

Abstract
While therapy-induced autophagy is conventionally conceived to be cytoprotective in nature, previous studies have identified multiple functions of autophagy, including a nonprotective form and the existence of a switch between the different forms of autophagy. The current work demonstrates the existence of the autophagic switch in response to the antitumor drug, cisplatin, in non-small cell lung cancer cells that are either wild-type (p53wt) or functionally null in p53 (crp53), the latter generated using CRISPR/Cas9 technology. Pharmacological and genetic inhibition of autophagy identified nonprotective autophagy in p53 wt cells and cytoprotective autophagy in crp53 cells. Furthermore, differences in cisplatin sensitivity between the two cell lines prove to be largely a function of the nature of the autophagy. Specifically, autophagy inhibition in the crp53 cells converts the temporal loss in cell viability in response to cisplatin to essentially parallel that observed in the p53 wt cells. This enhanced sensitivity is due to cisplatin-induced apoptosis that occurs without necessitating the restoration of functional p53. In contrast, inhibition of the autophagy has no observable impact on the temporal response to cisplatin or the extent of cisplatin-induced apoptosis in the p53 wt cells, consistent with the definition of nonprotective autophagy. Taken together, our current studies provide evidence that nonprotective autophagy in p53wt non-small cell lung cancer cells can be “switched” to protective autophagy in isogenic crp53 cells, and that furthermore inhibition of cytoprotective autophagy is sufficient to restore cisplatin sensitivity in these cells, largely through the increased promotion of apoptosis, despite the absence of functional p53.
Keywords: autophagy, autophagic switch, therapy-induced autophagy, cisplatin, p53, drug resistance, drug sensitivity, lung cancer
Graphic Abstract.
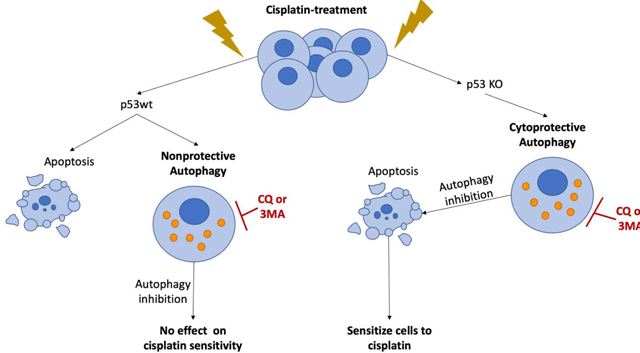
Cisplatin sensitivity in p53 wt and p53 deficient lung cancer cells appears to be a consequence of whether the drug-induced autophagy is functionally nonprotective or protective.
1. Introduction
Platinum-based drugs are widely used in the treatment of a number of malignancies, including but not limited to, lung, ovarian, head and neck, testicular, and cervical cancers (1). While cisplatin is a highly effective anti-cancer drug, resistance to treatment remains a frequent limitation to drug effectiveness (2). Several mechanisms have been proposed for cisplatin resistance, including drug efflux via the multi-drug resistance pump, DNA damage repair, and inhibition of apoptosis (3). In addition, it has been demonstrated that cells upregulate autophagy, a conventionally cytoprotective mechanism under conditions of cellular stress, in response to cisplatin treatment (4,5).
One paradigm that has currently achieved general acceptance is that macroautophagy (hereafter, autophagy) is an intrinsic mechanism of tumor cell resistance to therapy (6–9). However, it bears reiterating that although drug and radiation sensitivity can often be influenced by autophagy inhibition, this observation does not unequivocally prove that autophagy confers “resistance”, since clinical resistance is largely empirical and would be determined by lack of tumor responsiveness to concentrations of chemotherapeutic drugs or doses of radiation that can be tolerated by the patient (10,11). Furthermore, since virtually all experimental tumors undergo autophagy upon treatment with chemotherapy or radiation, it cannot be anticipated that experimental tumors will obligatorily demonstrate drug resistance as a consequence of autophagy induction.
Nevertheless, it is indisputable that chemosensitization and radiosensitization can occur when autophagy that takes the cytoprotective form is inhibited by pharmacological and/or genetic approaches (12). However, it is further critical to note that autophagy can have (at least) four different forms (13). In addition to the cytoprotective form, autophagy can be cytotoxic or cytostatic; of particular relevance to the current studies, autophagy can also be nonprotective. All of these forms of autophagy are identified by the functional outcomes of their inhibition by pharmacologic or genetic strategies. The nonprotective form of autophagy is particularly intriguing in that its inhibition does not alter drug or radiation sensitivity (12). Nonprotective autophagy was observed upon genetic autophagy inhibition in a seminal paper by Michaud et al and more recently in studies using A549 non-small cell lung cancer cells by Eng et al, although the “nonprotective” terminology has not yet been commonly accepted (14,15). Similar observations were reported in other studies involving non-small cell lung (NSCLC) cell lines (reviewed in (16)). Consequently, it cannot be predicted what function autophagy may take in response to a particular stress (i.e. chemotherapy or radiation) either in experimental tumor models or in cancer patients.
Studies both in our laboratory as well as that of other researchers have also identified the existence of an “autophagic switch”, where the function of autophagy can be altered in response to external or biological stresses (17,18). In our previous work, we demonstrated that ZR-75–1 breast cancer cells expressing wildtype p53 exhibited cytoprotective autophagy when treated with radiation alone but where the function of autophagy was converted to a cytotoxic form when radiation was administered in combination with 1,25-dihydroxy vitamin D3 (19). We have also reported on a switch between the cytoprotective and nonprotective forms of autophagy in response to ionizing radiation depending on whether cells expressed wild-type or mutated/null forms of p53 (20).
The current work was designed to extend our studies of the autophagic switch between cytoprotective and nonprotective autophagy to the cancer chemotherapeutic drug, cisplatin, utilizing an isogenic pair of H460 NSCLC cell lines that were either wild type or functionally null in p53. Cisplatin-induced DNA damage generally results in activation of p53 and, depending on the extent of damage, induces a variety of cellular responses including autophagy, apoptosis and senescence (21). Depending on the spatiotemporal localization of p53, it can both suppress and activate autophagy (22). Loss of p53 is also associated with increased tolerance to cisplatin-induced DNA adducts and replicative bypass contributing to cell survival and treatment resistance (3).
In the current studies, cisplatin induced autophagy to similar extents in H460 cells where p53 activity had been nullified by CRISP/Cas9 (H460 crp53) as in the parental p53 wt H460 (H460wt) NSCLC cells. However, this work confirmed the existence of what we have termed the “autophagic switch” in that cisplatin induced two different functional forms of autophagy, protective autophagy in the H460crp53 cells and nonprotective autophagy in the H460wt cells. This autophagic switch was associated with greater sensitivity to cisplatin in the p53 wt cells. Of particular relevance, with pharmacologic or genetic inhibition of the cytoprotective autophagy in the p53 crp cells, the temporal decline in cell viability in response to cisplatin became virtually identical to that in the p53 wt cells through increased susceptibility to the promotion of apoptosis. These observations suggest that sensitivity and resistance to cisplatin, and potentially other chemotherapeutics, can be influenced by the functional nature of drug-induced autophagy and furthermore cannot be predicted exclusively based on the p53 status of the tumor cell.
2. Materials and Methods
2.1. Antibodies and reagents:
Primary antibodies: SQSTM1/p62 (BD Biosciences, 610497), ATG5 (Cell Signaling Technology, 2630), LC3B (Cell Signaling Technology, 3868), TP53 (BD Biosciences, 554293), GAPDH (Cell Signaling Technology, 2118). Horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling, anti-mouse, 7076S; anti-rabbit, 7074S), and TRITC--conjugated secondary antibodies (Invitrogen, A21424) or FITC-conjugated secondary antibodies (Invitrogen, A11070) were used. 4’, 6-Diamidino-2-phenylindole (DAPI) was purchased from Thermo Fisher Scientific (P36931). Puromycin was purchased from Sigma (P7255), and cisplatin from the Cayman Chemical Company (13119).
2.2. Cell lines:
H460 NSCLC cells were obtained from ATCC (NCI-H460). p53 knockout H460 were generated by co-transfecting cells (3×106 in 10 cm dish) with 1 μg CRISPR-Cas9 plasmids targeting the p53 loci (Santa Cruz Biotechnologies; cat #sc-416469) and 1 μg a homology directed repair plasmid (Santa Cruz, cat. #sc-416469-HDR) expressing a puromycin selection marker. Cells were transfected using PolyJet reagent (Signagen) following the manufacturer’s guidelines. After 72 hr, cells were exposed to 2.5 μg/mL puromycin with daily media exchanges to replenish the selection agent. After all cells in control plates were killed (96 h), puromycin was removed and the cells allowed to recover and grow as individual colonies, which were then individually selected and examined for expression of p53. For the generation of shATG5 H460 cells, Mission shRNA bacterial stocks for ATG5 were purchased from Sigma Aldrich. Lentiviruses were produced in HEK 293TN cells co-transfected using EndoFectin™ Lenti Transfection Reagent (GeneCopoeia, 1001–01) with a packaging mixture of psPAX2 and pMD2.G constructs (Addgene). The media including viruses were used to infect H460 cells.
2.3. Cell culture and treatment:
H460wt cells and H460crp53 cells were maintained in DMEM media supplemented with 10% (v/v) fetal bovine serum (Thermo Scientific), 100 U/ml penicillin G sodium (Invitrogen), and 100 μg/ml streptomycin sulfate (Invitrogen). H460 shATG5 and H460 shControl cells were maintained with puromycin (1 μg/ml; Sigma) for selection. All cells were cultured at 37°C and under a humidified, 5% CO2 condition. For most experiments (unless stated otherwise), cells were seeded (day −2), treated with different dose of cisplatin (day −1), incubated for 24 h (day 0) followed by removal of cisplatin and incubation in fresh medium.
2.4. Growth inhibition and clonogenic survival assays:
Cell growth inhibition was measured by trypan blue exclusion at the indicated time points after treatment. Cells were stained with 0.4% trypan blue (Sigma, T10282), and counted using a hemocytometer. For the clonogenic assay, cells were plated in 6-well plates at the density of 200–300 per well, pre-treated with chloroquine (CQ, 0 or 10 μM) or 3-MA (0 or 1 mM) for 3 h, and then treated with 10 μM cisplatin. After an 11 day period of incubation following cisplatin exposure, colonies were washed with phosphate-buffered saline (PBS, Life Technologies), and then fixed with 100% methanol, stained with 0.1% crystal violet (Sigma), and counted.
2.5. Cell Viability Assay:
Cell viability was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay. Cells were plated at a density of 2,500 cells per well in a 96 well plate and treated with the indicated doses of cisplatin for 24 h. Cisplatin was removed and replaced with fresh medium. Cell viability was assessed 24 h post-treatment removal. Cells were washed with PBS and stained with MTT reagent (2 mg/ml: Sigma, M2128) in PBS at 37°C for 3 h, protected from light. The MTT solution was aspirated and dimethyl sulfoxide (DMSO) was added for 15 mins. Colorimetric change was analyzed at 490 nm with a spectrophotometer (ELx800UV, BioTeK).
2.6. Assessment of cell death:
Apoptosis was monitored by Annexin V-FITC and PI double staining (Annexin V-FITC apoptosis detection kit, BD Biosciences, 556547) according to the manufacturer’s instructions. At the indicated time points, cells were trypsinized, harvested and washed with cold PBS. Pellets were then resuspended in an annexinV/PI-containing buffer solution and incubated for 15 minutes before being analyzed by flow cytometry using BD FACSCanto II and BD FACSDiva software at the Flow Cytometry Core Facility at Virginia Commonwealth University.
2.7. Determination and quantification of acidic vesicles with acridine orange staining:
Cells were seeded in 6-well plates. After the indicated treatment, cells were stained with 1 μg/ml acridine orange at 37°C for 15 min and then washed with PBS, and observed under an inverted fluorescence microscope (Olympus, Tokyo, Japan). For quantification of autophagic vesicles (AVOs), cells were trypsinized, harvested and washed twice with PBS. Pellet fractions were resuspended in PBS and analyzed by BD FACSCanto II (excitation/emission: 488/610–620) and BD FACSDiva software at the Flow Cytometry Core Facility at Virginia Commonwealth University as described in our previous publications (23).
2.8. Western blot and immunofluorescence staining:
Western blotting was performed as described previously. Densitometry analysis on western blot was performed using Image J2 software. For immunofluorescence staining, cells were fixed with 100% methanol for 15 min, permeabilized by 0.1% Triton X-100 for 15 min, and then blocked with 5% bovine serum albumin (BSA). After incubation with primary antibody (1:100) overnight, cells were exposed to FITC- or TRITC-conjugated secondary antibody for 2 h at room temperature. Finally, nuclei were stained by DAPI. Immunofluorescence was detected by inverted fluorescence microscopy (Olympus, Tokyo, Japan).
2.9. DNA Damage assessment:
Cells were seeded in 6-well plates and incubated overnight. After exposure to the indicated treatment, cells were fixed with 70% ethanol for 15 minutes and then blocked with 5% bovine serum albumin (BSA). Cells were incubated with γH2AX antibody (1:1000; BD Pharmingen, 560445) for 2 h, and fluorescence was quantified using flow cytometry. For quantification of DNA damage, cells were trypsinized, harvested and washed with PBS. Pellet fractions were resuspended in PBS and analyzed by BD FACSCanto II and BD FACSDiva software. All experimental procedures were performed with cells protected from light.
2.10. Statistical analysis:
Data are shown as mean ± SD from at least three separate experiments. One-way ANOVA followed by Turkey’s post-hoc test was used to assess statistical differences between groups. Levels of significance were assessed by two-tailed t-test using Graphpad Prism 5.0 software; p-value < 0.05 was considered statistically significant.
3. Results
3.1. Cisplatin sensitivity in p53 wt and p53 knock-out H460 cells
In previous work, we reported that radiation-induced autophagy was nonprotective in the H460 non-small cell lung cancer cell line regardless of p53 status (24). In order to investigate whether autophagy is universally nonprotective in this cell line, the current studies utilized the antitumor drug, cisplatin, as the primary autophagy inducer. Furthermore, CRISPR/Cas9 technology was used to generate a set of isogenic NSCLC cell lines expressing wild-type p53 (H460wt) and a knockout of p53 (H460crp53) to discern the effects of p53 status on cisplatin sensitivity and autophagic function.
Figure 1A shows a Western blot confirming the knockdown of p53 in H460 non-small cell lung cancer cells (H460crp53) and, for comparison, wild-type p53 in the parental cell line. Figure 1B compares sensitivity to cisplatin in H460wt and H460crp53 cells based on clonogenic survival. The H460wt cells were clearly more sensitive to cisplatin than the cells that are null in p53, with apparent IC50 values of 1.2 ± 0.8 μM and 3.5 ± 1.6 μM, respectively. Sensitivity was also compared using a standard MTT assay. As with the clonogenic survival studies, the H460wt cells were more sensitive than the H460crp53 cells, with IC50 values of 1.0 ± 0.4 μM and 14.4 ± 6.4 μM respectively (Figure 1C). The differences in relative IC50 values likely reflect the different protocols used to generate the data.
Figure 1. Cisplatin sensitivity in H460wt cells and H460crp53 cells.
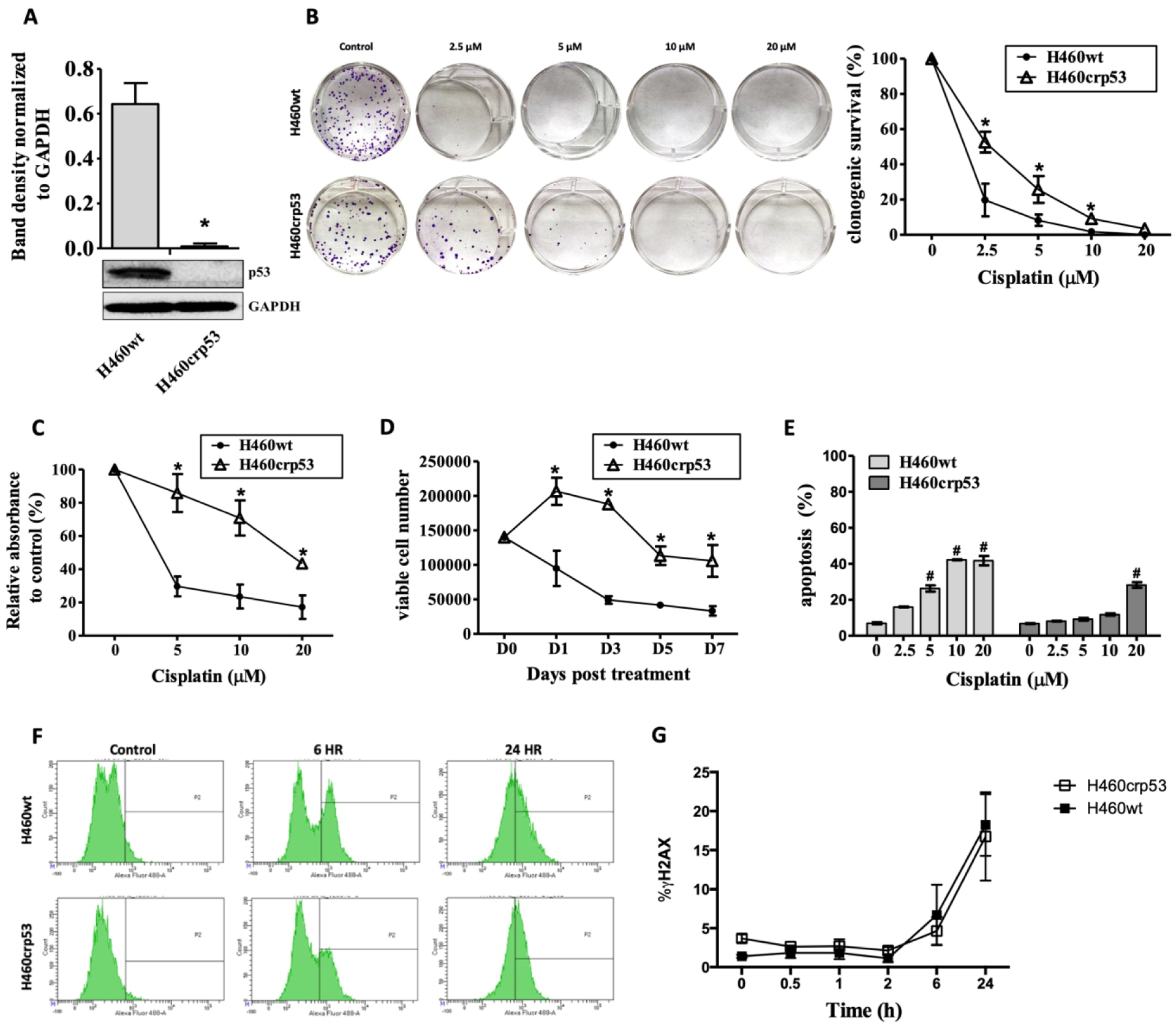
A. p53 knockout. Western blot indicative of p53 status of the H460 cells. The bar graph in each panel indicates the relative band intensity generated from densitometric scans of three independent experiments in arbitrary densitometric units. Cells were treated with cisplatin at the indicated doses for 24 h (day 0), after which cells were washed and incubated in fresh medium (n=3). B. Clonogenic survival assay. After exposure to cisplatin, cells were incubated in fresh medium for 11 days. Representative images of colony formation assay are presented and quantification of colonies are expressed as relative percentage compared to controls. C. MTT assay. After exposure to cisplatin, cells were incubated in fresh medium for 3 days. Results are expressed as relative percentage compared to controls (n=3). D. Cell viability. Cells were treated with cisplatin (10 μM) for 24 h, washed free of drug, incubated with fresh medium and stained with trypan blue (n=3). E. Apoptosis Dose Response. Cells were treated with cisplatin at the indicated doses for 24 h and apoptosis was assessed by Annexin V-FITC staining. Apoptosis was measured 24 h after cisplatin removal (n=3). F-G. DNA Damage. Temporal assessment of DNA damage in response to cisplatin. Extent of DNA damage was measured utilizing flow cytometry to quantify γH2AX staining in cells treated with cisplatin (10 μM). (F) Representative images of flow cytometry data and (G) quantification of fluorescence was graphed (n=3). Unless stated, otherwise data were from three independent experiments, *p < 0.05, cisplatin treated group vs untreated control group in each cell line.
Although the clonogenic survival assay is generally considered to be the gold standard for assessing drug and radiation sensitivity, it does not actually distinguish between “permanent” growth arrest and cell death. Accordingly, we evaluated the temporal response to 10 μM cisplatin (a clinically relevant concentration) in the two cell lines by monitoring viable cell number using trypan blue exclusion assay over a period of 7 days. Figure 1D indicates that the temporal response pattern largely reflects the outcome of the clonogenic survival assays in that the H460wt demonstrate a rapid growth decline, indicative of cell death, while the H460crp53 cells initially continue to proliferate and only begin to succumb to the drug effects after 3 days.
It is generally thought that lack of p53 function attenuates apoptosis (25,26). An evaluation of the extent of apoptosis by annexin V/PI staining demonstrated that H460wt cells exposed to different doses of cisplatin underwent a much more pronounced degree of apoptosis/necrosis than H460crp53 cells (Figure 1E). This did not appear to be a consequence of differential DNA damage since cisplatin promoted equivalent DNA damage in the H460p53 wt and H460crp53 cells, based on γH2AX staining performed over a 24 h time period (Figure 1F–G).
3.2. Cisplatin-induced autophagy in p53 wt and p53 knockout cells
Cytoprotective autophagy is generally considered to be a mechanism to ameliorate or evade apoptosis; hence, we assessed the capacity of cisplatin to promote autophagy in both cell lines. Figure 2A presents images of acridine orange staining of autophagic vacuoles, a rough but generally accurate indication of the extent of autophagy. Assessment of acidic vesicle formation by flow cytometry indicated that the extent of autophagy induced by cisplatin was similar in the two cell lines (Figure 2B). To further compare the extent of autophagy and whether cisplatin-induced autophagy is going to completion, degradation of p62/SQSTM1 was evaluated by western blotting. Figure 2C (and quantification of the band densities in Figure 2D) indicates that autophagic flux is clearly occurring in both cell lines. Moreover, both H460wt and H460crp53 cells demonstrate co-localization of LC3 and the lysosomal marker, LAMP-2, when exposed to cisplatin indicative of autophagic flux (Figure 2E). These studies indicate that autophagy appears to be induced to a similar extent in cisplatin-treated H460 cell, regardless of the status of p53, although the process is slightly more rapid in the H460wtp53 cells.
Figure 2. Cisplatin induces autophagy in H460wt cells and H460crp53 cells.

Cells were treated with cisplatin at the indicated doses for 24 h (day 0), after which cells were washed and incubated in fresh medium. A. Acridine orange staining was performed after exposure to 10 μM cisplatin. Cells were treated with cisplatin for 24 h (day 0) and stained 48 h post-drug removal (day 3, n=3). B. Quantification of acridine orange staining. Autophagy induction was quantified by flow cytometry in response to increasing concentration of cisplatin 2 days after drug exposure (n=3). C. Western blotting. Levels of p62 were determined by western blotting at the indicated times after 10 μM cisplatin exposure for 24 h (D0). Lysates were collected on indicated days. One of three representative experiments is shown (n=3). D. Western blot Densitometry. The bar graph in each panel indicates the relative band intensity generated from densitometric scans of two independent experiments in arbitrary densitometric units (n=3). E. Co-localization of LC3 and LAMP. Fluorescence microscopy showing LC3 and LAMP2 co-localization in response to 10 μM cisplatin exposure 2 days after cisplatin removal. (20X objective, n=2) Unless stated, otherwise data were from three independent experiments, *p < 0.05, cisplatin treated group vs untreated control group in each cell line.
3.2. Evidence for cytoprotective autophagy in the p53 knock-out cells and nonprotective autophagy in the p53 wild type H460 cells
We then evaluated the nature of the autophagy based on sensitization or lack of sensitization when autophagy was inhibited using the pharmacological autophagy inhibitors, chloroquine (CQ) or 3-methyl adenine (3-MA). Both H460 cell lines were pre-treated with CQ (10 μM) or 3-MA (1 mM) for 3 h followed by exposure to 10 μM cisplatin for 24 h. Autophagy was measured after 2 days of incubation with fresh medium. The increase in LC3-II puncta formation in the presence of CQ (due to inhibition of autolysosome formation and accumulation of autophagosomes) and decrease in LC3-II puncta with 3-MA (due to interference with autophagosome formation) indicated that CQ and 3-MA inhibited cisplatin-induced autophagy (Figure 3A). The inhibition of autophagy in cells exposed to CQ was further confirmed by the increased number and density of autophagic vacuoles shown in Figure 3B (i.e. autophagy is interrupted before the autophagosomes can fuse with the autolysosomes). Interference with p62/SQSTM1 degradation further confirmed that CQ and 3-MA inhibited cisplatin-induced autophagy (Figure 3C). Differential p62 degradation between the two cell lines is reflective and consistent with the differential rate of autophagic flux shown in Figure 2C, where p62 degradation occurred earlier in H460wt cells compared to H460crp53 cells. Temporal response studies were then performed to determine the impact of autophagy inhibition on sensitivity to cisplatin. Figures 3E and 3F–I shows that CQ and 3-MA increased cell death and apoptosis in response to cisplatin in the H460crp53 cells but failed to influence cisplatin-induced cell death and apoptosis in the H460wt cells (Figure 3D and 3F–I), indicating that autophagy was cytoprotective in function in the H460crp53 cells but nonprotective in the H460wt cells.
Figure 3. Influence of pharmacologic autophagy inhibitors on cisplatin sensitivity in H460wt cells and H460crp53 cells.
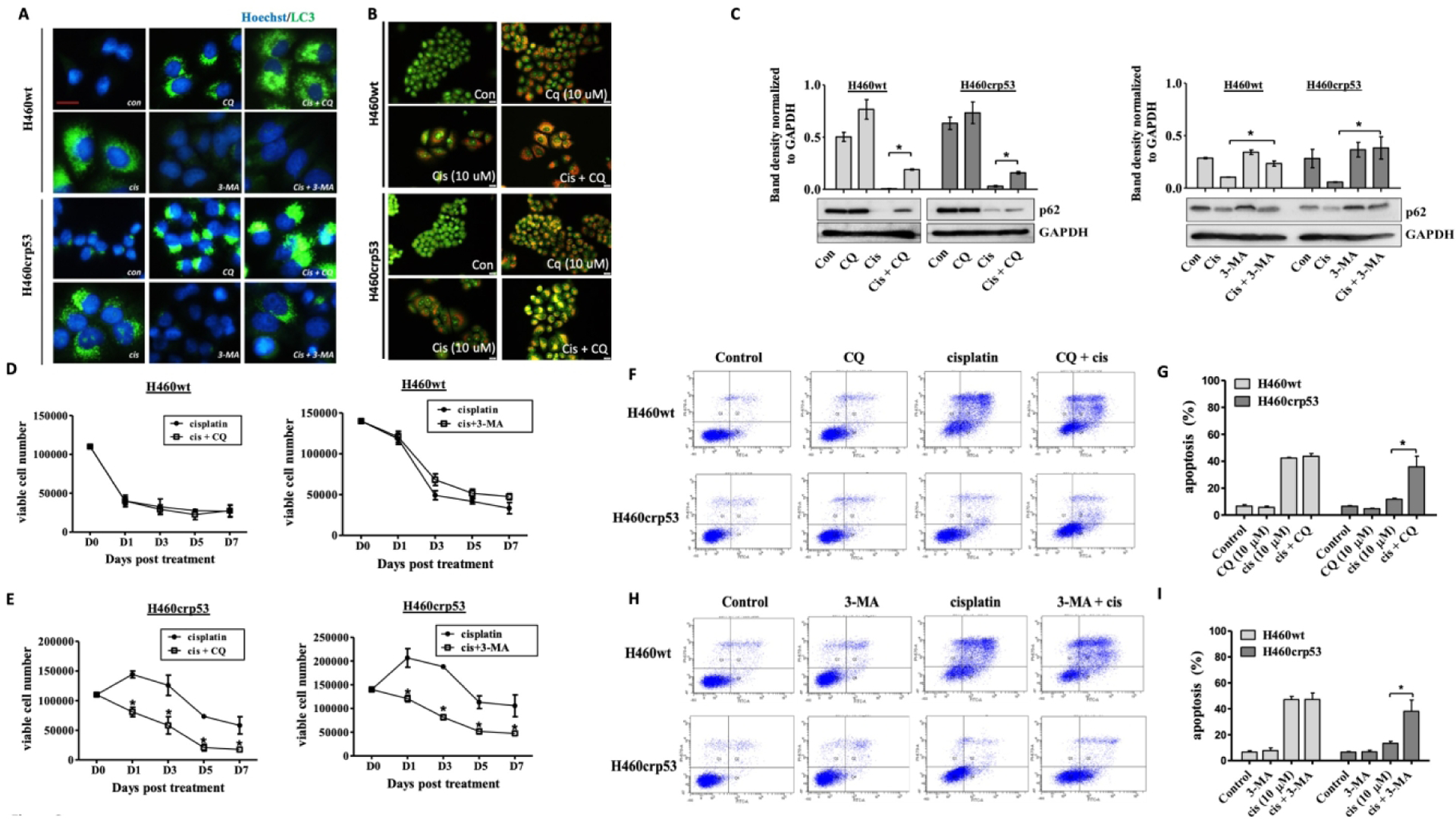
A. Inhibition of autophagy by CQ and 3-MA. Fluorescence microscopy showing increased LC3 puncta following CQ (10 μM) co-treatment with 10 μM cisplatin, and decreased LC3 puncta following 3-MA (1 mM) co-treatment with 10 μM cisplatin. Cells were pretreated with CQ (10 μM) and 3-MA (1 mM) followed by an additional 24 h with cisplatin. Images were taken 48 h after cisplatin removal. Nuclei were stained with Hoechst 33342 and vacuoles with LC3 antibody (20x objective, n=2). B. Inhibition of autophagy by CQ. Cells were pretreated with CQ (10 μM) followed by an additional 24 h with cisplatin. Acridine orange staining and imaging was performed taken 48 h after 10 μM cisplatin removal (20x objective, n=3). C. Inhibition of autophagy by CQ and 3-MA. Western blot showing autophagy blockade by CQ (10 μM) and 3-MA (1 mM) based on levels of p62/SQSTM1 (n=3). The bar graph in each panel indicates the relative band intensity generated from densitometric scans of two independent experiments in arbitrary densitometric units. D and E. Influence of autophagy inhibition on cisplatin sensitivity. Viability of H460wt cells and H460crp53 cells was monitored based on trypan blue exclusion at indicated days following 10 μM cisplatin exposure in combination with CQ (10 μM) or 3-MA (1 mM) (n=3). F-I. Influence of autophagy inhibition on cisplatin induced apoptosis. Annexin V-PI staining showing influence of CQ (10 μM) and 3-MA (1 mM) on apoptosis of H460 cells exposed to cisplatin (10 μM). Cells were pretreated with CQ or 3-MA for 3 h followed by co-treatment with cisplatin for 24 h. Apoptosis was measured 24 h after cisplatin removal. (E and G) Representative images of flow cytometry data and (F and H) quantification of fluorescence was graphed (n=3). Unless stated otherwise, data were from three independent experiments, *p<0.05, cisplatin versus cisplatin + CQ (3-MA).
To confirm the findings generated using pharmacological inhibition, short hairpin RNA (shRNA) was used to knock down Atg5, an autophagy regulatory gene, in both H460 cells lines. Figure 4A verifies the status of Atg5 by Western blotting in the two cell lines. The decrease in LC3 puncta (Figure 4B) indicates that shAtg5 effectively inhibited autophagy in both H460 cell lines. Inhibition of autophagy was confirmed by interference with cisplatin-induced degradation of p62/SQSTM1 (Figure 4C). Here, genetic interfere with autophagy yielded a similar outcome to that observed with the pharmacological autophagy inhibitors; specifically, autophagy inhibition failed to alter growth inhibition and apoptosis in response to cisplatin in H460wt cells (Figures 4D and 4E–F), but increased cell growth inhibition and apoptosis in the H460crp53 cells (Figures 4D and 4E–F).
Figure 4. Influence of genetic autophagy inhibition on cisplatin sensitivity in H460wt cells and H460crp53 cells.
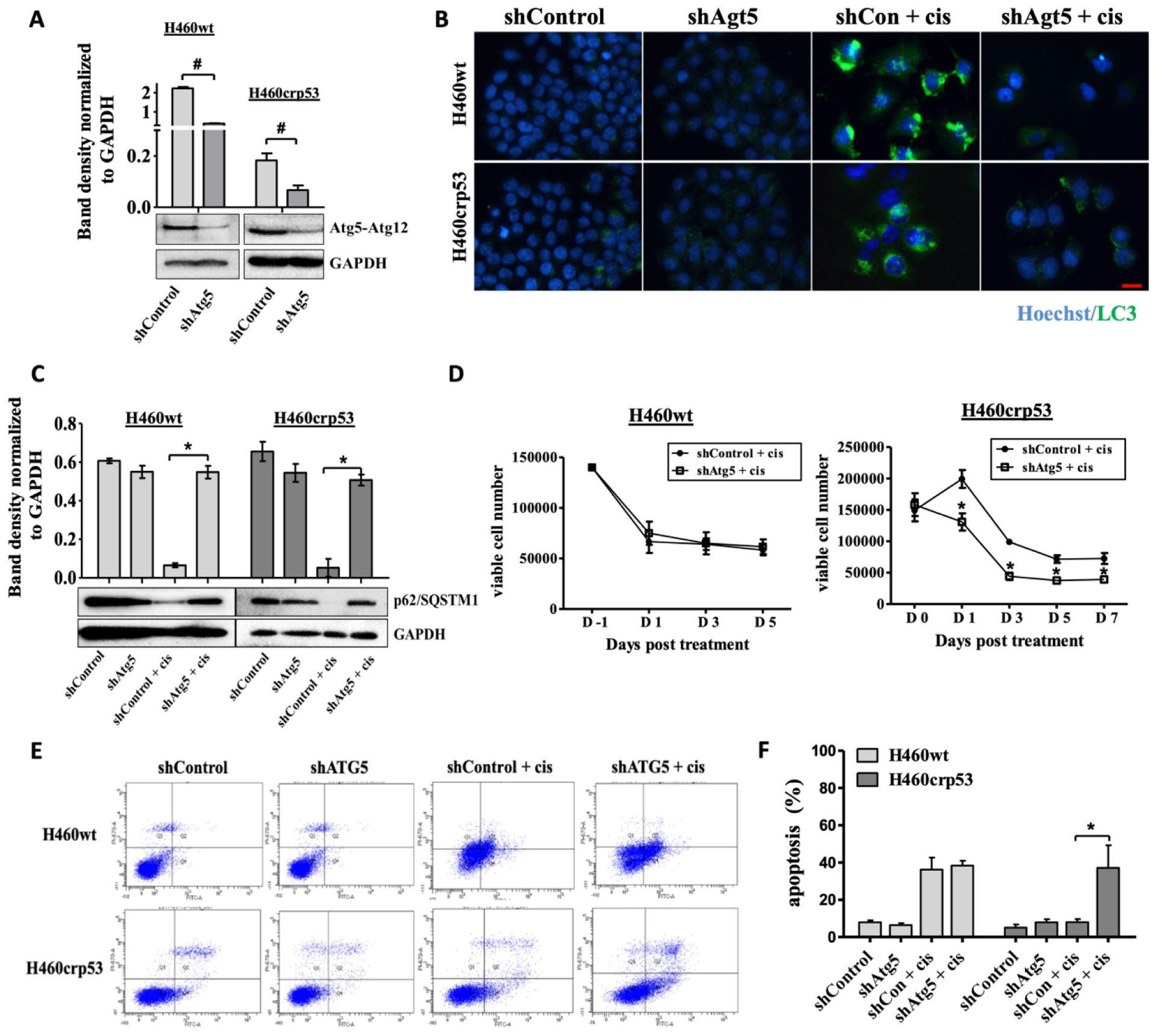
A. Western blot showing the silencing of Atg5 in H460wt cells and H460crp53 cells. B and C. Autophagy inhibition by Atg5 silencing. The bar graph in each panel indicates the relative band intensity generated from densitometric scans of three independent experiments in arbitrary densitometric units (n=3). B. Fluorescence microscopy showing decreased LC3 puncta following treatment with 10 μM cisplatin in shAtg5 H460 cells. Images were taken 48 h after cisplatin removal. Nuclei were stained with Hoechst 33324 (20x objective, n=2). C. Western blot showing autophagy blockade by Atg5 knockdown based on levels of p62/SQSTM1. Proteins were collected 48 h after cisplatin removal (n=2). The bar graph in each panel indicates the relative band intensity generated from densitometric scans of two independent experiments in arbitrary densitometric units. D. Influence of autophagy inhibition on cisplatin sensitivity. Viability of H460wt cells and H460crp53 cells was monitored based on trypan blue exclusion at indicated days following cisplatin exposure in shATG5 in H460wt cells and H460crp53 cells (n=3). E-F. Influence of autophagy inhibition on cisplatin induced apoptosis. Annexin V-PI staining showing apoptosis in H460 cells exposed to cisplatin (10 μM) with and without ATG5 silencing. Apoptosis was measured 24 h after cisplatin removal. (E) Representative images of flow cytometry data and (F) quantification of fluorescence was graphed (n=3). Unless stated otherwise, data were from three independent experiments, *p < 0.05, shControl + cisplatin versus shAtg5 + cisplatin, #p < 0.05, shControl versus shAtg5.
Taken together, these data demonstrate that in the H460wt cells the autophagy is nonprotective since there is no sensitization with autophagy inhibition and no increase in apoptosis. In contrast, autophagy “switches” to the cytoprotective form/function when p53 is knocked out, as autophagy inhibition increases sensitivity to cisplatin and results in enhanced apoptosis in the H460crp53 cells.
3.5. Inhibition of cytoprotective autophagy shifts the temporal response to cisplatin in the crp53 cells
To confirm the contributions of cytoprotective autophagy to cisplatin sensitivity, we compared the temporal responses shown in Fig. 3D and 3E with and without pharmacological autophagy inhibition by replotting these time courses in Figures 5A and 5B. The blockade of cytoprotective autophagy in H460crp53 cells exposed to cisplatin resulted in a temporal decline in cell viability that was essentially identical to that in the cisplatin-treated H460 p53wt cells. In agreement with these observations, Figure 5C demonstrates a similar relationship when cell viability data from Fig. 4D is plotted together to show overlap of the decline in cell viability in response to cisplatin when autophagy has been genetically inhibited in H460crp53 cells when compared to H460wt shcontrol cells exposed to cisplatin.
Figure 5. Inhibition of cytoprotective autophagy shifts the temporal response to cisplatin in the H460crp53 cells.

A-C. Influence of autophagy inhibition on cisplatin sensitivity in p53 wt and p53 KO cells. Cell viability data from figure 3D and 3E were overlaid to compare the functional role of autophagy in H460crp53 and H460wt cells using the pharmacological inhibitors, CQ (A) and 3MA (B). C. Cell viability data from figure 4D were overlaid to compare the functional role of autophagy on cisplatin sensitivity via genetic silencing of ATG5. p < 0.05: wtp53 shControl + cis vs crp53 shATG5 +cis. ns: wtp53 + cis vs crp53 + cis +CQ (or + 3-MA or shATG5)
4. Discussion
4.1. Protective and Nonprotective Autophagy and Drug Sensitivity/Resistance
This work addresses three separate but closely related questions in the autophagy field relating to the tumor cell response to chemotherapeutic drugs. One is the oft-stated paradigm suggesting that autophagy is a mechanism of therapeutic resistance (27–29). This premise is based on the frequently-reproduced observation in which inhibition of drug or radiation-induced autophagy can enhance sensitivity to the therapeutic challenge (30). These observations are indisputable as extensive evidence is provided for the cytoprotective function of therapy-induced autophagy. However, it is unclear whether these observations are proof that autophagy can actually be considered an intrinsic mechanism of resistance (31). The availability of isogenic cells that are either wild-type or null in p53 allowed us to directly address the question of whether tumor cells that undergo protective autophagy are intrinsically less sensitive (in this case to the chemotherapeutic drug, cisplatin) than the same tumor cells that fail to undergo protective autophagy?
Assessment of clonogenic survival and cell viability indicated that the p53 wt H460 cells, where autophagy was nonprotective, were more sensitive to cisplatin than the H460crp53 cells. This differential sensitivity to cisplatin was largely attributable to the relative extent of apoptosis and was not a consequence of differences in the extent of DNA damage induced by cisplatin in the two cell lines. Rather, differences in apoptosis seems to be related to the functional form of autophagy induced in response to cisplatin.
Autophagy is one mechanism thought to be induced by cancer cells to evade apoptosis (32). Cisplatin resulted in autophagy induction to a similar extent in both the H460wt and H460crp53 cells. Autophagy inhibition (by pharmacological and genetic interventions) increased cisplatin-induced cell death and apoptosis in H460crp53 cells (i.e. evidence of cytoprotective autophagy) but did not alter either outcome in the p53 wt H460 cells (i.e. evidence of nonprotective autophagy). As a result, the temporal decline in cell viability in the H460crp53 cells under conditions of autophagy inhibition essentially paralleled that observed in p53 wt H460 cells, suggesting cytoprotective autophagy was contributing to the differential sensitivity and apoptosis observed between the two cell lines when exposed to cisplatin. Consequently, these findings support the premise that cytoprotective autophagy can confer a relative degree of resistance to chemotherapy.
Whether cytoprotective autophagy is conserved in a dose-dependent manner with cisplatin treatment remains to be elucidated; however, the data presented in this work suggests that extensive injury incurred by cells exposed to higher doses of cisplatin is sufficient to overcome cytoprotective autophagy and induce cell death. This in evident in Figure 1B, where clonogenic survival studies failed to demonstrate differences in sensitivity between p53 knockout and p53 wt cells as well as Figure 1E showing significant apoptotic cell death at 20 μM cisplatin. Interestingly, this differs from our recent findings in studies involving ionizing radiation, where radiation sensitivity appeared to be a function primarily of the extent of senescence and appeared to be largely unrelated to autophagy (24). In these studies, the same set of isogenic cell lines, H460wt and H460crp53 cells, exhibited nonprotective autophagy in both cells when exposed to radiation. Moreover, autophagy inhibition failed to alter radiation sensitivity or radiation-induced apoptosis in either cell line. In this regard, we demonstrate that crp53 cells have the capacity to undergo nonprotective autophagy (in the case of radiation) and this response is “switched” to protective autophagy in the case of cisplatin treatment. Taken together, these studies suggest the existence of an autophagic switch, not only between different cell lines, but also depending on the therapeutic agent utilized. Please also see the additional discussion relating to the autophagic switch in section 4.3.
4.2. p53 function, autophagy and drug sensitivity/resistance
Secondly, this work further interrogates, albeit indirectly, the relative contributions of p53 status and autophagy to sensitivity and resistance to chemotherapy. Tasdemir et al and colleagues had reported that inhibition of cytoplasmic p53 led to autophagy in enucleated cells and conversely that cytoplasmic p53 was able to repress the enhanced autophagy of p53 null cells, providing evidence of a relationship between p53 and autophagy (33). Topotecan, a topoisomerase I inhibitor, induced cytoprotective autophagy in p53wt colon cancer cells in vitro and in vivo, but induced cytotoxic autophagy in p53 null colon cancer cells (34). Tripathi et al. demonstrated that cisplatin induced protective autophagy in p53 knockdown embryonal carcinoma cells, which would be consistent with the findings presented in this work (35). However, Maycotte et al reported on nonprotective autophagy in p53 null 4T1 breast tumor cells exposed to cisplatin (36). These differential outcomes indicate that it cannot be predicted, a priori, the nature that drug or radiation-induced autophagy will exhibit, based solely on the status of p53 in the cells.
In the current work, cisplatin induced similar levels of autophagy in both p53 wt and H460crp53 cells, indicating that the capacity to undergo cisplatin-induced autophagy is essentially p53-independent. However, as in many of the studies cited above, the nature of the autophagy changed in association with the different p53 status of the two cell lines. Inhibition of autophagy increased drug sensitivity and apoptosis in the H460crp53 cell to similar extents as p53wt cells exposed to cisplatin. This critical observation from the current work suggests that cells lacking functional p53 are capable of undergoing apoptosis to the same degree as p53 wild type cells. As was shown in Figure 5, autophagy inhibition in H460crp53 cells shifted the temporal response to cisplatin to be virtually identical to that in the H460wt cells, suggesting that cytoprotective autophagy and not p53 function was largely responsible for the reduced sensitivity to cisplatin of the crp53 cells.
4.3. The autophagic switch in cancer therapy
The last and closely linked question relates to the involvement of what we have termed the “autophagic switch”. In previous work we demonstrated an alteration in autophagy function from the cytoprotective form in response to radiation (in p53 wt cells) to either cytotoxic autophagy in p53wt breast tumor cells or cytostatic autophagy in non-small cell lung cancer cells upon the combination of radiation with vitamin D or vitamin D analogs (19,37). The change in radiation-induced autophagy from the cytoprotective to the nonprotective form was further observed in breast, head and neck, and NSCLC cell lines provides additional evidence of the autophagic switch (20). Moreover, utilizing a doxycycline-inducible p53 expression model of H1299 NSCLC cells (tp53 null), demonstrated that parental p53 null cells exhibited nonprotective autophagy in response to radiation, yet when p53 expression was induced, autophagic function was switched to protective (20). The current work demonstrates cisplatin-induced autophagy switches from the nonprotective form in wt p53 H460 cells to the cytoprotective form in the H460crp53 cells, suggesting p53 status does not obligate autophagy to a particular function.
Further juxtaposition of these data sets suggest that the functional form of autophagy elicited is in part individualized to the cell line and therapeutic modality utilized. The in vitro and in vivo study relating to topotecan-induced autophagy in p53 wt and p53 knockout colon cancer cells by Li also supports the premise that autophagy can switch from the cytoprotective to the nonprotective function (34). Moreover, the “autophagic switch” is not limited to p53 function as Shen and colleagues found that gemcitabine-induced autophagy switches from the cytoprotective form in ER− BCap37 breast cancer cell lines to the cytotoxic form in ER+ BCap37 cell lines (18). In these studies, Shen et al demonstrate that genetic silencing and pharmacological inhibition of ERα resulted in an “autophagic switch” from cytotoxic to cytoprotective autophagy. They provide evidence that the switch is mediated through ER-mediated non-canonical autophagy (38). In an osteosarcoma model, drug-resistant cells exhibited cytoprotective autophagy, with greater reliance on autophagy for metabolic maintenance, whereas, drug-sensitive cells exhibited cytotoxic autophagy in response to camptothecin (39). While the current studies provide an additional model of the “autophagic switch” in cancer therapy using a set of isogenic cell lines, the mechanistic basis for the autophagic switch, remains to be determined.
4.4. Conclusions
Taken together, these studies provide proof of concept that cytoprotective autophagy can confer intrinsic resistance to chemotherapy, based on a comparison of cisplatin sensitivity in two isogenic cell lines where autophagy demonstrated, respectively, cytoprotective and nonprotective functions. However, it is necessary to recognize that autophagy induced by chemotherapy or radiation may not always be cytoprotective in the clinic and the therapeutic benefit of autophagy inhibition may only be successful in scenarios where the autophagy is cytoprotective. Furthermore, it cannot be predicted whether therapy-induced autophagy will be protective or nonprotective based on functional p53 status. If autophagy inhibition is to be incorporated into therapeutic intervention, it will likely be necessary to identify the functional form(s) of autophagy for each therapeutic intervention in a particular patient (i.e. personalized medicine), reiterating the importance of screening prior to the inclusion of autophagic inhibitors to clinical regimens (40).
Acknowledgments
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Breast Cancer Research Program [grant # W81XWH-19-1-0490 (DAG)] and Massey Center Support Grant P30 CA016059. The authors gratefully acknowledge financial support (JX) from the China Scholarship Council. Services and products in support of the research project were generated by the VCU Massey Cancer Center Flow Cytometry Shared Resource, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
No potential conflict of interest was reported by any of the authors.
References
- 1.Dilruba S, Kalayda GV. Platinum-based drugs: past, present and future. Vol. 77, Cancer Chemotherapy and Pharmacology. Springer Verlag; 2016. p. 1103–24. [DOI] [PubMed] [Google Scholar]
- 2.Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Vol. 31, Oncogene. 2012. p. 1869–83. [DOI] [PubMed] [Google Scholar]
- 4.Lin JF, Lin YC, Tsai TF, Chen HE, Chou KY, Hwang TIS. Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des Devel Ther. 2017. May 16;11:1517–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Zhang L, Zhou H, Wang W, Luo Y, Yang H, et al. Inhibition of autophagy promotes cisplatin-induced apoptotic cell death through Atg5 and Beclin 1 in A549 human lung cancer cells. Mol Med Rep. 2018. May 1;17(5):6859–65. [DOI] [PubMed] [Google Scholar]
- 6.Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHAto overcome Bcr-Abl-mediated drug resistance. Blood. 2007. July 1;110(1):313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren J-HH, He W-SS, Nong L, Zhu Q-YY, Hu K, Zhang R-GG, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. TL - 25. Cancer Biother Radiopharm. 2010;25 VN-r(1):75–80. [DOI] [PubMed] [Google Scholar]
- 8.Hu YL, Jahangiri A, DeLay M, Aghi MK. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Vol. 72, Cancer Research. 2012. p. 4294–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JG, Shin JH, Shim HS, Lee CY, Kim DJ, Kim YS, et al. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir Res. 2015;16:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duffy A, Le J, Sausville E, Emadi A. Autophagy modulation: a target for cancer treatment development. Cancer Chemother Pharmacol. 2015. March;75(3):439–47. [DOI] [PubMed] [Google Scholar]
- 11.Gewirtz DA. The Challenge of Developing Autophagy Inhibition as a Therapeutic Strategy. 2016;76(19):5610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gewirtz DA. Cytoprotective and nonprotective autophagy in cancer therapy. Autophagy. 2013;9(9):1263–5. [DOI] [PubMed] [Google Scholar]
- 13.Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014. February;74(3):647–51. [DOI] [PubMed] [Google Scholar]
- 14.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science. 2011;334(6062):1573–8. [DOI] [PubMed] [Google Scholar]
- 15.Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci. 2016;113(1):182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saleh T, Cuttino L, Gewirtz DA. Autophagy is not uniformly cytoprotective: a personalized medicine approach for autophagy inhibition as a therapeutic strategy in non-small cell lung cancer. Biochim Biophys Acta. 2016;1860(10):2130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gewirtz DA. An autophagic switch in the response of tumor cells to radiation and chemotherapy. Biochem Pharmacol. 2014. August;90(3):208–11. [DOI] [PubMed] [Google Scholar]
- 18.Shen P, Chen M, He M, Chen L, Song Y, Xiao P, et al. Inhibition of ERα/ERK/P62 cascades induces “autophagic switch” in the estrogen receptor-positive breast cancer cells exposed to gemcitabine. Oncotarget. 2016;7(30):48501–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson EN, Bristol ML, Di X, Maltese WA, Koterba K, Beckman MJ, et al. A Switch Between Cytoprotective and Cytotoxic Autophagy in the Radiosensitization of Breast Tumor Cells by Chloroquine and Vitamin D. Horm Cancer. 2011. October;2(5):272–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakradeo S, Sharma K, Alhaddad A, Bakhshwin D, Le N, Harada H, et al. Yet another function of p53 - the switch that determines whether radiation-induced autophagy will be cytoprotective or nonprotective: Implications for autophagy inhibition as a therapeutic strategy. Mol Pharmacol. 2015;87(5):803–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Musich PR, Zou Y. Differential DNA damage responses in p53 proficient and deficient cells: cisplatin-induced nuclear import of XPA is independent of ATR checkpoint in p53-deficient lung cancer cells. Int J Biochem Mol Biol. 2011. April 15;2(2):138–45. [PMC free article] [PubMed] [Google Scholar]
- 22.Clairambault J, Eliaš J. Diverse spatio-temporal dynamical patterns of p53 and cell fate decisions. In: AIP Conference Proceedings. American Institute of Physics Inc.; 2016. [Google Scholar]
- 23.Goehe RW, Di X, Sharma K, Bristol ML, Henderson SC, Valerie K, et al. The Autophagy-Senescence Connection in Chemotherapy: Must Tumor Cells (Self) Eat Before They Sleep? J Pharmacol Exp Ther. 2012;343(3):763–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Patel NH, Saleh T, Cudjoe EK, Alotaibi M, Wu Y, et al. Differential Radiation Sensitivity in p53 Wild-Type and p53-Deficient Tumor Cells Associated with Senescence but not Apoptosis or (Nonprotective) Autophagy. Radiat Res. 2018;190(5):538–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuribayashi K, El-Deiry WS. Regulation of programmed cell death by the p53 pathway. Vol. 615, Advances in Experimental Medicine and Biology. 2008. p. 201–21. [DOI] [PubMed] [Google Scholar]
- 26.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993. September 24;74(6):957–67. [DOI] [PubMed] [Google Scholar]
- 27.Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Vol. 1806, Biochimica et Biophysica Acta - Reviews on Cancer. 2010. p. 220–9. [DOI] [PubMed] [Google Scholar]
- 29.Kumar A, Singh UK, Chaudhary A. Targeting autophagy to overcome drug resistance in cancer therapy. Vol. 7, Future Medicinal Chemistry. Future Science; 2015. p. 1535–42. [DOI] [PubMed] [Google Scholar]
- 30.Yun CW, Lee SH. The Roles of Autophagy in Cancer. Vol. 19, International Journal of Molecular Sciences. 2018. p. e3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Vol. 30, Genes and Development. Cold Spring Harbor Laboratory Press; 2016. p. 1913–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med. 2009. September;13(9B):3644–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008. June;10(6):676–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li D-D, Sun T, Wu X-Q, Chen S-P, Deng R, Jiang S, et al. The Inhibition of Autophagy Sensitises Colon Cancer Cells with Wild-Type p53 but Not Mutant p53 to Topotecan Treatment. Fimia GM, editor. PLoS One. 2012. September 14;7(9):e45058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tripathi R, Ash D, Shaha C. Beclin-1-p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J Cell Mol Med. 2014. November 1;18(11):2275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8(2):200–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma K, Goehe RW, Di X, Hicks MA, Torti SV, Torti FM, et al. A novel cytostatic form of autophagy in sensitization of non-small cell lung cancer cells to radiation by vitamin D and the vitamin D analog, EB 1089. Autophagy. 2014;10(12):2346–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Felzen V, Hiebel C, Koziollek-Drechsler I, Reißig S, Wolfrum U, Kögel D, et al. Estrogen receptor α regulates non-canonical autophagy that provides stress resistance to neuroblastoma and breast cancer cells and involves BAG3 function. Cell Death Dis. 2015. July 9;6:e1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hollomon MG, Gordon N, Santiago-O’Farrill JM, Kleinerman ES. Knockdown of autophagy-related protein 5, ATG5, decreases oxidative stress and has an opposing effect on camptothecin-induced cytotoxicity in osteosarcoma cells. BMC Cancer. 2013. December 26;13:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gewirtz DA. The Switch between Protective and Nonprotective Autophagy; Implications for Autophagy Inhibition as a Therapeutic Strategy in Cancer. Biology (Basel). 2020;9(12):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]