

Abstract
Immune cells identify and destroy tumors by recognizing cellular traits indicative of oncogenic transformation. In this study, we found that myocardin related transcription factors (MRTFs), which promote migration and metastatic invasion, also sensitize cancer cells to the immune system. Melanoma and breast cancer cells with high MRTF expression were selectively eliminated by cytotoxic lymphocytes in mouse models of metastasis. This immunosurveillance phenotype was further enhanced by treatment with immune checkpoint blockade (ICB) antibodies. We also observed that high MRTF signaling in human melanoma is associated with ICB efficacy in patients. Using biophysical and functional assays, we showed that MRTF overexpression rigidified the filamentous actin cytoskeleton and that this mechanical change rendered mouse and human cancer cells more vulnerable to cytotoxic T lymphocytes and natural killer cells. Collectively, these results suggest that immunosurveillance has a mechanical dimension, which we call mechanosurveillance, that is particularly relevant for the targeting of metastatic disease.
IN BRIEF
Myocardin related transcription factors promote metastatic colonization by inducing cell spreading and migration. Tello et al. show that the cellular stiffening that accompanies this morphologic change triggers a mechanical form of immunosurveillance in which cytotoxic lymphocytes destroy the metastatic cells.
INTRODUCTION
Immune cells detect and eliminate cancer by recognizing characteristic features that are indicative of oncogenic transformation. This process, known as immunosurveillance, is critical for the destruction of incipient tumors and plays a central role in anti-cancer immunotherapy (Finn, 2018). It is generally thought that immunosurveillance is mediated by molecular cues, such as stress-ligands, neoantigens, and danger associated molecular patterns, that trigger activating receptors on patrolling immune cells (Hernandez et al., 2016; Schumacher et al., 2019; Vesely et al., 2011). These biochemical features, however, are not exhibited by all cancer cells at all stages of disease, and they can be found in untransformed tissue, as well. Hence, effective immunosurveillance must utilize additional determinants. In that regard, it is intriguing that cancer progression also involves profound changes in cellular architecture and mechanics (Hall, 2009; Northcott et al., 2018; Suresh, 2007). These biophysical events are critical for promoting migration and invasion, but whether they also serve as a basis for immunosurveillance is not known.
Cytotoxic lymphocytes, comprising natural killer (NK) cells and cytotoxic T lymphocytes (CTLs), play a central role in anti-cancer immunosurveillance by destroying transformed cells (Finn, 2018). Their cytolytic activity is triggered by recognition of surface molecules characteristic of stress and transformation, including cognate peptide-major histocompatibility complex (MHC) and the UL16 binding proteins, which engage the T cell antigen receptor (TCR) and the activating NK receptor NKG2D, respectively (Lanier, 2005; Zhang and Bevan, 2011). The binding of these and other stimulatory ligands drives the formation of a stereotyped interface between the lymphocyte and its target, called the immune synapse (Dustin and Long, 2010). The lymphocyte then secretes toxic granzyme proteases and the pore forming protein perforin into the synaptic space, thereby inducing target cell apoptosis.
Immune synapses are physically active structures, exerting nanonewton scale mechanical forces that enhance the efficiency of perforin and granzyme-mediated killing (Bashour et al., 2014; Basu et al., 2016; Husson et al., 2011). These forces are also thought to facilitate lymphocyte activation by promoting mechanotransduction. Indeed, several activating immunoreceptors, including the TCR, only reach full signaling capacity under applied force (Friedland et al., 2009; Liu et al., 2014). This requirement places physical demands on the target cell surface, which must presumably be rigid enough to counterbalance the mechanical load placed upon receptor-bound ligands. Consistent with this idea, stiff surfaces bearing stimulatory ligands induce substantially stronger lymphocyte activation than softer surfaces coated with the same proteins (Blumenthal et al., 2020; Comrie et al., 2015; Judokusumo et al., 2012; Saitakis et al., 2017; Wan et al., 2013). Hence, it is not unreasonable to expect that the biophysical properties of cancer cells might regulate their susceptibility to cytotoxic lymphocyte-mediated attack.
Cytotoxic lymphocytes are particularly effective at combatting metastatic cancer cells (Dye, 1986; Eyles et al., 2010; Malladi et al., 2016; Pommier et al., 2018; Wei et al., 2018), which live alone or in small groups far from the immunosuppressive microenvironment of the primary tumor. Interestingly, metastasis is associated with dramatic remodeling of the filamentous actin (F-actin) cytoskeleton, which drives the morphological and biophysical changes required for local invasion from the primary tumor, intravasation into circulation, and subsequent extravasation into target organs (Bravo-Cordero et al., 2012). It is now becoming clear that F-actin dynamics are also critical for metastatic outgrowth in the new microenvironment, which typically occurs in the perivascular niche, a nutrient rich milieu on the abluminal surface of microvessels (Ghajar et al., 2013; Kienast et al., 2010). To expand successfully in this space, metastatic cells first establish strong adhesion to the microvascular basement membrane (Shibue and Weinberg, 2009; Valiente et al., 2014). This triggers a mechanotransduction response in which cell spreading and migration are coupled to the activation of myocardin-related transcription factors (MRTF) A and B (Er et al., 2018). In the steady state, MRTF isoforms are sequestered in the cytoplasm via binding to monomeric globular actin (G-actin). Cytoskeletal growth, often induced by Rho-family GTPases like RAC1, depletes G-actin, liberating MRTFA and MRTFB to enter the nucleus, where they form complexes with the DNA-binding protein serum response factor (SRF) to drive expression of more G-actin and cytoskeletal components. (Gau and Roy, 2018; Gualdrini et al., 2016; Kim et al., 2017; Lionarons et al., 2019; Medjkane et al., 2009; Olson and Nordheim, 2010). Morphoregulation by MRTF is absolutely required for metastatic invasion and subsequent proliferative expansion (Er et al., 2018). However, because analogous shape changes are known to increase cell stiffness (Kasza et al., 2009), it is tempting to speculate that MRTF signaling might also mechanically sensitize cancer cells to cytotoxic lymphocytes. In the present study, we explored this hypothesis by analyzing the effects of MRTF on the mechanical properties of cancer cells, their immune sensitivity, and their capacity to colonize tissues in vivo. Our results suggest a mechanical form of immunosurveillance that enables cytotoxic lymphocytes to target the specific biophysical features of metastatic cancer cells.
RESULTS
MRTF overexpression sensitizes metastatic cells to the immune system
Given the importance of MRTF isoforms for migration, metastasis, and the regulation of F-actin, we reasoned that manipulating MRTFA and MRTFB levels might reveal novel vulnerabilities associated with cancer cell architecture. To explore this idea, we employed an established model of metastatic colonization in which malignant cancer cells expressing luciferase are injected intravenously into congenic mice and their subsequent growth in the lung monitored by bioluminescent imaging (Fig. 1A). shRNA-mediated suppression of MRTFA and MRTFB reduced lung colonization by both B16F10 melanoma and E0771 breast cancer cells (Fig. 1B–1C and Fig. S1A), consistent with previous data showing that MRTF is required for metastatic seeding (Er et al., 2018; Kim et al., 2017; Medjkane et al., 2009). However, B16F10 and E0771 cell lines overexpressing MRTFB (B16F10-MRTFB and E0771-MRTFB) exhibited dramatically reduced lung colonization (Fig. 1D–1E and Fig. S1A), the opposite of what one would expect for cells with enhanced MRTF-induced invasiveness. E0771-MRTFA cells also metastasized poorly, although B16F10-MRTFA cells displayed increased colonization (Fig. S2A). Overexpression of MRTFA and MRTFB did not significantly alter cancer cell growth and division in vitro, as assessed by the acquisition of vital dye and the dilution of tracing stain, respectively (Fig. S1B–S1C). Proliferation in vivo, which we quantified by staining tumor sections for the Ki67 marker, was similarly unchanged (Fig. S1D). We also examined the effects of MRTF overexpression on steady state cell death, and found that only E0771-MRTFA cells exhibited increased apoptosis (Fig. S1E). These results suggested that the reduced metastatic capacity conferred by MRTF was not entirely intrinsic to the tumor cells and instead required component(s) of the metastatic microenvironment.
Fig. 1. MRTF overexpression sensitizes metastatic tumors to cytotoxic lymphocytes.
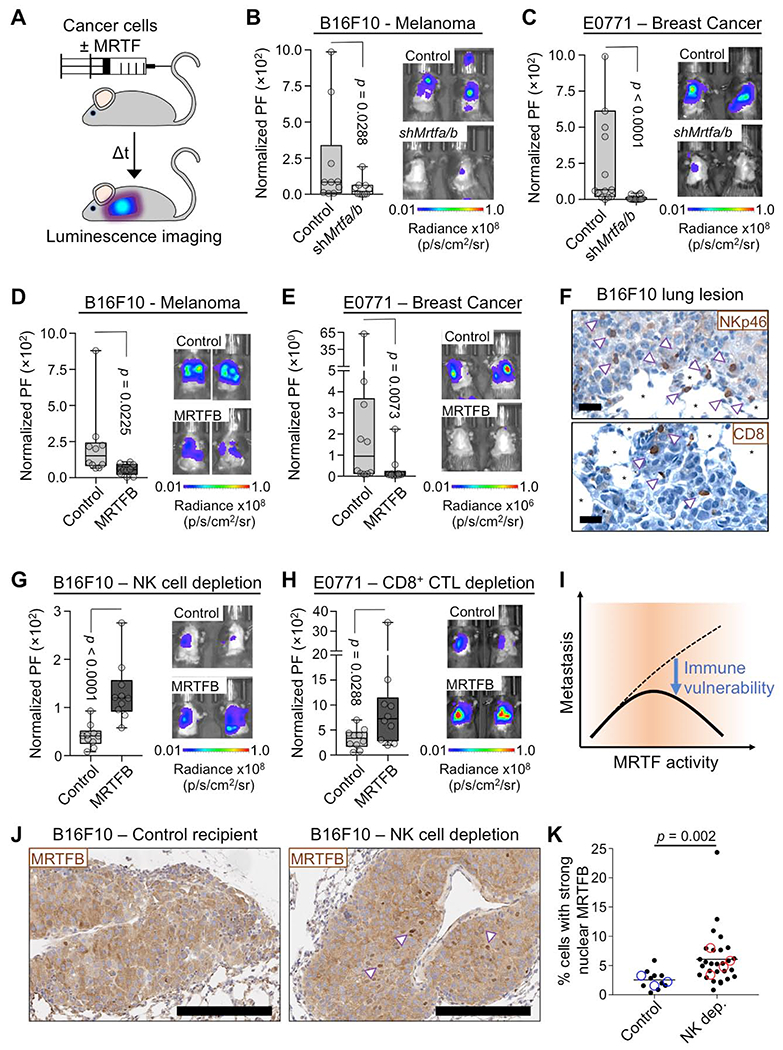
(A) Experimental design for the lung colonization model. (B-C) Metastatic burden in lungs of C57BL/6J mice injected with syngeneic control or Mrtfa/bRNAi B16F10 (B) or E0771 (C) cells, measured by bioluminescent imaging (BLI) 3 weeks after tail vein injection and normalized to the first day of injection. PF: photon flux (n = 10 mice per group). (D-E) BLI of mice 3 weeks post tail vein injection with B16F10 (D) or E0771 (E) cells overexpressing MRTFB or empty vector control (n = 10 mice per group). (F) Representative IHC images of NK cells (arrowheads, NKp46 staining) and CD8+ T cells (arrowheads, CD8 staining) in B16F10 lung metastases. *: alveolar space, scale bars: 20 μm. (G-H) BLI of mice pretreated with anti-asialo GM1 antibody (G) or anti-CD8 antibody (H) for NK and CD8+ T cell depletion, respectively, and imaged 2 weeks after injection of B16F10 (G) or E0771 (H) cells overexpressing MRTFB or empty vector control (n = 10 mice per group). (I) Increased MRTF activity promotes metastasis by enhancing invasiveness (dashed line), but it also sensitizes metastatic cells to cytotoxic lymphocytes (solid line). (J-K) B16F10 lung metastases from control and anti-asialo GM1 treated (NK cell depletion) mice were stained for MRTFB. (J) Representative IHC images, with arrowheads indicating examples of strong nuclear localization. Scale bars: 200 μm. (K) Percentage of cells in each type of tumor with MRTFB nuclear/cytoplasmic ratio > 2 (strong nuclear MRTFB). Closed symbols represent individual lesions (n ≥ 10 lesions of > 200 cells per group), and open symbols denote individual mice (n ≥ 3 per group). Statistical significance was evaluated using lesion data. All p values calculated by Mann-Whitney test. See also Fig. S1 and S2.
Histological analyses of B16F10 and E0771 lung lesions revealed a substantial number of infiltrating CD8+ CTLs and NK cells (Fig. 1F and Fig. S2B). To assess the importance of these lymphocytes for MRTF dependent tumor suppression, we treated mice with NK cell or CD8+ T cell depleting antibodies prior to injecting cancer cells. NK cell depletion dramatically enhanced colonization by B16F10-MRTFA and B16F10-MRTFB cells (Fig. 1G and Fig. S2C). In the case of B16F10-MRTFB cells, this effect led to a striking phenotypic reversal in which the overexpressing cells now exhibited stronger metastatic growth than B16F10 controls (compare Fig. 1D to 1G). Depletion of CD8+ T cells induced similarly remarkable increases in both E0771-MRTFB and E0771-MRTFA colonization (Fig. 1H and Fig. S2D). Interestingly, NK depletion did not rescue the metastatic activity of E0771-MRTFB cells (Fig. S2E), implying that these cells were primarily constrained by CTLs in vivo, whereas B16F10-MRTFB tumors were subject to NK-mediated control. Collectively, these results indicate that the increased metastatic potential conferred by MRTF signaling is curbed in vivo by cytotoxic lymphocytes (Fig. 1I). The selective targeting of cancer cells with high MRTF activity by the immune system would be expected to suppress this trait in the tumor, a process known as immunoediting (Schreiber et al., 2011). Consistent with this prediction, B16F10 metastases from mice lacking NK cells exhibited more MRTFB nuclear localization (an index of MRTF signaling) than analogous metastases from control animals (Fig. 1J–1K, Fig. S2F). Hence, MRTF pathway activation is a liability for cancer cells, but only in the context of cytotoxic lymphocytes.
MRTF makes cancer cells more stimulatory to CTLs and NK cells
Tumors overexpressing MRTF did not exhibit greater CTL and NK cell infiltration (Fig. S2G–H), implying that the immune-mediated suppression we observed in this context was not caused by altered lymphocyte recruitment but rather by enhanced cancer cell vulnerability to cytotoxic responses. To investigate this possibility, we measured the in vitro killing of B16F10 and E0771 cells by CTLs expressing the OT1 TCR, which recognizes OVA bound to the MHC protein H-2Kb. While B16F10 cells expressed less MHC than E0771 cells, both succumbed to CTLs in the presence of sufficient antigen. shRNA-induced suppression of MRTFA and MRTFB had modest effects on cytotoxicity, moderately reducing B16F10 killing and leaving E0771 killing unchanged (Fig. S3A). By contrast, overexpression of either MRTFA or MRTFB markedly enhanced CTL-mediated lysis of both B16F10 and E0771 cells (Fig. 2A–2B). This effect was specific for cellular cytotoxicity, as overexpression of MRTF did not, in general, augment apoptotic responses to staurosporine and tumor necrosis factor (TNF), although E0771-MRTFA cells were more vulnerable to these agents (Fig. S3B–C). To further characterize the MRTF overexpression phenotype, we examined the dynamic properties of individual CTL-cancer cell interactions by live microscopy. B16F10-MRTFA, B16F10-MRTFB, and control B16F10 cells were mixed with OT1 CTLs and imaged in the presence of propidium iodide (PI), a fluorescent dye that stains nuclei upon cell death (Fig. 2C, Movies S1–S3). Consistent with our bulk cytotoxicity experiments, we found that synapses between CTLs and B16F10-MRTFA or B16F10-MRTFB cells were more likely to result in cancer cell death than synapses between CTLs and B16F10 controls (Fig. 2D). MRTF overexpression also correlated with an increased speed of killing, defined by the time interval between synapse formation and target death (Fig. 2E, Movies S1–S3). Collectively, these results indicate that increased MRTF signaling sensitizes cancer cells to cytotoxic lymphocytes.
Fig. 2. MRTF expression sensitizes cancer cells to CTL-mediated lysis.
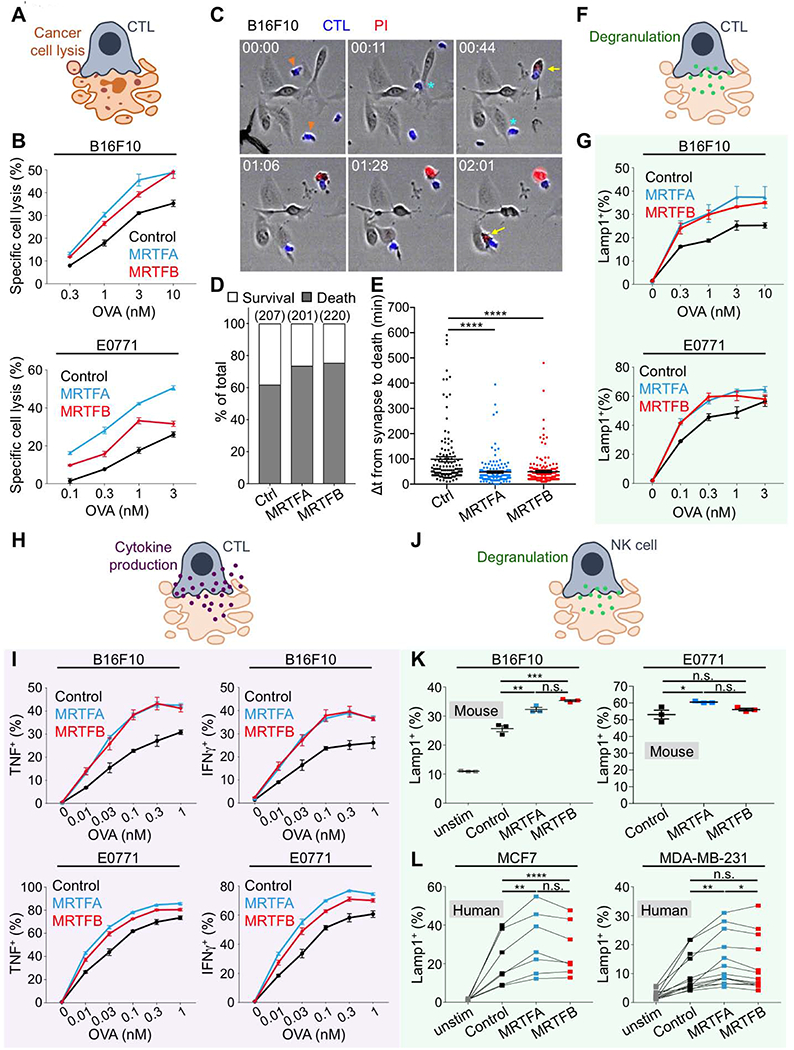
(A) Diagram of CTL-mediated cancer cell lysis. (B) B16F10 (top) or E0771 (bottom) cells overexpressing MRTFA, MRTFB, or empty vector were loaded with increasing concentrations of OVA and then mixed with OT1 CTLs. Specific lysis was quantified after 5 h. (C-E) CTLs labeled with cell trace violet were mixed with OVA-loaded B16F10 cells overexpressing MRTFA, MRTFB, or empty vector and then imaged in the presence of PI. (C) Time-lapse montage showing representative B16F10 killing events. CTLs of interest are denoted by orange arrowheads in the first image. Cyan asterisks indicate synapse formation, and yellow arrows the influx of PI into dying targets. Time in HH:MM is shown at the top left corner of each image. (D) Frequency of survival and death in 1:1 conjugates involving CTLs and the indicated B16F10 cells. The total number of conjugates analyzed is shown in parenthesis at the top of each bar. (E) The time delay between synapse formation and death, quantified for all 1:1 synapses (n ≥ 128 events per sample). ****p ≤ 0.0001, calculated by one-way ANOVA. Data in C-E are representative of two independent experiments. (F) Diagram of CTL degranulation after synapse formation. (G) Degranulation, measured by surface exposure of CTL Lamp1 90 min after mixing with B16F10 (top) or E0771 (bottom) cells overexpressing MRTFA, MRTFB, or empty vector. (H) Diagram of CTL cytokine secretion after synapse formation. (I) Production of TNF (left) and IFNγ (right), measured by intracellular staining of CTLs 4 h after mixing with B16F10 (top) or E0771 (bottom) cells overexpressing MRTFA, MRTFB, or empty vector. (J) Diagram showing NK cell degranulation after synapse formation. (K) Splenic murine NK cells were mixed with B16F10 (left) or E0771 (right) cells overexpressing MRTFA, MRTFB, or empty vector and degranulation quantified after 4 h. Data in B, G, I, and K are shown as mean ± SEM of technical triplicates, representative of 3 independent experiments. (L) PBMCs derived from peripheral blood were mixed with the indicated control or MRTFA/B overexpressing cell lines. After 5 h, NK cell degranulation was measured by surface exposure of Lamp1. Connecting lines indicate samples derived from the same donor. (n = 9 donors for MCF7 experiments, n = 10 donors for MDA-MB-231). ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05, and n.s.: not significant for p > 0.05, calculated by one-way ANOVA with sample pairing. See also Fig. S3 and Movies S1–S3.
To investigate the basis for this phenotype, we examined whether MRTF overexpression modulated cancer cell responses to the molecular components of cytotoxicity. Neither MRTFA nor MRTFB affected the sensitivity of B16F10 and E0771 cell lines to purified perforin and granzyme B (Fig. S3D), indicating that intrinsic cellular responses to these agents were unaffected by MRTF signaling. Cytotoxic lymphocytes also produce Fas ligand (FasL), which induces apoptosis in target cells expressing the death receptor Fas (Nagata, 1999). Although E0771-MRTFA cells expressed elevated levels of Fas and underwent apoptosis in response to soluble FasL, E0771-MRTFB, B16F10-MRTFA, and B16F10-MRTFB cells expressed little to no Fas and were resistant to FasL-mediated killing (Fig. S3E–F). Hence, differential sensitivity to perforin/granzyme or FasL did not broadly explain how MRTF signaling makes cancer cells more vulnerable to cytotoxic attack.
Next, we examined whether cancer cells overexpressing MRTFA and MRTFB induce stronger lymphocyte activation. Perforin and granzymes are stored in specialized secretory lysosomes called lytic granules, which fuse with the plasma membrane after synapse formation (Stinchcombe and Griffiths, 2007). To quantify granule exocytosis, also called degranulation, we monitored surface exposure of the lysosomal marker Lamp1 in OT1 CTLs cocultured with antigen-loaded cancer cells. B16F10-MRTFA/B and E0771-MRTFA/B cells induced stronger degranulation responses than did their respective controls (Fig. 2F–2G), implying that increased MRTF signaling in the target enables more effective CTL stimulation. Activated CTLs also generate and release the inflammatory cytokines interferon-γ (IFNγ) and TNF. Production of both cytokines was markedly enhanced in cocultures with cancer cells overexpressing MRTFA or MRTFB (Fig. 2H–I). Collectively, these data indicated that MRTFA and MRTFB render cancer cells more stimulatory to cytotoxic lymphocytes.
To explore the generality of this paradigm, we extended our studies to splenic NK cells derived from C57BL/6 mice. Similar to our results with CTLs, we found that cancer cells overexpressing MRTFA or MRTFB induced stronger NK cell degranulation than did their control counterparts (Fig. 2J–K), although these effects were more obvious for B16F10 cells than for E0771 cells. We also examined primary human NK cells, which recognize and destroy the breast carcinoma cells lines MDA-MB-231 and MCF7. Peripheral blood mononuclear cells (PBMCs, ~10% CD56+CD3− NK cells) from multiple donors were mixed with control MDA-MB-231 and MCF7 cells as well as lines overexpressing MRTFA and MRTFB. Degranulation responses were significantly stronger in cocultures with MRTF overexpressing cells (Fig. 2J, L). Furthermore, live imaging revealed that MRTF overexpression significantly increased the speed of NK-mediated killing (Fig. S3G). We conclude that MRTFA and MRTFB influence cytotoxic immune cell-cell interactions in multiple species and lymphocyte cell types.
MRTF signaling is associated with enhanced responses to checkpoint blockade therapy
The capacity of MRTF signaling to boost CTL responses against cancer cells implied that it might increase the efficacy of immune checkpoint blockade (ICB), a group of antibody-based therapies that function by derepressing tumor specific T cells (Lesokhin et al., 2015; Wei et al., 2018). To investigate this possibility, we injected mice with control B16F10 or B16F10-MRTFB cells while administering biweekly doses of blocking antibody against CTLA4, an inhibitory checkpoint receptor (Fig. 3A). On its own, anti-CTLA4 treatment moderately reduced lung colonization by control B16F10 cells, but this did not translate into a significant survival benefit relative to animals receiving isotype control antibody (Fig. 3B–C). By contrast, blocking CTLA4 significantly inhibited B16F10-MRTFB metastasis, leading to a marked increase in survival. Antibody targeting of the related checkpoint receptor PD1 yielded a similar pattern of results (Fig. 3D). These data indicate that MRTF overexpression sensitizes cancer cells in vivo to a therapeutically enhanced immune system.
Fig. 3. MRTF boosts therapeutic T cell responses in the context of ICB.
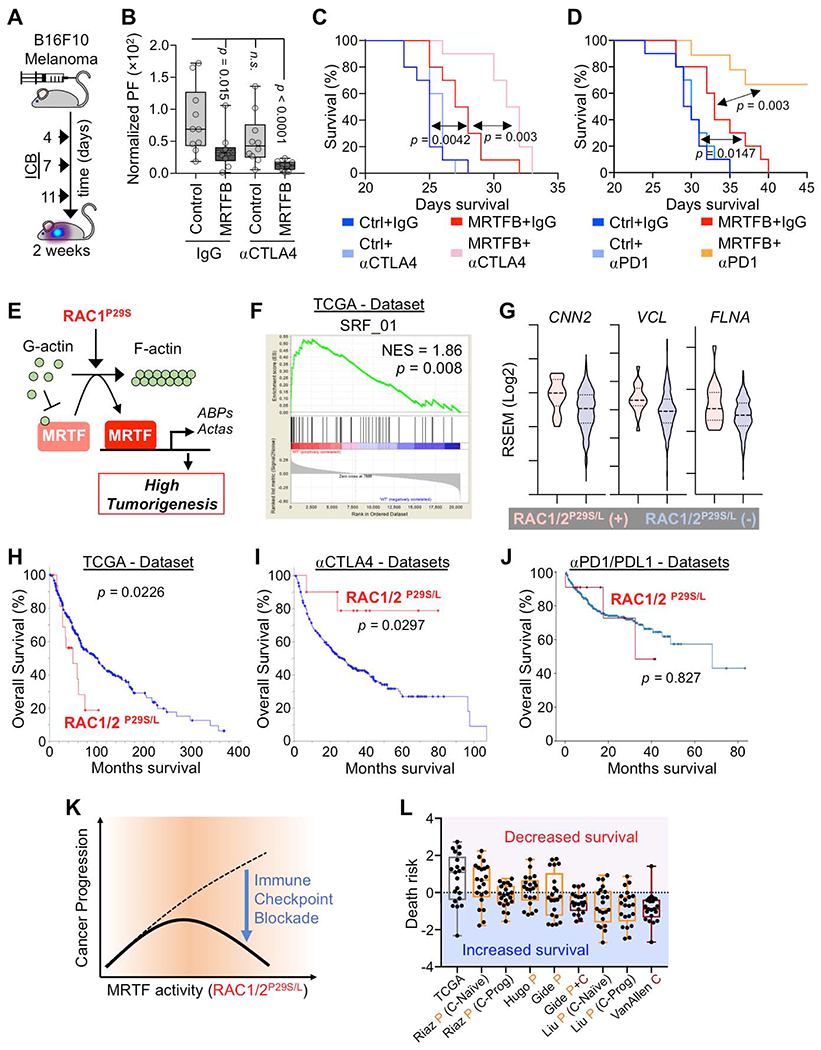
(A) Experimental design for ICB treatment of mice inoculated with B16F10 melanoma. (B) BLI of mice 2 weeks after injection with B16F10 cells overexpressing MRTFB or control vector and treatment with control IgG or anti-CTLA4 antibody (n = 10 mice per group). Box plots show upper and lower quartiles, median, maximum, and minimum values. p values calculated by Mann-Whitney test, n.s.: not significant for p = 0.1655. (C) Kaplan-Meier survival curves for percent survival of mice in B (n = 10 mice per group). (D) Kaplan-Meier survival curves for percent survival of mice injected with B16F10 cells overexpressing MRTFB or control vector and treated with control IgG or anti-PD1 antibody (n = 10 mice per group). (E) Diagram of pathway linking oncogenic RAC1P29S to MRTF (Lionarons et al., 2019). Acta: actin family of proteins, ABPs: actin binding proteins. (F) GSEA of MRTF-SRF target gene expression enrichment in RAC1/2P29S/L mutant skin cutaneous melanoma patients in The Cancer Genome Atlas (TCGA) dataset. NES: normalized enrichment score. (G) Violin plots showing increased expression of known MRTF-SRF target genes in RAC1/2P29S/L patients. Dashed lines medians, dotted lines upper and lower quartiles. (H-J) Overall survival of RAC1/2P29S/L patients in the TCGA dataset (H) and in melanoma patients treated with anti-CTLA4 (I) or anti-PD1/PDL1 (J). Anti-CTLA4 data was derived from Samstein et al., 2019 (75 patients); Miao et al., 2018 (144 patients); Van Allen et al., 2015 (20 patients); Catalanotti et al., 2017 (21 patients), and Liang et al., 2017 (14 patients). Anti PD-1 data was derived from Cheng et al., 2015 (375 patients). p values in C-D and H-J were calculated by Log-rank test. (K) MRTF activity induced by RAC1/2P29S/L potentiates melanoma progression (similar to Fig. 1I) but simultaneously sensitizes cancer cells to ICB. (L) Association between MRTF-SRF induced genes and survival for the TCGA data set and the indicated ICB data sets (n = 21 genes). Box plots show upper and lower quartiles, median, maximum, and minimum values. Anti-PD1 trials (letter P) are shown in orange and anti-CTLA4 trials (letter C) in maroon. See also Fig. S4 and supplemental tables 1–4.
Next, we examined clinical data for links between MRTF signaling and ICB responsiveness in humans. While there are no reported MRTF gain-of-function mutations in human solid tumors, MRTF signaling is strongly induced by the constitutively active P29S mutant form of RAC1 (Fig. 3E) (Lionarons et al., 2019). RAC1P29S and related mutations (e.g. RAC1P29L) are found in ~5 % of human melanomas, a particularly aggressive subset that exhibits resistance to BRAF inhibitors in the clinic (Lionarons et al., 2019; Van Allen et al., 2014; Watson et al., 2014). Using the TCGA database, we were able to corroborate a link between these mutations and the MRTF-SRF pathway in human melanoma. Gene Set Enrichment Analysis (GSEA) revealed that RAC1/2P29S/L tumors significantly increased the expression of genes containing SRF binding sites (gene set SRF_01, Fig. 3F and Fig. S4A–B), including CNN2 (calponin 2), VCL (vinculin), and FLNA (filamin A) (Fig. 3G). Based on our in vivo mouse experiments, we reasoned that these tumors might also be more sensitive to ICB. Analysis of TCGA data revealed that patients with RAC1/2P29S/L melanoma exhibited reduced overall survival (Fig. 3H and Fig. S4B, Supplemental Table 1). In patients receiving anti-CTLA4 therapy, however, RAC1/2P29S/L mutations correlated with significantly improved outcomes (Fig. 3I and Fig. S4D, Supplemental Table 2), a complete reversal of the non-ICB treated phenotype. This effect was less pronounced in ICB trials targeting PD1 or its ligand, PDL1, where the survival of RAC1P29S/L patients essentially overlapped with that of wild type RAC1 controls (Fig. 3J and Fig. S4B, Supplemental Table 3). We conclude that constitutively active RAC1 both induces MRTF signaling in human melanoma and sensitizes tumors to ICB treatment, particularly anti-CTLA4 (Fig. 3K).
The RAC1P29S mutation has been associated with UV damage (Cancer Genome Atlas, 2015), raising the possibility that the pro-survival effect we observed in the context of anti-CTLA4 did not result specifically from MRTF signaling but rather from T cell recognition of UV-induced neoantigens. To explore this alternative hypothesis, we examined whether melanoma patients with UV-associated driver mutations other than RAC1/2P29S/L also responded better to anti-CTLA4 therapy. UV-induced mutations in PPP6C, IDH1, or FBXW7 did not correlate with increased survival, and while patients with NF1 truncation did exhibit modestly improved responses, this effect was not statistically significant and, furthermore, it was primarily attributable to patients with co-occurring RAC1P29S/L mutations (Fig. S4C–E). We also investigated MRTF-SRF signature genes and found that melanomas with UV-induced NF1, PPP6C, IDH1, or FBXW7 mutations failed to increase the expression of this gene set (Fig. S4D). These results suggest that MRTF-SRF-induced gene expression, rather than UV damage alone, promotes increased responsiveness to anti-CTLA4 in RAC1/2P29S/L melanoma.
Finally, to evaluate the relationship between MRTF signaling and ICB responsiveness in human melanoma more directly, we measured the association between MRTF-SRF target genes and clinical outcomes. Our analysis focused on the 21 members of the SRF_01 gene set that were most highly correlated with RAC1/2P29S/L melanoma. Most of these genes were associated with reduced survival in the TCGA dataset. In the context of ICB, however, they were more predictive of improved outcomes (Fig. 3L). These data suggest that melanoma cells with high MRTF activity are particularly vulnerable to ICB. Interestingly, we have found that subsets of breast and lung cancer cells exhibit strong nuclear MRTFB staining in both primary tumors and metastases (Fig. S4F, Supplemental Table 4), indicating that enhanced MRTF signaling is a feature found in other malignancies, one that could potentially be exploited by immunotherapy.
MRTF signaling increases cancer cell rigidity
To investigate how MRTFA and MRTFB render cancer cells more stimulatory to cytotoxic lymphocytes, we performed whole transcriptome RNA-sequencing of B16F10 and E0771 cells overexpressing each isoform. MRTFA induced hundreds of changes in both cell lines, the majority of which were gene expression increases (Fig. 4A–C). Some of the most strongly activated genes (Acta1, Actg2, Fhl1, and Myh11) encoded actin proteins and cytoskeletal regulators. Although MRTFB generated more modest expression changes, nevertheless it induced many of the same cytoskeletal genes (Fig. 4A–C), implying that these genes were the ones responsible for the shared effects of MRTFA and MRTFB on immune sensitization. To further explore this idea, we performed Gene Ontology analysis using gene sets induced by one MRTF isoform in both cells lines and also gene sets induced by both MRTF isoforms in each cell line (Fig. 4D and Fig. S5). The results did not reveal substantial induction of immune related pathways. Processes and components pertaining to the cytoskeleton and cellular architecture were dramatically induced, however, in line with the known functions of MRTF (Gau and Roy, 2018; Olson and Nordheim, 2010). Consistent with these results, B16F10, E0771, MDA-MB-231, and MCF7 cells overexpressing MRTFA and MRTFB contained copious amounts of F-actin, which in some cases formed dense arrays of stress fibers (Fig. 4E–F).
Fig. 4. MRTF overexpression augments the F-actin cytoskeleton.
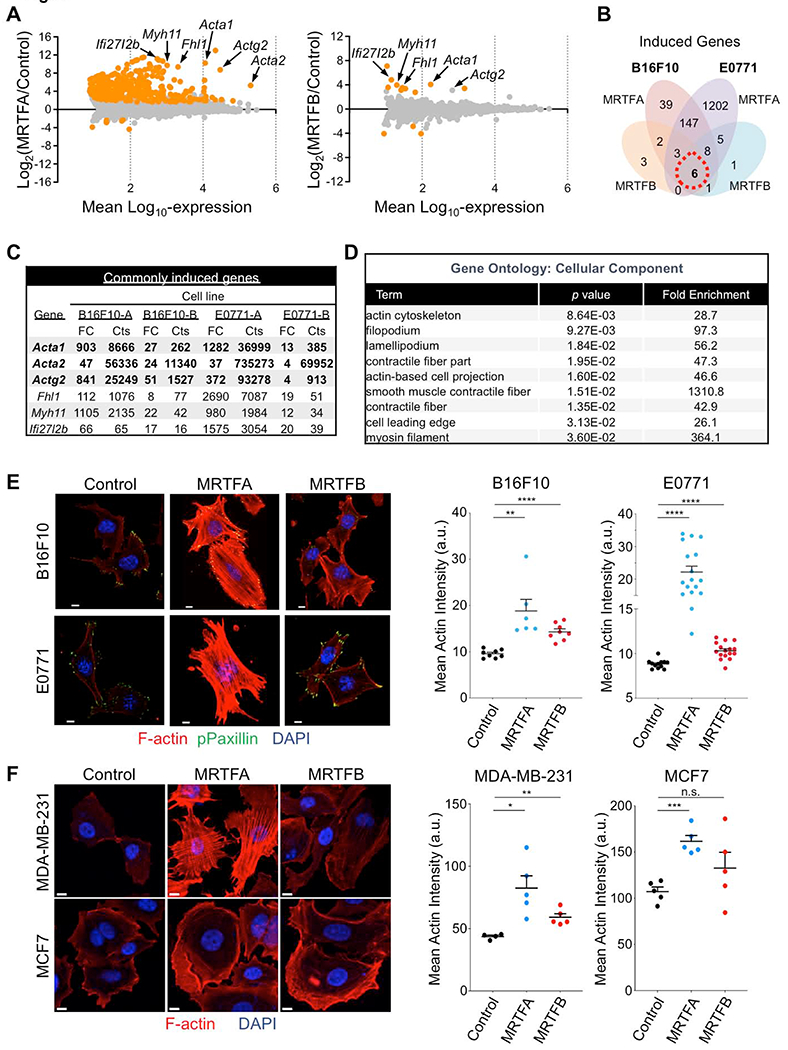
(A) Mean-difference plots showing gene expression changes induced by MRTFA (left) and MRTFB (right). Graphs incorporate data from both B16F10 and E0771 cells. Statistically significant gene expression changes are colored orange (p ≤ 0.05). Strongly induced genes encoding actin isoforms and F-actin regulators are indicated. (B) Venn diagram of induced genes exclusive to or shared by B16F10 and E0771 cells overexpressing MRTFA or MRTFB in culture. (C) Left, table of commonly induced genes, in which genes with over 50 RNA sequence counts in all data sets are shown in bold. FC: Fold Change, Cts: RNA sequencing read counts. (D) Gene Ontology (GO) analysis using the set of genes induced by MRTFA or MRTFB in all cell lines (red dashed circle in Venn diagram). Statistically significant GO terms are shown with reported p values. (E-F) Left, confocal images of representative murine (E) and human (F) cancer cells. Control and MRTFA/B overexpressing lines were stained with with DAPI (blue), phalloidin (F-actin, red) and anti-phospho-paxillin (green, E only). Right, graphs showing mean actin intensity for each cell line. Error bars denote SEM, n.s.: not significant for p > 0.05, **p ≤ 0.01, ****p ≤ 0.0001; two-tailed Student’s t test; n ≥ 8 images per cell line in E, n = 5 images per cell line in F; representative of 3 independent experiments. All scale bars: 10 μm. See also Fig. S5.
These striking architectural phenotypes raised the possibility that MRTF signaling might modulate immune activation by altering the biophysical properties of cancer cells. Given that the stiffness of the opposing surface controls mechanotransduction through the immune synapse (Blumenthal et al., 2020; Comrie et al., 2015; Judokusumo et al., 2012; Saitakis et al., 2017; Wan et al., 2013), and knowing the importance of the actin cytoskeleton for cellular architecture, we hypothesized that MRTF signaling might render cancer cells more stimulatory by increasing their rigidity (Fig. 5A). Using atomic force microscopy (AFM), we profiled the effects of MRTFA and MRTFB on the deformability of B16F10, E0771, MDA-MB-231, and MCF7 cells (Fig. 5B). Overexpression of either MRTF isoform significantly increased the average and peak stiffness of every cell line examined (Fig. 5C–F). Cell stiffness decreased dramatically in the presence of the F-actin depolymerizing agent Latrunculin A (Fig. 5G), confirming the importance of the actin cytoskeleton for this property (Rotsch and Radmacher, 2000). These results establish strong correlations between the biophysical effects of MRTF overexpression and the capacity of cancer cells to activate cytotoxic lymphocytes.
Fig. 5. MRTF signaling increases cell stiffness.
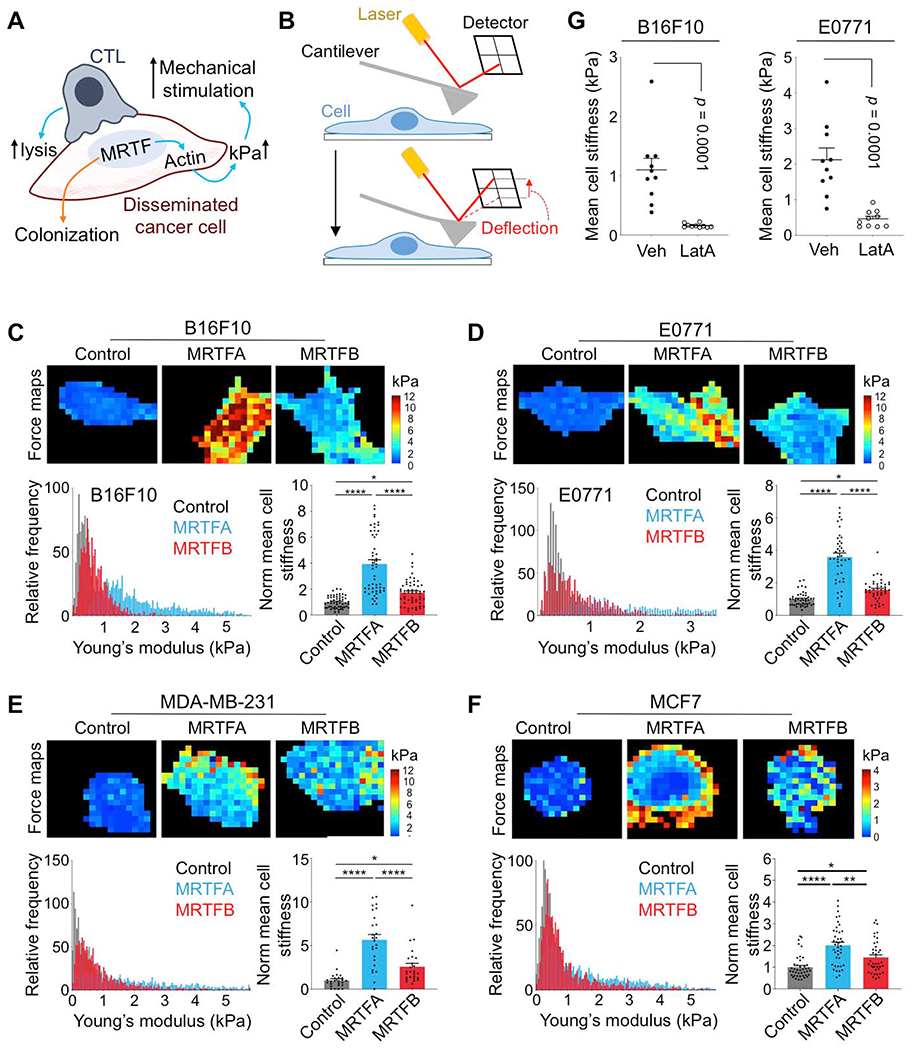
(A) MRTF activity promotes metastatic colonization but also sensitizes cancer cells to cytotoxic lymphocytes by boosting actin polymerization and cell stiffness (kPa). (B) The AFM indentation approach: cantilever deflection is proportional to the loading force, which is used to calculate the Young’s modulus of a cell. (C-F) AFM stiffness measurements comparing B16F10 (C), E0771 (D), MDA-MB-231 (E), and MCF7 (F) cells overexpressing MRTFA or MRTFB with respective control cell lines. Above, force maps of representative cells, with Young’s modulus value (kPa) indicated in pseudocolor. Below left, probability histograms of pooled Young’s modulus measurements (kPa) from representative experiments, n = 10 cells. Below right, graphs of mean cell stiffness values normalized to control. Data from 4 independent experiments are shown as mean ± SEM (One-way ANOVA with Tukey’s multiple comparisons test; *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001; n ≥ 40 cells per cell line). (G) Graphs showing mean cell stiffness of B16F10 (left) and E0771 (right) cells treated with DMSO vehicle (Veh) or with 100 ng/ml Latrunculin A (LatA) for 20 min. Data shown as mean ± SEM (two-tailed Student’s t test; n = 10 cells per condition), representative of 2 independent experiments.
MRTF controls lymphocyte activation through the target cell cytoskeleton
Although the data above are consistent with a biophysical mechanism for MRTF-induced immune vulnerability, they do not rule out the possibility that enhanced MRTF signaling might sensitize cancer cells to cytotoxic lymphocytes by changing the surface expression of a critical immunoreceptor ligand. To address this issue rigorously, it was necessary to evaluate the relative importance of the cancer cell surface vis-à-vis the underlying cytoskeleton. We initially examined the expression of activating ligands for the TCR and NKG2D. We were particularly interested in NKG2D as an NK receptor because it was downregulated by human NK cells cocultured with MDA-MB-231 or MCF7 targets (Fig. S6A), implying the presence of cognate ligands on the target surface. MRTF overexpression had little to no effect on NKG2D ligands in both mouse and human cell lines, and although MHC levels were higher in E0771-MRTFA cells, we did not observe increased MHC expression in B16F10-MRTFA cells or in any of the cell lines overexpressing MRTFB (Fig. S6B–C). Hence, changes in MHC or NKG2D ligands did not explain how MRTFA and MRTFB rendered cancer cells more stimulatory to cytotoxic lymphocytes.
Next, we assessed the stimulatory capacity of giant plasma membrane vesicles (GPMVs) derived from cancer cells of interest (Fig. 6A) (Schneider et al., 2017; Sezgin et al., 2012). We reasoned that GPMVs would contain all of the molecular machinery present on the cell surface, but not the cytoskeleton, enabling us to delineate the effects of the former from the latter. Consistent with previous work (Schneider et al., 2017), we found that GPMVs contained little to no cortical F-actin and also that they were enriched in the plasma membrane markers H-2Kb and ATP1A1 while lacking the nuclear protein histone-H3 and the Golgi marker GM130 (Fig. S7A–B). Furthermore, GPMVs derived from antigen-loaded B16F10 cells stimulated CTL calcium flux and cytokine production (Fig. 6B–C), indicating that they contained the surface ligands required for T cell activation. Importantly, whereas B16F10-MRTFA and B16F10-MRTFB cells induced stronger CTL cytokine production than control B16F10 cells (Fig. 2 and Fig. 6C), GPMVs derived from MRTF overexpressing cells were not more stimulatory than GPMVs derived from controls (Fig. 6C), suggesting that cell surface molecules are not responsible for MRTF-induced immune activation. Using IFNγ, which drives higher MHC expression (Fig. S6B), we were able to enhance lymphocyte activation by both B16F10 cells and the GPMVs they generated. This treatment, however, did not alter the consequences of MRTF signaling, which continued to affect only lymphocyte responses to intact cells, but not to GPMVs (Fig. 6C). Based on these results, we surmised that the increased stimulatory capacity of MRTF overexpressing cells resulted from components beneath the plasma membrane, most likely the F-actin cytoskeleton.
Fig. 6. Cytoskeletal stiffness underlies the immune vulnerability of metastatic cells.
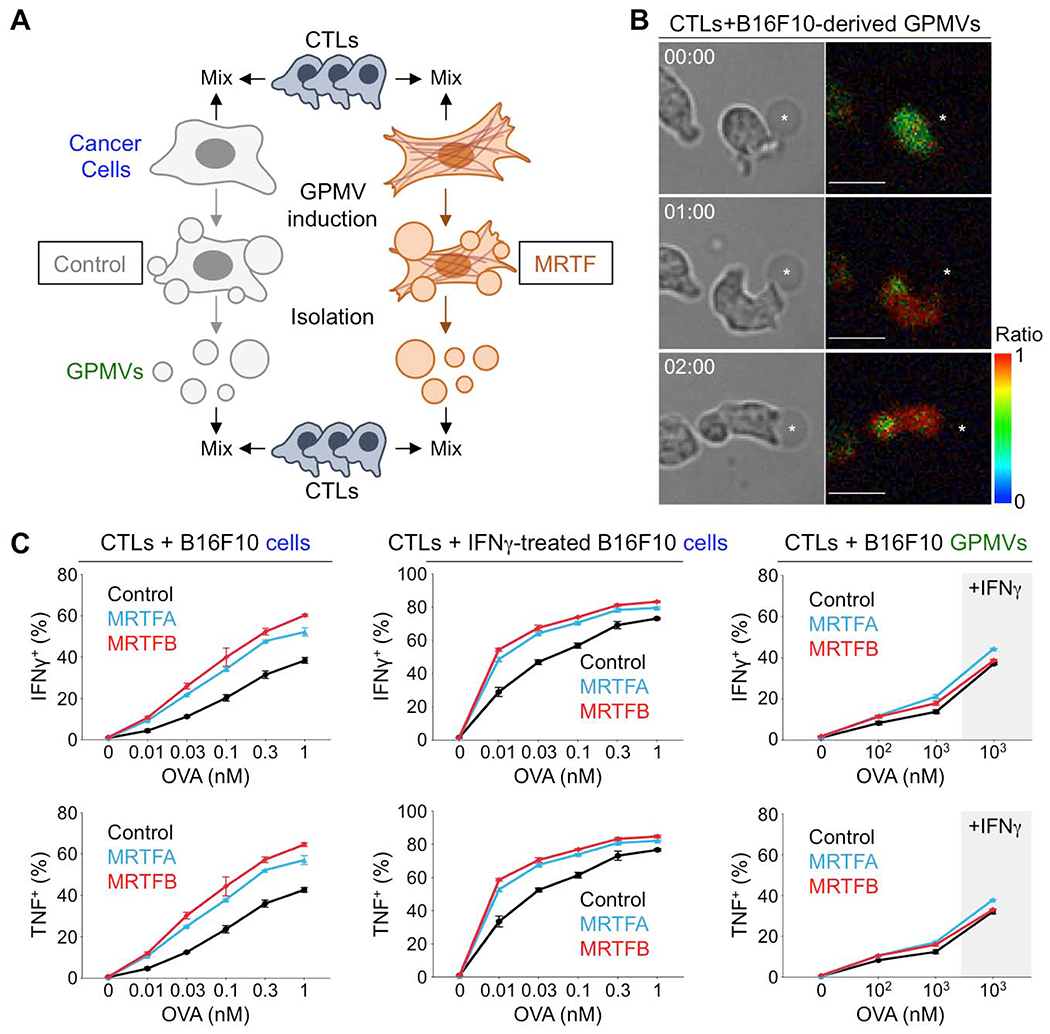
(A) CTLs were stimulated using OVA-loaded B16F10 cells or GPMVs derived from these cells. (B) Time-lapse montage of a representative OT1 CTL Ca2+ response during contact with an OVA-loaded B16F10-derived GPMV (asterisk). Paired bright field (left) and pseudocolored Fura-2 ratio images (right) are shown. Time in MM:SS is indicated in the upper left corner of each bright field image. Scale bars: 10 μm. Color bar indicates the value of the Fura-2 ratio. (C) Graphs showing the percentage of CTLs producing IFNγ or TNF, measured by intracellular staining 4 h after mixing with B16F10 cells (left), B16F10 cells pretreated with IFNγ (center), or GPMVs derived from untreated or IFNγ-treated B16F10 cells (right). Data shown as mean ± SEM of technical triplicates, representative of 3 independent experiments. See also Fig. S6 and S7.
To confirm the importance of the cytoskeleton more directly, we required an approach capable of disrupting cancer cell F-actin while leaving lymphocyte F-actin intact. Overexpression of the Salmonella enterica SpvB actin-binding domain (also called DeAct) induces profound F-actin severing in both mouse and human cells (Harterink et al., 2017) (Fig. 7A). In our hands, transient transfection with DeAct strongly depleted F-actin in cancer cell lines, leading to architectural collapse and cell rounding (Fig. S7C). DeAct expressing cells, which we identified via a Cherry marker, also exhibited dramatic reductions in stiffness that correlated with the level of marker expression (Fig. 7B–7D, S7D). Importantly, DeAct reversed the effects of MRTF overexpression on cancer cell killing by CTLs (Fig. 7E). Indeed, DeAct+ B16F10-MRTFA and B16F10-MRTFB cells were no more sensitive to cellular cytotoxicity than DeAct+ B16F10 controls. DeAct transfection also eliminated the capacity of B16F10-MRTFA and B16F10-MRTFB cells to drive stronger CTL degranulation and cytokine production (Fig. 7F–G, S7E). We observed similar results in studies of human NK cells, where DeAct largely reversed the ability of target cell MRTF to potentiate NK cell degranulation (Fig. 7H). We conclude that MRTF signaling boosts the stimulatory capacity of cancer cells via the F-actin cytoskeleton.
Fig. 7. MRTF-induced immune sensitization requires the F-actin cytoskeleton.
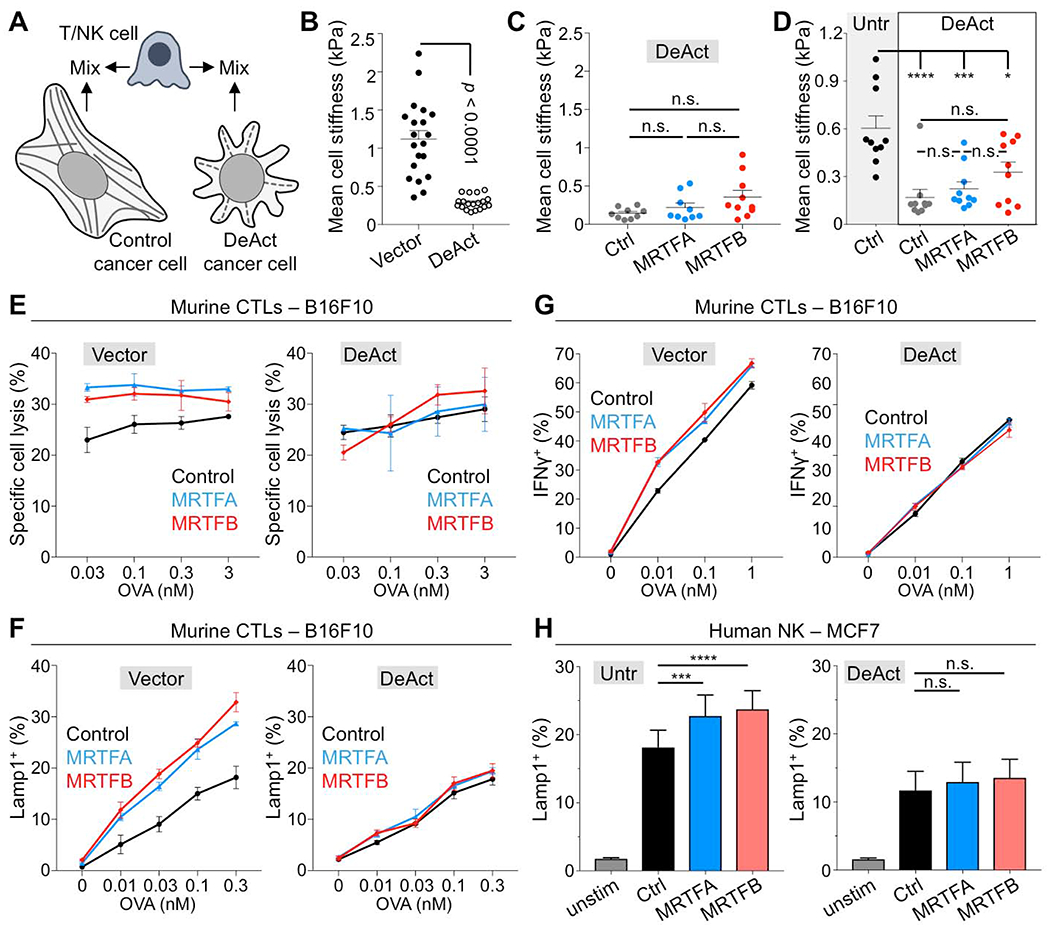
(A) Using DeAct to evaluate the role of cancer cell F-actin in the stimulation of cytotoxic lymphocytes. (B) Mean cell stiffness of B16F10 cells transfected with DeAct or vector control. Data shown as mean ± SEM (p: two-tailed Student’s t test; n ≥ 19 cells per condition), representative of 2 independent experiments. (C) Mean cell stiffness of B16F10, B16F10-MRTFA, and B16F10-MRTFB cells transfected with DeAct. Data shown as mean ± SEM (n.s.: not significant for p > 0.05, calculated by one-way ANOVA; n ≥ 9 cells per condition), representative of 2 independent experiments. (D) Mean cell stiffness of untransfected (Untr) control MCF7 cells, control MCF7 cells transfected with DeAct, MCF7-MRTFA cells transfected with DeAct, and MCF7-MRTFB cells transfected with DeAct. Data shown as mean ± SEM (n.s.: not significant for p > 0.05, **p ≤ 0.01, calculated by one-way ANOVA; n ≥ 9 cells per condition), representative of 2 independent experiments. (E-G) B16F10 cells overexpressing MRTFA, MRTFB, or empty vector were transfected with DeAct (right) or vector control (left), loaded with increasing concentrations of OVA and then mixed with OT1 CTLs. (E) Specific lysis, measured after 5 h. (F) CTL degranulation, measured by surface Lamp1 exposure after 90 min. (G) IFNγ production, measured by intracellular staining after 4 h. Data in E-G are shown as mean ± SEM of technical triplicates, representative of 2 independent experiments. (H) MCF7, MCF7-MRTFA, and MCF7-MRTFB cells were transfected with DeAct or left untransfected (Untr) and then mixed with human PBMCs. After 5 h, NK cell degranulation was measured by surface exposure of Lamp1. (n = 9 donors). ****p ≤ 0.0001, ***p ≤ 0.001, and n.s.: not significant for p > 0.05, calculated by one-way ANOVA with sample pairing. See also Fig. S6 and S7.
DISCUSSION
Characteristic genetic and biochemical traits enable cancer cells to grow in an unregulated manner, but they also create vulnerabilities, such as oncogene addiction and sensitivity to genotoxic agents, that can be targeted by appropriate therapeutic modalities (Behan et al., 2019; Bryant et al., 2005; DeBerardinis and Chandel, 2016; Farmer et al., 2005; Weinstein, 2002). Our present study extends this paradigm into the biophysical domain by demonstrating that the mechanical properties that enable metastatic growth also serve as an Achilles heel for destruction by cytotoxic lymphocytes. Isolated cancer cells are typically less rigid than untransformed cells from the same parent tissue (Guck et al., 2005; Hou et al., 2009; Xu et al., 2012). To occupy the metastatic niche, however, cancer cells must spread on the microvascular basement membrane (Er et al., 2018; Valiente et al., 2014), thereby increasing their rigidity to the point where they trigger robust lymphocyte activation. This coupling of colonization with biophysical vulnerability provides an explanation for why metastasis is so inefficient, and it also identifies mechanosensing of cancer cell rigidity as a mode of immunosurveillance. In principle, mechanical immunosurveillance, or mechanosurveillance, would enable the immune system to target cellular dysfunction that does not detectably alter the biochemical recognition of cell surface proteins or secreted factors. In practice, however, it seems more likely that both biochemical and biophysical features will control immune vulnerability in a combinatorial manner. For instance, our observation that MRTF-induced suppression of E0771 cells requires CD8+ CTLs in vivo, while the anti-B16F10 response is dominated by NK cells, probably reflects the fact that E0771 cells express higher levels of class I MHC, which would activate T cells and inhibit NK cells. Hence, the contexts within which mechanosurveillance operates will be dictated by specific molecular interactions between cytotoxic lymphocytes and target cells of interest. Deciphering this crosstalk will be a fascinating area of future study.
Certain features of the MRTFA overexpression phenotype, such as enhanced B16F10 metastasis and increased Fas expression in E0771 cells, did not conform with the mechanosurveillance model. It is important to note, however, that MRTFA overexpression activated a much larger list of genes than MRTFB, a list that included not just the targets of MRTFB but also many other genes that were not affected by MRTFB at all. Given that MRTFB overexpression was sufficient to generate in vitro and in vivo effects consistent with mechanosurveillance, it seems likely that the discordant aspects of the MRTFA phenotype resulted from physiological changes that were independent of cytoskeletal remodeling and stiffness.
It is generally thought that increasing target rigidity amplifies lymphocyte activation via mechanosensitive cell surface receptors (Huse, 2017; Zhu et al., 2019). Certain immunoreceptors, including the TCR, integrins, and the NK receptor CD16, form “catch bonds” with their ligands that increase in affinity under applied force (Gonzalez et al., 2019; Kong et al., 2009; Liu et al., 2014). There are also indications that synaptic forces induce conformational changes in the TCR and within integrin adhesion complexes that are required for optimal signal transduction (del Rio et al., 2009; Friedland et al., 2009; Lee et al., 2015). Actin dependent stiffening of the target cell could facilitate all of these processes by restraining deformation orthogonal to the cell surface. Increased cortical F-actin could also restrict the lateral mobility of ligands in the plasma membrane by strengthening interactions between the membrane and the cytoskeleton or by altering the confinement properties of membrane corrals (Gauthier et al., 2012; Jacobson et al., 2019). Reduced lateral mobility is known to facilitate integrin mechanotransduction at T cell-dendritic cell synapses (Comrie et al., 2015), and thus it is an intriguing potential mechanism for mechanosurveillance.
Although the immune sensitization mechanism characterized in this study resulted from a cell intrinsic mechanical trait (cellular stiffness), tumor progression induces cell extrinsic biophysical changes, as well, such as increased ECM adhesiveness and rigidity (Kai et al., 2019; Levental et al., 2009), which could also be coupled to immune vulnerabilities. ECM remodeling is generally thought to promote malignancy by stimulating cancer cell migration, epithelial to mesenchymal transition (EMT), and transcriptional programs important for tumorigenesis (Kai et al., 2019). ECM stiffness, in particular, has been shown to drive cancer cell proliferation via the transcription factor YAP (Yes-associated protein) (Albrengues et al., 2018; Panciera et al., 2020). These same changes in the ECM, however, could also promote anti-tumor immunity via increased MRTF signaling (Er et al., 2018), which we now know triggers mechanosurveillance. Further study of how the ECM and other aspects of tumor architecture affect both the movement and the functionality of infiltrating immune cells could identify additional therapeutic opportunities.
The idea that metastatic cells must fine-tune MRTF signaling to balance the benefits of cell spreading with the drawbacks of immune activation conceptually parallels work on the regulation of EMT during cancer progression (Alderton, 2013; Ocana et al., 2012; Tsai et al., 2012). Although EMT supports migration, invasion, multipotency, and resistance to certain therapies (Zhang and Weinberg, 2018), it can also hinder proliferation and promote apoptosis (David et al., 2016; Kong et al., 2017). Hence, to metastasize effectively, cancer cells must employ transcriptional programs of both EMT and mesenchymal to epithelial transition (MET), balancing the proliferative and invasive properties of each cellular state. The importance of fine tuning EMT in this way is highlighted by the observation of partial EMT signatures in patients with metastatic disease (Puram et al., 2018). Interestingly, the loss of some, but not all, epithelial characteristics during partial EMT is thought to be important for the acquisition of stem cell-like properties and metastatic dormancy, a state of prolonged quiescence in which cancer cells evade the immune system (Lawson et al., 2015; Malladi et al., 2016; Pommier et al., 2018). Dormancy ends when metastatic colonies engage the ECM, thereby activating YAP and MRTF to drive outgrowth (Albrengues et al., 2018; Er et al., 2018; Shibue and Weinberg, 2009). Determining how these various cellular states affect not only the biochemical but also the biophysical properties of cancer cells will provide for a better understanding of how metastatic tumors balance partial EMT, stem cell-like behavior, and awakening from metastatic dormancy in order to grow in the face of immunosurveillance.
The RAC1P29S/L allele is generally associated with enhanced melanoma malignancy and resistance to targeted therapies (Lionarons et al., 2019; Van Allen et al., 2014; Watson et al., 2014), including the BRAF inhibitor vemurafenib. Our results, however, indicate that this mutation actually increases tumor responsiveness to ICB, particularly anti-CTLA4, implying that RAC1P29S/L may be useful as a positive predictive indicator for this class of treatments. Although RAC1 activates multiple downstream signaling pathways, MRTF has been shown to be vital for the specific effects of RAC1P29S on melanoma physiology (Lionarons et al., 2019), and our mechanistic results in vitro and in mouse models support a role for MRTF in triggering anti-tumor immunity via mechanosurveillance. Critically, we have found that MRTF-SRF induced genes exhibit increased expression in human RAC1/2P29S/L melanoma, and we have also documented associations between these genes and improved ICB outcomes, independent of RAC1 mutation. These results provide direct evidence that MRTF signaling enhances ICB-induced immunosurveillance of human tumors. Further study of this pathway could reveal additional strategies for identifying patients likely to benefit from immunotherapy.
Although anti-PD1/PDL1 therapy did improve the survival of RAC1/2P29S/L melanoma patients, the beneficial effects were less pronounced than what was observed with anti-CTLA4. These results are perplexing in light of our experiments showing that both anti-CTLA4 and anti-PD1 were effective at combating B16F10-MRTFB metastasis. This discrepancy could reflect differences between the B16F10 lung colonization model and human melanoma. It is also possible, however, that aspects of the RAC1/2P29S/L mutant phenotype other than increased MRTF signaling may negatively affect therapeutic responses to anti-PD1/PDL1. This second explanation is consistent with our observation that MRTF-SRF induced gene expression is a reasonable predictor of anti-PD1/PDL1 responsiveness when evaluated independently of the RAC1/2P29S/L mutation.
A number of intracellular pathogens, including HIV, Chlamydia, and Listeria, dramatically remodel the host cell cytoskeleton to enable intracellular motility, proliferation, and the infection of neighboring cells (Bhavsar et al., 2007; Taylor et al., 2011; Wesolowski and Paumet, 2017). The capacity of these architectural changes to modulate the immune response, both positively and negatively, has not been examined. It has been shown, however, that CD4+ T cells latently infected with HIV contain high levels of viral integration in the Mrtfa and Mrtfb loci, implying that disruption of the MRTF pathway enables infected cells to elude the immune system (Maldarelli et al., 2014). Furthermore, the HIV virulence factor Nef, which disrupts cytoskeletal polarity, was recently found to enhance the survival of infected cells in an immunocompetent mouse model (Usmani et al., 2019). Therefore, defining the genetic and molecular bases of mechanosurveillance will likely illuminate cellular immunity against not only cancer but also infectious disease.
LIMITATIONS OF THE STUDY
Although our results strongly suggest that MRTF overexpressing cells trigger more powerful lymphocyte responses because of their enhanced rigidity, it remains possible that the cytoskeletal remodeling driven by MRTF might also boost lymphocyte activation by altering the lateral mobility and/or clustering of cell surface immunoreceptor ligands. Similarly, while we have provided compelling links between MRTF signaling, cancer cell rigidity, and immunosurveillance, we have not definitively shown that MRTF dependent immunosurveillance depends on cancer cell stiffening in vivo. This will require an experimental system capable of measuring and modulating the biophysical properties of tumor cells in intact tissues. Finally the strong relationship between cell-intrinsic stiffness and mechanosurveillance that we have observed in experimental metastases may be less apparent in large primary tumors, where cell-extrinsic properties such as hypercellularity, angiogenesis, and dysregulated ECM also make substantial contributions to the biophysical landscape.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Morgan Huse (husem@mskcc.org).
Materials availability.
The materials associated with this study are available upon request, with the exception of the human breast and lung tissues, which are unique biological resources. Histological images of these tissues are available for distribution, however.
Data and code availability.
RNA-seq data has been deposited in the Gene Expression Omnibus (GEO) under the accession number GSE164860. All other source data is available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
The animal protocols used for this study were approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center and the Animal Care and Use Committee of the University of Illinois at Chicago. For metastasis assays, 4-6 week old female recipient C57BL/6J mice were purchased from the Jackson Laboratory. Mice were assigned randomly into groups for in vivo experiments. 2-6 month old male and female OT1 αβTCR transgenic mice (obtained from Jackson Laboratory) were used to generate OT1 CTLs for in vitro assays. 2-4 month old C57BL/6J mice (obtained from Jackson Laboratory) were used to generate NK cells for in vitro assays. All animals were housed under specific pathogen free conditions.
Human studies.
Human studies were performed using patient samples and data from MSKCC cohorts and the TCGA. Patient samples of breast and lung cancer with matched brain metastases were obtained under an MSKCC Institutional Review Board (IRB) approved biospecimen protocol (Protocol Number: IRB #15-068) from the MSK Department of Pathology. Information about individual samples may be found in Supplemental Tables 1–4. Human NK cells were isolated from peripheral blood samples obtained from healthy volunteer donors via the New York Blood Center (NYBC, http://nybloodcenter.org/). The Memorial Sloan Kettering Cancer Center Institutional Review Board (MSKCC IRB) waived the need for additional research consent for anonymous NYBC samples.
Cell culture.
B16F10 cells (male) and E0771 cells (female) were cultured in RPMI at 37 °C, while MCF7 cells (female) and MDA-MB-231 cells (female) were cultured in DMEM at 37 °C. Media were supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM L-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. All MDA-MB-231 experiments utilized the brain metastatic subline MDA-231Br (Bos et al., 2009). HEK-293T cells (female, used to generate ecotropic retrovirus) were cultured at 37 °C in DMEM supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM L-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. B16F10 and E0771 cells were authenticated by STR profiling, conducted by IDEXX Bioanalytics, and were negative for mycoplasma and viral pathogens.
METHOD DETAILS
Constructs.
Retroviral vectors for LifeAct-GFP, GFP, and TGL expression have been described (Le Floc’h et al., 2013; Ponomarev et al., 2004). Constructs for MRTFA/B silencing and overexpression were gifts from Ron Prywes (Addgene # 27161, #19846, and #27175)(Lee et al., 2010; Muehlich et al., 2008; Selvaraj and Prywes, 2003). To generate inducible expression constructs, the coding sequences of MRTFA and MRTFB were PCR amplified and subcloned into the pRetroX-Tight-Hygro vector (Takara Bio, 631034). Doxycycline inducible expression was achieved by pLVX-TetON-Advanced (Takara Bio, 632162) expression. DeAct-SpvB was expressed using the vector pTetON-DHFRdd-SpvB; CMV-Cherry (Addgene Plasmid #89463). An MSCV vector expressing Cherry was employed as a transfection control to pair with DeAct-SpvB.
Mice and cell culture.
To generate OT1 CTLs, T cells from OT1 αβTCR transgenic mice were mixed with congenic splenocytes pulsed with 100 nM OVA and cultured in RPMI medium containing 10 % (vol/vol) FBS. Cells were supplemented with interleukin 2 (IL-2, 30 IU/ml, NIH BRB Repository) after 24 h and were split as needed in RPMI containing IL-2 and used for functional assays after 7 days in culture. Murine NK cells were isolated from C57BL/6J splenocytes by negative selection using an NK cell isolation kit (MACS, 130-115-818) and incubated overnight in 1000 U/ml IL-2. For human NK cell experiments, peripheral blood mononuclear cells (PBMCs) were purified from buffy coats by density gradient centrifugation (Ficoll-Paque Plus; GE Healthcare) and then cryopreserved in FBS with 10% DMSO. One day prior to the experiment, cells were thawed and incubated in clone media (DMEM, 30% Ham’s F-12, 10% human serum, 1 mM sodium pyruvate, 1% MEM nonessential amino acids, 2 mM L-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin) supplemented with 200 U/ml IL-2 (Proleukin, Prometheus, Cat # 65483011607) at 37 °C. For certain experiments, human NK cells were purified by negative selection using magnetic beads (RosetteSep™ Human NK Cell Enrichment Cocktail, Stem Cell Technologies) and cultured at 106 cells/ml in clone media with 200 U/ml IL-2. Inducible MRTFA/B overexpression cell lines (e.g. B16F10-MRTFB cells) were prepared by sequential transduction with rTTA, TGL, and either control or MRTF overexpression vectors, followed by culture in G418 and hygromycin. MRTF expression was induced by treating cells with 500 ng/ml doxycycline hyclate (Sigma) 24-48 h prior to the experiment. Cell lines expressing Cherry-DeAct or Cherry alone were prepared 48 h before the experiment by transient transfection of the DeAct or Cherry control plasmids using lipofectamine 2000 (ThermoFisher) following the manufacturer’s recommended protocol. 24h before the experiment, DeAct and MRTF were induced together using 500 ng/ml doxycycline. shMrtfa/b B16F10 or E0771 cells and accompanying control cells lines were prepared by transduction with pLKO.1 shMrtfa/b and control pLKO.1, respectively, and then selection with 2 μg/mL puromycin. Knockdown/overexpression of MRTF was confirmed by immunoblotting. Cells were lysed in 1× Laemmli buffer, separated on a NuPAGE® Novex® 10% Bis-Tris Protein Gel, and transferred to nitrocellulose. Membranes were incubated with antibodies against MRTFA (Santa Cruz Biotechnology, sc-390324), MRTFB (Bethyl Laboratories, Cat # A302-768A), or GAPDH (Cell Signaling, Cat# 2118L) overnight at 4 °C followed by IRDye conjugated secondary antibodies (Licor Biosciences, Cat# 926-68071 and 926-32210).
Metastasis assays.
2 × 105 B16F10 or E0771 cells were injected into the tail vein of 4-6 week old C57BL/6J mice (Jackson Labs, 000664). Doxycycline was included in the mouse food (2500 mg/kg) to maintain MRTF expression for the duration of the experiment. Mouse hair was removed using clippers to prevent interference with bioluminescent imaging (BLI). Lung metastasis burden was quantified weekly using retro-orbital D-luciferin (150 mg kg−1) injection followed by imaging via the IVIS Spectrum Xenogen instrument (Caliper Life Sciences) installed with Living Image software v.2.50. Metastatic load per mouse was calculated by dividing the total photon flux signal at the end point of the experiment by the total photon flux measured immediately after cancer cell delivery on the day of injection. NK cell depletion was performed by injecting mice intraperitoneally (i.p.) with anti-asialo GM1 antibody (Wako Chemicals, 986-10001), as previously described (Er et al., 2018), 6 days and 1 day before tail vein injection of cancer cells and once a week thereafter. CD8 positive T cell depletion was achieved using 250 μg InVivoMab anti-mouse CD8α antibody (clone 53-6.7, BioXCell, BE0004-1) or IgG2a control (BioXCell, BE0089), injected 2 days and 1 day before tumor delivery and once every week thereafter. Immune checkpoint blockade was achieved by injecting mice i.p. with 125 μg anti-CTLA-4 (clone 9D9), anti PD-1 (clone RPM1-14), or mouse isotype control (clone MPC-11 or clone 2A3) antibodies 4 and 7 days after delivery of cancer cells followed by bi-weekly injections thereafter.
Killing, lytic granule secretion, and cytokine production assays.
For CTL functional assays, cancer cell targets were cultured overnight on fibronectin-coated 96-well plates. They were then loaded with varying concentrations of OVA for 2 h and washed three times in medium. To assess killing, OT1 CTLs were added at a 4:1 effector to target (E:T) ratio and incubated for 5 h at 37 °C in culture medium. Cells were then labeled with APC conjugated anti-CD8a antibody (Tonbo Biosciences, 20-0081), and specific lysis of target cells (GFP+, CD8−) was determined by propidium iodide (PI, Thermo Fisher Scientific) incorporation using flow cytometry. Killing of B16F10 cells was quantified with a lactate dehydrogenase cytotoxicity assay kit (Clontech) using the manufacturer’s recommended protocol. All measurements were performed in triplicate. To assess lytic granule secretion, the E:T ratio was 2:1, and cells were incubated for 90 min at 37 °C in the presence of eFluor660 conjugated anti-Lamp1 antibody (1 μg/ml, Clone 1D4B, eBiosciences). Cells were then labeled with anti-CD8a antibody, and the percentage of CTLs (CD8+) with positive Lamp1 staining was quantified by flow cytometry. To assess cytokine production, the E:T ratio was 2:1, and cells were incubated for 4 h at 37 °C in the presence of BD GolgiPlug™ protein transport inhibitor (BD Biosciences). Cells were then labeled with anti-CD8a antibody and a dead cell marker (Live/Dead Fixable Aqua Dead Cell Stain Kit), fixed, and permeabilized using the BD Cytofix/Cytoperm™ kit. After labeling with PE conjugated anti-TNF (BioLegend, 506306) and PE/Cy7 conjugated anti-IFNγ (BioLegend, 505826) antibodies, the percentage of CTLs (CD8+) expressing TNF and IFNγ was determined by flow cytometry. All functional assays were performed in triplicate. For mouse NK cell functional assays, cancer cell targets were cultured overnight on fibronectin-coated 96-well plates. They were then mixed with NK cells at a 1:1 ratio and incubated for 3-6 h at 37 °C in the presence of eFluor660 conjugated anti-Lamp1 antibody. Subsequently, mixtures were labeled with PerCP-Cy5.5 conjugated anti-NK1.1 antibody (eBioscience, 45-5941-82) and the percentage of NK1.1+ cells with positive Lamp1 staining was quantified by flow cytometry. For human NK cell functional assays, cancer cell targets were cultured overnight on fibronectin-coated 96-well plates. To assess NK cell degranulation, 2 × 105 PBMCs were added to each well (NK cells comprise 5-15 % of PBMCs) in the presence of monensin (GolgiSTOP™; 1:1,000 dilution; BD) and Brilliant Violet 786-labeled anti-Lamp1 mAb (clone SJ25C1, BD Horizon) for 5 h at 37 °C. After incubation, cells were collected in a 96-well V-bottom plate, washed and stained with dead cell marker (Live/Dead Fixable Near IR Dead Cell Stain Kit), ECD-labeled anti-CD56 mAb (Beckman Coulter), BV650-labeled anti-CD3 mAb (clone UCHT1, BD Horizon), and PE-labeled anti-NKG2D (clone 1D11, BioLegend) . Finally, cells were washed in 1% FBS/PBS and subjected to flow cytometry (LSR Fortessa). All flow cytometric analysis was performed using FlowJo software.
In vitro cell growth and proliferation assays.
To assess cell viability, the CellTiter-Glo® Luminescent Cell Viability Assay Kit (Promega, G7570) was used according to manufacturer instructions. To assess cell proliferation, cells were labeled with CellTrace Violet (CTV, Thermo Fisher) according to manufacturer instructions, and CTV dilution was quantified by flow cytometry.
Cell death assays.
Cell death was induced by treating cells seeded on fibronectin-coated 96 well plates with varying concentrations of staurosporine (Cell Signaling Technology), FasL (PeproTech), TNF (PeproTech), and granzyme B (BioLegend). Granzyme B was activated using Cathepsin C/DPPI (R&D Systems) according to manufacturer instructions, and applied to cells in combination with a sublytic concentration of perforin, which was purified as previously described (Basu et al., 2016). Cell death after 5 h treatment was quantified by PI incorporation or using the Caspase Glo system (Promega) according to manufacturer instructions.
Transcriptome sequencing and analysis.
RNA was collected using the RNeasy Mini Kit (Qiagen, 74106) according to manufacturer instructions. After RiboGreen quantification and quality control by Agilent BioAnalyzer, 500 ng of total RNA underwent polyA selection and TruSeq library preparation according to instructions provided by Illumina (TruSeq Stranded mRNA LT Kit), with 8 cycles of PCR. Samples were barcoded and run on a HiSeq 4000 in a 50bp/50bp paired end run, using the HiSeq 3000/4000 SBS Kit (Illumina). An average of 41 million paired reads was generated per sample and the average fraction of mRNA bases was 74%.
Histology
To quantify in vivo proliferation, mouse lungs bearing metastatic tumors were paraffin embedded, sectioned into 5 μm thick slices, stained for Ki67 (Cell Signaling Technologies, 9129) together with DAPI, and imaged using a 3DHISTECH Pannoramic Scanner with a 20× objective lens. For imaging of murine lungs, tissue was PFA perfused and fixed overnight at 4 °C, followed by paraffin embedding. Sections were stained for NKp46 (R&D Systems, Cat # AF2225), CD8 (Cell Signaling Technology, Cat # 98941), and MRTFB (Bethyl Laboratories, Cat # A302-768A). Metastatic lesions were analyzed and CD8+ and NKp46+ cells counted using CaseViewer (3D Histech) or Aperio Imagescope (Leica Biosystems) software. Patient samples of breast and lung cancer with matched brain metastases were immunostained using the Ventana Discovery with and anti-MRTFB antibody (Bethyl Laboratories, Cat # A302-768A), followed by imaging using CaseViewer.
Confocal and live imaging.
Confocal images were acquired on a Leica Sp8 machine equipped with a white light excitation laser and a 40× objective lens. For imaging of CTL-mediated killing, B16F10 targets were cultured overnight on fibronectin-coated 8-well chambers (Lab-tek) and then pulsed with 100 nM of OVA for 2 h. After washing, cancer cells were mixed with CTV-labeled OT1 CTLs in the presence of 1.5 μM PI. Chambers containing cells of interest were imaged at 5.5 min intervals using a Zeiss Zen epifluorescence microscope at 10× magnification for 11 hours. For imaging of human NK cell-mediated killing, MCF7 cells (1 × 104 per imaging chamber) were plated on fibronectin and then stained with 1 μM Calcein Red-Orange (Invitrogen). Time-lapse imaging commenced upon the addition of 3 × 104 CTV-labeled NK cells. Images were collected using an Sp8 confocal microscope with 63× 1.4 NA objective at 37°C for 8 h, with an image acquired every 6 min.
Cell surface proteins.
Target cells were gently detached using Trypsin/EDTA and washed prior to antibody staining. B16F10 and E0771 cells were stained with APC-labeled anti-Fas (clone SA367H8, BioLegend), PE-labeled anti-H2Kb (clone AF6-88.5, BioLegend), PE-labeled anti-H2Db (clone 28-14-8, eBioscience), and mouse NKG2D-Fc (Madera et al., 2016)(kind gift from J. C. Sun) followed by a PE-labeled secondary antibody. MDA-MB-231 and MCF7 cells were stained with PE-labeled anti-MICA/B (clone 6D4, BD Pharmingen) and PE-Cy5-labeled anti-HLA-ABC (G46-2.6, BD Pharmingen). Surface expression was then quantified by flow cytometry.
Atomic Force Microscopy (AFM).
Cells were seeded on glass-bottom petri dishes (FluoroDish FD5040) coated with fibronectin (from bovine plasma, Millipore Sigma) and then kept in complete RPMI medium with 10 mM HEPES pH 7.0 during the acquisition of stiffness maps. Experiments were performed at 37 °C with an MFP-3D-BIO AFM microscope (Oxford Instruments) using cantilevers with 5 μm diameter colloidal borosilicate probes (nominal spring constant k = 0.1 N/m, Novascan). Before each experiment, the exact spring constant of the cantilever was determined using the thermal noise method and its optical sensitivity determined using a PBS-filled glass bottom petri dish as an infinitely stiff surface. 10-12 cells from each experimental group were tested in each session. Bright field images of each cell were collected during AFM measurements using an inverted optical objective (Zeiss AxioObserver Z1) integrated with the AFM. Stiffness maps of 60 × 60 μm2 (18 × 18 points) were collected in areas containing both cells and substrate at 1.5 Hz for a single approach/withdraw cycle. A trigger point of 1 nN was used to ensure sample penetration of 1-2 μm. Force curves in each map were fitted according to the Hertz model (Igor Pro, Wavemetrics). Data fitting was performed in the range from 0 to 50% of the maximum applied force to consider only measurements within the first 1 μm of indentation. The following settings were used: tip Poisson vtip = 0.19, tip Young’s modulus Etip = 68 GPa, and sample Poisson vsample = 0.45. Stiffness histograms were obtained by identifying the stiffness values belonging to each cell (and not the substrate values, shown in black on force maps) through a mask and plotting the results from each cell line as a single population. All measurements made < 500 nm above the substrate were excluded. Extraction of the stiffness from the raw Igor Binary Wave (.ibw) data with an overlapped mask was obtained by means of a home-built routine implemented in Igor (Igor Pro, Wavemetrics). Stiffness distribution histograms were obtained using the histogram analysis tool in Excel (Microsoft) after normalizing for the total number of data points.
Giant plasma membrane vesicle (GPMV) isolation and purification.
GPMVs were generated as previously described (Sezgin et al., 2012) with minor modifications for scale and cell type. 1.5 × 106 cells were seeded in a 10 cm2 dish and incubated for 18 h with or without 25 ng/ml IFNγ and the indicated concentrations of OVA peptide. Cells were then transferred into 5 ml of GPMV buffer (10 mM HEPES, 150 mM NaCl, 2 mM CaCl2, 25 mM PFA, 2 mM DTT, pH 7.4) for 1 h at 37 °C. To purify GPMVs, the suspension was centrifuged at 100 × g for 10 min to pellet cell debris, and then the supernatant was centrifuged at 2000 × g for 1 h at 4 °C to pellet the GPMVs. For lymphocyte stimulation, GPMVs were washed and then resuspended in 500 μl of cell culture medium. 100 μl of this purified GPMV sample was then mixed with 104 CTLs in a v-bottom 96-well plate and incubated for the indicated times. For immunoblot analysis of GPMV protein content, GPMV pellets were resuspended in 1 × Alfa Aesar Laemmli SDS Sample Buffer (Fisher Scientific, AAJ61337AC) prior to gel electrophoresis. For imaging studies (Fig. S7A), B16F10 cells transiently transfected with GFP or LifeAct-GFP were labeled with CellMask™ Orange Plasma Membrane Stain according to manufacturer instructions, and then either imaged or used to generate GPMVs for imaging.
Ca2+ imaging of CTLs and GPMVs.
CTLs were loaded with 5 μg/ml Fura-2AM and then added to poly-L-lysine-coated chambers containing GPMVs derived from OVA-loaded B16F10 cells. Fura-2 images using 340 nm and 380 nm excitation were acquired every 30 seconds for 20 min, using a 20× objective lens fitted to an IX-81 microscope stage (Olympus).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics.
Analyses were carried out using either representative experiments or pooled data as indicated (n is defined in the figure legends for each experiment). Statistical tests (two-tailed Mann-Whitney, two-tailed ANOVA, paired and unpaired two-tailed t tests and Log-rank Mantel-Cox tests) were performed using GraphPad Prism. Log-rank tests for patient survival were implemented in cBioportal (Cerami et al., 2012; Gao et al., 2013). Unless otherwise indicated, error bars denote SEM. No statistical methods were used to determine sample size prior to experiments.
Transcriptome analysis.
Output data were mapped to the target genome with the rnaStar aligner (Dobin et al., 2013) using the 2 pass mapping method (Engstrom et al., 2013). After postprocessing with PICARD, the expression count matrix was computed using HTSeq (www-huber.embl.de/users/anders/HTSeq). The raw count matrix generated by HTSeq was then processed in DESeq (www-huber.embl.de/users/anders/DESeq) to normalize the full dataset and analyze differential expression between sample groups. Gene Ontology analysis of Biological Processes and Cellular Components was performed using the DAVID 6.8 Functional Annotation Tool (Huang da et al., 2009) with Benjamini correction for multiple hypothesis testing and a cut-off of 2 genes minimum per cluster. Each Gene Ontology table in Fig. 4 and Fig. S5 lists the 10 annotation clusters with the highest enrichment scores and lowest p values below p ≤ 0.05.
Analysis of clinical data.
Patient analysis was carried out using cBioportal (Cerami et al., 2012; Gao et al., 2013). The TCGA PanCancer Atlas database (Ellrott et al., 2018; Gao et al., 2018; Hoadley et al., 2018; Liu et al., 2018; Sanchez-Vega et al., 2018; Taylor et al., 2018) was used for survival analysis of RAC1/2P29S/L skin cutaneous melanoma patients (Supplemental Table 1). Gene set enrichment analysis (GSEA) of SRF target gene expression enrichment was performed using data from TCGA skin cutaneous melanoma patients that were positive or negative for the following mutations: RAC1/2P29S/L, NF1 truncation mutant, PPP6CR264C, FBXW7 mutant, and IDH1R132C/L. RNA-sequencing read count estimation values using RNA-Seq by Expectation Maximization (RSEM) were downloaded from the Genomics Data Commons Data Portal. The SRF_01 gene set (M12047) from the Molecular Signatures Database (MSigDB v7.0) was used for GSEA analysis. For survival analysis of RAC1/2P29S/L, NF1 truncation mutant, PPP6CR264C, FBXW7 mutant , and IDH1R132C/L patients treated with anti-CTLA4, patients that underwent anti-CTLA4 therapy were selected from reference studies (Catalanotti et al., 2017; Liang et al., 2017; Miao et al., 2018; Samstein et al., 2019) (Supplemental Table S2). Melanoma patients receiving anti-PD1 and anti-PDL1 therapy were selected from the MSK-IMPACT pool (Cheng et al., 2015) (Supplemental Table S3). The overall survival of patients with RAC1/2P29S/L mutation were compared to patients with no such mutation using the python lifelines package. Death risk associated with individual MRTF-SRF target genes was obtained using the 21 genes that significantly contributed to SRF_01 gene signature enrichment in the GSEA analysis of TCGA melanoma. Z-scores for each gene were calculated within the Tumor Immune Dysfunction and Exclusion framework using the Cox proportional hazard model (Fu et al., 2020; Jiang et al., 2018), with input data from the TCGA and several ICB trials (Gide et al., 2019; Hugo et al., 2016; Liu et al., 2019; Riaz et al., 2017; Van Allen et al., 2015).
Image analysis.
To quantify F-actin intensity in fixed cells, each image was subjected to intensity thresholding in Imaris (Bitplane) to establish the space occupied by cells, after which the average intensity of Alexa Fluor 594-labeled phalloidin within the cellular volume was determined. The same procedure was used to quantify F-actin intensity in Cherry and Cherry-DeAct fixed cells, except that ROIs were established around individual Cherry+ cells after initial intensity thresholding, and then the average intensity of Alexa Fluor 647-labeled phalloidin within each cell ROI was determined. Cancer cell proliferation in sections was quantified by determining the total number of cell nuclei (DAPI) and Ki67+ nuclei via automated segmentation. MRTFB nuclear:cytoplasmic ratios were calculated by automated segmentation using Halo image analysis software (Indica Labs). Fig. S2F shows the results from every cell. To calculate the percentage of cells with strong nuclear MRTFB localization (Fig. 1K), the number of cancer cells with nuclear:cytoplasmic ratio larger than 2 were divided by the total number of cancer cells in metastatic lesions containing more than 200 cancer cells. In live imaging experiments, target cell death was scored either by PI influx and cellular collapse (B16F10 cells) or by Calcein Red-Orange leakage (MCF7 cells). To assess the speed of killing, the time difference between synapse formation and target cell death was determined. To assess killing frequency, all contacts made between one lymphocyte and one target cell were scored, excluding contacts formed within 30 min of the end of the time-lapse.
Supplementary Material
Supplemental Table 1. Related to Fig. 3. The TCGA melanoma data set analyzed for Fig. 3H and Fig. S4A and S4D.
Supplemental Table 2. Related to Fig. 3. Anti-CTLA4 clinical trial data used for Fig. 3I and Fig. S4C, S4D, and S4E.
Supplemental Table 3. Related to Fig. 3. Anti-PD1/PDL1 clinical trial data used for Fig. 3J.
Supplemental Table 4. Related to Fig. 3. Human lung and breast cancer material used for the histological studies shown in Fig. S4F.
Movie S1. CTL-mediated killing of a B16F10 cell. Related to Fig. 2. A 1650× time-lapse movie of a CTV-labeled OT1 CTL (blue fluorescence) attacking an OVA-loaded B16F10 cell (Target) in the presence of PI. Target cell death is associated with structural collapse and PI influx (red fluorescence). Time in HH:MM is indicated in the upper left corner. Scale bar = 50 μm.
Movie S2. CTL-mediated killing of a B16F10-MRTFA cell. Related to Fig. 2. A 1650× time-lapse movie of a CTV-labeled OT1 CTL (blue fluorescence) attacking an OVA-loaded B16F10-MRTFA cell (Target) in the presence of PI. Target cell death is associated with structural collapse and PI influx (red fluorescence). Time in HH:MM is indicated in the upper left corner. Scale bar = 50 μm.
Movie S3. CTL-mediated killing of a B16F10-MRTFB cell. Related to Fig. 2. A 1650× time-lapse movie of a CTV-labeled OT1 CTL (blue fluorescence) attacking an OVA-loaded B16F10-MRTFB cell (Target) in the presence of PI. Target cell death is associated with structural collapse and PI influx (red fluorescence). Time in HH:MM is indicated in the upper left corner. Scale bar = 50 μm.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-MRTFB antibody | Bethyl Laboratories | Cat#A302-768A |
| anti-MRTFA antibody | Santa Cruz Biotechnology | Cat#sc-390324 |
| Anti-GAPDH antibody | Cell Signaling | Cat#2118L |
| IRDye 680RD Goat anti-Rabbit IgG Secondary Antibody | Licor Biosciences | Cat#926-68071 |
| IRDye 800CW Goat anti-Mouse IgG Secondary Antibody | Licor Biosciences | Cat#926-32210 |
| APC conjugated anti-CD8a antibody | Tonbo Biosciences | Cat#20-0081 |
| eFluor660 conjugated anti-Lamp1 antibody | eBiosciences | Clone: 1D4B; Cat#50-1071-82 |
| PE conjugated anti-TNF | BioLegend | Cat#506306 |
| PE/Cy7 conjugated anti-IFNγ | BioLegend | Cat#505826 |
| PerCP-Cy5.5 conjugated anti-NK1.1 antibody | eBioscience | Cat#45-5941-82 |
| Brilliant Violet 786-labeled anti-Lamp1 mAb | BD Horizon | Cat#563869; Clone: SJ25C1 |
| ECD-labeled anti-CD56 mAb | Beckman Coulter | Cat#A82943 |
| BV650-labeled anti-CD3 mAb | BD Horizon | Cat#563852; clone:UCHT1 |
| PE-labeled anti-NKG2D | BioLegend | Cat#320806; clone:1D11 |
| Anti-ATP1A1 antibody | Cell Signaling Technology | Cat#3010S |
| Anti-Histone H3 antibody | Cell Signaling Technology | Cat#9715 |
| Anti-H2Kb antibody | Abcam | Cat#ab93364 |
| Anti-GM130 antibody | BD Biosciences | Cat#610822 |
| Phospho Paxillin (Tyr118) Antibody | Cell Signaling Technology | Cat#2541S |
| APC-labeled anti-Fas | Biolegend | Cat#152603 |
| PE anti-mouse H-2Kb Antibody | Biolegend | Cat#116508; Clone: AF6-88.5 |
| PE Anti-Mouse MHC Class I (H-2Db) | eBiosciece | Cat#12-5999-81 |
| mouse NKG2D-Fc | Joseph Sun (Madera et al., 2016) | N/A |
| PE anti-human IgG | Jackson | Cat#109-116-098 |
| PE-labeled anti-MICA/B | BD Pharmingen | Cat#558352; clone: 6D4 |
| PE-Cy5-labeled anti-HLA-ABC | BD Pharmingen | Cat#555554; clone: G46-2.6 |
| InVivoMab anti-mouse CD8α | Bio X Cell | Cat#BE0004-1; Clone: 53-6.7 |
| Anti-Asialo GM1 | Wako Chemicals | Cat#986-10001 |
| InVivoPlus rat IgG2a isotype control | Bio X Cell | Cat#BP0089; Clone: 2A3 |
| InVivoPlus anti-mouse PD-1 (CD279) | Bio X Cell | Cat#BP0146: Clone: RMP1-14 |
|
InVivoPlus anti-mouse CTLA-4 (CD152) Anti-CTLA-4 Mouse Monoclonal Antibody |
Bio X Cell | Cat#BP0164; Clone: BP0164 |
| InVivoPlus mouse IgG2b isotype control | Bio X Cell | Cat#BP0086; Clone: MPC-11 |
| Anti-CD8 rabbit monoclonal | Cell Signaling Technology | Cat#98941 |
| Anti-NKp46 goat polyclonal | R&D Systems | Cat#AF2225 |
| Biological Samples | ||
| Human breast and lung cancer samples | MSK Department of Pathology | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human sFas Ligand (FasL) | PeproTech | Cat#310-03H |
| Live/Dead Fixable Near IR Dead Cell Stain Kit | Molecular Probes | Cat#L34976 |
| GolgiSTOPTM | BD | Cat#554724 |
| CellTrace Violet | Thermo Fisher | Cat#C34557 |
| Staurosporine | Cell Signaling Technology | Cat#9953 |
| Recombinant Murine TNF-α | PeproTech | Cat#315-01A |
| Recombinant Mouse Granzyme B | Biolegend | Cat#554804 |
| R&D Systems™ Mouse Active Cathepsin C/DPPI Recombinant Protein | R&D Systems™ | Cat#2336CY010 |
| Alexa Fluor™ 594 Phalloidin | Thermo Fisher Scientific, Invitrogen | Cat#A12381 |
| DAPI | Thermo Fisher Scientific | Cat#D1306 |
| CellMask™ Orange Plasma membrane Stain | Invitrogen | Cat#C10045 |
| mouse interferon gamma | BioLegend | Cat#575304 |
| Live/Dead Fixable Aqua Dead Cell Stain Kit | ThermoFisher | Cat#L34957 |
| BD Cytofix/Cytoperm Plus Kit (with BD GolgiPlug) | BD | Cat#555028 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| Human interleukin 2 | NCI Biological Resources Branch Repository | N/A |
| NK Cell Isolation Kit, mouse | Miltenyi Biotec MACS | Cat#130-115-818 |
| D-Luciferin, potassium salt | Goldbio | Cat#LUCK-1G |
| Fibronectin, Bovine Plasma | Millipore Sigma | Cat#341631-5MG |
| Fura-2, AM, cell permeant | Thermo Fisher Scientific | Cat#F1221 |
| Doxycycline hyclate | Sigma-Aldrich | Cat#PHR1145-1G |
| Ki67 | Cell Signaling Technologies | Cat#9129 |
| RosetteSepTM Human NK Cell Enrichment Cocktail | Stem Cell Technologies | Cat#15025 |
| IL-2 | Proleukin, Prometheus | Cat#65483011607 |
| Critical Commercial Assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay Kit | Promega | Cat#G7570 |
| LDH Cytotoxicity Detection Kit | Takara | MK401 |
| Caspase-Glo® 3/7 Assay System | Promega | Cat#G8090 |
| Deposited Data | ||
| GEO | NCBI | GSE164860 |
| cBioportal | Cerami et al., 2012; Gao et al., 2013 | |
| TCGA PanCancer Atlas database | Ellrott et al., 2018; Gao et al., 2018; Hoadley et al., 2018; Liu 821 et al., 2018; Sanchez-Vega et al., 2018; Taylor et al., 2018 | |
| Genomics Data Commons Data Portal | https://portal.gdc.cancer.gov/ | |
| Tumor Immune Dysfunction and Exclusion | http://tide.dfci.harvard.edu/ | |
| Experimental Models: Cell Lines | ||
| E0771 | CH3 Biosystems | 94A001 |
| B16F10 | Wolchok Lab | N/A |
| MCF-7 | ATCC | HTB-22 |
| MDA-MB-231 | Massagué Lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse:C57BL/6J | The Jackson Laboratory | Cat#000664 |
| Mouse:OT1:B6.129S6-Rag2tm1Fwa Tg(TcraTcrb)1100Mjb | Taconic Farms | Cat#2334 |
| Recombinant DNA | ||
| p3XFLAG-MKL2 | Ron Prywes (Selvaraj et al., 2003) | Addgene#27175 |
| pRetroX-Tight-Hygro vector | Takara Bio | Cat#631034 |
| pLVX-TetON-Advanced | Takara Bio | Cat#632162 |
| pTetON-DHFRdd-SpvB; CMV-Cherry | Brad Zuchero (Harterink et al., 2017) | Addgene#89463 |
| pLKO.1-MKL1/2 shRNA | Ron Prywes (Lee et al., 2010) | Addgene#27161 |
| MSCV-LifeAct-GFP | Le Floc’h et al., 2013 | N/A |
| MSCV-GFP | Le Floc’h et al., 2013 | N/A |
| TGL | Ponomarev et al., 2004 | N/A |
| pRevTRE MKL1 | Ron Prywes (Muehlich et al., 2008) | Addgene#19846 |
| Software and Algorithms | ||
| Living Image software v.2.50 | Perkin Elmer | N/A |
| FlowJo software v10.7.1 | BD | N/A |
| Imaris software v8 | Bitplane | N/A |
| CaseViewer | 3D Histech | N/A |
| Aperio Imagescope | Leica Biosystems | N/A |
| Igor Pro | Wavemetrics | N/A |
| Prism v8 | GraphPad | N/A |
| rnaStar aligner | Dobin et al., 2013 | N/A |
| HTSeq | EMBL | www-huber.embl.de/users/anders/HTSeq |
| DESeq | EMBL | www-huber.embl.de/users/anders/DESeq |
| DAVID 6.8 Functional Annotation Tool | Huang da et al., 2009 | N/A |
| Halo image analysis software | Indica Labs | N/A |
HIGHLIGHTS.
Metastatic cells with high MRTF activity are vulnerable to cytotoxic lymphocytes in vivo
Strong MRTF signaling is associated with responsiveness to immune checkpoint blockade
Cancer cells overexpressing MRTF induce stronger lymphocyte activation and cytotoxicity
MRTF makes cancer cells more stimulatory to lymphocytes by increasing their rigidity
ACKNOWLEDGEMENTS
We thank C. Firl, L. Stafford, C. Jeronimo, and X. Zheng for technical support; A. Barlas, B. Wang, E. Chan, and the MSKCC Molecular Cytology Core Facility for assistance with imaging and AFM; the MSKCC Animal Imaging Core for bioluminescence imaging; the Integrated Genomics Operation (IGO), Bioinformatics Core, Center for Molecular Oncology, and the Molecular Diagnostics Service at MSKCC for RNA sequencing and analysis; Z.A. Lei and the UIC Research Informatics Core for assistance with GSEA analysis; R.J. Deaton, P.L. Nguyen, and the UIC Department of Pathology Research Imaging Core for image analysis; K. Pham and A. Rudensky for critical reading of the manuscript; N. Biais, F. Paumet, D. Yuan, L. C. Kam, and members of the M. H., J. M., K. C. H., and J. C. Sun labs for advice and assistance. Supported in part by the NIH (R01-AI087644 to M. H., R35-CA252978 to J. M., R01-AI125651 to K. C. H., and P30-CA008748 to MSKCC), the NSF (CMMI-1562905 to M. H.), the Parker Institute for Cancer Immunotherapy (M. H.), the Gerry Metastasis and Tumor Ecosystems Center (J.M.), NCATS (UL1TR002003 to UIC, RIC), Cycle for Survival (IGO), the Kravis Center for Molecular Oncology, the Leukemia and Lymphoma Society (M. H.), the Ramon Areces Foundation (M. T.-L.), and UIC (Department of Physiology and Biophysics funds to E. E. E.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Kuttner V, et al. (2018). Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton GK (2013). Metastasis: Epithelial to mesenchymal and back again. Nat Rev Cancer 13, 3. [DOI] [PubMed] [Google Scholar]
- Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, Hone JC, and Kam LC (2014). CD28 and CD3 have complementary roles in T-cell traction forces. Proc Natl Acad Sci U S A 111, 2241–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Whitlock BM, Husson J, Le Floc’h A, Jin W, Oyler-Yaniv A, Dotiwala F, Giannone G, Hivroz C, Biais N, et al. (2016). Cytotoxic T Cells Use Mechanical Force to Potentiate Target Cell Killing. Cell 165, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. (2019). Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516. [DOI] [PubMed] [Google Scholar]
- Bhavsar AP, Guttman JA, and Finlay BB (2007). Manipulation of host-cell pathways by bacterial pathogens. Nature 449, 827–834. [DOI] [PubMed] [Google Scholar]
- Blumenthal D, Chandra V, Avery L, and Burkhardt JK (2020). Mouse T cell priming is enhanced by maturation-dependent stiffening of the dendritic cell cortex. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, et al. (2009). Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Hodgson L, and Condeelis J. (2012). Directed cell invasion and migration during metastasis. Curr Opin Cell Biol 24, 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalanotti F, Cheng DT, Shoushtari AN, Johnson DB, Panageas KS, Momtaz P, Higham C, Won HH, Harding JJ, Merghoub T, et al. (2017). PTEN Loss-of-Function Alterations Are Associated With Intrinsic Resistance to BRAF Inhibitors in Metastatic Melanoma. Jco Precis Oncol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. (2015). Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 17, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comrie WA, Li S, Boyle S, and Burkhardt JK (2015). The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. The Journal of cell biology 208, 457–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, and Massague J. (2016). TGF-beta Tumor Suppression through a Lethal EMT. Cell 164, 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, and Sheetz MP (2009). Stretching single talin rod molecules activates vinculin binding. Science 323, 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, and Long EO (2010). Cytotoxic immunological synapses. Immunol Rev 235, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye SE (1986). The antimetastatic function of concomitant antitumor immunity. I. Host Ly-1+2+ effector T cells prevent the enumeration of metastatic tumor cells in a biological assay. J Immunol 136, 1504–1509. [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 6, 271–281 e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom PG, Steijger T, Sipos B, Grant GR, Kahles A, Ratsch G, Goldman N, Hubbard TJ, Harrow J, Guigo R, et al. (2013). Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods 10, 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, Griscom B, Rosenblum M, Boire A, Brogi E, et al. (2018). Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol 20, 966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y, et al. (2010). Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 120, 2030–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Finn OJ (2018). A Believer’s Overview of Cancer Immunosurveillance and Immunotherapy. J Immunol 200, 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland JC, Lee MH, and Boettiger D. (2009). Mechanically activated integrin switch controls alpha5beta1 function. Science 323, 642–644. [DOI] [PubMed] [Google Scholar]
- Fu JX, Li KR, Zhang WB, Wan CX, Zhang J, Jiang P, and Liu XS (2020). Large-scale public data reuse to model immunotherapy response and resistance. Genome Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, Liao WW, Reynolds SM, Wyczalkowski MA, Yao L, et al. (2018). Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep 23, 227–238 e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau D, and Roy P. (2018). SRF’ing and SAP’ing - the role of MRTF proteins in cell migration. J Cell Sci 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier NC, Masters TA, and Sheetz MP (2012). Mechanical feedback between membrane tension and dynamics. Trends Cell Biol 22, 527–535. [DOI] [PubMed] [Google Scholar]
- Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, et al. (2013). The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15, 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, Madore J, Lim SY, Velickovic R, Wongchenko M, et al. (2019). Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 35, 238-+. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Chames P, Kerfelec B, Baty D, Robert P, and Limozin L. (2019). Nanobody-CD16 Catch Bond Reveals NK Cell Mechanosensitivity. Biophys J 116, 1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualdrini F, Esnault C, Horswell S, Stewart A, Matthews N, and Treisman R. (2016). SRF Co-factors Control the Balance between Cell Proliferation and Contractility. Mol Cell 64, 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guck J, Schinkinger S, Lincoln B, Wottawah F, Ebert S, Romeyke M, Lenz D, Erickson HM, Ananthakrishnan R, Mitchell D, et al. (2005). Optical deformability as an inherent cell marker for testing malignant transformation and metastatic competence. Biophys J 88, 3689–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. (2009). The cytoskeleton and cancer. Cancer Metastasis Rev 28, 5–14. [DOI] [PubMed] [Google Scholar]
- Harterink M, da Silva ME, Will L, Turan J, Ibrahim A, Lang AE, van Battum EY, Pasterkamp RJ, Kapitein LC, Kudryashov D, et al. (2017). DeActs: genetically encoded tools for perturbing the actin cytoskeleton in single cells. Nature Methods 14, 479-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez C, Huebener P, and Schwabe RF (2016). Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35, 5931–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. (2018). Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou HW, Li QS, Lee GY, Kumar AP, Ong CN, and Lim CT (2009). Deformability study of breast cancer cells using microfluidics. Biomedical microdevices 11, 557–564. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song CY, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M. (2017). Mechanical forces in the immune system. Nat Rev Immunol 17, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husson J, Chemin K, Bohineust A, Hivroz C, and Henry N. (2011). Force generation upon T cell receptor engagement. PloS one 6, e19680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K, Liu P, and Lagerholm BC (2019). The Lateral Organization and Mobility of Plasma Membrane Components. Cell 177, 806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu SQ, Pan D, Fu JX, Sahu A, Hu XH, Li ZY, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nature Medicine 24, 1550-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judokusumo E, Tabdanov E, Kumari S, Dustin ML, and Kam LC (2012). Mechanosensing in T lymphocyte activation. Biophys J 102, L5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai F, Drain AP, and Weaver VM (2019). The Extracellular Matrix Modulates the Metastatic Journey. Dev Cell 49, 332–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasza KE, Nakamura F, Hu S, Kollmannsberger P, Bonakdar N, Fabry B, Stossel TP, Wang N, and Weitz DA (2009). Filamin A is essential for active cell stiffening but not passive stiffening under external force. Biophys J 96, 4326–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, and Winkler F. (2010). Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16, 116–122. [DOI] [PubMed] [Google Scholar]
- Kim T, Hwang D, Lee D, Kim JH, Kim SY, and Lim DS (2017). MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J 36, 520–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, Garcia AJ, Mould AP, Humphries MJ, and Zhu C. (2009). Demonstration of catch bonds between an integrin and its ligand. The Journal of cell biology 185, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Kuilman T, Shahrabi A, Boshuizen J, Kemper K, Song JY, Niessen HWM, Rozeman EA, Geukes Foppen MH, Blank CU, et al. (2017). Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 550, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL (2005). NK cell recognition. Annu Rev Immunol 23, 225–274. [DOI] [PubMed] [Google Scholar]
- Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, et al. (2015). Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Floc’h A, Tanaka Y, Bantilan NS, Voisinne G, Altan-Bonnet G, Fukui Y, and Huse M. (2013). Annular PIP3 accumulation controls actin architecture and modulates cytotoxicity at the immunological synapse. J Exp Med 210, 2721–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Glassman CR, Deshpande NR, Badgandi HB, Parrish HL, Uttamapinant C, Stawski PS, Ting AY, and Kuhns MS (2015). A Mechanical Switch Couples T Cell Receptor Triggering to the Cytoplasmic Juxtamembrane Regions of CD3zetazeta. Immunity 43, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Vasishtha M, and Prywes R. (2010). Activation and Repression of Cellular Immediate Early Genes by Serum Response Factor Cofactors. J Biol Chem 285, 22036–22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesokhin AM, Callahan MK, Postow MA, and Wolchok JD (2015). On being less tolerant: enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Science translational medicine 7, 280sr281. [DOI] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Hendricks W, Kiefer J, Schmidt J, Sekar S, Carpten J, Craig DW, Adkins J, Cuyugan L, Manojlovic Z, et al. (2017). Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res 27, 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionarons DA, Hancock DC, Rana S, East P, Moore C, Murillo MM, Carvalho J, Spencer-Dene B, Herbert E, Stamp G, et al. (2019). RAC1(P29S) Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 36, 68–83 e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Chen W, Evavold BD, and Zhu C. (2014). Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell 157, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, Zimmer L, Gutzmer R, Satzger I, Loquai C, et al. (2019). Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nature Medicine 25, 1916-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madera S, Rapp M, Firth MA, Beilke JN, Lanier LL, and Sun JC (2016). Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. Journal of Experimental Medicine 213, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, et al. (2014). HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345, 179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, and Massague J. (2016). Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 165, 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, and Treisman R. (2009). Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol 11, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Vokes NI, Liu D, Taylor-Weiner A, Wankowicz SM, Adeegbe D, Keliher D, Schilling B, Tracy A, et al. (2018). Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet 50, 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muehlich S, Wang RG, Lee SM, Lewis TC, Dai C, and Prywes R. (2008). Serum-induced phosphorylation of the serum response factor coactivator MKL1 by the extracellular signal-regulated kinase 1/2 pathway inhibits its nuclear localization. Mol Cell Biol 28, 6302–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S. (1999). Fas ligand-induced apoptosis. Annu Rev Genet 33, 29–55. [DOI] [PubMed] [Google Scholar]
- Northcott JM, Dean IS, Mouw JK, and Weaver VM (2018). Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front Cell Dev Biol 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, and Nieto MA (2012). Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724. [DOI] [PubMed] [Google Scholar]
- Olson EN, and Nordheim A. (2010). Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panciera T, Citron A, Di Biagio D, Battilana G, Gandin A, Giulitti S, Forcato M, Bicciato S, Panzetta V, Fusco S, et al. (2020). Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat Mater. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, Auffray C, Albrengues J, Egeblad M, Iacobuzio-Donahue CA, et al. (2018). Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev V, Doubrovin M, Serganova I, Vider J, Shavrin A, Beresten T, Ivanova A, Ageyeva L, Tourkova V, Balatoni J, et al. (2004). A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur J Nucl Med Mol Imaging 31, 740–751. [DOI] [PubMed] [Google Scholar]
- Puram SV, Parikh AS, and Tirosh I. (2018). Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol Cell Oncol 5, e1448244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotsch C, and Radmacher M. (2000). Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys J 78, 520–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitakis M, Dogniaux S, Goudot C, Bufi N, Asnacios S, Maurin M, Randriamampita C, Asnacios A, and Hivroz C. (2017). Different TCR-induced T lymphocyte responses are potentiated by stiffness with variable sensitivity. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. (2019). Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Waithe D, Clausen MP, Galiani S, Koller T, Ozhan G, Eggeling C, and Sezgin E. (2017). Diffusion of lipids and GPI-anchored proteins in actin-free plasma membrane vesicles measured by STED-FCS. Mol Biol Cell 28, 1507–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, and Smyth MJ (2011). Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, Scheper W, and Kvistborg P. (2019). Cancer Neoantigens. Annu Rev Immunol 37, 173–200. [DOI] [PubMed] [Google Scholar]
- Selvaraj A, and Prywes R. (2003). Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum response factor, is required for skeletal myogenic differentiation. J Biol Chem 278, 41977–41987. [DOI] [PubMed] [Google Scholar]
- Sezgin E, Kaiser HJ, Baumgart T, Schwille P, Simons K, and Levental I. (2012). Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat Protoc 7, 1042–1051. [DOI] [PubMed] [Google Scholar]
- Shibue T, and Weinberg RA (2009). Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A 106, 10290–10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe JC, and Griffiths GM (2007). Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol 23, 495–517. [DOI] [PubMed] [Google Scholar]
- Suresh S. (2007). Biomechanics and biophysics of cancer cells. Acta Biomater 3, 413–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. (2018). Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 33, 676–689 e673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MP, Koyuncu OO, and Enquist LW (2011). Subversion of the actin cytoskeleton during viral infection. Nat Rev Microbiol 9, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, and Yang J. (2012). Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usmani SM, Murooka TT, Deruaz M, Koh WH, Sharaf RR, Di Pilato M, Power KA, Lopez P, Hnatiuk R, Vrbanac VD, et al. (2019). HIV-1 Balances the Fitness Costs and Benefits of Disrupting the Host Cell Actin Cytoskeleton Early after Mucosal Transmission. Cell Host Microbe 25, 73–86 e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, et al. (2014). Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. (2014). The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 4, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely MD, Kershaw MH, Schreiber RD, and Smyth MJ (2011). Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29, 235–271. [DOI] [PubMed] [Google Scholar]
- Wan Z, Zhang S, Fan Y, Liu K, Du F, Davey AM, Zhang H, Han W, Xiong C, and Liu W. (2013). B cell activation is regulated by the stiffness properties of the substrate presenting the antigens. J Immunol 190, 4661–4675. [DOI] [PubMed] [Google Scholar]
- Watson IR, Li L, Mahdavi M, Gutschner T, Genovese G, Wang G, Fang Z, Tepper JM, Stemke-Hale K, et al. (2014). The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res 74, 4845–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Duffy CR, and Allison JP (2018). Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086. [DOI] [PubMed] [Google Scholar]
- Weinstein IB (2002). Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Wesolowski J, and Paumet F. (2017). Taking control: reorganization of the host cytoskeleton by Chlamydia. F1000Res 6, 2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Mezencev R, Kim B, Wang L, McDonald J, and Sulchek T. (2012). Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PloS one 7, e46609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, and Bevan MJ (2011). CD8(+) T cells: foot soldiers of the immune system. Immunity 35, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Weinberg RA (2018). Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front Med 12, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Chen W, Lou J, Rittase W, and Li K. (2019). Mechanosensing through immunoreceptors. Nat Immunol 20, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Related to Fig. 3. The TCGA melanoma data set analyzed for Fig. 3H and Fig. S4A and S4D.
Supplemental Table 2. Related to Fig. 3. Anti-CTLA4 clinical trial data used for Fig. 3I and Fig. S4C, S4D, and S4E.
Supplemental Table 3. Related to Fig. 3. Anti-PD1/PDL1 clinical trial data used for Fig. 3J.
Supplemental Table 4. Related to Fig. 3. Human lung and breast cancer material used for the histological studies shown in Fig. S4F.
Movie S1. CTL-mediated killing of a B16F10 cell. Related to Fig. 2. A 1650× time-lapse movie of a CTV-labeled OT1 CTL (blue fluorescence) attacking an OVA-loaded B16F10 cell (Target) in the presence of PI. Target cell death is associated with structural collapse and PI influx (red fluorescence). Time in HH:MM is indicated in the upper left corner. Scale bar = 50 μm.
Movie S2. CTL-mediated killing of a B16F10-MRTFA cell. Related to Fig. 2. A 1650× time-lapse movie of a CTV-labeled OT1 CTL (blue fluorescence) attacking an OVA-loaded B16F10-MRTFA cell (Target) in the presence of PI. Target cell death is associated with structural collapse and PI influx (red fluorescence). Time in HH:MM is indicated in the upper left corner. Scale bar = 50 μm.
Movie S3. CTL-mediated killing of a B16F10-MRTFB cell. Related to Fig. 2. A 1650× time-lapse movie of a CTV-labeled OT1 CTL (blue fluorescence) attacking an OVA-loaded B16F10-MRTFB cell (Target) in the presence of PI. Target cell death is associated with structural collapse and PI influx (red fluorescence). Time in HH:MM is indicated in the upper left corner. Scale bar = 50 μm.
Data Availability Statement
RNA-seq data has been deposited in the Gene Expression Omnibus (GEO) under the accession number GSE164860. All other source data is available upon request.