

Abstract
Alcohol‐associated liver disease (ALD) is a major cause of mortality. Gut barrier dysfunction–induced bacterial translocation and endotoxin release contribute to the pathogenesis of ALD. Probiotic Lactobacillus rhamnosus GG (LGG) is known to be beneficial on experimental ALD by reinforcing the intestinal barrier function. In this study, we aim to investigate whether the protective effects of LGG on intestinal barrier function is mediated by exosome‐like nanoparticles (ELNPs) released by LGG. Intestinal epithelial cells and macrophages were treated with LGG‐derived ELNPs (LDNPs) isolated from LGG culture. LDNPs increased tight junction protein expression in epithelial cells and protected from the lipopolysaccharide‐induced inflammatory response in macrophages. Three‐day oral application of LDNPs protected the intestine from alcohol‐induced barrier dysfunction and the liver from steatosis and injury in an animal model of ALD. Co‐administration of an aryl hydrocarbon receptor (AhR) inhibitor abolished the protective effects of LDNPs, indicating that the effects are mediated, at least in part, by intestinal AhR signaling. We further demonstrated that LDNP administration increased intestinal interleukin‐22‐Reg3 and nuclear factor erythroid 2‐related factor 2 (Nrf2)–tight junction signaling pathways, leading to the inhibition of bacterial translocation and endotoxin release in ALD mice. This protective effect was associated with LDNP enrichment of bacterial tryptophan metabolites that are AhR agonists. Conclusions: Our results suggest that the beneficial effects of LGG and their supernatant in ALD are likely mediated by bacterial AhR ligand–enriched LDNPs that increase Reg3 and Nrf2 expression, leading to the improved barrier function. These findings provide a strategy for the treatment of ALD and other gut barrier dysfunction–associated diseases.
Abbreviations
- AF
alcohol‐fed
- AhR
aryl hydrocarbon receptor
- ALD
alcohol‐associated liver disease
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BMDM
bone marrow–derived macrophage
- Cyp1a1
cytochrome P450 family 1 subfamily A member 1
- ELNP
exosome‐like nanoparticle
- EtOH
ethanol
- H&E
hematoxylin and eosin
- I3A
indole‐3‐aldehyde
- IA
indoleacrylic acid
- Il‐1β
interleukin‐1β
- Il‐6
interleukin‐6
- Il‐22
interleukin‐22
- LC‐MS
liquid chromatography–mass spectrometry
- LDNP
LGG‐derived ELNP
- LGG
Lactobacillus rhamnosus GG
- LGGs
LGG culture supernatant
- LGGs(np‐d)
nanoparticle‐depleted LGGs
- LPL
lamina propria lymphocyte
- LPS
lipopolysaccharide
- mRNA
messenger RNA
- MRS
deMan, Rogosa and Sharpe
- NP
nanoparticle
- Nqo1
NAD(P)H: quinone acceptor oxidoreductase 1
- Nrf2
nuclear factor erythroid 2‐related factor 2
- ns
not significant
- PBS
phosphate‐buffered saline
- PF
pair‐fed
- PM
peritoneal macrophage
- Reg3
regenerating islet‐derived protein 3
- ROS
reactive oxygen species
- TNFα
tumor necrosis factor alpha
Probiotics have been used safely as a food supplement for over 100 years. In the last decade, there has been increasing interest in using probiotics to prevent or treat specific diseases in experimental animals and humans.( 1 ) Multiple clinical trials have been conducted to investigate the effects of probiotics in the management of disease states, such as gastrointestinal disorders( 2 ) and metabolic liver diseases.( 3 , 4 ) However, the results of these clinical trials are mixed, likely for multiple reasons. It is clear that viable probiotics must colonize the gut to exert their function. Host factors, including baseline microbiota, may cause “colonization resistance” to exclude the potential invading probiotics.( 5 ) The compositional patterns of baseline microbiota may not respond well to incoming probiotic bacteria.( 6 ) In addition, the patient’s underlying medical condition may negatively affect the ability of the probiotic to survive and colonize the gut; in particular, conditions such as an immunocompromised state and/or consumption of medications such as antibiotics are problematic. In addition, safety outcomes of probiotics in disease management are inconsistent.( 7 )
Recently, we demonstrated that the culture supernatant of a well‐characterized probiotic strain, Lactobacillus rhamnosus GG (LGG), was effective in the treatment of both experimental alcohol‐ associated liver disease (ALD)( 8 , 9 ) and nonalcoholic liver disease,( 10 ) indicating the benefits of the LGG fermentation products. However, the mechanisms of action of the LGG supernatant (LGGs) are yet to be defined.
Soluble proteins produced by LGG have been shown to regulate intestinal epithelial cell survival and growth,( 11 ) and molecules produced by LGG with different effects on the host have been described.( 12 , 13 , 14 ) How these individual products are released from bacteria and interact with the host cells/bacteria is not clear. Bacteria, along with mammalian cells, release exosome‐like nanoparticles (ELNPs), which are implicated in bacterial cell–cell interactions and bacterial cell–host cell communications.( 15 , 16 ) ELNPs carry a variety of genetic materials (microRNA [miRNA], messenger RNA [mRNA], and other noncoding RNAs), proteins, and metabolites.( 17 ) Recent studies suggest that ELNPs function as natural effectors of signaling between cells and across various tissues through the transfer of their cargos.( 18 ) In the past, it was believed that only Gram‐negative bacteria can produce ELNPs, and that Gram‐positive bacteria are not able to produce ELNPs due to their thick cell wall. However, recent studies showed that Gram‐positive bacteria can also release ELNPs.( 16 , 19 , 20 ) It has been shown that Lactobacillus‐derived ELNPs have multiple functions, including stimulating the host nervous system, inducing hepatic cancer cell death, and enhancing the immune response against pathogenic bacteria.( 16 , 21 , 22 )
We therefore hypothesize that ELNPs released from LGG (LGG‐derived exosome‐like nanoparticles, LDNPs) contribute to the protective effects of LGG against experimental ALD. The effects of LDNPs may be mediated through the delivery of the specific cargo material to intestinal cells to protect barrier integrity. In this report, we provide evidence that ELNPs are released from LGG, and that these LDNPs are protective against alcohol‐induced liver injury. We showed that LDNP treatment increased the expression of antimicrobial peptides (Reg3β and Reg3γ) and intestinal tight junction proteins in cultured intestinal epithelial cells and in intestinal tissue of mice fed alcohol. We further showed that the LDNPs were enriched in bacterial tryptophan metabolites, indole derivatives, which are endogenous aryl hydrocarbon receptor (AhR) agonists. We also showed that LDNPs protected against experimental ALD through intestinal AhR‐IL22‐Reg3‐related and nuclear factor erythroid 2‐related factor 2 (Nrf2)–mediated signaling pathways, leading to reduced bacterial translocation and lipopolysaccharide (LPS) release.
Material and Methods
LGG Culture and LDNP Isolation
LGG was purchased from American Type Culture Collection (ATCC 53103, Rockville, MD) and cultured in autoclaved deMan, Rogosa and Sharpe (MRS) broth at 37°C for 40 hours. The culture density was measured with a spectrophotometer at OD600. The culture suspension (2 × 109 CFU/mL) was centrifuged at 2,000g for 10 minutes, at 5,000g for 20 minutes, and then at 10,000g for 30 minutes to eliminate debris including dead cells and other waste materials. The obtained supernatant was filtered and ultracentrifuged at 150,000g for 70 minutes (Optima L‐100XP Ultra Centrifuge; Bechman Coulter, Atlanta, GA). After ultracentrifugation, the supernatants were collected and stored (nanoparticle‐depleted LGGs, or LGGs[np‐d]), and the pellet containing LDNPs was washed in phosphate‐buffered saline (PBS), ultracentrifuged, resuspended in PBS, and stored at −80°C for later use.
Animals and Treatments
C57BL/6J mice (6‐8 weeks of age) from Jackson Laboratory (Bar Harbor, ME) were maintained at 22°C with a 12‐hour light/dark cycle and initially had free access to a normal chow diet and tap water. Mice were then fed the Lieber DeCarli diet containing 5% alcohol (vol/vol) (alcohol‐fed, AF) or isocaloric maltose dextrin (pair‐fed, PF). For the AF groups, mice were initially fed the control Lieber‐DeCarli liquid diet (Bio‐Serve, Flemington, NJ) for 5 days, to acclimate to the liquid diet. The content of alcohol in the liquid diet was gradually increased from 1.6% (vol/vol) to 5% (vol/vol) in the next 6 days and remained at 5% for the subsequent 10 days. Mice in the PF group were fed isocaloric maltose dextrin in substitution for alcohol in the liquid diet. On experimental day 10, a bolus of ethanol (EtOH) (5 g/kg body weight) was given to AF mice by gavage 9 hours before harvesting, whereas mice in the PF groups received a gavage of isocaloric maltose dextrin (10d+1b model). LDNPs were administered to mice in the last 3 days by daily gavage of 200 µL of LDNPs (50 µg protein content). AhR inhibitor, CH223191 (Sigma‐Aldrich, St. Louis, MO), was gavaged at 10 mmol/kg in the last 3 days. Control mice were gavaged with an equal volume of control vehicle (PBS). All mice were treated according to the protocols reviewed and approved by the Institutional Animal Care and Use Committee of the University of Louisville.
Statistics
Statistical analyses were performed using the statistical computer package, GraphPad Prism version 6, (GraphPad Software Inc., San Diego, CA). Results are expressed as means ± SEM. Statistical comparisons were made using two‐way analysis of variance with Tukey’s post hoc test or Student t test, where appropriate. Differences were considered to be significant at P < 0.05. Significance is noted as *P < 0.05, **P < 0.01, and ***P < 0.001 among groups.
Additional material and methods are found in the Supporting Information.
Results
LGG Produces Exosome‐Like Nanoparticles
We characterized the sizes and concentrations of nanoparticles produced by LGG. LDNPs were isolated by ultracentrifugation from bacteria culture (2 × 109 CFU/mL). The mean diameter of LDNPs was 75 ± 12.7 nm, and the protein concentration of LDNP preparation was 2.43 ± 0.45 μg/mL. We also analyzed the nanoparticles (NPs) from culture medium without bacterial inoculation. MRS medium (for LGG culture) contained NPs with a diameter of 115 ± 26.4 nm, which is larger than LDNPs isolated from LGG‐conditioned culture supernatant. In addition, the protein concentration of NPs from the medium (0.30 ± 0.028 μg/mL) were significantly lower compared with LDNPs (2.43 ± 0.45 µg/mL) (Fig. 1A,B). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis showed that there were no specific protein bands in MRS‐derived NPs. In contrast, LDNPs showed significant protein bands at sizes of 75 kDa and 40 kDa (Fig. 1C). Proteomics analysis of LDNPs identified 60 proteins consisting of those involved in metabolism, cell wall component/peptidoglycan remodeling, transporters, structure components of ribosomes, nucleic acid binding proteins, phage‐related proteins, and amidases (data not shown), of which, p75 and p40 abundantly existed in the LDNPs. A previous study showed that LGG produced the signature protein p75 and p40.( 11 ) The strong Coomassie blue staining of the proteins from LDNPs at 75 kDa and 40 kDa are therefore likely p75 and p40 proteins, respectively. Proteins in LDNPs appear stable in the solution with pH of 2.2, because incubation of the LDNPs in a pH 2.2 solution at 37°C for 2 hours did not cause degradation or aggregation of p75 and p40 proteins (Supporting Fig. S1). Furthermore, there was no positive immune‐staining of the CD63 band in LDNPs (Fig. 1D), indicating that the LDNPs do not contain eukaryotic ELNPs.
FIG. 1.
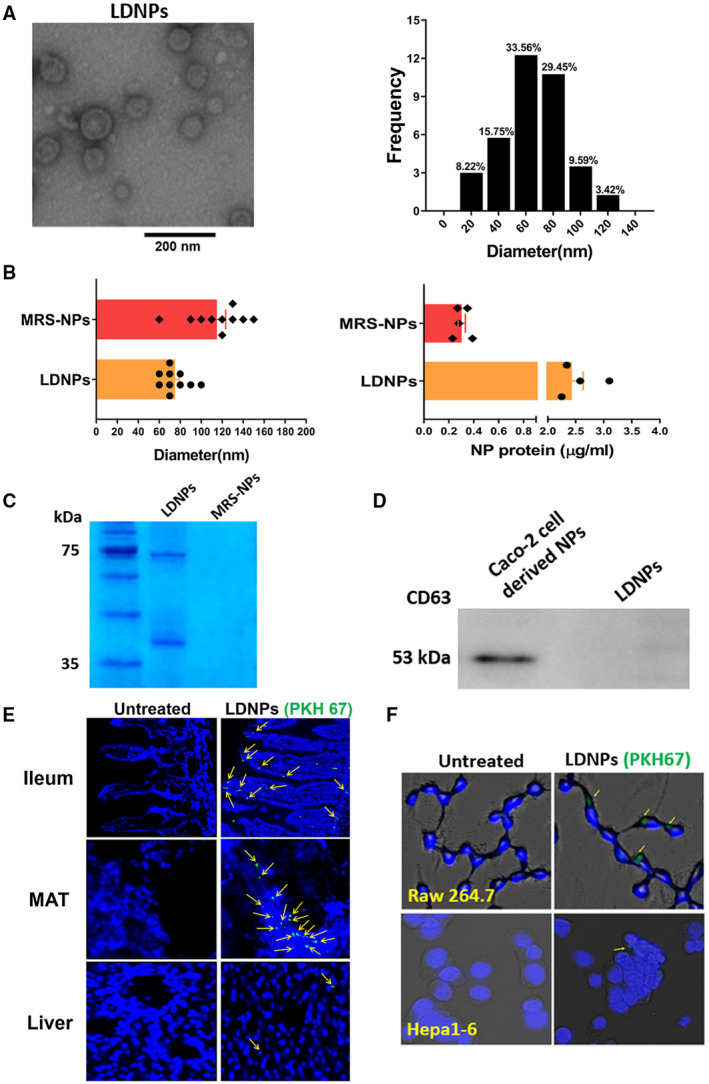
Characterization of LDNPs. (A) Representative transmission electron microscopy image of LDNPs and the frequency of observed NPs by diameter. (B) Size (left panel) and protein concentration (right panel) comparison between LDNPs and MRS‐derived NPs. (C) Coomassie blue staining of protein bands on sodium dodecyl sulfate–polyacrylamide gel electrophoresis. (D) Western blot for CD63 protein in LDNPs and Caco‐2‐derived NPs. (E) Uptake of (PKH67‐labeled) LDNPs in the ileum, mesenteric adipose tissue, and liver tissue. (F) Uptake of LDNPs in macrophages RAW264.7 and hepatocytes Hepa1‐6. Yellow arrows indicate PKH67‐positive staining of LDNPs. 4´,6‐diamidino‐2‐phenylindole (blue) was used for nucleic counter staining. Abbreviations: MAT, mesenteric adipose tissue; and MRS‐NP, MRS‐derived NP.
We next examined which mouse organs and/or cells took up LDNPs when administered orally. LDNPs were labeled with fluorescent dye PKH67 and gavaged to mice. Sliced tissues showed that LDNPs localized abundantly in the intestinal villi, lamina propria of ileum, and mesenteric adipose tissue, but rarely in the liver (Fig. 1E). We then incubated macrophage RAW264.7 cells and mouse hepatocyte Hepa1‐6 cells with PKH67‐labeled LDNPs. PKH67‐positive LDNPs were found in macrophages but not hepatocytes (Fig. 1F). These data suggest that LDNPs are taken up primarily by the intestine immune and epithelial cells when given orally.
LDNPs Inhibit LPS‐Induced Inflammation in Macrophage
To investigate the inflammatory response, RAW264.7 cells, peritoneal macrophages (PMs), and bone marrow–derived macrophages (BMDMs) were pretreated with LDNPs and then stimulated by LPS. LPS significantly up‐regulated the mRNA expression of tumor necrosis factor alpha (Tnf ). LDNP at 0.02 µg/mL slightly, and at 0.2 µg/mL significantly, suppressed Tnf mRNA expression in RAW264.7 cells. A higher concentration of LDNPs (2 μg/mL) had no further effect (Fig. 2A, left panel). We therefore used 0.2 μg/mL LDNPs in subsequent experiments. In addition to Tnf , LDNP pretreatment also suppressed LPS‐induced mRNA expression of pro‐inflammatory mediators: interleukin‐6 (Il‐6), interleukin‐1b (Il‐1 ), and monocyte chemoattractant protein 1 (Mcp‐1) (Fig. 2A, right panel). LPS‐induced increases in TNFα and IL‐1β proteins were also significantly suppressed by LDNP treatment (Fig. 2B). Previously, we showed that LGG culture supernatant suppressed LPS‐induced TNFα expression.( 23 ) To determine whether this effect is mediated by LDNPs, we treated RAW264.7 cells with LGGs, LDNPs, or LGGs(np‐d). LPS‐induced Tnf mRNA expression was markedly reduced by LDNPs and LGGs, but not by LGGs(np‐d) (Fig. 2C), suggesting that LDNPs mediated the inhibitory effect of LGGs on LPS‐induced TNFα expression. Furthermore, as shown in Fig. 2D, the LDNPs‐mediated reduction of the LPS‐induced mRNA expression of Tnf and Il‐1 was time‐dependent. Maximal inhibition was achieved at 24 hours after the addition of LDNPs for Tnf and at 48 hours for Il‐1 . We therefore used a 24‐hour incubation time in the subsequent experiments. Next, we extended our experiments in RAW264.7 cells to PMs and BMDMs. Consistent with the findings in RAW264.7 cells, LDNPs significantly reduced Tnf and Il‐1 mRNA expression in PM and BMDM (Fig. 2E,F). It is possible that nanoparticles from other bacteria could also interact with LPS directly, thus having an effect on the LPS‐induced inflammatory response. To confirm that the inhibition of inflammation in macrophages by LDNP was LGG‐specific, we performed same experiments using Bilophila wadsworthia–derived nanoparticles, and as expected, no such effects were observed (data not shown).
FIG. 2.
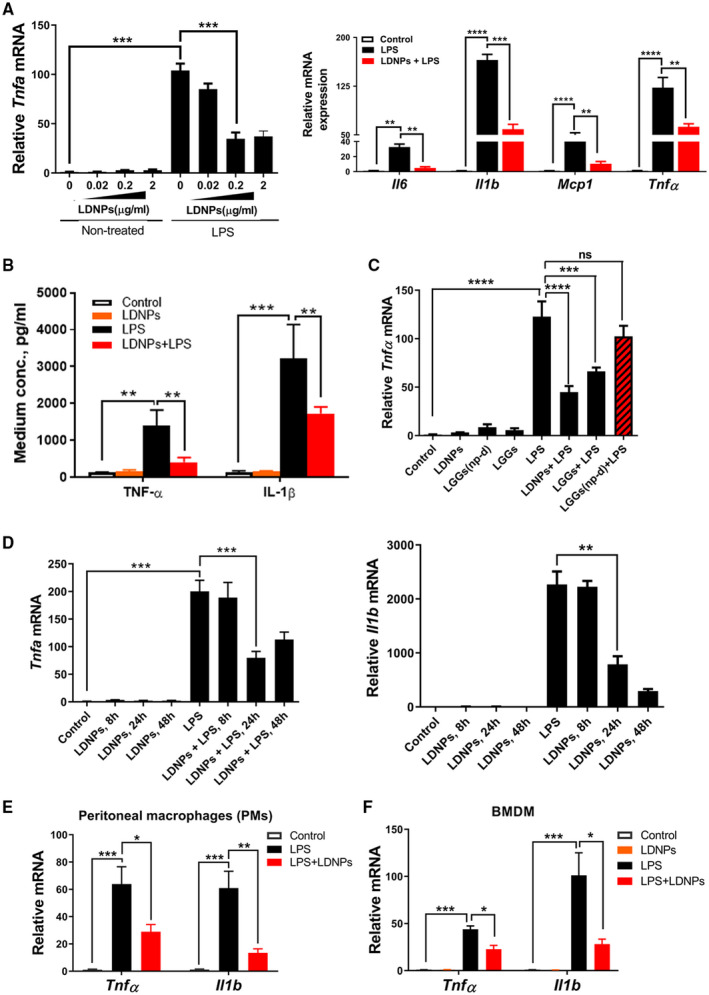
LDNPs inhibited LPS‐induced inflammation in macrophages. (A‐D) RAW264.7 cells. Dose‐dependent effects of LDNPs on relative Tnfα mRNA expression with or without LPS stimulation (A, left panel); relative mRNA expression of inflammatory mediators (A, right panel) and protein levels of TNF‐α and IL‐1β (B) after LDNPs and LPS treatment. (C) Effects of LDNPs depletion in LGGs on LPS‐induced Tnf mRNA expression. (D) Effects of LDNPs on Tnf and Il‐1 mRNA expression are time‐dependent. LDNPs inhibited LPS‐induced Tnf and Il‐1 mRNA expression in PMs (E) and BMDMs (F). Data shown represent the mean ± SEM of at least three independent experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviations: Mcp1, monocyte chemoattractant protein 1; ns, not significant.
LDNPs Increase AhR Reporter Activity
To investigate the potential mechanisms underlying this inhibitory effect of LDNPs on LPS induced inflammation in macrophages, we analyzed the metabolite cargo composition of LDNPs by liquid chromatography–mass spectrometry (LC‐MS)–based metabolomics technologies. We identified over 2,000 metabolites (data not shown). Interestingly, tryptophan‐derived AhR ligands exist abundantly in the LDNP cargo (Fig. 3A). Luciferase reporter analysis showed that both LDNPs and LGGs increased AhR activity. However, when the LDNPs were depleted from LGGs, the induction of AhR reporter activity was significantly decreased (Fig. 3B). Moreover, when LDNPs were depleted, the concentrations of indoleacrylic acid (IA) and indole‐3‐aldehyde (I3A) in LGGs were markedly reduced (Fig. 3C). Taken together, these data suggest that LNDPs are enriched in AhR ligands from bacterial tryptophan metabolism.
FIG. 3.
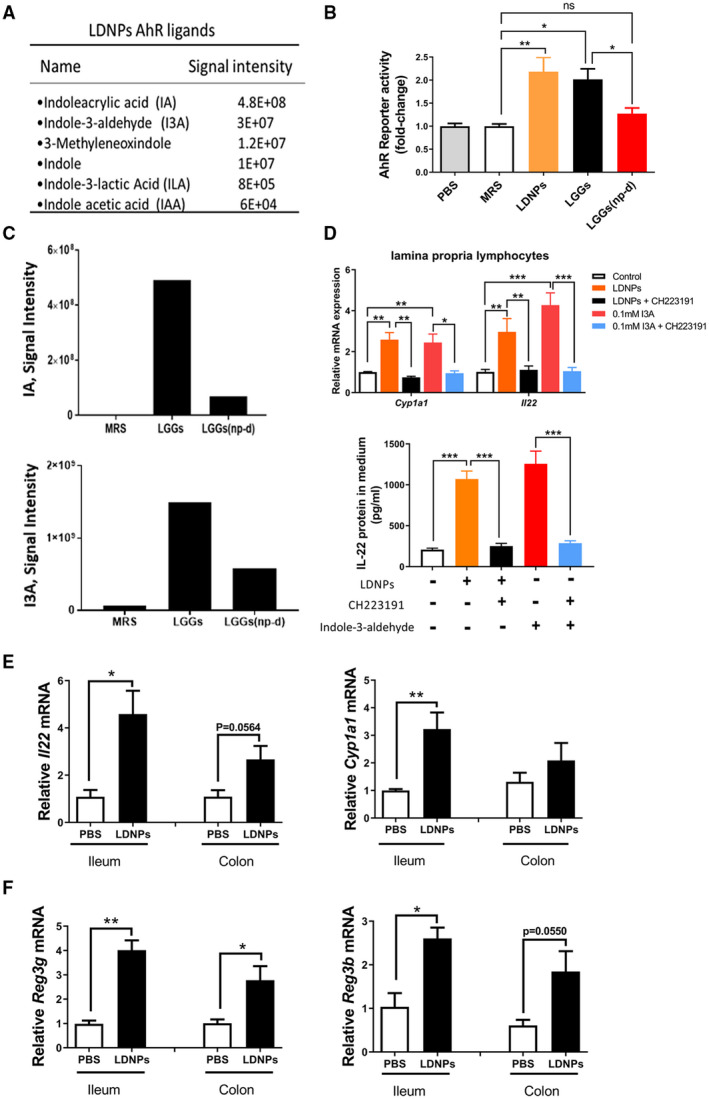
LDNPs increased AhR reporter activity and intestinal downstream signaling. (A) Signal intensity of representative enriched bacterial metabolites of tryptophan in LDNPs analyzed by LC‐MS. (B) AhR reporter activity of MRS, LDNPs, LGGs, and LGGs(np‐d). (C) Signal intensity of IA and I3A. The standard curve study by LC‐MS showed a linear representation in the signal range shown in the y‐axes. (D) Upper panel: The effects of the AhR inhibitor, CH229131, on LDNPs‐induced up‐regulation of Cyp1a1 and Il‐22 mRNA expression in lamina propria lymphocytes; lower panel: IL‐22 protein level in the culture medium of LPLs. AhR ligand I3A: positive control. (E) Relative mRNA expression of Il‐22 and Cyp2E1 in mouse ileum and colon. (F) Relative mRNA expression of Reg3γ and Reg3β in mouse ileum and colon. Data are presented as the mean ± SEM (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: IAA, indole acetic acid; LPL, lamina propria lymphocyte.
LDNPs Increase Intestinal AhR Downstream Signaling
AhR is a ligand‐activated nuclear receptor and is expressed in many cell types, including intestinal type 3 innate lymphoid (ILC‐3) cells. To examine whether LDNPs increase intestinal AhR signaling, we isolated ileum lamina propria lymphocytes (LPLs) from mice. Incubation of the LPLs with LDNPs produced a significant induction of mRNA expression of cytochrome P450 family 1 subfamily A member 1 (Cyp1a1) and Il‐22, which are two transcriptional targets of AhR.( 24 ) Importantly, the effects of LDNPs on the expression of Cyp1a1 and Il‐22 were completely inhibited by the AhR inhibitor, CH223191 (Fig. 3D, upper panel; I3A was used as a positive control ligand for the analysis). In addition, we found that the protein level of IL‐22 in the medium was increased about five‐fold by LDNPs, and the up‐regulation of IL‐22 was completely inhibited by CH223191 (Fig. 3D, lower panel). To determine whether oral administration of LDNPs increases intestinal AhR signaling, we gavaged mice with LDNPs. LDNPs administration markedly increased mRNA expression of Il‐22 and Cyp1a1 in the ileum, but not in the colon (Fig. 3E). It is known that intestinal IL‐22 mediates the expression of Reg3b and Reg3g, two major antimicrobial peptides expressed in intestinal Paneth cells. As shown in Fig. 3F, LDNP administration markedly increased ileum Reg3b and Reg3g mRNA expression. In the colon, Reg3g expression was significantly increased, whereas Reg3b was marginally increased.
LDNPs Increase Intestinal Tight Junction Protein Expression
Intestinal barrier function plays a crucial role in a variety of disease conditions. To investigate whether LDNPs modulate intestinal barrier function, we evaluated tight junction protein expression in intestinal epithelial cells (Caco‐2). LDNP treatment significantly increased three major tight junction proteins, ZO‐1, occludin and claudin‐1, in Caco‐2 cells (Fig. 4A). To determine whether this effect was AhR‐mediated, I3A, a ligand of AhR as a positive control, and the AhR inhibitor, CH223191, were added to Caco‐2 cell culture. I3A markedly increased Cyp1a1 mRNA expression and Cyp1a1 activity, which was blocked by CH223191, indicating an AhR regulation. Similar to I3A, LDNPs induced a significant upregulation of Cyp1a1 mRNA expression and increased Cyp1a1 activity, which was blocked by CH223191, indicating an AhR‐dependent effect of LDNPs (Fig. 4B). Importantly, both I3A and LDNPs increased the protein expression of tight junction proteins, which was inhibited by CH223191 (Fig. 4C). These data suggest that LDNPs have AhR agonist‐like activity and that the up‐regulation of intestinal epithelial cell tight junctions by LDNPs is AhR‐dependent.
FIG. 4.
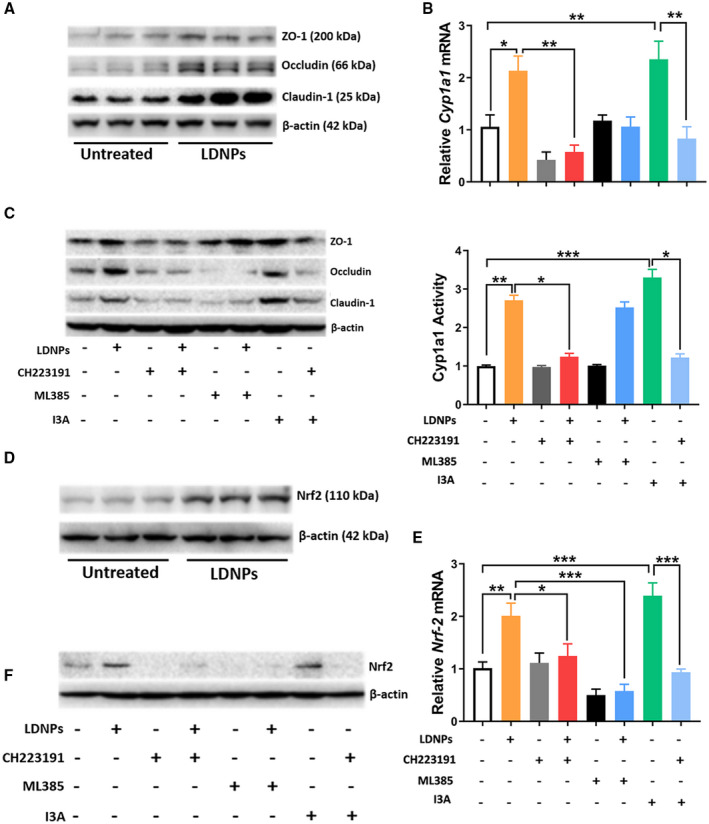
LDNPs increased intestinal tight junction expression in Caco‐2 cells. (A) Western blot for ZO‐1, occludin, and claudin‐1 protein in Caco‐2 cell lysates. (B) Relative Cyp1a1 mRNA expression (upper panel) and Cyp1a1 activity (lower panel) in Caco‐2 cells treated with LDNPs, CH223191 (AhR inhibitor), ML385 (Nrf2 inhibitor), and I3A. (C) Western blot for tight junction proteins in Caco‐2 cell lysates. (D) Western blot for Nrf2 protein in cell lysates of LDNPs‐treated Caco‐2 cells. (E) Relative mRNA expression of Nrf‐2 in Caco‐2 cells treated with LDNPs, CH223191, ML385, and I3A. I3A was used as an AhR ligand control. (F) Effects of CH223191 or ML385 on Nrf2 protein level in Caco‐2 cells treated with LDNPs. Data shown represent the mean ± SEM of at least three independent experiments performed in triplicate for cell culture studies. *P < 0.05, **P < 0.01, ***P < 0.001.
Previous studies demonstrated that tight junction proteins were regulated by cellular oxidative stress,( 25 ) and Nrf2 is important in anti‐oxidant regulation in the intestine.( 26 , 27 ) To determine the role of oxidative stress signaling in the up‐regulation of tight junction proteins, a Nrf2 inhibitor, ML385, was co‐administered with LDNPs. Indeed, LDNP treatment significantly increased Nrf2 expression at both mRNA and protein levels in Caco‐2 cells, and this up‐regulation was completely inhibited by ML385 (Fig. 4D,F). Interestingly, LDNPs‐induced Nrf2 expression was also blocked by CH223191 (Fig. 4E,F), suggesting that AhR is required for the up‐regulation of Nrf2 by LDNPs. Importantly, LDNP‐induced tight junction protein up‐regulation was inhibited by ML385 (Fig. 4C). It should be noted that Nrf2 inhibition blocked the LDNP‐induced Cyp1a1 mRNA expression, but not activity (Fig. 4B), for reason(s) not yet known.
LDNPs Prevent ALD
Increased serum endotoxin levels and hepatic bacterial translocation are manifestations of gut barrier dysfunction and hallmarks of ALD. To examine whether LDNP treatment could protect against alcohol‐induced liver injury, we fed mice with alcohol in a binge‐on‐chronic alcohol exposure model. Specifically, mice were fed the Lieber DeCarli diet containing 5% EtOH (vol/vol) for 10 days, and a bolus of EtOH (10d+1b) was gavaged to mice on the last day, 9 hours before sacrifice. LDNPs were orally gavaged at a dose of 50 μg/mouse once a day for the last 3 days (Fig. 5A). Alcohol feeding increased hepatic fat accumulation, as determined by hematoxylin and eosin (H&E) and Oil Red O staining, which was markedly decreased by LDNP treatment (Fig. 5B). The histological observations of hepatic steatosis were confirmed by hepatic triglyceride measurement (Fig. 5C). Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were increased by alcohol and decreased by LDNP treatment (Fig. 5D). LDNP treatment prevented the apoptotic cell death by alcohol, as demonstrated by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling assay (Fig. 5E). Alcohol feeding–induced hepatic inflammation was significantly reduced by LDNPs treatment, as shown by hepatic mRNA expression of pro‐inflammatory mediators, Tnfα and Il‐1 (Fig. 5F).
FIG. 5.
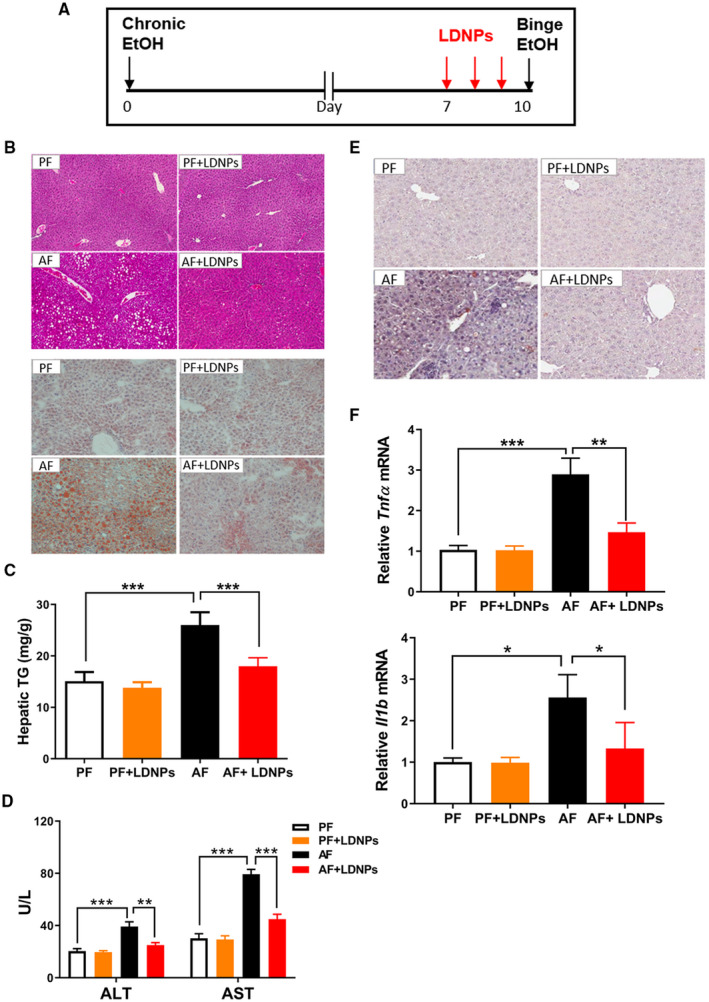
LDNPs reversed/prevented ALD. (A) Experimental design of animal treatment. (B) Representative microphotographs of H&E (upper panel) and Oil Red O (lower panel) stained mouse liver sections. (C) Hepatic triglyceride levels. (D) Serum ALT and AST levels. (E) Representative microphotographs of terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling stained mouse liver sections. (F) Hepatic Tnf and Il‐1 mRNA expression. Data are expressed as mean ± SEM (n = 5‐7 mice/group). *P < 0.05, **P < 0.01, ***P < 0.001.
LDNPs Increase Intestinal AhR Activity and Decrease Hepatic Bacterial Translocation
We next sought to delineate the intestinal signaling pathway linked to AhR activation in mice with experimental ALD. Alcohol feeding significantly decreased intestinal Cyp1a1 mRNA expression and Cyp1a1 activity (Fig. 6A,B), indicating an attenuated AhR activation by alcohol. LDNP treatment significantly increased both mRNA expression and activity of Cyp1a1 in mice under both PF and AF conditions (Fig. 6A,B). Ileum mRNA and serum protein levels of IL‐22 were significantly decreased by alcohol (Fig. 6C), which agrees with a previous study.( 28 ) Notably, LDNP treatment markedly increased ileum IL‐22 expression (Fig. 6C). Importantly, we did not observe any effects of either alcohol or LDNPs on Il‐22 and Cyp1a1 mRNA expression in hepatic tissues (Supporting Fig. S2), indicating that the effect of LDNPs on the AhR pathway is likely intestine‐specific.
FIG. 6.
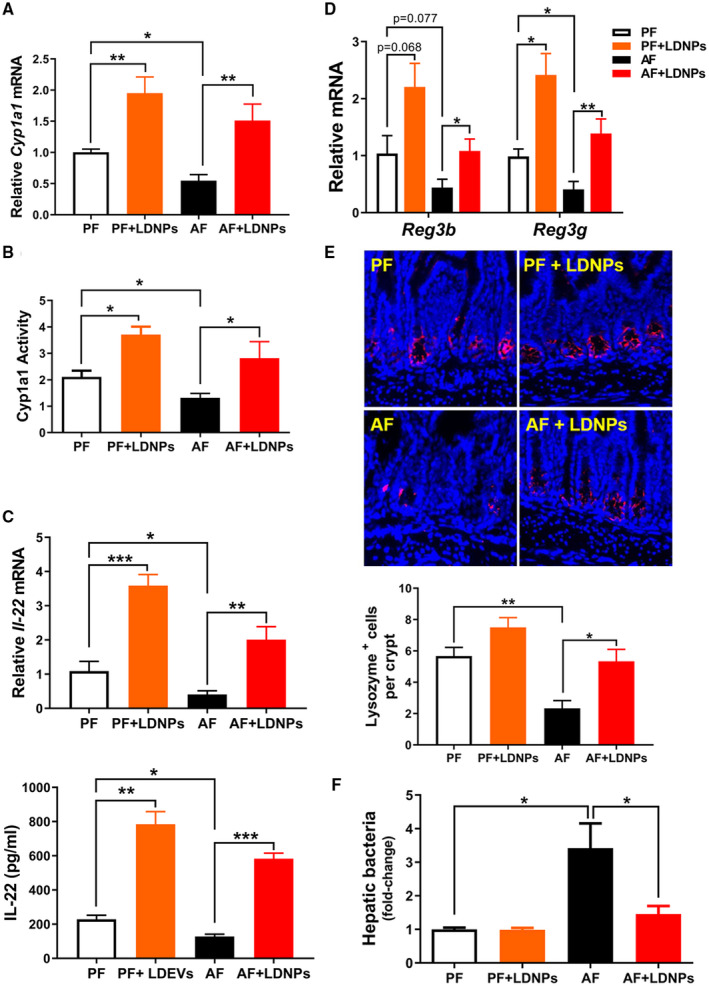
LDNPs increased intestinal AhR activity and decreased hepatic bacterial translocation. Relative ileum Cyp1a1 mRNA expression (A) and activity (B) in LDNPs‐treated PF‐fed or AF‐fed mice. (C) Relative ileum Il‐22 mRNA expression (upper panel) and serum IL‐22 protein level (lower panel). (D) Effects of LDNPs on ileum mRNA expression of Reg3b and Reg3g. (E) Upper panel: Representative microphotographs of immunofluorescence staining for lysozyme on mouse ileum tissue. Lower panel: Quantification of lysozyme‐positive stained Paneth cells (red). 4´,6‐diamidino‐2‐phenylindole (blue): nucleic counter stain. (F) Fold change of bacteria load in the livers of LDNPs‐treated PF‐fed or AF‐fed mice. Data are expressed as the mean ± SEM (n = 5‐7 mice/group). *P < 0.05, **P < 0.01, ***P < 0.001.
Reg3β and Reg3γ are produced in Paneth cells and play a critical role in maintaining bacterial homeostasis and inhibiting bacterial translocation. Ileum mRNA levels of Reg3b or Reg3g were slightly or significantly decreased by alcohol, respectively. LDNP treatment increased mRNA expression of Reg3b and Reg3g under both PF and AF conditions (Fig. 6D). Furthermore, lysozyme staining of ileum tissue showed decreased Paneth cell numbers, which were increased by LNDPs treatment (Fig. 6E). Elevation of Reg3β and Reg3γ should result in reduced bacterial translocation. As expected, we found that LDNP treatment markedly decreased AF‐increased hepatic bacteria load (Fig. 6F). Taken together, our results demonstrate that LDNPs increased intestinal AhR‐IL‐22‐Reg3 signaling pathways, leading to reduced bacterial translocation in the livers of mice fed alcohol.
LDNP Treatment Decreases Circulating Endotoxin Levels Through Nrf2 Activation
We further sought to investigate whether LDNP treatment affects the intestinal Nrf2 pathway, leading to up‐regulation of intestinal tight junction protein expression and reduction of endotoxemia. AF significantly decreased ileum nuclear Nrf2 protein levels (Fig. 7A) and slightly decreased Nrf2 mRNA expression (Fig. 7B). LDNP treatment increased intestinal Nrf2 expression in PF mice and prevented the reduction in AF mice (Fig. 7B). NAD(P)H : quinone acceptor oxidoreductase 1 (Nqo1) is an inducible enzyme that is regulated by the Nrf2 pathway and plays an important role in combating oxidative stress.( 29 ) AF significantly decreased intestinal Nqo1 mRNA expression. LDNP treatment markedly increased Nqo1 mRNA in PF mice and prevented the reduction in AF mice (Fig. 7C). We further examined cellular reactive oxygen species (ROS) status. Dihydroethidium staining of the ileum tissues showed that AF increased ROS, which was significantly reduced by LDNP treatment (Fig. 7D).
FIG. 7.
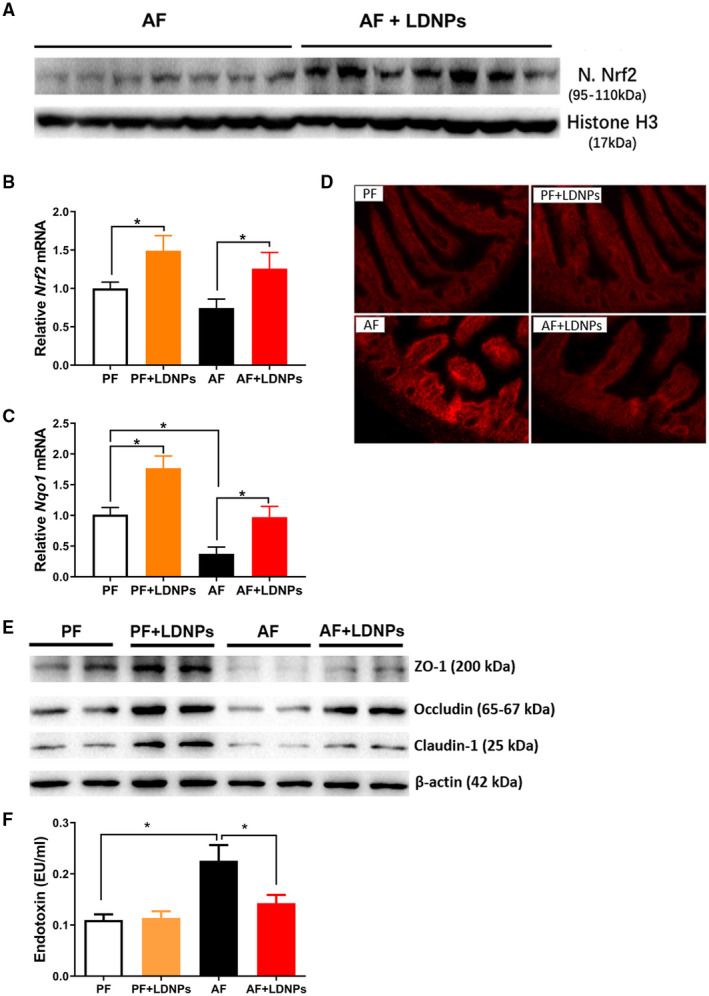
LDNP treatment decreased circulating endotoxin level through Nrf2 activation. (A) Western blot for nuclear Nrf2 protein in ileum tissues of LDNPs‐treated mice fed with alcohol. Histone H3 serves as a loading control. (B,C) Relative mRNA level of Nrf2 and Nqo1 in mouse ileum. (D) Dihydroethidium staining for the measurement of ROS in the ileum tissues. (E) Western blots for tight junction proteins in the ileum tissue. (F) Serum endotoxin levels. Data are expressed as the mean ± SEM (n = 5‐7 mice/group). *P < 0.05.
We then evaluated intestinal leakiness by examining the intestinal tight junction protein expression and serum endotoxin (LPS) levels. Agreeing with a previous study,( 23 ) AF decreased ileum protein expression of ZO‐1, occludin and claudin‐1. LDNP treatment prevented the down‐regulation of these proteins (Fig. 7E). AF increased serum LPS levels, which was inhibited by LDNP treatment (Fig. 7F). Taken together, our data demonstrate that LDNPs administration inhibits alcohol exposure–induced oxidative stress through the up‐regulation of Nrf2 expression, resulting in improved intestinal barrier function.
Effects of LDNPs Are Regulated by the AhR Signaling Pathway
We next sought to determine whether the beneficial effects of LDNPs in ALD are mediated by the intestinal AhR pathway by administrating the AhR inhibitor, CH223191, to AF mice. As shown in Fig. 8A, CH223191 significantly increased hepatic fat accumulation and liver triglyceride assay confirmed the histological findings. Importantly, the suppressive effect on hepatic fat by LDNP treatment was blunted when CH223191 was co‐administered. CH223191 slightly increased the serum levels of ALT and AST and blocked the effects of LDNPs when the mice were co‐fed with CH223191.
FIG. 8.
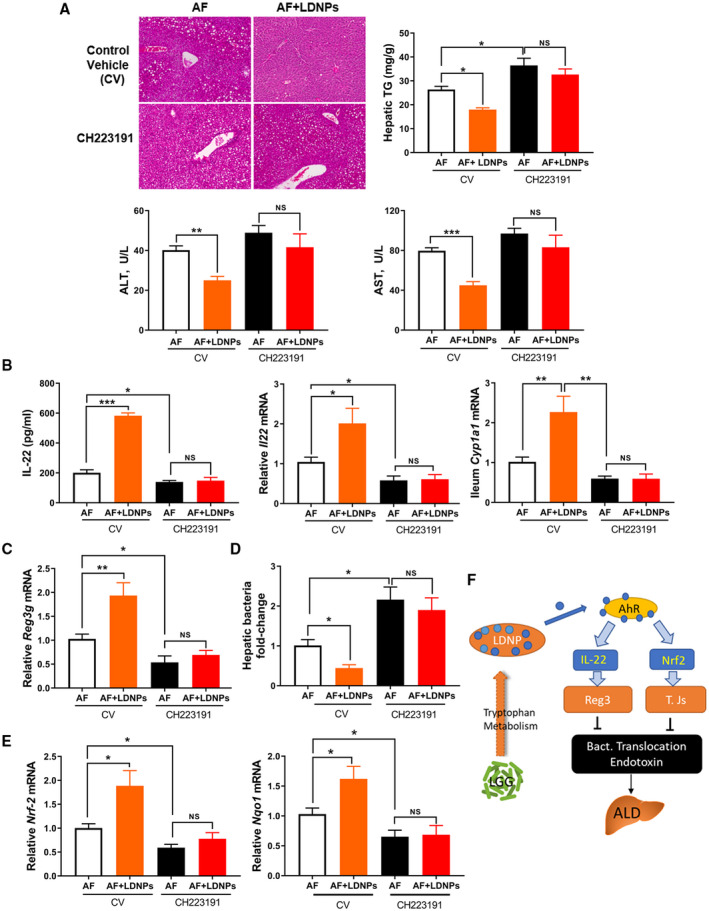
Effects of LDNPs in ALD are regulated by the AhR signaling pathway. (A) Upper panel: H&E staining of liver tissues (left) and hepatic triglyceride levels (right) of alcohol‐fed mice that co‐administered with LDNPs and control vehicle or AhR inhibitor CH223191. Lower panel: serum ALT and AST. (B) Serum IL‐22 protein levels (left panel); relative ileum mRNA expression of Il‐22 and Cyp1a1 (middle and right panel). (C) Ileum Reg3g mRNA expression. (D) Fold change of hepatic bacterial load. (E) Ileum mRNA expression of Nrf2 and Nqo1. Data are expressed as the mean ± SEM (n = 5‐7 mice/group). (F) Proposed model of LDNP action on intestinal AhR signaling in ALD. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: CV, control vehicle.
We then determined the effects of this AhR inhibitor on downstream signaling. Ileum Il‐22 mRNA and protein expression was reduced by CH223191 in AF mice. LNDPs treatment alone significantly increased IL‐22 expression in AF mice, and this effect was completely inhibited when CH223191 was co‐administered (Fig. 8B, left and middle panel). Cyp1a1 mRNA expression was increased by LDNP treatment and reduced by CH223191, and no change was found when LNDPs were gavaged with the inhibitor (Fig. 8B, right panel). Similar to the IL‐22 regulation, we found that Reg3g mRNA expression was increased by LDNPs and blocked by CH223191 (Fig. 8C). As a result, bacterial translocation, as determined by hepatic bacteria load, was significantly increased by the inhibitor, and the effect of LDNPs was blocked when the inhibitor was co‐administered (Fig. 8D).
Additionally, we found that ileum Nrf2 and Nqo1 mRNA expression was reduced by the AhR inhibitor in AF mice, and again, LDNPs were not able to increase the expression of these anti‐oxidant molecules when administered together with CH223191 (Fig. 8E). These data suggest that the preventive effects of LDNPs on the alcohol‐induced bacterial translocation into the liver is mediated by an AhR‐regulated signaling pathway.
Discussion
Intestinal bacterial microbiome homeostasis is maintained under physiological conditions. Interruption of this balance is often associated with disease development and/or progression. Administration of probiotics in preclinical studies and clinical practice has shown beneficial effects in restricting the overgrowth of pathogenic bacteria and control of the pathophysiological processes in the host. As we noted, probiotics have been used for the management of ALD to normalize the gut microbiota dysbiosis and attenuate liver injury.( 30 )
The action of bacteria on host cells is a complex process that is incompletely understood. Recent studies suggest that the ELNP is one of the important mediators of cell–cell interaction. These kinds of nanoparticles are produced by almost all organisms, including bacteria.( 31 ) While it is well known that Gram‐negative bacteria produce ELNPs, recent studies further demonstrate that Gram‐positive bacteria can also release ELNPs, despite having a thick cell wall.( 16 , 19 , 20 ) LGG is one of the best‐characterized Gram‐positive probiotics, and we hypothesized that LGG’s function was mediated through its ELNPs to regulate intestinal function in mice with experimental ALD.
ALD is characterized by gut barrier dysfunction, which leads to increased bacterial translocation and endotoxin release into circulation. Previously, we demonstrated that administration of LGG‐viable bacteria and LGGs prevented ALD in mice. In the present study, we showed that 3‐day administration of LDNPs was able to reverse/prevent alcohol‐induced hepatic fat accumulation, liver enzyme elevation, inflammation, and apoptotic cell death in mice using the binge‐on‐chronic alcohol (10d+1b) model.
We showed that LGG produces ELNPs (LDNPs) with an average size of 75 nm. The culture medium, MRS, also contains NPs, but with a bigger size and in a much smaller amount. LDNPs do not contain the eukaryotic EV marker (CD63) but have LGG‐derived proteins (p75 and p40). We showed that LDNPs are pH2.2‐resistant, which makes LDNPs suitable for oral administration. Indeed, it has been shown that ELNPs protect their cargo, including proteins, metabolites, and genetic material such as miRNA and mRNA from enzymatic degradation.( 17 ) We further demonstrated that the LDNPs localize primarily in the intestine tissue and immune cells when orally gavaged. This unique feature of intestinal targeting of LDNPs provides a strategy suitable for the treatment of diseases with intestinal dysfunction as an etiology.
The fact that macrophages take up LDNPs led us first to examine LDNP anti‐inflammatory activity. We showed that LDNPs reduced LPS‐stimulated expression of pro‐inflammatory mediators in RAW264.7 macrophages, mouse PMs, and BMDMs. Analyzing the cargo composition showed that LDNPs contain abundant IA, I3A, and indol‐3‐lactic acid, which are tryptophan bacterial metabolites and endogenous AhR ligands. Indeed, treatment with LDNPs significantly increased AhR reporter activity and downstream AhR signaling. Importantly, our results demonstrate that those AhR ligands are enriched in LDNPs, as depleting LDNPs decreased AhR ligand content in LGGs and blunted the AhR reporter activity of LGGs. However, the mechanisms underlying the LNDPs biogenesis in AhR ligand enrichment need future investigation.
AhR is a ligand‐activated nuclear receptor. Intestinal AhR activation in RAR‐related orphan receptor gamma–expressing ILC‐3 cells leads to increased IL‐22 expression and usually confers protection from bacterial infection and translocation by increasing Paneth cell–produced antimicrobial peptides, REG3γ and REG3β.( 28 , 32 ) Here, we demonstrated that LDNP treatment led to an increased AhR activity, as reflected by increased expression of Cyp1a1 and IL‐22 in LPLs. The role of AhR in the LDNP‐mediated IL‐22 increase was further demonstrated by using an AhR inhibitor, which completely blocked the effects of LDNPs on IL‐22 production. We found that LDNP treatment resulted in an increase of Reg3 expression in the ileum. It is therefore clear that oral LDNPs administration up‐regulates the intestinal AhR‐IL22‐Reg3 signaling pathway, which may provide protection against bacterial translocation under disease conditions. This notion was confirmed by our data in mice with experimental ALD. Three‐day LDNPs oral gavage reduced alcohol‐induced hepatic bacteria load, which was associated with an up‐regulation of Paneth cell numbers and the expression of Reg3 and IL‐22, and increased AhR activity. As a result, LDNP treatment reversed/prevented ALD in mice fed alcohol.
Intestinal tight junction–mediated barrier integrity plays a key role in alcohol‐induced endotoxin release. We found that LDNPs administration to intestinal epithelial Caco‐2 cells led to an increased expression of tight junction proteins. Strikingly, this increase was blocked by an AhR inhibitor, indicating that AhR mediates this effect of LDNPs. It is well‐known that intestinal tight junctions are damaged by disease‐initiated oxidative stress, and Nrf2 is an important mediator. Here, we found that LDNP treatment increased Nrf2 expression, which was blocked by the AhR inhibitor. These data suggest that the effect of LDNPs on intestinal tight junction expression is mediated by AhR through the up‐regulation of Nrf2. It should be noted that a Nrf2 inhibitor blocked LDNP‐induced Cyp1a1 mRNA expression. Whether Nrf2 regulates AhR activity in the intestine is currently unclear.
We further confirmed the role of the AhR‐mediated effects of LDNP in ALD mice using an AhR inhibitor. Co‐administration of CH223191 abolished the beneficial effects of LDNPs on alcohol‐induced fatty liver and liver injury, which were associated with the blockade of IL22‐Reg3‐mediated reduction of bacterial translocation and Nrf2‐mediated LPS release.
The current study has several limitations. Although an AhR inhibitor was used, more inhibitors with various characteristics and intestinal‐specific AhR knockout mouse model would further confirm the requirement of AhR in the action of LDNPs in ALD. The inhibitors could be delivered to the intestine by LDNPs, because we showed that LDNPs primarily target the intestine. We used the National Institute on Alcohol Abuse and Alcoholism binge‐on‐chronic model in the study, but our results need to be evaluated in other animal models of ALD. In addition, because of the short period of alcohol exposure that may not be sufficient to change microbiota, the effect of LDNPs on microbiota needs to be studied in other relevant models. Finally, although the AhR‐Nrf2‐mediated signaling pathway is crucial, there could be other mechanisms by which LDNPs protect against ALD. LDNPs could change the susceptibility of cells to alcohol‐induced alterations by delivering other cargo molecules, such as proteins and genetic material, into intestinal cells. LDNPs could interact with bacterial cells and affect gut microbiota homeostasis. LDNPs could also stimulate the production of neurochemicals, hormones, and growth factors that are known to promote liver regeneration. Whether any of these possibilities underlie the protective effects of LDNPs remains to be studied.
In conclusion, we demonstrate that LDNPs protect against alcohol exposure–induced fatty liver and injury through intestinal AhR‐Nrf2 signaling pathways to increase antimicrobial peptide (Reg3γ and Reg3β) and tight junction protein expression, leading to reduced bacterial translocation and endotoxemia (Fig. 8F). Our results suggest that the beneficial effects of probiotics and their supernatant are likely mediated by its exosome‐like NPs released from the probiotic bacteria. These findings may lead to a strategy for the treatment of ALD and other gut barrier dysfunction–associated diseases.
Supporting information
Supplementary Material
Acknowledgment
We thank Mrs. Marion McClain for the manuscript proofreading.
Supported by the U.S. Department of Veterans Affairs (1I01BX002996), National Institutes of Health (P20GM113226, P50AA024337, R01AA021434, R01AA023190, R01AA023681, R01AA028435, S10OD020106, U01AA026934, U01AA026936, and U01AA026980).
Potential conflict of interest: Nothing to report.
References
- 1. Sanders ME, Merenstein DJ, Reid G, Gibson GR, Rastall RA. Probiotics and prebiotics in intestinal health and disease: from biology to the clinic. Nat Rev Gastroenterol Hepatol 2019;16:605‐616. [DOI] [PubMed] [Google Scholar]
- 2. Brussow H. Probiotics and prebiotics in clinical tests: an update. F1000Res 2019;8:1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knudsen C, Neyrinck AM, Lanthier N, Delzenne NM. Microbiota and nonalcoholic fatty liver disease: promising prospects for clinical interventions? Curr Opin Clin Nutr Metab Care 2019;22:393‐400. [DOI] [PubMed] [Google Scholar]
- 4. Han SH, Suk KT, Kim DJ, Kim MY, Baik SK, Kim YD, et al. Effects of probiotics (cultured Lactobacillus subtilis/Streptococcus faecium) in the treatment of alcoholic hepatitis: randomized‐controlled multicenter study. Eur J Gastroenterol Hepatol 2015;27:1300‐1306. [DOI] [PubMed] [Google Scholar]
- 5. Sorbara MT, Pamer EG. Interbacterial mechanisms of colonization resistance and the strategies pathogens use to overcome them. Mucosal Immunol 2019;12:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clarke G, Sandhu KV, Griffin BT, Dinan TG, Cryan JF, Hyland NP. Gut reactions: breaking down xenobiotic‐microbiome interactions. Pharmacol Rev 2019;71:198‐224. [DOI] [PubMed] [Google Scholar]
- 7. Land MH, Rouster‐Stevens K, Woods CR, Cannon ML, Cnota J, Shetty AK. Lactobacillus sepsis associated with probiotic therapy. Pediatrics 2005;115:178‐181. [DOI] [PubMed] [Google Scholar]
- 8. Zhao H, Zhao C, Dong Y, Zhang M, Wang Y, Li F, et al. Inhibition of miR122a by Lactobacillus rhamnosus GG culture supernatant increases intestinal occludin expression and protects mice from alcoholic liver disease. Toxicol Lett 2015;234:194‐200. [DOI] [PubMed] [Google Scholar]
- 9. Wang Y, Liu Y, Sidhu A, Ma Z, McClain C, Feng W. Lactobacillus rhamnosus GG culture supernatant ameliorates acute alcohol‐induced intestinal permeability and liver injury. Am J Physiol Gastrointest Liver Physiol 2012;303:G32‐G41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu QI, Liu Y, Li F, Gu Z, Liu M, Shao T, et al. Probiotic culture supernatant improves metabolic function through FGF21‐adiponectin pathway in mice. J Nutr Biochem 2020;75:108256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan F, Cao H, Cover TL, Whitehead R, Washington MK, Polk DB. Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology 2007;132:562‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Segawa S, Fujiya M, Konishi H, Ueno N, Kobayashi N, Shigyo T, et al. Probiotic‐derived polyphosphate enhances the epithelial barrier function and maintains intestinal homeostasis through integrin‐p38 MAPK pathway. PLoS One 2011;6:e23278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu R, Fasano S, Madayiputhiya N, Morin NP, Nataro J, Fasano A. Isolation, identification, and characterization of small bioactive peptides from Lactobacillus GG conditional media that exert both anti‐Gram‐negative and Gram‐positive bactericidal activity. J Pediatr Gastroenterol Nutr 2009;49:23‐30. [DOI] [PubMed] [Google Scholar]
- 14. Segers ME, Lebeer S. Towards a better understanding of Lactobacillus rhamnosus GG–host interactions. Microb Cell Fact 2014;13(Suppl. 1):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi E‐B, Hong S‐W, Kim D‐K, Jeon SG, Kim K‐R, Cho S‐H, et al. Decreased diversity of nasal microbiota and their secreted extracellular vesicles in patients with chronic rhinosinusitis based on a metagenomic analysis. Allergy 2014;69:517‐526. [DOI] [PubMed] [Google Scholar]
- 16. Ñahui Palomino RA, Vanpouille C, Laghi L, Parolin C, Melikov K, Backlund P, et al. Extracellular vesicles from symbiotic vaginal lactobacilli inhibit HIV‐1 infection of human tissues. Nat Commun 2019;10:5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 2018;19:213‐228. [DOI] [PubMed] [Google Scholar]
- 18. Yáñez‐Mó M, Siljander P‐M, Andreu Z, Bedina Zavec A, Borràs FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 2015;4:27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown L, Wolf JM, Prados‐Rosales R, Casadevall A. Through the wall: extracellular vesicles in Gram‐positive bacteria, mycobacteria and fungi. Nat Rev Microbiol 2015;13:620‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee E‐Y, Choi D‐Y, Kim D‐K, Kim J‐W, Park JO, Kim S, et al. Gram‐positive bacteria produce membrane vesicles: proteomics‐based characterization of Staphylococcus aureus‐derived membrane vesicles. Proteomics 2009;9:5425‐5436. [DOI] [PubMed] [Google Scholar]
- 21. Behzadi E, Mahmoodzadeh Hosseini H, Imani Fooladi AA. The inhibitory impacts of Lactobacillus rhamnosus GG‐derived extracellular vesicles on the growth of hepatic cancer cells. Microb Pathog 2017;110:1‐6. [DOI] [PubMed] [Google Scholar]
- 22. Al‐Nedawi K, Mian MF, Hossain N, Karimi K, Mao Y‐K, Forsythe P, et al. Gut commensal microvesicles reproduce parent bacterial signals to host immune and enteric nervous systems. FASEB J 2015;29:684‐695. [DOI] [PubMed] [Google Scholar]
- 23. Wang Y, Liu Y, Kirpich I, Ma Z, Wang C, Zhang M, et al. Lactobacillus rhamnosus GG reduces hepatic TNFalpha production and inflammation in chronic alcohol‐induced liver injury. J Nutr Biochem 2013;24:1609‐1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng Y, Jin UH, Allred CD, Jayaraman A, Chapkin RS, Safe S. Aryl hydrocarbon receptor activity of tryptophan metabolites in young adult mouse colonocytes. Drug Metab Dispos 2015;43:1536‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rao R. Oxidative stress‐induced disruption of epithelial and endothelial tight junctions. Front Biosci 2008;13:7210‐7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun 2019;10:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wen Z, Liu W, Li X, Chen W, Liu Z, Wen J, et al. A protective role of the NRF2‐Keap1 pathway in maintaining intestinal barrier function. Oxid Med Cell Longev 2019;2019:1759149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hendrikx T, Duan YI, Wang Y, Oh J‐H, Alexander LM, Huang W, et al. Bacteria engineered to produce IL‐22 in intestine induce expression of REG3G to reduce ethanol‐induced liver disease in mice. Gut 2019;68:1504‐1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lau WL, Liu S‐M, Pahlevan S, Yuan J, Khazaeli M, Ni Z, et al. Role of Nrf2 dysfunction in uremia‐associated intestinal inflammation and epithelial barrier disruption. Dig Dis Sci 2015;60:1215‐1222. [DOI] [PubMed] [Google Scholar]
- 30. Kirpich IA, Solovieva NV, Leikhter SN, Shidakova NA, Lebedeva OV, Sidorov PI, et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol‐induced liver injury: a pilot study. Alcohol 2008;42:675‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun 2012;80:1948‐1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L, Fouts D, Stärkel P, Hartmann P, Chen P, Llorente C, et al. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa‐associated microbiota and preventing bacterial translocation. Cell Host Microbe 2016;19:227‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material