., Scully K., Zhu X., Cai L., Zhang J., Prefontaine G. G., Krones A., Ohgi K. A., Zhu P., Garcia-Bassets I., Liu F., Taylor H., Lozach J., Jayes F. L., Korach K. S., Glass C. K., Fu X. D., Rosenfeld M. G., Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446, 882–887 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Long H. K., Blackledge N. P., Klose R. J., ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem. Soc. Trans. 41, 727–740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horsefield S., Burdett H., Zhang X., Manik M. K., Shi Y., Chen J., Qi T., Gilley J., Lai J. S., Rank M. X., Casey L. W., Gu W., Ericsson D. J., Foley G., Hughes R. O., Bosanac T., von Itzstein M., Rathjen J. P., Nanson J. D., Boden M., Dry I. B., Williams S. J., Staskawicz B. J., Coleman M. P., Ve T., Dodds P. N., Kobe B., NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 365, 793–799 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Siklenka K., Erkek S., Godmann M., Lambrot R., McGraw S., Lafleur C., Cohen T., Xia J., Suderman M., Hallett M., Trasler J., Peters A. H., Kimmins S., Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 350, aab2006 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Nishiyama A., Sharov A. A., Piao Y., Amano M., Amano T., Hoang H. G., Binder B. Y., Tapnio R., Bassey U., Malinou J. N., Correa-Cerro L. S., Yu H., Xin L., Meyers E., Zalzman M., Nakatake Y., Stagg C., Sharova L., Qian Y., Dudekula D., Sheer S., Cadet J. S., Hirata T., Yang H. T., Goldberg I., Evans M. K., Longo D. L., Schlessinger D., Ko M. S., Systematic repression of transcription factors reveals limited patterns of gene expression changes in ES cells. Sci. Rep. 3, 1390 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Z., Li C., Kang B., Gao G., Zhang Z., GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norman T. M., Horlbeck M. A., Replogle J. M., Ge A. Y., Xu A., Jost M., Gilbert L. A., Weissman J. S., Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 365, 786–793 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lees A. M., Deconinck A. E., Campbell B. D., Lees R. S., Atherin: A newly identified, lesion-specific, LDL-binding protein in human atherosclerosis. Atherosclerosis 182, 219–230 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mermoud J. E., Costanzi C., Pehrson J. R., Brockdorff N., Histone macroH2A1.2 relocates to the inactive X chromosome after initiation and propagation of X-inactivation. J. Cell. Biol. 147, 1399–1408 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volkel S., Stielow B., Finkernagel F., Stiewe T., Nist A., Suske G., Zinc finger independent genome-wide binding of Sp2 potentiates recruitment of histone-fold protein Nf-y distinguishing it from Sp1 and Sp3. PLOS Genet. 11, e1005102 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramirez F., Dundar F., Diehl S., Gruning B. A., Manke T., deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orlando D. A., Chen M. W., Brown V. E., Solanki S., Choi Y. J., Olson E. R., Fritz C. C., Bradner J. E., Guenther M. G., Quantitative ChIP-seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Gruning B. A., Guerler A., Hillman-Jackson J., Hiltemann S., Jalili V., Rasche H., Soranzo N., Goecks J., Taylor J., Nekrutenko A., Blankenberg D., The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu T., Ortiz J. A., Taing L., Meyer C. A., Lee B., Zhang Y., Shin H., Wong S. S., Ma J., Lei Y., Pape U. J., Poidinger M., Chen Y., Yeung K., Brown M., Turpaz Y., Liu X. S., Cistrome: An integrative platform for transcriptional regulation studies. Genome Biol. 12, R83 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huber W., Carey V. J., Gentleman R., Anders S., Carlson M., Carvalho B. S., Bravo H. C., Davis S., Gatto L., Girke T., Gottardo R., Hahne F., Hansen K. D., Irizarry R. A., Lawrence M., Love M. I., MacDonald J., Obenchain V., Oles A. K., Pages H., Reyes A., Shannon P., Smyth G. K., Tenenbaum D., Waldron L., Morgan M., Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu P., Xiao S., Xin X., Song C. X., Huang W., McDee D., Tanaka T., Wang T., He C., Zhong S., Spatiotemporal clustering of the epigenome reveals rules of dynamic gene regulation. Genome Res. 23, 352–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dennis G. Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A., DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4, R60 (2003). [PubMed] [Google Scholar]
- 56.Letunic I., Bork P., Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C. L., Haase J., Janes J., Huss J. W. III, Su A. I., BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10, R130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown D. A., Di Cerbo V., Feldmann A., Ahn J., Ito S., Blackledge N. P., Nakayama M., McClellan M., Dimitrova E., Turberfield A. H., Long H. K., King H. W., Kriaucionis S., Schermelleh L., Kutateladze T. G., Koseki H., Klose R. J., The SET1 complex selects actively transcribed target genes via multivalent interaction with CpG island chromatin. Cell Rep. 20, 2313–2327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Otwinowski Z., Minor W., [20] Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 63.Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf2229/DC1