

Abstract
The short telomere syndromes are the most common premature aging disorders. Although studies in genetically modified cells and animal models have suggested telomere dysfunction may promote genome instability, only a minority of humans with inherited loss-of-function mutations in telomerase and related genes develop cancer. Solid tumors are relatively rare and the vast majority of cancers are bone marrow-derived with myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) comprising three-quarter of cases. In contrast to young short telomere syndrome patients who develop aplastic anemia, MDS and AML are usually diagnosed in adults who have milder short telomere defects. Here, we dissect the mechanisms by which these two bone marrow failure states, aplastic anemia and MDS-AML, evolve in the setting of varying degrees of telomere shortening. We will define the implications of these observations for patient care as well as for understanding the genetics and biology of age-related myeloid clonal evolution.
Keywords: bone marrow failure, clonal hematopoiesis of indeterminate potential (CHIP), somatic reversion
Telomere length is a heritable trait; it is determined by the parental telomere length as well as variants that affect the integrity and abundance of telomerase and its regulators [1–4]. Telomere length naturally shortens with age, and this association has raised the question as to whether telomere shortening may contribute to age-associated cancer risk [5–7]. However, several lines of evidence have supported a tumor suppressive role for telomere shortening [8], and tumor prone mouse models have consistently shown improved outcomes and survival in the setting of short telomeres (reviewed in [8]). These models have included cancers driven by oncogenic mutations, such as Myc over-expression, as well as loss of tumor suppressors, such as p16/Ink4a [9,10]. There is also increasing clarity in the genetic epidemiology literature regarding a role for long, rather than short, telomere length in promoting cancer risk [11,12]. We recently proposed some inherited telomere lengthening mutations provoke a familial cancer prone ‘long telomere syndrome’ that includes common age-related malignancies such as melanoma and chronic lymphocytic leukemia and reviewed the literature linking long telomere length to cancer risk [8,13]. Here, we focus on the spectrum and biology of cancers that arise in the short telomere syndromes and discuss their implications for understanding the mechanisms underlying myeloid clonal evolution with aging.
Telomere length predicts the onset and severity of short telomere syndrome phenotypes
The short telomere syndromes are a group of disorders caused by germline mutations in telomerase and other telomere maintenance genes [13,14]. Affected patients have short telomere lengths that fall below or overlap with the lower tail of the population’s normal distribution [14]. The cancer prevalence in these patients allows for a clinically relevant context, beyond genetically modified cell-based and animal models, to understand the causal role of short telomeres as a driver of human cancer. Short telomere syndromes have variable manifestations; the inherited telomere length as well as the mutation type determine their onset and severity [14,15]. Early-onset disease, in infants and children with extremely short telomere lengths, appears in high turnover tissues and is recognized usually as immunodeficiency and/or aplastic anemia [14–16] (Figure 1). Adults with short telomere syndromes who have less severe short telomere defects predominantly present with disease in slow turnover tissues, primarily in the liver and lung [17]. Idiopathic pulmonary fibrosis (IPF), emphysema and other telomere-related lung disease, by virtue of their high prevalence, are estimated to account for 90% of short telomere presentations [18,19]. These adult-onset presentations also make the short telomere syndromes likely the most common genetic disorders of premature aging [18].
Figure 1. Age-dependent phenotypes of the short telomere syndromes.
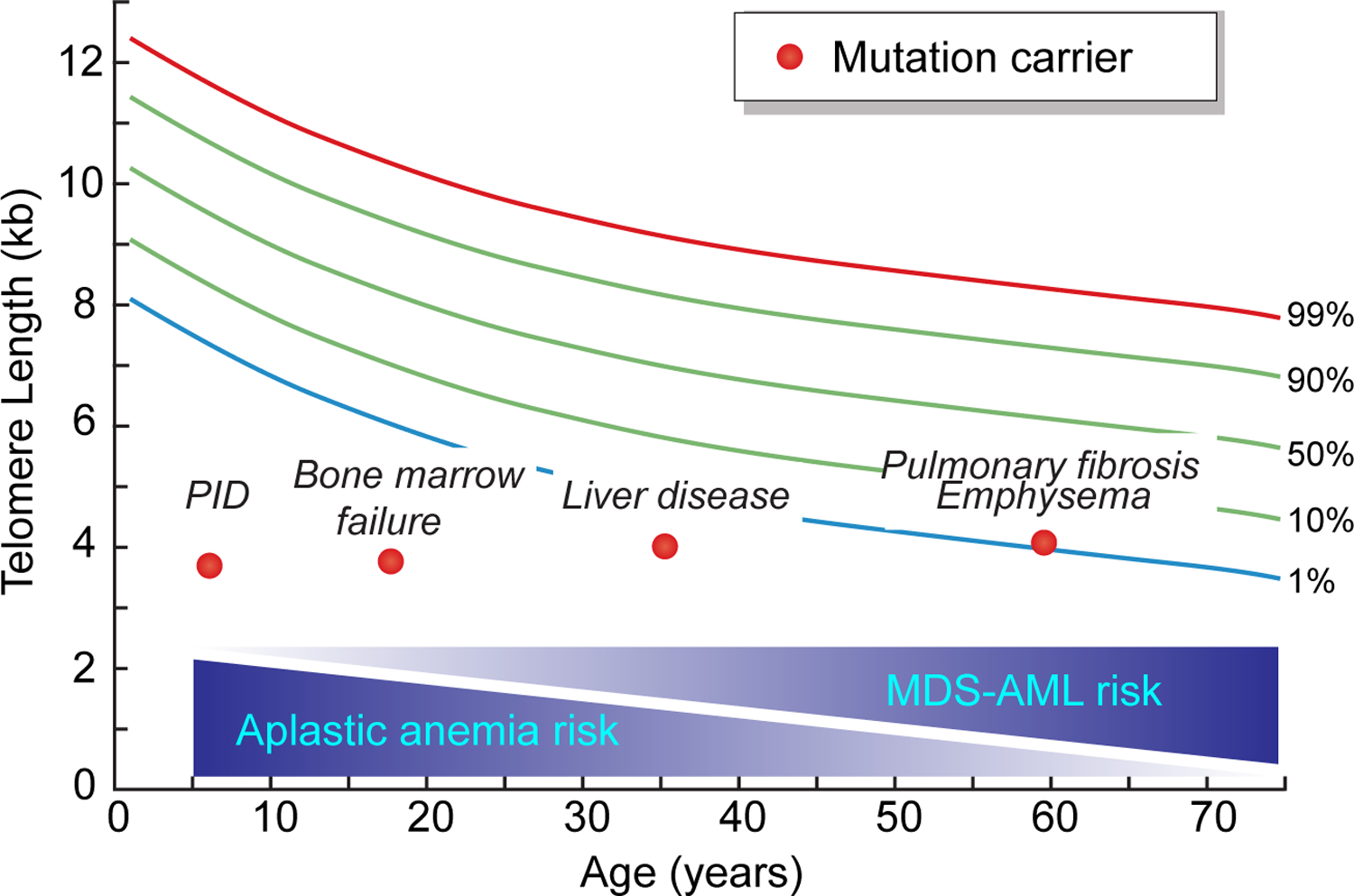
The four short telomere syndrome presentations are shown on a schematic telogram relative to the normal population percentiles. Each dot refers to an individual patient at the typical age at diagnosis. The gradient schemes below show the decreasing risk of aplastic anemia with age along with the concurrent increase in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) risk with age.
Short telomere syndrome mutations affect telomerase activity and biogenesis
To date, 14 genes have been linked to the human short telomere syndrome phenotype. Mutations in these genes disrupt telomerase enzyme activity, telomerase biogenesis, telomerase recruitment or other telomere maintenance functions [8]. Their most common mode of inheritance is autosomal dominant [14]. The known disease-associated genes are estimated to explain 50–75% of short telomere syndrome presentations [20,21]. Loss-of-function mutations in TERT, the telomerase reverse transcriptase gene, are most common and they explain more than 40% of cases in cohorts that include children and adults [14]. Mutations in the pseudouridylase dyskerin, encoded by the X-linked gene DKC1, were the first to be identified [22]. Loss-of-function of dyskerin may be associated with classic mucocutaneous findings that define the rare genodermatosis dyskeratosis congenita [23]. Dyskerin is involved in the stability and biogenesis of a group of RNAs that contain a 3’ end box H/ACA motif, including the telomerase RNA component, TR, as well as a number of non-coding RNAs [24,25]. The most recently identified short telomere syndrome gene is ZCCHC8, a component of the nuclear RNA exosome targeting complex; it is required for TR 3’ end RNA maturation and telomerase function [20].
The short telomere syndromes are a low penetrance cancer prone syndrome
An increasing clinical recognition as well as the availability of genetic and molecular testing has allowed several cohorts to quantify cancer rates in the short telomere syndromes. They have varied in their makeup of pediatric and adult cases, but altogether, estimates have been consistent reporting an overall non-cutaneous cancer diagnosis in 10–15% [23,26,27]. Therefore, as we discuss below, while cancer rates may be up to 200-fold higher over population rates for some cancers, their overall risk in the short telomere syndromes is relatively low especially when compared to high penetrance cancer predisposition syndromes such as those related to BRCA1/2 or TP53 mutations.
Solid tumors are overall rare with classic dyskeratosis congenita males being at highest risk
The rate of solid cancers is relatively low. In a recent study of 180 Johns Hopkins patients with a median age of 50, 3% had a non-cutaneous solid tumor diagnosis [27]. The most common histology was squamous affecting the upper aerodigestive tract (oral cavity, esophagus) and the anal mucosa [26–28] (Figure 2). Readily resectable squamous and basal skin cancers are also common and have been estimated to affect ~10% in one study of children and young adults [26], but the overall prevalence of these cancers may be higher in older adults based on our clinical experience. The solid cancer spectrum is thus narrow and, in general, solid tumors arising de novo, outside of the lung or liver transplant setting, appear to have a favorable outcome.
Figure 2. Pie graph shows relative distribution of cancers of the most common non-cutaneous malignancies in the short telomere syndromes with their estimated respective percentages.
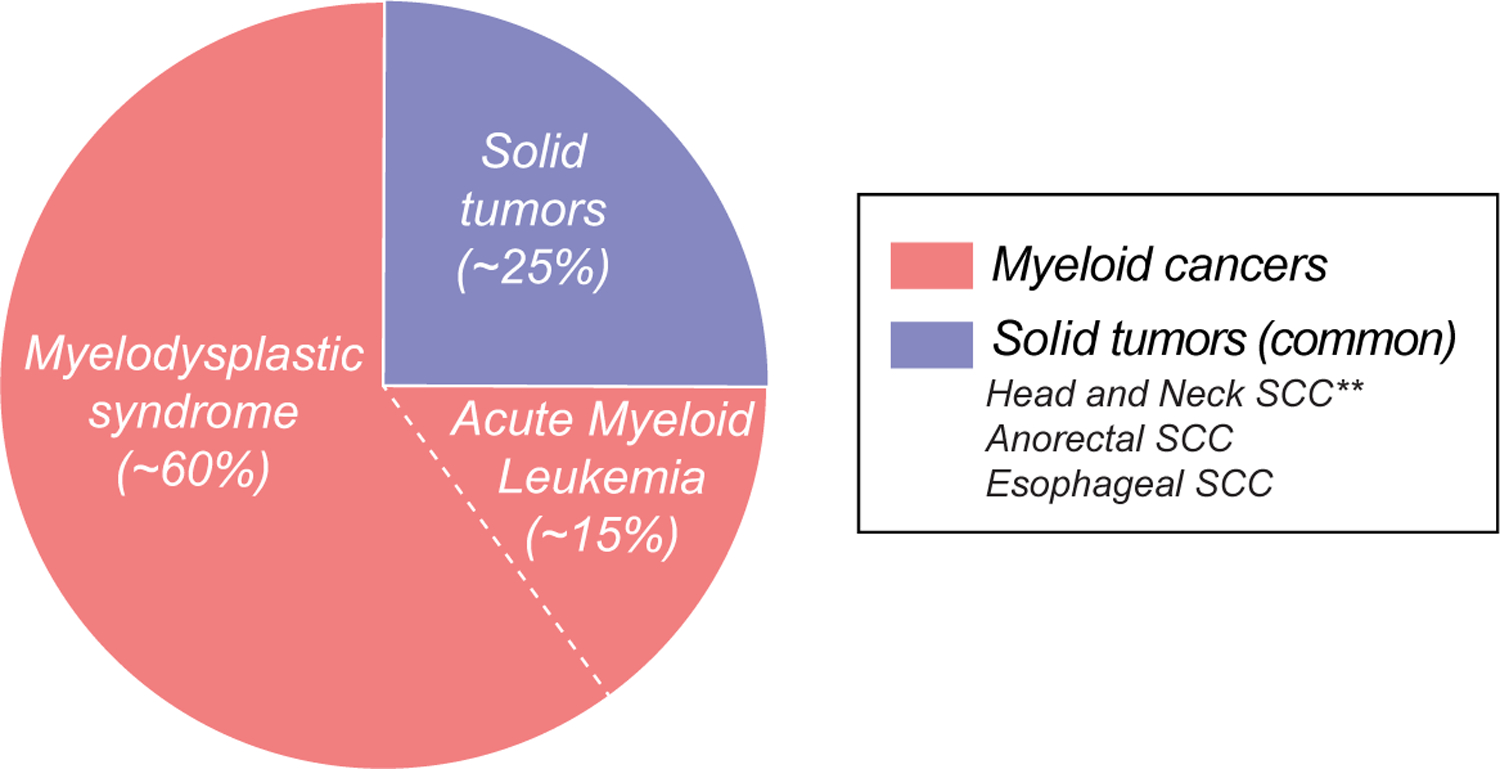
The most common solid tumors are listed.
In contrast to nearly all other phenotypes heretofore described where telomere length is the predictor of onset and severity, there is some evidence of a genotype-phenotype correlation in with respect to solid cancers [27]. The highest risk is in young adult males who have classic mucocutaneous features of dyskeratosis congenita and DKC1 mutations [27]. These observations, if verified in other studies, suggest that disturbances related to TR-independent functions of dyskerin may play a role in promoting carcinogenesis [27,29]. They also indicate that classic dyskeratosis congenita patients, especially males, are likely to derive the most benefit from screening for mucosal solid tumors.
Myeloid cancers are the most common short telomere syndrome malignancies
The most common short telomere syndrome cancers are myelodysplastic syndrome (MDS, ~60%) followed by acute myeloid leukemia (AML, ~15%) [27] (Figure 2). These two cancers fall on a continuum of age-related myeloid clonal processes. They are diagnosed on average in the sixth decade, two decades younger than unselected MDS-AML [27]. Overall, short telomere syndrome patients have at least a 150-fold higher risk for developing MDS; yet only a small subset, 8–10%, develop it [26,27]. The risk for AML is lower but still 50-fold higher than the population [26,27]. Importantly, relative to aplastic anemia, MDS and AML are diagnosed on average three decades older [27] (Figure 1). And, unlike squamous tumors, there is no genotype-phenotype association with their risk [27]. Since lung disease is also age-dependent, short telomere MDS-AML patients often have the diagnosis of pulmonary fibrosis, concurrently or soon after diagnosis, and morality related to end-stage lung and/or liver disease is the primary cause of mortality in two-thirds of these cancer patients [27] (Figure 1).
Clinical features of short telomere MDS-AML and treatment implications
The short telomere syndromes are one of several known MDS-AML prone genetic syndromes (reviewed in [30,31]). Among families with MDS-AML, the short telomere syndrome disease genes are thought to be one of the most common causes; they account for 3–20% of familial cases depending on the cohort studied ([32] and Dr. Jane Churpek, personal communication). In unselected cohorts of patients with MDS-AML, the exact percentage of patients with telomere-related germline mutations has not been studied using contemporary gene panels. Most but not all of short telomere MDS-AML patients have distinguishing clinical histories suggestive of the short telomere syndrome diagnosis (e.g. personal or family history of pulmonary fibrosis) [27,32]. There are also histopathologic features of telomere-mediated MDS-AML that may distinguish them from unselected cases; these features are relevant for understanding the role of short telomere defect in driving the evolution of these cancers. For example, there is a greater proportion of short telomere MDS-AML patients who have hypoplastic MDS-AML; this is generally a rare finding in unselected cases [27] (Figure 3). Regardless of bone marrow cellularity, the heretofore short telomere-mediated MDS-AML cases studied all have hypoproliferative disease (i.e. associated with low blood counts at diagnosis) [27]. Their cancers are also often indolent with a slowly progressive MDS course [27], likely reflecting the proliferative constraint caused by short telomeres.
Figure 3. Short telomere MDS tends to be hypoplastic and hypoproliferative.
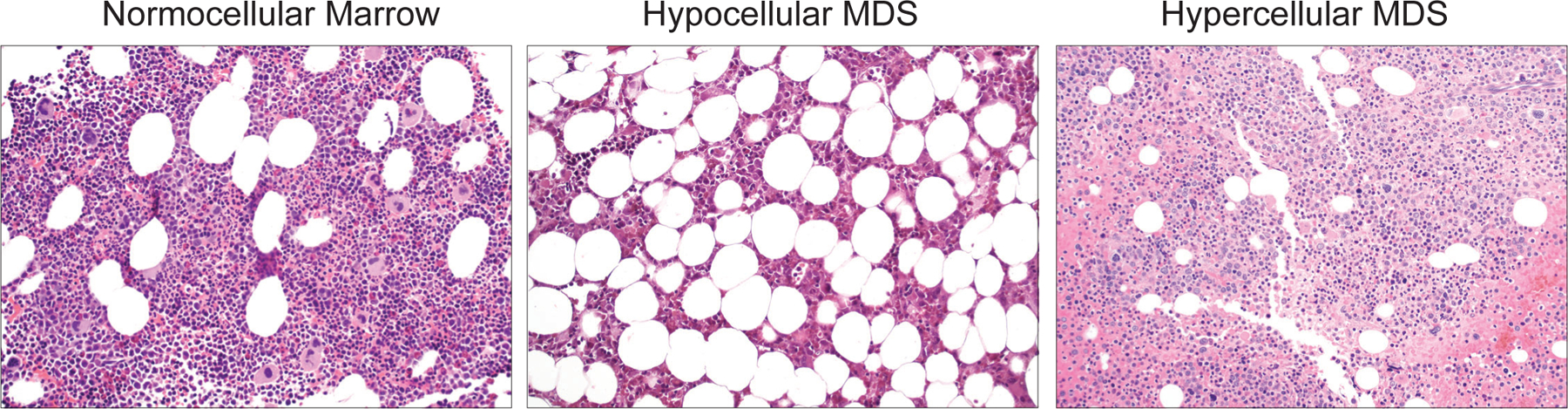
Low power (20X) photomicrographs of bone marrow biopsies from a young adult showing normocellular marrow, a short telomere patient with a hypocellular marrow from hypoplastic myelodysplastic syndrome (MDS), and an adult with a hypercellular marrow in a patient with hyperplastic MDS as labeled above. Hypoplastic MDS is rare in unselected populations but represents more than half of short telomere MDS cases [27]. Images generously provided by Dr. Amy S. Duffield of the Johns Hopkins Division of Hematopathology.
Impact of clinical diagnosis of short telomere MDS-AML
Making the genetic diagnosis of telomere-mediated MDS-AML influences treatment decisions in several settings. For example, it informs the choice of hematopoietic stem cell transplant (HSCT) regimen because these patients are susceptible to pulmonary toxins. However, in contrast to aplastic anemia, the published experience with HSCT regimens in MDS-AML is limited. Like with other inherited forms of MDS-AML [33], this genetic diagnosis also influences the choice of related transplant donor given the familial, usually autosomal dominant nature of mutations. The evaluation and selection of potentials donors who may be asymptomatic also requires an extensive consent process and experienced genetic counseling. The short telomere syndrome diagnosis may additionally influence the need and duration of antibiotic prophylaxis, such as against cytomegalovirus, given the underlying immune vulnerability and T cell immunodeficiency which may co-occur with bone marrow failure [34]. The extrahematopoietic pulmonary and hepatic complications of telomere shortening also complicate the clinical course of MDS-AML [27,35].
Mechanisms of clonal evolution with aging
What distinguishes the short telomere-mediated bone marrow failure state of MDS-AML from that of aplastic anemia? We recently compared telomere lengths of these respective patient subsets and found that, relative to aplastic anemia, MDS-AML patients have significantly longer telomere length, and on average, they are three decades older at diagnosis [27] (Figures 1 and 4). These observations lead us to propose a model for how these states evolve differently given the differential starting point of telomere length. In the case of aplastic anemia, short telomeres limit hematopoietic stem cell self-renewal, as has been shown extensively in animal models [34,36]. Thus under the replicative pressures of hematopoiesis, even if clonal mutations arise, the short telomere length will limit their evolution because of stem cell dropout (Figure 4). In contrast, an overall short but relatively longer stem-progenitor can acquire compensatory mutations in response to less severe stem cell dropout pressures. The longer replicative potential in these stem-progenitors would in turn be permissive for evolution to the clonal redundancy seen in overt cancer (Figure 4). It is notable that the somatic mutational landscape of short telomere MDS-AML mirrors what is seen in unselected cases of these same cancers [27]. However, their clinical course is marked by hypoproduction of blood components.
Figure 4. Model for the evolution of aplastic anemia and clonal hematopoiesis in MDS-AML in the short telomere syndromes.
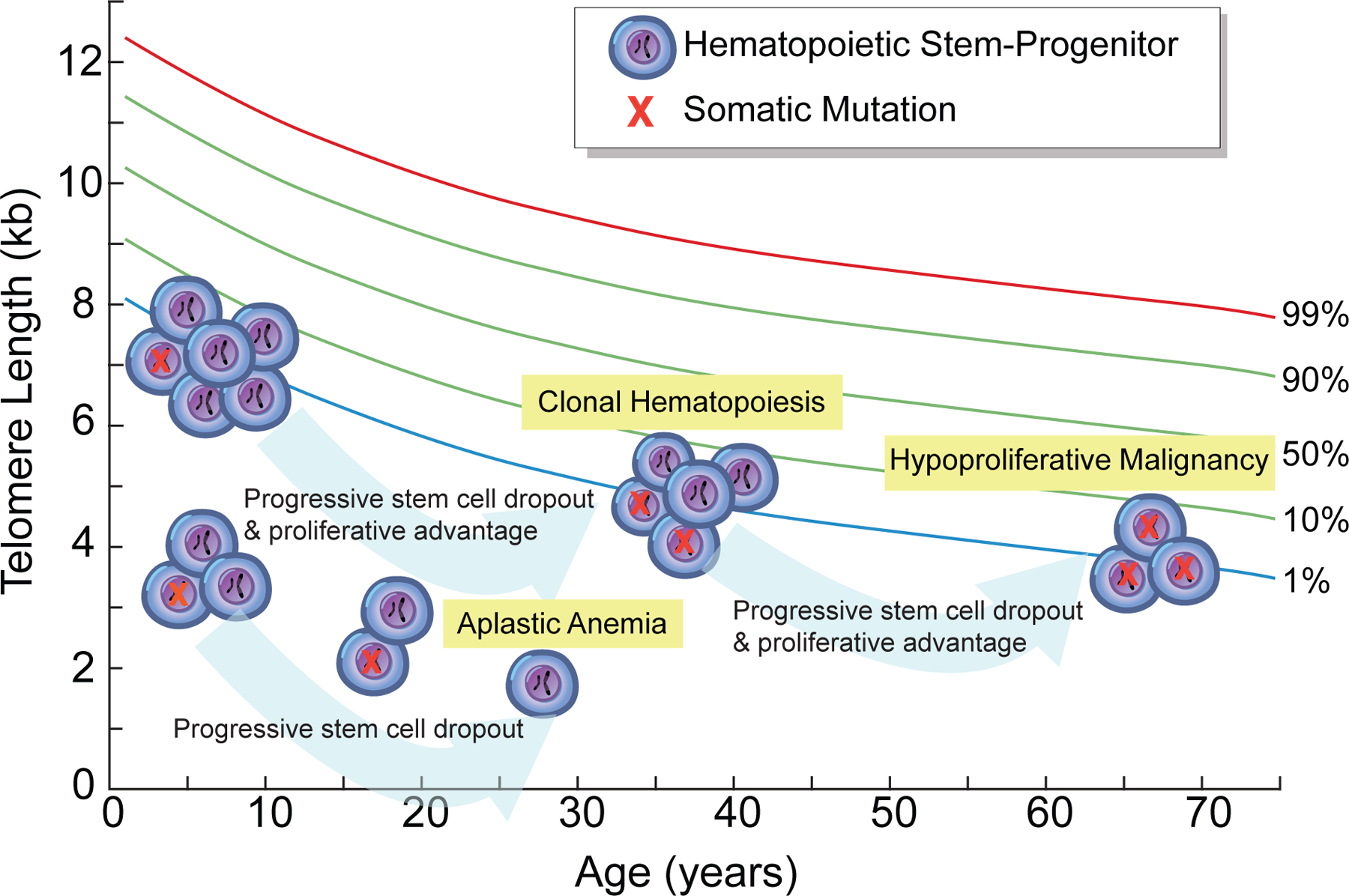
Short telomere syndrome aplastic anemia patients have shorter telomere length at birth. They are thus more prone to stem-progenitor cell dropout and eventual loss of clonal mutations that may arise under the pressure of telomere shortening and with age. In contrast, short telomere MDS-AML patients have relatively longer telomere length (albeit shorter than age-matched controls) and are predicted to sustain clonal events that may arise with stem cell dropout, especially as they age. This would ultimately be predicted to lead to an age-dependent increase in MDS-AML.
Clonal hematopoiesis in adults with short telomere syndromes
Hematopoiesis is normally oligoclonal but it becomes increasingly clonal with age. The mechanism driving this age-related clonality are not known but they manifest in a recognizable pattern of recurrent somatic mutations in the same genes seen in MDS-AML and other hematologic malignancies [37,38]. These mutations are thought to precede malignant transformation of these cancers albeit in a very small subset [37,38]. Among adults with short telomere syndrome mutations, who do not have MDS-AML, our group recently found that the rate of detectable age-related clonal mutations is at least 3-fold higher than populations that are significantly older [27,39–41]. These mutations fall in the same genes that define the emerging state of clonal hematopoiesis of indeterminate potential (CHIP) [37]. Our model would suggest that the stem cell dropout that takes place in the setting of short telomeres, with aging, drives the clonal evolution and selection rather than and increase mutation rate per se (Figure 4). The premature clonal evolution we documented in these patients suggests that telomere shortening, at least in the context of germline mutations in telomerase and related genes, is sufficient to drive the CHIP phenotype.
The risk of clonal evolution with androgens
The high rate of CHIP mutations along with the MDS-AML prone state of adults with telomerase and other telomere related genes begs pause and raises caution for suggested attempts to use androgens in adults with short telomere syndromes. Since the 1960s, androgens have been used to temporize cytopenias in aplastic anemia [42]. In a small retrospective study of children with dyskeratosis congenita-associated aplastic anemia, approximately half had short-term improvements in blood counts. Among those responding, the duration of treatment was variable with a median of 2–3 years, and patients were reported to discontinue androgens because of tolerability/toxicity as well as lack of efficacy [43]. An exploratory phase 1/2 study of adults with germline mutations in telomerase and telomere-related genes examined the safety of the androgen danazol and its effect on blood counts. While improvements in blood counts were seen, nearly half the patients had no follow-up at the 24-month endpoint raising questions regarding the safety of this approach [44]. This study also measured telomere length using a quantitative PCR method in a limited subset and reported a decrease in the rate of telomere attrition [44]. However, subsequent analyses, using flow cytometry and fluorescence in situ hybridization, a more reproducible method, found that androgens do not alter the rate of telomere attrition [45]. One concern beyond the robustness of measurement tools used in that study, in light of the data we review here, is that the effect of androgens may be related to the output of mutation-containing clones without reversing the telomere length defect per se. Indeed, the literature contains one example where an aplastic anemia patient’s blood counts ‘improved’ and telomeres ‘lengthened’, but the patient died from an aggressive and treatment refractory AML (of note, the common variant reported in TERT in this case is now appreciated to be a benign polymorphism [46]). The MDS-AML prone state and the high rate of somatic clonal cancer predisposing mutations raise important questions as to the safety of androgens in short telomere patients, beyond limited indications such as with transfusion-dependence, and especially in adults.
Somatic reversion in the short telomere syndromes
Beyond CHIP mutations, the selective pressures of hematopoiesis in the short telomere bone marrow also promote somatic reversion ([47–49] and reviewed in [50]). For example, in TR heterozygous mutation carriers, mitotic recombination causes the acquisition and selection of clones with isodisomy of the wild-type allele. There is also the example of TERT promoter gain-of-function mutations, which are acquired somatically in some solid tumors, and are detectable in 5% of IPF patients who carry loss-of-function mutations in telomerase [49]. These patients do not have any hematologic malignancy and some have had long-term followup, up to 10 years [49]. These observations indicate that these somatic reversion events, which in some cases may ‘correct’ the underlying genetic defect, are also not sufficient to drive cancer. Whether these somatic reversion events protect or promote clonal expansion is not currently known.
Summary and Perspective
If clonal mutations are so common in adults with short telomere syndromes, what is the reason underlying the still relatively low rate of MDS-AML? One answer may be that IPF is highly penetrant, and given its relatively limited treatment options, it is the primary cause of mortality in most short telomere adults including those with myeloid cancer promoting mutations. It is also possible that there are other genetic modifiers of the MDS-AML phenotype that are yet-to-be identified. Overall, the study of humans with germline defects in telomerase and other telomere maintenance genes provides a highly compelling and relevant context for reconsidering the role of short telomeres in cancer with aging. The new findings suggest that hypoproliferative myeloid cancers are the most prevalent malignant consequence of telomere shortening in humans.
Highlights.
Clonal hematopoiesis is common in adult short telomere syndrome patients.
Longer telomeres may allow clonal evolution to MDS, in contrast to aplastic anemia.
Acknowledgements.
Work in the Armanios group is supported by the United Statses National Institutes of Health grants CA225027 and HL119476, and support from the S&R, Gary Williams, and Commonwealth Foundations (to M.A.). We acknowledge a gift in the name of Mrs. P. Godrej (M.A.). KES is supported by NIH T32 HL007525 and the Turock Scholars Fund at the Telomere Center at Johns Hopkins.
Footnotes
Conflict of Interest. The authors declare no conflict of interest.
References
- 1.Armanios M, Blackburn EH: The telomere syndromes. Nat Rev Genet 2012, 13:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I, et al. : Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet 2013, 45:422–427, 427e421–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aviv A: Genetics of leukocyte telomere length and its role in atherosclerosis. Mutat Res 2012, 730:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hao LY, Armanios M, Strong MA, Karim B, Feldser DM, Huso D, Greider CW: Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 2005, 123:1121–1131. [DOI] [PubMed] [Google Scholar]
- 5.Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S: Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. Embo J 1992, 11:1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW: Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91:25–34. [DOI] [PubMed] [Google Scholar]
- 7.Artandi SE, DePinho RA: Telomeres and telomerase in cancer. Carcinogenesis 2010, 31:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNally EJ, Luncsford PJ, Armanios M: Long telomeres and cancer risk: the price of cellular immortality. J Clin Invest 2019, 130.**This paper synthesizes an argument, from genetic and animal model studies, supporting a strong role for long telomere length as a genetic driver of a subset of familial cancers.
- 9.Feldser DM, Greider CW: Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 2007, 11:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA: Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 1999, 97:515–525. [DOI] [PubMed] [Google Scholar]
- 11.Rode L, Nordestgaard BG, Bojesen SE: Long telomeres and cancer risk among 95 568 individuals from the general population. Int J Epidemiol 2016, 45:1634–1643. [DOI] [PubMed] [Google Scholar]
- 12.Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, Wade KH, Timpson NJ, Evans DM, Willeit P, et al. : Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanley SE, Armanios M: The short and long telomere syndromes: paired paradigms for molecular medicine. Curr Opin Genet Dev 2015, 33:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alder JK, Hanumanthu VS, Strong MA, DeZern AE, Stanley SE, Takemoto CM, Danilova L, Applegate CD, Bolton SG, Mohr DW, et al. : Diagnostic utility of telomere length testing in a hospital-based setting. Proc Natl Acad Sci U S A 2018, 115:E2358–E2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parry EM, Alder JK, Qi X, Chen JJ, Armanios M: Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood 2011, 117:5607–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonassaint NL, Guo N, Califano JA, Montgomery EA, Armanios M: The gastrointestinal manifestations of telomere-mediated disease. Aging Cell 2013, 12:319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armanios M: Telomeres and age-related disease: how telomere biology informs clinical paradigms. J Clin Invest 2013, 123:996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanley SE, Merck SJ, Armanios M: Telomerase and the Genetics of Emphysema Susceptibility. Implications for Pathogenesis Paradigms and Patient Care. Ann Am Thorac Soc 2016, 13 Suppl 5:S447–S451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merck SJ, Armanios M: Shall we call them “telomere-mediated”? Renaming the idiopathic after the cause is found. Eur Respir J 2016, 48:1556–1558. [DOI] [PubMed] [Google Scholar]
- 20.Gable DL, Gaysinskaya V, Atik CC, Talbot CC Jr., Kang B, Stanley SE, Pugh EW, Amat-Codina N, Schenk KM, Arcasoy MO, et al. : ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev 2019, 33:1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savage SA, Alter BP: Dyskeratosis congenita. Hematol Oncol Clin North Am 2009, 23:215–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I: X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet 1998, 19:32–38. [DOI] [PubMed] [Google Scholar]
- 23.Dokal I: Dyskeratosis congenita in all its forms. Br J Haematol 2000, 110:768–779. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell JR, Wood E, Collins K: A telomerase component is defective in the human disease dyskeratosis congenita. Nature 1999, 402:551–555. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell JR, Cheng J, Collins K: A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3’ end. Mol Cell Biol 1999, 19:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alter BP, Giri N, Savage SA, Rosenberg PS: Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schratz KE, Haley L, Danoff SK, Blackford AL, DeZern AE, Gocke C, Duffield AS, Armanios M: Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood, in press.**This study defines the clinical-pathologic features and natural history of short telomere syndrome cancers including novel genotype-phenotype correlations. It also reports a high rate of CHIP mutations in adults with short teloemre syndromes.
- 28.Alter BP, Giri N, Savage SA, Rosenberg PS: Cancer in dyskeratosis congenita. Blood 2009, 113:6549–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, Cordon-Cardo C, Pandolfi PP: Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 2003, 299:259–262. [DOI] [PubMed] [Google Scholar]
- 30.Brown AL, Churpek JE, Malcovati L, Dohner H, Godley LA: Recognition of familial myeloid neoplasia in adults. Semin Hematol 2017, 54:60–68. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy AL, Shimamura A: Genetic predisposition to MDS: clinical features and clonal evolution. Blood 2019, 133:1071–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holme H, Hossain U, Kirwan M, Walne A, Vulliamy T, Dokal I: Marked genetic heterogeneity in familial myelodysplasia/acute myeloid leukaemia. Br J Haematol 2012, 158:242–248. [DOI] [PubMed] [Google Scholar]
- 33.DiNardo CD, Bannon SA, Routbort M, Franklin A, Mork M, Armanios M, Mace EM, Orange JS, Jeff-Eke M, Churpek JE, et al. : Evaluation of Patients and Families With Concern for Predispositions to Hematologic Malignancies Within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagner CL, Hanumanthu VS, Talbot CC Jr., Abraham RS, Hamm D, Gable DL, Kanakry CG, Applegate CD, Siliciano J, Jackson JB, et al. : Short telomere syndromes cause a primary T cell immunodeficiency. J Clin Invest 2018, 128:5222–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorgy AI, Jonassaint NL, Stanley SE, Koteish A, DeZern AE, Walter JE, Sopha SC, Hamilton JP, Hoover-Fong J, Chen AR, et al. : Hepatopulmonary syndrome is a frequent cause of dyspnea in the short telomere disorders. Chest 2015, 148:1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL: Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447:725–729. [DOI] [PubMed] [Google Scholar]
- 37.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL: Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaiswal S, Ebert BL: Clonal hematopoiesis in human aging and disease. Science 2019, 366.**This review summarizes current understanding of the mechanims underlying age-related mutagenesis, the somatic mutational landscape that defines age-related clonal hematopoiesis and its implications for cancer risk.
- 39.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. : Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014, 371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. : Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014, 371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. : Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014, 20:1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shahidi NT, Diamond LK: Testosterone-induced remission in aplastic anemia of both acquired and congenital types. Further observations in 24 cases. N Engl J Med 1961, 264:953–967. [DOI] [PubMed] [Google Scholar]
- 43.Khincha PP, Wentzensen IM, Giri N, Alter BP, Savage SA: Response to androgen therapy in patients with dyskeratosis congenita. Br J Haematol 2014, 165:349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Townsley DM, Dumitriu B, Liu D, Biancotto A, Weinstein B, Chen C, Hardy N, Mihalek AD, Lingala S, Kim YJ, et al. : Danazol Treatment for Telomere Diseases. N Engl J Med 2016, 374:1922–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khincha PP, Bertuch AA, Gadalla SM, Giri N, Alter BP, Savage SA: Similar telomere attrition rates in androgen-treated and untreated patients with dyskeratosis congenita. Blood Adv 2018, 2:1243–1249.**This study, which used robust telomere length measurement tools found that androgen-treated patients had no change in telomere length raising questions as to the safety of androgens on promoting clonal evolution.
- 46.Ziegler P, Schrezenmeier H, Akkad J, Brassat U, Vankann L, Panse J, Wilop S, Balabanov S, Schwarz K, Martens UM, et al. : Telomere elongation and clinical response to androgen treatment in a patient with aplastic anemia and a heterozygous hTERT gene mutation. Ann Hematol 2012, 91:1115–1120. [DOI] [PubMed] [Google Scholar]
- 47.Jongmans MC, Verwiel ET, Heijdra Y, Vulliamy T, Kamping EJ, Hehir-Kwa JY, Bongers EM, Pfundt R, van Emst L, van Leeuwen FN, et al. : Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet 2012, 90:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, Armanios M: Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest 2015, 147:1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maryoung L, Yue Y, Young A, Newton CA, Barba C, van Oers NS, Wang RC, Garcia CK: Somatic mutations in telomerase promoter counterbalance germline loss-of-function mutations. J Clin Invest 2017, 127:982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Revy P, Kannengiesser C, Fischer A: Somatic genetic rescue in Mendelian haematopoietic diseases. Nat Rev Genet 2019, 20:582–598.**This review summarizes current knowledge of somatic reversion in inherited bone marrow failure and primary immunodeficiency disorders.