

Abstract
Embryonic development in mammals is highly sensitive to changes in gene expression within the placenta. The placenta is also highly enriched for genes showing parent-of-origin or imprinted expression, which is predicted to evolve rapidly in response to parental conflict. However, little is known about the evolution of placental gene expression, or if divergence of placental gene expression plays an important role in mammalian speciation. We used crosses between two species of dwarf hamsters (Phodopus sungorus and Phodopus campbelli) to examine the genetic and regulatory underpinnings of severe placental overgrowth in their hybrids. Using quantitative genetic mapping and mitochondrial substitution lines, we show that overgrowth of hybrid placentas was primarily caused by genetic differences on the maternally inherited P. sungorus X chromosome. Mitochondrial interactions did not contribute to abnormal hybrid placental development, and there was only weak correspondence between placental disruption and embryonic growth. Genome-wide analyses of placental transcriptomes from the parental species and first- and second-generation hybrids revealed a central group of co-expressed X-linked and autosomal genes that were highly enriched for maternally biased expression. Expression of this gene network was strongly correlated with placental size and showed widespread misexpression dependent on epistatic interactions with X-linked hybrid incompatibilities. Collectively, our results indicate that the X chromosome is likely to play a prominent role in the evolution of placental gene expression and the accumulation of hybrid developmental barriers between mammalian species.
Keywords: gene expression, reproductive isolation, genomic imprinting, Phodopus
Introduction
Developing mammalian embryos depend on the extra-embryonic placenta for a broad array of functions including hormone production, immunity, and as a conduit for maternal nutrients and gas exchange (Reik et al. 2003; Levy 2007). Normal intrauterine development in humans and mice depends on the tightly controlled placental expression of a diverse set of genes (Constancia et al. 2002; Levy 2007; Plasschaert and Bartolomei 2014). Placental gene expression has also likely played an important role in the evolution of mammalian development (Haig 1996; Capellini et al. 2011; Kaneko-Ishino and Ishino 2019). Indeed, much of the phenotypic diversity across mammalian species is thought to have evolved by changes in gene expression during critical stages of development (King and Wilson 1975; Carroll 2008; Sears et al. 2015). However, relatively little is known about the broader genomic organization, functional integration, and evolution of placental gene expression networks across species (Al Adhami et al. 2015).
The placenta is characterized by two unusual regulatory phenomena that likely play critical roles in its evolution. First, the placenta is highly enriched for genes showing monoallelic expression due to epigenetic silencing of one parental allele (i.e., genomic imprinting, Morison et al. 2005; Hudson et al. 2010; Babak et al. 2015). Genomic imprinting is thought to have evolved to help resolve fitness conflicts between maternally and paternally inherited alleles (i.e., kinship or parental conflict theory, Haig 2000). While perhaps only approximately 100–200 autosomal genes showed strongly imprinted expression across tissues (Babak et al. 2015), disruption of genomic imprinting has emerged as an important cause of congenital disorders in humans (Hirasawa and Feil 2010; Lee and Bartolomei 2013) and as a potential driver of reproductive barriers between species (Crespi and Nosil 2013; Wolf and Brandvain 2014). Second, some mammals also show imprinted paternal X chromosome inactivation in extra-embryonic tissues (i.e., imprinted XCI; Heard and Disteche 2006), representing a striking deviation from random XCI found in most somatic cells of placental mammals (Lyon 1961; Dupont and Gribnau 2013). The X chromosome in general, and imprinted XCI in particular, has been shown to play important roles in placental development (Hemberger 2002; Mcgraw et al. 2013). Moreover, the mouse X chromosome appears enriched for genes preferentially expressed in the placenta (Khil et al. 2004).
Resolution of genetic conflict may explain the origin of genomic imprinting in mammals (Haig 2000). However, theory also predicts that once imprinting is established adaptation among interacting genes can drive the evolutionary expansion of regulatory networks of similarly imprinted genes (Wolf and Hager 2006; Wolf and Brandvain 2014; Patten et al. 2016; O'Brien and Wolf 2017). Given the relative scarcity of autosomal imprinting overall (Babak et al. 2015), the X chromosome is expected to harbor the majority of genes showing imprinted (maternal) placental expression in species with imprinted XCI. Thus, co-evolutionary interactions between the maternal X chromosome and maternally expressed autosomal genes should be relatively common within placental regulatory pathways in imprinted XCI species. Despite these predictions, the overall importance of the X chromosome to the evolution of placental gene expression remains unclear. Many molecular genetic studies on the placenta have focused on established disease models or targeted genetic manipulations of imprinted autosomal genes (e.g., gene knockdowns or knockouts; Sanli and Feil 2015), revealing fundamental insights into the mechanisms and functional consequences of genomic imprinting in the placenta and other tissues. In parallel, meta-analyses of expression data have revealed that clusters of imprinted autosomal genes appear to fall within larger networks of co-expressed genes that include both imprinted and bi-allelically expressed loci (Varrault et al. 2006; Al Adhami et al. 2015). The extent and functional relevance of such regulatory networks remain unclear, but the emerging model of genome-wide networks of imprinted and nonimprinted genes represents a conceptual shift from the view of imprinting controlled primarily through local cis-regulatory effects (Patten et al. 2016).
Parent-of-origin effects for abnormal embryonic and placental growth are common in mammalian hybrids (Vrana 2007; Brekke and Good 2014), suggesting that hybrid systems may provide powerful models for understanding how the evolution of gene expression impacts placental development. Here, we focus on crosses between two closely related species of dwarf hamsters (Phodopus sungorus and Phodopus campbelli) that yield strong parent-of-origin growth effects in reciprocal F1 crosses (Brekke and Good 2014). Extensive placental overgrowth and disrupted embryonic development occur in hybrid crosses when the mother is P. sungorus (female P. sungorus × male P. campbelli; hereafter S × C with uppercase used to denote placental overgrowth), often resulting in hybrid and maternal death during birth. The reciprocal cross results in normal embryonic development (female P. campbelli × male P. sungorus; hereafter c × s with lowercase used to denote normal placental growth), although adult hybrid males are smaller (Brekke and Good 2014) and completely sterile (Safronova et al. 1999; Bikchurina et al. 2018). Intrinsic postzygotic reproductive isolation (i.e., hybrid inviability or sterility) generally tends to be asymmetric in reciprocal crosses between closely related species due to incompatible interactions at sex-linked or imprinted loci (Turelli and Moyle 2007). Although the genetic architecture of hybrid placental growth has not been determined in dwarf hamsters, massively overgrown F1 hybrid S × C placenta do show extensive disruption of gene expression pathways that are highly enriched for embryonic growth and genomic imprinting (Brekke et al. 2016). Building on these previous studies, we combine quantitative genetic and transcriptomic analyses to test the hypothesis that the X chromosome plays a central role in the evolution of placental gene expression, embryonic development, and reproductive barriers between species.
Materials and methods
Animals and experimental crosses
Experimental animals were drawn from established colonies of wild-derived P. campbelli and P. sungorus at the University of Montana (Brekke and Good 2014), which were originally established by Kathy Wynne-Edwards (Scribner and Wynne-Edwards 1994). Colonies were maintained as outbred, though overall inbreeding levels are high (Brekke et al. 2018). All breeding experiments were done in compliance with the University of Montana Institutional Animal Care and Use Committee regulations (animal use protocols 039-13JGDBS-090413 & 050-16JGDBS-082316).
We previously reported results from experimental crosses within and between P. campbelli and P. sungorus used to examine late-term placental and embryonic phenotypes (Brekke and Good 2014) and placental transcriptomes (n = 40 placental transcriptomes, five males and five females for each cross type; Brekke et al. 2016). Here we combined these results with new data from two additional genetic crossing experiments used to evaluate the role of the mitochondrial and nuclear genomes in the genetic basis of asymmetric hybrid placental and embryonic overgrowth (Figure 1). First, we generated mitochondrial substitution lines wherein c × s hybrid females were crossed to P. sungorus males for 10 additional backcross (BC) generations. This crossing scheme is predicted to recover hamsters that are ∼99.9% P. sungorus across the nuclear genome but retain the mitochondria of P. campbelli (P. sungorusmtC). Tenth-generation P. sungorusmtC females were crossed to P. campbelli males to test for F1 overgrowth (SmtC×C), thereby mimicking the overgrown S × C hybrid across the nuclear genome while substituting the P. sungorus mitochondria for P. campbelli mitochondria. Second, we crossed F1 c × s hybrid females to P. campbelli males to generate a BC panel of late-term embryos and placentas. In the context of genetic elements with sex-limited inheritance or expression, these BC hybrids have the same paternal contribution found in overgrown S × C F1 hybrids, while varying the species origin of maternally inherited alleles.
Figure 1.

Summary of genetic crosses and experiments. Reciprocal species intercrosses between P. sungorus (gray chromosomes) and P. campbelli (white chromosomes) result in asymmetric placental overgrowth and inviability. Only females with a P. campbelli mother and P. sungorus father are both viable and fertile (c × s, indicated with a dashed box). The first generation cross with normal sized placentas is indicated with lowercase letters (c × s), and the reciprocal cross with overgrown placentas is indicated with uppercase letters (S × C). A mitochondrial substitution line was created through 10 generations of backcrossing hybrid females to a P. sungorus male, resulting in females with P. sungorus nuclear genomes and P. campbelli mitochondria (SmtC) that were then crossed to P. campbelli males. The BC mapping panel was created by a single generation of backcrossing a fertile hybrid female to a P. campbelli male, resulting in offspring showing a range of placenta phenotypes. Data types collected for each experiment are reported in the corresponding panels.
All advanced crosses were conducted through c × s hybrid females as S × C F1 hybrids generally do not survive birth (Brekke and Good 2014) and c × s males are completely sterile (Safronova et al. 1999; Ishishita et al. 2015; Bikchurina et al. 2018). For both crossing experiments, females were sacrificed at late gestation and offspring placentas and embryos were collected, weighed, and snap-frozen on dry ice. Embryos were developmentally scored following (Brekke and Good 2014) to ensure that all offspring were in the final 4 days of gestation corresponding to Theiler’s Stages 24–27 (Theiler 1972). Developmental abnormalities such as embryonic fluid accumulation (edema) and embryo reabsorption were noted, and embryo and placenta weights were assessed with stepwise model selection and adjusted for variation in edema, litter size and Theiler stage using simple linear models as implemented in JMP (v12).
Genotyping
We genotyped 189 BC individuals (91 females and 98 males) and our original colony founders (14 P. campbelli and 11 P. sungorus) using double digest restriction site-associated DNA sequencing (ddRAD-seq; Peterson et al. 2012) with minor modifications. Genomic DNA was extracted from frozen embryos with a Machery-Nagel Nucleospin Tissue DNA extraction kit (catalog number 740952) following the manufacturer’s protocol, except that 5 µl RNase-A was added to the column and incubated for 15 minutes at room temperature. We then digested 1 µg of genomic DNA using the high-fidelity restriction enzyme SbfI-HF (New England BioLabs, catalog number R3642L), followed by MspI (New England BioLabs, catalog number R0106L) both with the CutSmart buffer (New England BioLabs). Libraries were prepared with a dual barcoding scheme incorporating both Illumina indexes and inline barcodes to uniquely identify each sample (Peterson et al. 2012). Size selection of adapter-ligated fragments (200–500 bp) was done with Agencourt AMPure XP beads. Both the size selection and PCR amplification were done prior to sample pooling to assure more even representation across samples. Combined sample pools were sequenced on 50% of an Illumina HiSeq 2500 lane in rapid-run mode and then on 50% of a lane of Illumina Hiseq 2500 in normal mode at the University of Oregon Genomics and Cell Characterization Core Facility. All samples were sequenced in both lanes and combined for subsequent analyses. We also independently determined the sex of embryos using a PCR assay of the Y-linked Sry gene as described previously (Brekke et al. 2016).
RAD libraries were cleaned and demultiplexed with Stacks (v1.20; process_radtags parameters -e sbfI –renz_2 mspI -r -c -q; Catchen et al. 2013). An initial list of unique RADtags from both read pairs was generated using ustacks (-H -r -d) using data from one female founder from each species. RADtag reference libraries were then generated using cstacks (-n 4). Reads from all the colony founders were aligned to the RADtag reference library with bwa-mem (v0.7.9a; Li and Durbin 2009) and single-nucleotide variants (SNVs) were called with the GATK HaplotypeCaller (v3.1-1, -stand_call_conf 30; Van Der Auwera et al. 2013). All SNVs that were polymorphic within a species in our colony founders were filtered out using GATK selectVariants (v3.1-1). BC individuals were genotyped at the ascertained SNVs sites using GATK UnifiedGenotyper (v3.1-1; -stand_call_conf 30; Van Der Auwera et al. 2013).
We also used an exon capture experiment to anchor placental-expressed genes onto the Phodopus genetic map (described below). We designed custom hybridization probes to target 9,756 fixed SNVs between P. campbelli and P. sungorus ascertained from species-specific transcriptomes (Genbank BioProject PRJNA306772 and DDBJ/EMBL/GenBank Accessions GEVA00000000 and GEVB00000000; Brekke et al. 2016). Exon boundaries were annotated for each transcript with a local BLAT search (Kent 2002) against the golden hamster (Mesocricetus auratus) draft reference genome (The Broad Institute Genome Assembly & Analysis Group, MesAur1.0). For each gene, we selected 1-2 SNVs located furthest from inferred exon boundaries and included probes matching both alternative bases to avoid species bias. Capture baits were manufactured by MYcroarray.
We selected 94 individuals (44 males and 50 females) from the BC panel and prepared Illumina sequencing libraries following the Meyer-Kircher protocol (Meyer and Kircher 2010). Ten cycles were used during the indexing PCR step. The indexed libraries were then combined into four pools and target enriched following the MyBaits-1 Custom Target Capture protocol and using mouse CoT-1 DNA supplied by the manufacturer as a blocking agent. The four captured pools were then reamplified with 20 PCR cycles, quantified with a Kappa qPCR quantification kit (catalog number KK4824), and pooled for Illumina sequencing. Enriched libraries were initially sequenced at the University of Montana Genomics Core on an Illumina MiSeq 75-bp paired-end sequencing, and followed by one lane of Illumina HiSeq 2500 100-bp single-end sequencing at the University of Oregon Genomics and Cell Characterization Core Facility. Raw sequence reads were adapter trimmed with Cutadapt (v1.6; -O 5 and -e 0.1; Martin 2011) and quality-filtered with Trimmomatic (v0.3.2; LEADING : 5, SLIDINGWINDOW : 4:15, MINLEN: 36, and HEADCROP : 13; Bolger et al. 2014). Filtered reads were then aligned to published transcriptome assemblies (Brekke et al. 2016) and the targeted SNVs were genotyped with GATK HaplotypeCaller (v3.1-1; -stand_call_conf 30.0 -stand_emit_conf 30) and filtered (selectvariants –restrictAllelesTo BIALLELIC -select “QD > 10.0”) so that only high-quality genotypes were used for estimating the location of each gene.
Quantitative genetic analysis
We constructed a genetic map using RADtag SNVs identified between the P. campbelli and P. sungorus colony founders and the program R/qtl (v1.45; Broman and Sen 2009). Putative X-linked RADtags were identified as markers that were heterozygous or homozygous for P. campbelli genotypes in females and always homozygous P. campbelli or P. sungorus in males. To estimate the map, we removed two BC individuals with low-sequencing coverage, identified putative X-linked markers based on Hardy–Weinberg expectations, and dropped all autosomal markers that were genotyped in fewer than 177 individuals (95%). We formed linkage groups and ordered the markers on each linkage group with the ripple(), compareorder(), and switch.order() functions until each linkage group was a short as possible. Then we sequentially dropped each marker to see if the likelihood of the map improved. Once all poor quality markers were removed, we repeated the ripple(), compareorder(), and switch.order() functions until the likelihood was maximized. The linkage groups in the final map were ordered by descending length in centiMorgans (cM).
We then used R/qtl to test for single-quantitative trait loci (QTL) associated with the variation in BC embryo and placental weight using the extended Haley–Knott method with imputation (Haley and Knott 1992; Feenstra et al. 2006). Next, we incorporated sex as a covariate and re-estimated QTL for both phenotypes. For all of single-QTL scans, we used 10,000 permutations to estimate genome-wide significance thresholds for autosomes and 337,364 permutations for the X chromosome. Finally, we used the QTL identified in the first two analyses as additive cofactors and re-scanned for additional QTL that were contingent on the presence of the earlier identified QTL (Broman and Sen 2009). We used 1000 permutations for autosome–autosome interactions, 1687 permutations for autosome–X interactions, and 113,815 permutations for X–X interactions to establish significance thresholds. QTL intervals were established based on 95% Bayesian confidence intervals, and the proportion of phenotypic variance explained by QTL was estimated as (Broman and Sen 2009).
Expressed genes were then integrated onto the genetic map by comparing captured SNV genotypes to RADtag genotypes. Following Brekke et al. (2019), we counted shared genotypes between each RADtag and each gene across all individuals, placing genes at the location of the RADtag with which they shared the most genotypes. In the event of a tie between multiple RADtags, the gene was placed at the proximal map location and only genes sharing at least 90% of genotypes with at least one RADtag were placed on the map. Given the low number of recombination events and the high number of genes, these RADtag-anchored positions represent coarse genetic locations for each gene. Instances where multiple genes were associated with a single RADtag were treated as a single unordered linkage block. Once integrated, likely genotyping errors in the capture data were identified using calc.errorlod() and the highest errors were extracted with top.errorlod() and a very strict cutoff of 1 such that even moderately questionable genotypes were identified. The genotypes of these sites were removed and then filled in with the fill.geno() function using the imputation method, which imputes the missing genotypes from the surrounding sites for each individual. These corrected genotypes were used for evaluating imprinting status of select candidate genes (see below).
Gene expression analyses
We chose 24 placentas from our BC mapping panel for genome-wide expression analysis using RNA-seq (Wang et al. 2009), including six males and six females with large placentas (mean = 0.232 ± 0.010 g) and six males and six females with normal sized placentas (mean = 0.140 ± 0.008 g). RNA was extracted from whole frozen placenta with an Omega Bio-tek E.Z.N.A. Total RNA Kit I (catalog number R6834) including a DNase digestion following the manufacturer’s protocol. All RNA samples were checked for quality and concentration on the bioanalyzer and all samples used had RNA integrity numbers >8.0. RNA-seq libraries were constructed from 2 µg of input RNA with the Agilent Sure-Select Strand-Specific RNA-seq Kit (catalog number G9691B) following the manufacturer’s recommendations. Libraries were amplified with 14 cycles of PCR, and pooled based on a Kappa qPCR Quantification Kit (catalog number KK4824). The pooled libraries were sequenced with two lanes of Illumina HiSeq2500 100-bp single-end sequencing.
RNA-seq data were processed as described previously (Brekke et al. 2016). Briefly, Illumina adapters were trimmed off reads with Cutadapt (v1.6; -O 5 -e 0.1; Martin 2011) and quality trimmed with Trimmomatic (v0.3.2; SE -phred 33 LEADING: 5 SLIDINGWINDOW: 4:15 HEADCROP: 13; Bolger et al. 2014). While an initial draft of the P. sungorus genome has been generated using Illumina shotgun sequencing, current annotation and assembly quality remains insufficient for reference-guided transcriptome analyses (Bao et al. 2019). Therefore, reads were aligned with bowtie2 (v2.2.3; Langmead and Salzberg 2012) to a published de novo placental transcriptome assembly (Genbank BioProject PRJNA306772 and DDBJ/EMBL/GenBank Accessions GEVA00000000 and GEVB00000000; Brekke et al. 2016), and filtered for potentially chimeric transcripts using draft genomic resources by excluding 1422 “genes” with exons that multiply mapped to different contigs. To evaluate expression level, we created a table of counts at the gene level using featureCounts (v1.4.2; Liao et al. 2014), which counted fragments (-p) and discarded reads with too long an insert (-P) or are chimeric (-C) or have a mapping quality (-Q) below 20. This table of counts was normalized with the trimmed mean of m-values (TMM) method (Robinson and Oshlack 2010).
We used the WGCNA package (version 1.68; Langfelder and Horvath 2008) to infer weighted gene co-expression networks using expression data from previously published parental and F1 genotypes (Brekke et al. 2016) and the newly generated BC. This network approach uses adjacency correlation matrices to identify hierarchical clusters of co-expressed genes (Zhang and Horvath 2005), enabling the reduction of complex clusters into representative expression profiles (i.e., module eigengenes) defined as the first component of a principle component analysis. A scale-free topology index was determined by soft thresholding (Supplementary Figure S1), which was then used to automatically detect signed, Pearson correlated modules via dynamic cutting. The signed module method splits otherwise correlated genes with increases in expression into separate modules from those with decreases in expression, which allowed us to evaluate upregulated gene sets independently from downregulated gene sets. We merged similar modules using a threshold of 0.25, which combines modules with a correlation of 0.75 or greater into a single module. For each module, we tested for correlations between the module eigengene and placental and embryonic weights. For modules showing significant correlations after correction for multiple testing, we then retested associations using a more stringent ANOVA model that controlled for developmental stage and sex.
Each module was assessed with a binomial exact test (R/stats package 3.6.1) for enrichment of candidate imprinted genes previously identified based on patterns of allele-specific expression in the placenta (Brekke et al. 2016) and for X-linked genes. Network connectivity was determined through pairwise correlation between genes, with P-values corrected with the q-value package (version 2.18.; Storey et al. 2019), using a false discovery rate threshold of 0.05. We then counted the number of additional genes significantly correlated with each gene within the module. Hub genes were defined as the top 5% most connected genes in each module. Overlap between F1 and BC modules was determined by comparing gene lists to get counts of shared genes. We also evaluated module conservation by comparing how strongly each gene was correlated to each module across data sets. To connect module conservation to phenotypes, we compared the concordance between each gene and placenta weight across F1 and BC datasets using a bivariate correlation as implemented in JMP (v12).
Genotypes at each gene placed on the genetic map were inferred by evaluating genotypes at flanking RAD markers. If a gene was placed in the same linkage block (cM location) as a RADtag, the marker genotype was used for the gene. Likewise, if the gene was placed between two markers with the same genotype, the concordant marker genotypes were used for the gene. If flanking RADtag genotypes were discordant, the genotype at the gene was treated as unknown. We then tested for expression interactions (EIs) between the X chromosome and diploid autosomal genotypes within the BC as follows. Autosomal genotypes were imputed for the 23 BC individuals with transcriptome data. EI scores were then calculated for all mapped autosomal genes by comparing the difference in fold change in expression of that gene for the two possible autosomal genotypes (i.e., homozygous for the P. campbelli allele or heterozygous) dependent on the genotype of the maternal X chromosome (P. campbelli or P. sungorus). To generate EI scores for each gene, normalized gene expression count tables generated during WGCNA were used to calculate log2-fold changes between alternative maternal X chromosome genotypes and the two genotypes at each autosomal locus. We excluded unmapped autosomal genes and genes with imputed genotypes for fewer than three individuals for each of the four X-by-autosome genotypic classes. We calculated the absolute value of EI scores, where a value of zero indicates no difference between the two autosomal genotypes when inheriting different maternal X chromosomes and a value of one indicates a onefold difference in expression between the two autosomal genotypes (i.e., a candidate X-autosome EI). We also considered a polarized (i.e., nonabsolute value) version of the statistic where positive values reflected greater change when maternal X and autosomal alleles genotypes were discordant.
EI was calculated as follows:
where
xsun = samples with a P. sungorus maternal X chromosome;
xcam = samples with a P. campbelli maternal X chromosome;
GeneCC = samples that are homozygous for the P. campbelli allele at the gene for which the EI score is calculated;
GeneSC = samples that are heterozygous at the gene for which the EI score is calculated.
Results
Mitochondrial interactions were not a major factor in extreme parent-of-origin hybrid overgrowth
If hybrid placental overgrowth is caused by deleterious interactions between the maternal P. sungorus mitochondrial genome and the P. campbelli nuclear genome, then crossing females from the P. sungorusmtC mitochondrial substitution lines to P. campbelli males should recover normal development. Alternatively, if heterospecific mitochondria have no effect on hybrid growth, then SmtC×C hybrids should be similar in size to overgrown S × C hybrids. In support of the second hypothesis, SmtC×C placentas were found to be extremely large (F4,213 = 106, P < 0.001, ANOVA), but not statistically different from S × C hybrids based on a Tukey’s honest significant difference (HSD) test (Supplementary Figure S2). We note that low levels of residual autosomal heterozygosity could theoretically mask mitochondrial incompatibilities in the 10th generation mitochondrial substitution P. sungorusmtC lines. However, such residual variation cannot also explain why the substitution lines did not recover normal placental development in the F1 hybrid crosses. On balance, these results strongly indicate that heterospecific mitochondrial interactions are not a major factor in extreme parent-of-origin hybrid overgrowth in dwarf hamsters.
The quantitative genetic basis of extreme parent-of-origin hybrid overgrowth
We generated a BC panel of 189 late-term embryos and placentas sampled from 38 litters. Using 1215 ddRAD SNVs, we constructed a 1231.7 cM genetic map that comprised 14 major linkage groups (Supplementary Figure S3 and Supplementary Table S1). The karyotype of P. sungorus comprised 14 chromosomes (2 N = 28; Romanenko et al. 2007), suggesting that each of our major linkage groups correspond to an individual chromosome. The X chromosome was inferred to have the shortest overall genetic distance (35.5 cM or 2.9% of the genetic map), while it appears medium-sized in ranked karyotype analyses (i.e., middle 30%; Gamperl et al. 1977; Romanenko et al. 2007) and comprises ∼10% of the haploid female karyotype (Haaf et al. 1987). The Phodopus X chromosome is metacentric with an Xp arm that has been described as heterochromatic (Gamperl et al. 1977) and nonrecombinant in females of both species and in c × s F1 hybrids (Bikchurina et al. 2018). Our inferred genetic map was consistent with strong repression of recombination on one end of the X chromosome in c × s females, albeit not complete repression as suggested in recent study that quantified signals of mismatch repair through immunolocalization of MLH1 (Bikchurina et al. 2018).
Using this genetic map, we then tested for QTL associated with variation in BC embryo and placenta weights (Figure 2). We observed a single QTL of large effect for placental weight on the X chromosome, with 52.3% of the observed variation in BC placental weight determined by the genotype of the maternally inherited X chromosome (Figure 2C). We estimated a QTL peak at 31.1 cM with a 95% Bayesian confidence interval between 29.6 and 32.6 cM. This X-linked QTL localized near the proximal boundary of where we also observed repressed recombination (Supplementary Figure S4), although the entire X chromosome exceeded a permutation-based significance threshold (P = 0.01). Male and female embryos inheriting a maternal P. sungorus X chromosome genotype at this QTL showed an ∼60% increase in average placenta weight (F1,179 = 178.4, P < 0.0001, ANOVA; Figure 2C inset). No additional placental QTL was uncovered when using sex, developmental stage, and litter size as covariates, nor when using the X-linked QTL as a cofactor.
Figure 2.
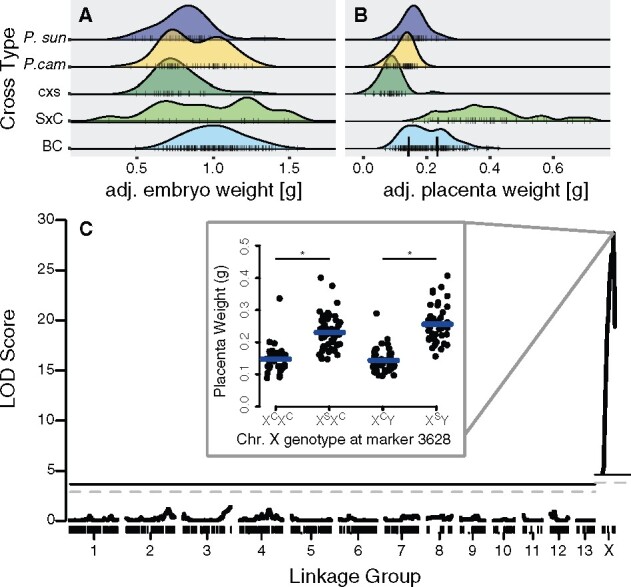
Phenotype distributions and QTL for placental size. Late term embryo size adjusted for Theiler stage and edema (A) and placenta size adjusted for Theiler stage (B) for P. sungorus, P. campbelli, reciprocal F1s and 189 BC conceptuses. (C) Genetic mapping revealed a single QTL for placenta weight on the X chromosome. The P. sungorus X chromosome increases placenta weight by ∼60% (inset, F1,179 = 178.4, P ≪ 0.0001, stars indicate significant differences assigned by a Tukey’s HSD test). Placenta weights were grouped by the genotype at marker 3,628. Genotypes are denoted with the maternal allele first followed by the paternal allele. Significance thresholds are denoted by solid (P = 0.01) and dashed (P = 0.05) horizontal lines.
No QTL for embryo weight was recovered in our experiment (P = 0.05 permutation-based significance threshold; Supplementary Figure S5), despite considerable variation in BC embryo weights (Figure 2A) and significant overgrowth of S × C embryos when compared to normally developing cross-types (Brekke and Good 2014). Severe embryonic swelling or edema is common in S × C hybrids and appears to drive overall differences in embryo weights between overgrown S × C hybrids and either species or c × s hybrids (stepwise best model: embryo weight ∼ developmental stage + edema; adjusted r2 = 0.40, F2,182 = 61.6, P ≪ 0.0001; adjusted embryo weight ∼ cross-type F3,182 = 0.74, P = 0.5312, ANOVA; Supplementary Figure S6). However, BC placenta and embryo weights remained moderately correlated after adjusting for developmental stage and edema (adjusted r2 = 0.159, F1,184 = 36.0, P ≪ 0.0001, ANOVA; Supplementary Figure S7A), with males showing a stronger correlation than females (males: adjusted r2 = 0.257, F1,95 = 33.8, P ≪ 0.0001; females: adjusted r2 = 0.065, F1,88 = 7.15, P = 0.0090, ANOVA). When we expanded our analysis of embryonic weights among genotypes to include the BC, we also detected a small but significant increase in embryo weight in the overgrown crosses after controlling for age and edema (adjusted r2 = 0.176, F4,376 = 21.1, P ≪ 0.0001, ANOVA; Supplementary Figure S7B). These apparent differences in embryonic growth were likely too subtle to detect in our QTL mapping experiment.
Networks of placental gene expression in first-generation reciprocal hybrids
We previously demonstrated extensive parent-of-origin dependent disruption of hybrid gene expression, with hundreds of genes significantly up- or downregulated in overgrown S × C hybrid placenta relative to both species (i.e., transgressive expression; Brekke et al. 2016). In this previous study, examination of allele-specific expression revealed 88 candidate imprinted genes in the placenta overall, with 79 genes showing strong bias toward maternal expression (i.e., paternally imprinted). Notably, 68% of candidate imprinted genes (60 genes) showed transgressive expression in overgrown hybrids, suggesting a link between misexpression of autosomal genes with biased parent-of-origin expression and placental overgrowth. Imprinted XCI was not disrupted in F1 hybrids. In contrast to a predominantly autosomal regulatory phenotype in F1 hybrid placenta, our BC experiments indicated that S × C F1 hybrid placental overgrowth was likely caused by genetic incompatibilities exposed on the maternally inherited P. sungorus X chromosome (Figure 2C).
Motivated by these parallel observations, we sought to identify groups of co-expressed genes associated with overgrowth phenotypes exposed in both of our first- and second-generation hybrid models of placental overgrowth. We first used our published late-term placental expression data from the parental species and reciprocal F1 hybrids to construct weighted gene co-expression networks, removing one female S × C sample during filtering (n = 39 placental transcriptomes, 5 males and 5 females per cross-type; Supplementary Figure S8). We placed 11,392 genes (including 70 candidate imprinted genes) into 29 signed clusters, or “modules,” of nonoverlapping gene sets. For each module, expression values were summarized with an “eigengene,” or the principal component capturing the largest proportion of the variance in gene expression. We then assessed each module for mode of eigengene inheritance and association with placental phenotypes (Supplementary Table S2 and Supplementary Figure S9). Two key gene networks emerged from this analysis. One module was comprised of 565 genes that tended to be highly expressed in S × C hybrid placenta relative to all other genotypes. The eigengene value for this module was positively correlated with placental weights (Figure 3A), and included only one candidate imprinted gene (Figure 3B).
Figure 3.

F1 placenta size-associated gene expression modules. Eigengene gene expression values summarize the group of genes that are upregulated (A and B) and downregulated (C and D) in overgrown S × C placentas, shown as differing by cross-type (transgressive expression; stars indicate significance groups at P < 0.05 based on a Tukey’s HSD test) and placental size (association with hybrid incompatibility phenotype controlling for Theiler stage and sex, P < 0.001, ANOVA, see Supplementary Table S2). The upregulated F1 module was not enriched for imprinted genes (C; total genes, N = 565; imprinted genes, N = 1), whereas the downregulated F1 module was enriched for highly connected candidate imprinted genes (Brekke et al. 2016) showing maternally biased expression (D; total genes, N = 1160; imprinted genes, N = 36). Data points rendered only for imprinted genes in (B and D).
The other module comprised 1,160 genes that tended to show a lower eigengene value in S × C hybrid placenta (Figure 3C). Expression of this downregulated set was negatively correlated with placental weights (Figure 3C), and included nearly half (44%) of the downregulated transgressive genes identified by Brekke et al. (2016). Eigengene values for this module exhibited a stronger parent-of-origin mode of inheritance than the upregulated set (Figure 3C), and was positively correlated with candidate imprinted gene expression (Figure 3D). The downregulated module included over 50% of the candidate placental imprinted genes overall (36 of 70 genes, P ≪ 0.0001; Supplementary Table S2). These findings mirror results from pairwise contrasts (Brekke et al. 2016) where overgrown S × C placentas showed an overall reduction in the expression levels of several putatively imprinted genes (Figure 4).
Figure 4.

Reduction in candidate imprinted gene expression in overgrown F1 and BC placentas. Each gray line represents the relative change in expression of a candidate imprinted gene between normal sized and large hybrid placentas. Genes were standardized by mean expression per gene (dotted line), and centered to display direction rather than magnitude of gene expression change. Both F1 and BC experiments demonstrate reduced gene expression of genes showing parent-of-origin bias accompanying placental overgrowth (grand mean indicated by colored line).
Networks of placental gene expression in second-generation backcrossed hybrids
To test for links between our BC QTL mapping experiment and emergent patterns of placental expression in F1 hybrid models, we next analyzed 24 transcriptomes from BC placentas (12 large and 12 normal sized placentas; 11,396 genes placed in the network). One large female placenta was removed during outlier filtering (Supplementary Figure S10). WGCNA analysis of the BC transcriptomes revealed seven modules that were correlated with placenta weights. No clusters were significantly associated with embryo weights after controlling for developmental stage and sex (Supplementary Table S3). The recombinant genotypes within this BC sample allow us to more clearly differentiate disrupted expression in overgrown hybrid placenta versus species differences (P. sungorus versus P. campbelli) or interspecific hybridization per se (hybrids versus parental species). Consistent with this, only about one-third of the core genes included in the up- and downregulated F1 modules were captured in the seven placenta-associated BC modules (563 of 1,725 genes). However, most of these overlapping genes (>90%, 513 of 563) were derived from the downregulated F1 S × C module (Supplementary Table S3). The BC module that was most strongly associated with placental weights (432 genes) tended to show lower summary expression in overgrown BC placenta (Figure 5A). This downregulated BC module was also highly enriched for autosomal imprinting (33 candidate imprinted genes, binomial exact test P ≪ 0.0001) and for genes in the downregulated F1 S × C module (206 genes, binomial exact test P ≪ 0.0001; Figure 4; Supplementary Table S3 and Supplementary Figure S11). In concordance with the overlap between gene lists, genes that were positively correlated with the F1 downregulated module eigengene were also positively correlated with the BC downregulated module eigengene (Supplementary Figure S12), indicating module conservation across first- and second-generation hybrids.
Figure 5.

BC downregulated module is associated with placenta size and candidate imprinted genes. (A) Summary expression of the BC downregulated module was significantly associated with placenta size controlling for Theiler stage and sex (P < 0.001, ANOVA, see Supplementary Table S3). (B) This module was enriched for highly connected, candidate imprinted genes (µimprinted=0.8489 ± 0.015; µnon imprinted=0.6370 ± 0.008; Welch’s two-sided t-test, t49.315 = 12.255, P < 0.0001; total genes, N = 432; imprinted genes, N = 33). Data points rendered only for imprinted genes. (C) The top 500 pairwise connections within this network largely involved the top 5% most connected genes, with interactions involving hub genes denoted by a thicker line and a solid black circumference notation at the interaction partner. Interactions involving a candidate imprinted gene are indicated in blue.
Connectivity within scale-free expression networks is commonly defined by the extent to which the expression level of a given gene is correlated with the expression of other genes. We found that candidate imprinted genes were much more highly connected than nonimprinted genes within the downregulated, placenta-associated BC module (Figure 5B). Co-expression modules are usually characterized by a few highly connected “hub” genes (Ghazalpour et al. 2006; Mack et al. 2019). We found that the top 5% most connected (hub) genes in the downregulated module were involved in 60% (300) of the top 500 pairwise interactions (Figure 5C, hub interactions indicated by thicker lines and larger circles). Candidate imprinted genes were highly overrepresented as hub genes in this network—one-third of hub genes (8 of 21 genes, binomial exact test P ≪ 0.0001) were candidate imprinted loci with maternally biased expression, including the top five most highly connected genes (Plxdc2, ProcR, Scara5, CD68, and Wnt4). Indeed, nearly half (238) of the top 500 pairwise correlations (Figure 5C, blue lines) involved at least one candidate imprinted gene (binomial exact test P ≪ 0.0001). These highly connected, downregulated genes represented many core biological functions of the placenta, ranging from broadly expressed genes involved in growth and development to those with specialized placental function (Table 1).
Table 1.
Hub genes in the downregulated, placenta-associated BC module
| Gene name | No. of interactions in top 500 | Module membership | Imprinting status | Function (UniProtKB) | Clotting/ angiogenesis | Immune | Growth and development | References |
|---|---|---|---|---|---|---|---|---|
| Plxdc2 | 32 | 0.9259 | Candidate | Cell surface signaling | Maybe | — | Yes | Cheng et al. 2014 |
| Procr | 28 | 0.9451 | Candidate | Cell surface signaling, blood coagulation | Yes | Yes | — | Sood et al. 2006; Bouwens et al. 2013 |
| Scara5 | 23 | 0.9218 | Candidate | Iron transport, blood coagulation | Yes | — | Yes | Li et al. 2009 |
| Cd68 | 20 | 0.9133 | Candidate | Immune function | — | Yes | — | Chistiakov et al. 2017 |
| Wnt4 | 20 | 0.9451 | Candidate | Developmental signaling | — | — | Yes | Sonderegger et al. 2010; Knöefler and Pollheimer 2013 |
| Ppard | 19 | 0.9181 | — | Transcription factor; steroid hormone receptor | — | — | Yes | Schmidt et al. 1992 |
| Adm | 18 | 0.9515 | — | Immune function | Yes | Yes | — | Li et al. 2013 |
| Boc | 18 | 0.9217 | — | Cell–cell adhesion, tissue differentiation | — | — | Yes | Zakaria et al. 2019 |
| Igsf11 | 17 | 0.8696 | — | Cell–cell adhesion, cell growth, neurogenesis | — | — | Yes | Jang et al. 2016 |
| Erv3 | 17 | 0.9463 | Candidate | Tissue identity and immunomodulation | — | Yes | Yes | Mangeney et al. 2007 |
| Tbc1d2b | 16 | 0.8934 | — | Protein modification and transport | — | — | Yes | Manshouri et al. 2019 |
| Olfml3 | 15 | 0.8884 | Candidate | Developmental signaling | Yes | — | — | Miljkovic-Licina et al. 2012 |
| Lmcd1 | 13 | 0.9115 | — | Transcription factor | — | — | Yes | Ferreira et al. 2019 |
| Fn3krp | 12 | 0.8801 | — | Protein modification/glycation | Maybe | Maybe | Yes | Karabag et al. 2007 |
| Numbl | 11 | 0.879 | — | Cell–cell adhesion, neurogenesis | — | — | Yes | Wilson et al. 2007 |
| Ipmk | 11 | 0.8049 | — | Lipid metabolism | — | — | Yes | Malabanan and Blind 2016 |
| F13a1 | 10 | 0.888 | — | Metal binding, blood coagulation | Yes | — | — | Muszbek et al. 2011 |
| Pdpn | 10 | 0.8987 | — | Cell–cell adhesion, developmental signaling | — | Yes | — | Astarita et al. 2012 |
| Larp6 | 10 | 0.824 | — | Collagen biosynthesis, fibrosis | — | — | Yes | Stefanovic et al. 2019 |
| Gypc | 10 | 0.8474 | — | Cell membrane stability, red blood cells | Yes | — | — | Wilder et al. 2009 |
| Fut4 | 10 | 0.8666 | — | Cell–cell adhesion, immune function | Maybe | Yes | — | Wang et al. 2013 |
The top 5% most connected genes in the downregulated module, with connection defined as the number of times the gene was included in the top 500 strongest pairwise correlations in gene expression between genes in the module. Gene imprinting status (Brekke et al. 2016) and functions are indicated.
Our transcriptome analyses revealed a central link between the X chromosome and the disruption of autosomal regulatory pathways in the placenta. To integrate our expression and placental phenotypes more directly, we next tested for QTL that explained expression variation in the overall module eigengene. Despite a small sample size (n = 23), the X chromosome was a significant predictor of the downregulated BC module expression after permutation (Supplementary Figure S13A). In principle, this signal could represent a predominant contribution of X-linked genes to the eigengene summary of expression within this parent-of-origin module, or a genome-wide trans-regulatory signal dependent on the species origin of the X chromosome. Consistent with the latter hypothesis, only ∼6% of genes in downregulated BC module were X-linked (25 of 432 genes, binomial exact test P = 0.006, not significant after correction for multiple testing). These genes were significantly under-represented in the correlation network with only 7 of the top 500 pairwise correlations including an X-linked gene (56 pairs expected; binomial exact test P ≪ 0.0001). It is also possible that the strong X-linked signal could be a correlated side effect of overall placental phenotypes on expression levels caused by X-linked hybrid incompatibilities. However, we found no significant QTL for the six other expression modules correlated with placental weight (Supplementary Figure S13, B and C), thus this signal seems unlikely to be a consequence of a spurious phenotypic correlation.
Integration of the placental transcriptome onto the Phodopus genetic map
These results strongly support the hypothesis that X-linked incompatibilities interacting with a hybrid autosomal background are the primary determinant of disrupted autosomal expression observed in both F1 and BC hybrids, but the architecture of underlying X-autosome incompatibilities remain unresolved. The current genome assembly for dwarf hamsters is highly fragmented and has not been arranged into an ordered physical map (Bao et al. 2019), limiting our ability to fully integrate our transcriptome and quantitative genetic analyses. To begin to overcome these limitations, we used a custom exon capture experiment to anchor 3,616 placenta-expressed genes onto the Phodopus genetic map, including 159 X-linked and 34 autosomal imprinted genes (Supplementary Table S13, Supplementary Figure S14). An additional 212 X-linked genes were identified based on patterns of inheritance and orthology with mouse, but were not ordered on the genetic map (371 X-linked genes total). We placed approximately one-third of the BC downregulated network genes (162 of 432 genes) on to the genetic map, including 17 of the 21 hub genes (Table 1). Genes in this anchored network were distributed across 12 of the 13 autosomes and the X chromosome (Supplementary Figure S15).
A scan for genetic interactions that influence gene expression
If there was a hybrid interaction between the X chromosome and a specific diploid genotype at an autosomal locus that influenced the expression of that gene, then individuals that inherited a maternal P. sungorus X should show a larger change in gene expression for one of the autosomal genotypes than for the other. To test for such interactions, we calculated EI scores by comparing the fold change difference in expression between the two possible autosomal genotypes of all mapped genes (i.e., homozygous for the P. campbelli allele or heterozygous) dependent on the genotype of the maternal X chromosome (P. campbelli or P. sungorus). Mapped genes from the BC downregulated module (n = 124) showed higher EI scores on average when compared to genes not placed in any WGCNA module (µdownregulated=0.229 ± 0.008, µnull=0.165 ± 0.006, F1,870 = 14.13, P = 0.0002, ANOVA). Next, we used a linear model to assess the relationship between gene connectivity and EI score in the downregulated module. Hub genes disproportionately overlap with male sterility loci in hybrid mice (Morgan et al. 2020), suggesting that incompatibilities may be more common among highly connected genes. However, we found support for only a slight increase in EI score for genes that were the most connected to the module (EI ∼ module correlation, adjusted r2 = 0.0243, F1,122 = 4.063, P = 0.046; Figure 6A). Interestingly, much of the signal for increased EI scores appeared to be driven by candidate imprinted genes rather than hub gene status per se (EI ∼ candidate imprinting status, adjusted r2 = 0.059, F1,122 = 8.75, P = 0.0037; EI ∼ hub, F1,122 = 0.005, P = 0.942; Figure 6B and Supplementary Figure S16), again underscoring the central role of genes with maternally biased expression within the downregulated module. One such candidate imprinted (nonhub) gene was Tfpi2 (Figure 6C). Individuals with an interspecific mismatch between the maternally inherited X chromosome (P. sungorus) and maternal allele at Tfpi2 (P. campbelli) showed more than a twofold (2.26×) decrease in expression compared to individuals with matching maternal genotypes.
Figure 6.

Expression interactions exposed in BC hybrids. (A) Greater disruption in gene expression with X chromosome–autosome mismatch (EI score) was weakly associated with genes more connected in the downregulated BC network module (F1,122 = 4.063, P = 0.046, ANOVA). Shown are comparisons between the gray module (gray; genes passing filter, N = 733) and the downregulated BC module (blue; genes passing filter, N = 124). The gray module includes all genes that were not placed in any module in the network analysis, and was used to generate a null expectation for the distribution of the EI score. (B) Increased EI score was explained by candidate imprinted genes, rather than highly connected hub genes (F1,122 = 8.75, P = 0.0037, ANOVA; downregulated BC module not imprinted nonhub genes, N = 102; imprinted nonhub genes, N = 9; not imprinted hub genes, N = 8; imprinted hub genes, N = 5). (C) EI expression pattern is illustrated with top gene Tfpi, where mismatch between maternal X genotype and maternal Tfpi genotype results in an average reduction in gene expression in addition to inheritance of a maternal P. sungorus X chromosome. Gene expression is reported as counts per million (CPM) transcripts. Letters and star indicate significant differences at P < 0.01, assigned by a Tukey’s HSD test.
We next polarized EI scores to evaluate specific X-autosome combinations with the downregulated module for candidate hybrid incompatibilities. Within the BC architecture, hybrid incompatibilities involving autosomal recessive or imprinted genes could manifest when the maternally inherited P. sungorus X chromosome was combined with maternally inherited P. campbelli autosomal alleles (all paternal alleles were P. campbelli). Consistent with this prediction, we observed positive polarized EI scores for imprinted genes within the downregulated module demonstrating that these EIs were driven largely by mismatches between the maternal autosomal and X chromosome genotypes (P = 0.043, Tukey’s HSD test, nonhub genes, imprinted vs. null module; Supplementary Figure S17A). Furthermore, we found that maternally mismatched imprinted genes in the downregulated module showed larger fold changes when compared to nonimprinted genes regardless of their status as a hub gene (P = 0.045, hub imprinted vs. nonimprinted; P < 0.0001, nonhub imprinted vs. nonimprinted, Tukey’s HSD test; Supplementary Figure S17B). Collectively, these patterns suggest that imprinted autosomal genes with biased maternal expression within the downregulated module are more likely to be involved in incompatible hybrid interactions with the maternal X chromosome.
Finally, we tested for parent-of-origin bias in expression for candidate imprinted genes in the BC. We found 31 genes previously identified as showing either maternal or paternal bias in expression in F1 placentas (Brekke et al. 2016) with expression data for at least seven heterozygous individuals in the BC. Of these, 22 genes show significant parent-of-origin bias in expression in the direction shown previously (Supplementary Figure S18, one-sample t-test, Bonferroni corrected P < 0.05). Although limited to a subset of BC individuals, these results suggest that imprinting status was at least partially maintained at many genes despite large changes in overall expression levels. We also confirmed a strong bias in expression of the maternal allele on the X chromosome (adjusted r2 = 0.17, F1,3560 = 930.3, P ≪ 0.0001; Supplementary Figure S19), indicating that paternal imprinted XCI is likely intact in BC females as has been previously reported for F1 females (Brekke et al. 2016).
Discussion
By combining quantitative genetic mapping of placental overgrowth with transcriptomic data, we uncovered genome-wide networks of gene expression that were disrupted as a consequence of incompatible genetic interactions with the X chromosome. These data indicate that genetic interactions between the X chromosome and networks of imprinted and nonimprinted autosomal genes are critical for proper placental development in dwarf hamsters. Below we compare our results with findings from other systems to examine the contribution of the X chromosome to the accumulation of reproductive barriers between species and the evolution of placental gene expression.
The evolution of hybrid placental overgrowth
A positive parent-of-origin correlation between hybrid placental and embryonic growth effects has been observed in some mammal hybrids (e.g., Vrana et al. 2000; Brown et al. 2012), providing a possible mechanistic link between the disruption of early development and extreme adult parent-of-origin growth effects observed in many mammal hybrids (Brekke and Good 2014). Our quantitative results underscore that the relationship between placental and embryonic growth may be weak early in development, especially given the difficulty of differentiating the effects of edema and other developmental defects from overall embryonic growth (Supplementary Figures S6 and S7). Consistent with this, parent-of-origin placental growth effects described in some mouse (Zechner et al. 1996; Kurz et al. 1999) and equine hybrids (Allen et al. 1993) also were not strongly correlated with hybrid embryonic growth.
Regardless of the unclear functional relationship between gross placental and embryonic growth phenotypes, our results show that the species origin of the maternal X chromosome is the major genetic factor responsible for placental overgrowth in Phodopus hybrids. Reproductive barriers between species often evolve through negative epistatic interactions between two or more loci, generally referred to Dobzhansky–Muller incompatibilities (DMIs; Dobzhansky 1937; Muller 1942; Orr and Turelli 2001, see also Bateson 1909). DMIs disproportionately involve X-linked loci, a phenomenon known as the large X-effect (Coyne and Orr 1989b). Hybrid incompatibilities presumably occur between the maternally inherited P. sungorus X chromosome and P. campbelli autosomal loci (e.g., dominant, recessive, or imprinted incompatibilities). However, paternal loci were invariant in our BC and thus paternal contributions to putative X-autosome mismatches could not be mapped in this experiment. Large-X effects for disrupted placental development are perhaps not surprising given the central role that the X chromosome tends to play in the evolution of reproductive isolation (Coyne and Orr 1989b; Turelli and Orr 2000; Masly and Presgraves 2007; Turelli and Moyle 2007) and the parent-of-origin dependent nature of placental hybrid inviability in dwarf hamsters (Figure 2). However, we had previously associated placental overgrowth with widespread disruption of autosomal gene expression (Brekke et al. 2016); a regulatory phenotype that could manifest entirely from hybrid incompatible interactions between imprinted autosomal genes. Our mapping results rule out this possibility.
Large effect X-linked QTL also underlie placental overgrowth in hybrid deer mice (Peromyscus maniculatus × P. polionotus; Vrana et al. 2000) and house mice (Mus spretus × M. musculus; Zechner et al. 1996; Hemberger et al. 1999). In all three rodent systems, imprinted XCI occurs in the placenta and incompatibilities on the maternally inherited X chromosome emerge as a central genetic determinant of placental overgrowth in hybrids. However, broader connections between these developmental phenotypes and the disruption of placental gene expression have remained unclear. Studies in deer mice have linked the X chromosome with disrupted imprinted placental pathways, including a putative interaction with the autosomal maternally imprinted gene Peg3 (Loschiavo et al. 2007). X-linked incompatibilities are also the primary cause of hybrid placental overgrowth growth in some hybrid crosses of house mice (M. spretus × M. musculus; Zechner et al. 1996; Hemberger et al. 1999), but no direct link between disrupted expression of candidate imprinted genes and placental overgrowth has been established (Shi et al. 2004; Zechner et al. 2004). However, a recent genome-wide study on the same Mus hybrid system showed transgressive autosomal expression in undergrown hybrid placentas (reciprocal F1 crosses were not performed), including disruption of the imprinted Kcnq1 cluster (Arévalo et al. 2021). In these experiments, males showed more severe placental undergrowth and disrupted gene expression, which the authors proposed may involve interactions with the imprinted X chromosome (Arévalo et al. 2021). Artificial insemination experiments between more divergent Mus species pairs (M. musculus × M. caroli) resulted in massively abnormal placenta showing local demethylation and overexpression of an X-linked retroelement (Brown et al. 2012). Similarly, loss of genomic imprinting has been correlated with overgrowth in fetuses derived from assisted reproduction between divergent cattle breeds (Chen et al. 2015, 2016). Most of these works have focused on F1 crosses or candidate gene approaches, limiting further insights into the role that X-autosome interactions play in the broader disruption regulatory pathways in hybrid placenta. Building on these previous studies, we show that X-linked hybrid incompatibilities underlie the disruption of placental growth, with widespread effects on the misexpression of imprinted autosomal pathways.
Gene expression plays a central role in organismal development and morphological evolution (King and Wilson 1975; Carroll 2008; Sears et al. 2015), but overall importance of regulatory incompatibilities to species formation has remained unclear (Butlin et al. 2012; Guerrero et al. 2016). In mammals, progress has been made in linking disruption of specific epigenetic regulatory mechanisms on the X chromosome (e.g., meiotic sex chromosome inactivation) to the evolution of hybrid male sterility during the relatively early stages of speciation (Bhattacharyya et al. 2013; Campbell et al. 2013; Davis et al. 2015; Larson et al. 2017). However, it has remained unclear if other developmental pathways are also predisposed to disruption in animal hybrids (Coyne and Orr 2004; Brekke and Good 2014). We suggest that placental development may represent a second developmental hotspot for the evolution of postzygotic reproductive isolation through the widespread disruption of gene expression.
There are several interesting parallels between the evolution hybrid male sterility and abnormal hybrid placental development in mammals. The X chromosome appears to play a central role in the genetic basis of both hybrid male sterility (Storchová et al. 2004; Good et al. 2008; Davis et al. 2015) and placental overgrowth (Figure 2; Zechner et al. 1996; Hemberger et al. 1999; Vrana et al. 2000). Both forms of postzygotic isolation manifest during developmental stages where the X chromosome is subject to chromosome-wide epigenetic silencing. Whereas meiotic sex chromosome inactivation may often be disrupted in the case of hybrid male sterility (Lifschytz and Lindsley 1972; Bhattacharyya et al. 2013; Larson et al. 2017), imprinted XCI appears to be maintained in hybrid hamster placentas (Supplementary Figure S19; Brekke et al. 2016). Although the specific mechanisms underlying abnormal placental development are yet to be determined, tissues with imprinted XCI may select for the evolution of co-adapted epistatic networks of gene expression that are subsequently prone to disruption in hybrids (see below). Finally, aspects of placental development and spermatogenesis are both thought to evolve rapidly in response to genomic conflict (Haig 2000; Larson et al. 2018), which may contribute to the rapid evolution of hybrid incompatibilities (Crespi and Nosil 2013). It is well-established that hybrid male sterility tends to evolve rapidly in animals (Coyne and Orr 1989a; Wu et al. 1996), while hybrid growth effects have been detected across a broad range of evolutionary divergence in mammals (Brekke and Good 2014). All three systems where X-linked placental incompatibilities have been found (Phodopus, Mus, Peromyscus) involve crosses between relatively closely related species pairs (Brekke and Good 2014), suggesting that placental hybrid inviability can evolve rapidly and contribute to the early stages of reproductive isolation.
Through analysis of these parallel systems of hybrid sterility and inviability, a trend is emerging where sex chromosome evolution and genetic conflict within regulatory systems appears to fuel divergence within these key developmental processes (Crespi and Nosil 2013; Larson et al. 2018), ultimately leading to the formation of reproductive barriers between species. Striking parallels also exist in plants, where hybrid seed inviability also evolves rapidly (Garner et al. 2016; Coughlan and Matute 2020) and has been linked to the intensity of parental conflict (Coughlan et al. 2020) and the disruption of imprinted gene expression in the extra-embryonic endosperm (Wolff et al. 2015).
The evolution of placental gene expression networks
In both rodents and humans, fetal-derived trophoblast cells shape the vasculature at the maternal–fetal interface, allowing for nutrient transport and immune modulation (Gris et al. 2019). Notably, nine of the most connected genes in the BC downregulated module had functions related to coagulation and/or angiogenesis. Endothelial protein C receptor (ProcR) is an important anti-coagulant receptor in the trophoblast coagulation cascade (Bouwens et al. 2013). Allelic variants that result in under-expression of ProcR are associated with fetal loss in humans (Cochery-Nouvellon et al. 2009), and there is some evidence that maternal and fetal ProcR genotypes can interact to either prevent or induce placenta-mediated adverse pregnancy outcomes (Sood et al. 2006). In a healthy rodent placenta, the coagulation initiating tissue factor is counterbalanced by anti-coagulation proteins produced in differentiated syncytiotrophoblast tissue (Sood et al. 2006). Development fails without the early expression of the coagulation cascade in the placenta (Isermann et al. 2003). However, low levels of anticoagulants later in development are associated with preeclampsia and pregnancy loss (Ebina et al. 2015).
Several of the hub genes (Table 1) are known to contribute to differentiation of placental layers. For example, Wnt signaling is broadly important in placentation and embryonic development, and Wnt4 specifically may be involved in signaling between the fetal and maternal placental layers (Sonderegger et al. 2010; Knöefler and Pollheimer 2013). Another such specialized hub gene, Erv3, is part of a family of genes co-opted from endogenous retroviruses and are involved in immunomodulation, fusion, and differentiation of trophoblasts (Mangeney et al. 2007), and are increasingly recognized for their role in regulating placental gene expression (Pavlicev et al. 2015; Chuong 2018). Similarly, the Plexin domain containing 2 gene (Plxdc2) encodes an endothelial cell-surface transmembrane receptor (Cheng et al. 2014) that is often co-expressed with Wnt signaling genes (Miller et al. 2007). Other candidate hub genes play roles in cell–cell adhesion and differentiation (Wilson et al. 2007; Jang et al. 2016; Zakaria et al. 2019), immune function (Astarita et al. 2012; Li et al. 2013; Wang et al. 2013; Chistiakov et al. 2017), nutrient metabolism and delivery (Schmidt et al. 1992; Li et al. 2009; Malabanan and Blind 2016), and transcriptional regulation (Ferreira et al. 2019).
Overall, our expression data suggest a strong connection between placental overgrowth, the maternally expressed (P. sungorus) X chromosome, and the imprinted expression of autosomal genes. Our candidate imprinted gene set included several genes known to be maternally (e.g., Igf2, Mest, and Peg3) or paternally imprinted (e.g., Axl, H19, Tfpi2, and Wt1) in mice, as well as several novel candidates including most of the hub genes (Table 1). Confirmation that these candidates reflect the evolution of novel parent-of-origin epigenetic silencing in Phodopus (e.g., through DNA methylation or other mechanisms) awaits detailed functional validation beyond the scope of the current study. Others have argued that contamination of maternal blood or tissue may often bias patterns of allele-specific expression in the post-embryonic placenta (Wang et al. 2011, but see Finn et al. 2014). We previously found little evidence for extensive maternal contamination of dissected placental tissue in Phodopus (i.e., genome-wide paternal: maternal allele ratios were ∼1:1 in overgrown S × C placentas), but it is possible that maternally biased expression of some of these candidates reflects a large maternal contribution to overall placental expression levels. Indeed, hub genes such as Wnt4 are thought to be directly involved in signaling between the fetal and maternal placental layers (Sonderegger et al. 2010; Knöefler and Pollheimer 2013). Regardless of the underlying regulatory mechanisms—epigenomic imprinting or fetal–maternal transcript sharing—our results suggest that X-linked and autosomal genes with maternally biased expression play a central role in the evolution of placental development and the disruption of placental pathways in hybrids.
The existence of placental networks of maternally biased gene expression is consistent with some predictions of the co-adaptation theory of gene expression, whereby maternal expression at one gene can select for maternally biased expression at other positively interacting genes (Wolf and Hager 2006; Wolf 2013; Wolf and Brandvain 2014; O'Brien and Wolf 2017). Such a co-evolutionary process should result in the broader integration of imprinted gene networks, the evolution of separate co-expressed networks of maternally and paternally expressed genes, and the exposure of epistatic DMIs in second- (or later) generation hybrids with recombinant genotypes (Wolf and Brandvain 2014; Patten et al. 2016). We uncovered evidence for many such interactions in our preliminary screen for genetic interactions that influence gene expression levels in BC placenta (Figure 6; Supplementary Figures S16 and S17). For example, the maternally expressed Tfpi2 gene showed a greater than twofold decrease in expression when a maternally inherited P. campbelli allele was combined with a maternally inherited P. sungorus X chromosome (Figure 6C). Tfpi2 is imprinted in the placenta where its expression may limit trophoblast invasion (Jin et al. 2001) and also downregulated in several types of cancer (i.e., a potential tumor suppressor; Konduri et al. 2001; Ribarska et al. 2010). More data are needed to determine if this and other genes showing significant EIs contribute directly to abnormal placental growth phenotypes in hybrids.
More generally, we propose that the X chromosome is likely to play a central role in the evolution of maternally biased placental networks in many eutherian mammals. The strength of this prediction is dependent on patterns of X inactivation in the placenta and other extra-embryonic tissues of females as males only have a maternally derived X. The paternal X chromosome appears to be silenced in the placenta and other extra-embryonic tissues in at least four genera of rodents (i.e., Mus, Takagi and Sasaki 1975; Rattus, Wake et al. 1976; Phodopus, Brekke et al. 2016; Peromyscus, Vrana et al. 2000), resulting in predominantly maternal expression of X-linked genes in the placenta of males and females (Dupont and Gribnau 2013; Lee and Bartolomei 2013). Imprinted XCI is expected to contribute the vast majority of maternally expressed genes in the rodent placenta, which as a consequence should favor the evolution of maternal expression at interacting autosomal genes. In contrast, XCI appears to be random in extra-embryonic tissues of humans (Moreira De Mello et al. 2010), cattle (Chen et al. 2016), pigs (Zou et al. 2019), horses (Wang et al. 2012), and possibly rabbits (Okamoto et al. 2011). The more frequent occurrence of random XCI in this limited sample suggests that imprinted XCI may have evolved more recently in rodent extraembryonic tissues (Okamoto et al. 2011), and therefore may not apply broadly across the radiation of placental mammals. However, it is worth noting that rodents comprise over 40% of all placental mammal species (Burgin et al. 2018). The current sample of XCI in extraembryonic tissues is also biased toward a just few major lineages (i.e., ungulates, primates, rodents, and lagomorphs) and thus insufficient for accurate reconstruction of the ancestral state of extra-embryonic XCI in placental mammals. Moreover, male hemizygosity in species with random XCI may still favor the evolution of maternal expression at interacting autosomal genes under some conditions (Wolf and Brandvain 2014). In addition, the physiological integration of maternal blood supply and trophoblast-generated fetal vasculature is a particularly compelling biological context that could favor the evolution of coordinated maternal–fetal gene expression networks, regardless of the pattern of XCI. Given our data and these general theoretical predictions, the broader relevance of X-autosomal gene expression networks to placental evolution and development warrants further consideration.
Data Availability
All raw sequencing reads are archived at NCBI under BioProject PRJNA306772. Accession numbers for individual libraries are provided in Supplementary Table S5. Supplementary material is available at figshare: https://doi.org/10.25386/genetics.14171075.
Acknowledgments
Ryan Bracewell, Zak Clare-Salzler, Ted Cosart, Kris Crandell, Doug Emlen, Mafalda Ferreira, Lila Fishman, Evgueny Kroll, Lindy Henry, Matt Jones, Sara Keeble, Erica Larson, John McCutcheon, Colin Prather, Brice Sarver, Vanessa Stewart, Dan Vanderpool, Jon Velotta, Paul Vrana, and the UNVEIL network provided helpful comments on data analysis and interpretation. Kelly Carrick, Jess Wexler, and the University of Montana LAR staff for helping with animal care.
Funding
This research was supported by grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health (R01-HD073439, R01-HD094787 to J.M.G.), the National Science Foundation (EPSCoR OIA-1736249 to J.M.G. and Z.A.C.), a National Science Foundation Postdoctoral Fellowship in Biology (DBI-1612283 to S.C.S.), a National Science Foundation Doctoral Dissertation Improvement Grant (DEB-1406754 to T.D.B.), a Society for the Study of Evolution Rosemary Grant Award (to T.D.B.), and a David Nicholas Award (to T.D.B.). This study includes research conducted in the University of Montana Genomics Core, supported by a grant from the M. J. Murdock Charitable Trust.
Conflicts of interest
The authors declare no competing financial interests.
Literature cited
- Al Adhami H, Evano B, Le Digarcher A, Gueydan C, Dubois E, et al. 2015. A systems-level approach to parental genomic imprinting: the imprinted gene network includes extracellular matrix genes and regulates cell cycle exit and differentiation. Genome Res. 25:353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen WR, Skidmore JA, Stewart F, Antczak DF.. 1993. Effects of fetal genotype and uterine environment on placental development in equids. J Reprod Fertil. 98:55–60. [DOI] [PubMed] [Google Scholar]
- Arévalo L, Gardner S, Campbell P.. 2021. Haldane's rule in the placenta: sex-biased misregulation of the Kcnq1 imprinting cluster in hybrid mice. Evolution 75:86–100. [DOI] [PubMed] [Google Scholar]
- Astarita JL, Acton SE, Turley SJ.. 2012. Podoplanin: emerging functions in development the immune system and cancer. Front Immunol. 3:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babak T, DeVeale B, Tsang EK, Zhou YQ, Li X, et al. 2015. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat Genet. 47:544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao R, Onishi KG, Tolla E, Ebling FJP, Lewis JE, et al. 2019. Genome sequencing and transcriptome analyses of the Siberian hamster hypothalamus identify mechanisms for seasonal energy balance. Proc Natl Acad Sci USA. 116:13116–13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateson W. 1909. Heredity and variation in modern lights. In: Seward AC, editor. Darwin and Modern Science. Cambridge: Cambridge University Press. p. 85–101. [Google Scholar]
- Bhattacharyya T, Gregorova S, Mihola O, Anger M, Sebestova J, et al. 2013. Mechanistic basis of infertility of mouse intersubspecific hybrids. Proc Natl Acad Sci USA. 110:E468–E477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikchurina TI, Tishakova KV, Kizilova EA, Romanenko SA, Serdyukova NA, et al. 2018. Chromosome synapsis and recombination in male-sterile and female-fertile interspecies hybrids of the dwarf hamsters (Phodopus, Cricetidae). Genes 9:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwens EAM, Stavenuiter F, Mosnier LO.. 2013. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. J Thromb Haemost. 11:242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekke T, Henry L, Good J.. 2016. Genomic imprinting, disrupted placental expression, and speciation. Evolution 70:2690–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekke TD, Good JM.. 2014. Parent-of-origin growth effects and the evolution of hybrid inviability in dwarf hamsters. Evolution 68:3134–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekke TD, Steele KA, Mulley JF.. 2018. Inbred or outbred? Genetic diversity in laboratory rodent colonies. G3 (Bethesda). 8:679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekke TD, Supriya S, Denver MG, Thom A, Steele KA, et al. 2019. A high-density genetic map and molecular sex-typing assay for gerbils. Mamm Genome 30:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Sen S.. 2009. A Guide to QTL Mapping with R/Qtl. New York, NY: Springer. [Google Scholar]
- Brown JD, Piccuillo V, O'Neill RJ.. 2012. Retroelement demethylation associated with abnormal placentation in Mus musculus x Mus caroli hybrids. Biol Reprod. 86:88. [DOI] [PubMed] [Google Scholar]
- Burgin CJ, Colella JP, Kahn PL, Upham NS.. 2018. How many species of mammals are there? J Mamm. 99:1–14. [Google Scholar]
- Butlin R, Debelle A, Kerth C, Snook RR, Beukeboom LW, et al. 2012. What do we need to know about speciation? Trends Ecol Evol. 27:27–39. [DOI] [PubMed] [Google Scholar]
- Campbell P, Good JM, Nachman MD.. 2013. Meiotic sex chromosome inactivation is disrupted in sterile hybrid male house mice. Genetics 193:819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capellini I, Venditti C, Barton RA.. 2011. Placentation and maternal investment in mammals. Am Nat. 177:86–98. [DOI] [PubMed] [Google Scholar]
- Carroll SB. 2008. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134:25–36. [DOI] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA.. 2013. Stacks: an analysis tool set for population genomics. Mol Ecol. 22:3124–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Hagen DE, Elsik CG, Ji T, Morris CJ, et al. 2015. Characterization of global loss of imprinting in fetal overgrowth syndrome induced by assisted reproduction. Proc Natl Acad Sci USA. 112:4618–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Hagen DE, Wang JB, Elsik CG, Ji TM, et al. 2016. Global assessment of imprinted gene expression in the bovine conceptus by next generation sequencing. Epigenetics 11:501–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Zhong M, Kawaguchi R, Kassai M, Al-Ubaidi M, et al. 2014. Identification of PLXDC1 and PLXDC2 as the transmembrane receptors for the multifunctional factor PEDF. Elife 3:e05401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov DA, Killingsworth MC, Myasoedova VA, Orekhov AN, Bobryshev YV.. 2017. CD68/macrosialin: not just a histochemical marker. Lab Invest. 97:4–13. [DOI] [PubMed] [Google Scholar]
- Chuong EB. 2018. The placenta goes viral: retroviruses control gene expression in pregnancy. PLoS Biol. 16:e3000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochery-Nouvellon E, Chauleur C, Demattei C, Mercier E, Fabbro-Peray P, et al. 2009. The A6936G polymorphism of the endothelial protein C receptor gene is associated with the risk of unexplained foetal loss in Mediterranean European couples. Thromb Haemost. 102:656–667. [DOI] [PubMed] [Google Scholar]
- Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, et al. 2002. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 417:945–948. [DOI] [PubMed] [Google Scholar]
- Coughlan JM, Brown MW, Willis JH.. 2020. Patterns of hybrid seed inviability in the Mimulus guttatus sp. complex reveal a potential role of parental conflict in reproductive isolation. Curr Biol. 30:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan JM, Matute DR.. 2020. The importance of intrinsic postzygotic barriers throughout the speciation process. Philos Trans R Soc Lond B Biol Sci. 375:20190533. [10.1098/rstb.2019.0533] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Orr HA.. 1989a. Patterns of speciation in Drosophila. Evolution 43:362–381. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Orr HA.. 1989b. Two rules of speciation. In: Otte D, Endler J., editors. Speciation and Its Consequences. Sunderland, MA: Sinauer Associates. p. 180–207. [Google Scholar]
- Coyne JA, Orr HA.. 2004. Speciation. Sunderland, MA: Sinauer Associates, Inc. [Google Scholar]
- Crespi B, Nosil P.. 2013. Conflictual speciation: species formation via genomic conflict. Trends Ecol Evol. 28:48–57. [DOI] [PubMed] [Google Scholar]
- Davis BW, Seabury CM, Brashear WA, Li G, Roelke-Parker M, et al. 2015. Mechanisms underlying mammalian hybrid sterility in two feline interspecies models. Mol Biol Evol. 32:2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T. 1937. Genetics and the Origin of Species. New York, NY: Columbia University Press. [Google Scholar]
- Dupont C, Gribnau J.. 2013. Different flavors of X-chromosome inactivation in mammals. Curr Opin Cell Biol. 25:314–321. [DOI] [PubMed] [Google Scholar]
- Ebina Y, Ieko M, Naito S, Kobashi G, Deguchi M, et al. 2015. Low levels of plasma protein S, protein C and coagulation factor XII during early pregnancy and adverse pregnancy outcome. Thromb Haemost. 114:65–69. [DOI] [PubMed] [Google Scholar]
- Feenstra B, Skovgaard IM, Broman KW.. 2006. Mapping quantitative trait loci by an extension of the Haley-Knott regression method using estimating equations. Genetics 173:2269–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira DMS, Cheng AJ, Agudelo LZ, Cervenka I, Chaillou T, et al. 2019. LIM and cysteine-rich domains 1 (LMCD1) regulates skeletal muscle hypertrophy, calcium handling, and force. Skelet Muscle 9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn EH, Smith CL, Rodriguez J, Sidow A, Baker JC.. 2014. Maternal bias and escape from X chromosome imprinting in the midgestation mouse placenta. Dev Biol. 390:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamperl R, Vistorin G, Rosenkranz W.. 1977. New observations on karyotype of Djungarian hamster, Phodopus sungorus. Experientia 33:1020–1021. [DOI] [PubMed] [Google Scholar]
- Garner AG, Kenney AM, Fishman L, Sweigart AL.. 2016. Genetic loci with parent-of-origin effects cause hybrid seed lethality in crosses between Mimulus species. New Phytol. 211:319–331. [DOI] [PubMed] [Google Scholar]
- Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, et al. 2006. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2:e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Dean MD, Nachman MW.. 2008. A complex genetic basis to X-linked hybrid male sterility between two species of house mice. Genetics 179:2213–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris JC, Bouvier S, Cochery-Nouvellon E, Mercier E, Mousty E, et al. 2019. The role of haemostasis in placenta-mediated complications. Thromb Res. 181:S10–S14. [DOI] [PubMed] [Google Scholar]
- Guerrero RF, Posto AL, Moyle LC, Hahn MW.. 2016. Genome-wide patterns of regulatory divergence revealed by introgression lines. Evolution 70:696–706. [DOI] [PubMed] [Google Scholar]
- Haaf T, Weis H, Schmid M.. 1987. A comparative cytogenetic study on the mitotic and meiotic chromosomes in hamster species of the genus Phodopus (Rodentia, Cricetinae). Z Säugetierkd. 52:281–290. [Google Scholar]
- Haig D. 1996. Placental hormones, genomic imprinting, and maternal-fetal communication. J Evolution Biol. 9:357–380. [Google Scholar]
- Haig D. 2000. The kinship theory of genomic imprinting. Annu Rev Ecol Syst. 31:9–32. [Google Scholar]
- Haley CS, Knott SA.. 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity (Edinb). 69:315–324. [DOI] [PubMed] [Google Scholar]
- Heard E, Disteche CM.. 2006. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 20:1848–1867. [DOI] [PubMed] [Google Scholar]
- Hemberger M. 2002. The role of the X chromosome in mammalian extra embryonic development. Cytogenet Genome Res. 99:210–217. [DOI] [PubMed] [Google Scholar]
- Hemberger MC, Pearsall RS, Zechner U, Orth A, Otto S, et al. 1999. Genetic dissection of X-linked interspecific hybrid placental dysplasia in congenic mouse strains. Genetics 153:383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa R, Feil R.. 2010. Genomic imprinting and human disease. Essays Biochem Epigenet Disease Behav. 48:187–200. [DOI] [PubMed] [Google Scholar]
- Hudson QJ, Kulinski TM, Huetter SP, Barlow DP.. 2010. Genomic imprinting mechanisms in embryonic and extraembryonic mouse tissues. Heredity (Edinb). 105:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isermann B, Sood R, Pawlinski R, Zogg M, Kalloway S, et al. 2003. The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat Med. 9:331–337. [DOI] [PubMed] [Google Scholar]
- Ishishita S, Tsuboi K, Ohishi N, Tsuchiya K, Matsuda Y.. 2015. Abnormal pairing of X and Y sex chromosomes during meiosis I in interspecific hybrids of Phodopus campbelli and P. sungorus. Sci Rep. 5:9435–9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Oh D, Lee Y, Hosy E, Shin H, et al. 2016. Synaptic adhesion molecule IgSF11 regulates synaptic transmission and plasticity. Nat Neurosci. 19:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Udagawa K, Miyagi E, Nakazawa T, Hirahara F, et al. 2001. Expression of serine proteinase inhibitor PP5/TFPI-2/MSPI decreases the invasive potential of human choriocarcinoma cells in vitro and in vivo. Gynecol Oncol. 83:325–333. [DOI] [PubMed] [Google Scholar]
- Kaneko-Ishino T, Ishino F.. 2019. Evolution of viviparity in mammals: what genomic imprinting tells us about mammalian placental evolution. Reprod Fertil Dev. 31:1219–1227. [DOI] [PubMed] [Google Scholar]
- Karabag T, Kaya A, Temizhan A, Koc F, Yavuz S, et al. 2007. The influence of homocysteine levels on endothelial function and their relation with microvascular complications in T2DM patients without macrovascular disease. Acta Diabetol. 44:69–75. [DOI] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT - The BLAST-like alignment tool. Genome Res. 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khil PP, Smirnova NA, Romanienko PJ, Camerini-Otero RD.. 2004. The mouse X chromosome is enriched for sex-biased genes not subject to selection by meiotic sex chromosome inactivation. Nat Genet. 36:642–646. [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC.. 1975. Evolution at two levels in humans and chimpanzees. Science 188:107–116. [DOI] [PubMed] [Google Scholar]
- Knöfler M, Pollheimer J.. 2013. Human placental trophoblast invasion and differentiation: a particular focus on Wnt signaling. Front Genet. 4:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konduri SD, Tasiou A, Chandrasekar N, Rao JS.. 2001. Overexpression of tissue factor pathway inhibitor-2 (TFPI-2), decreases the invasiveness of prostate cancer cells in vitro. Int J Oncol. 18:127–131. [PubMed] [Google Scholar]
- Kurz H, Zechner U, Orth A, Fundele R.. 1999. Lack of correlation between placenta and offspring size in mouse interspecific crosses. Anat Embryol (Berl). 200:335–343. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S.. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. [10.1186/1471-2105-9-559] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson E, Vanderpool D, Keeble S, Dean M, Good J.. 2017. The composite regulatory basis of the large X-effect in mouse speciation. Mol Biol Evol. 34:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EL, Kopania EEK, Good JM.. 2018. Spermatogenesis and the evolution of mammalian sex chromosomes. Trends Genet. 34:722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Bartolomei MS.. 2013. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152:1308–1323. [DOI] [PubMed] [Google Scholar]
- Levy O. 2007. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol. 7:379–390. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Paragas N, Ned RM, Qiu A, Viltard M, et al. 2009. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev Cell 16:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Schwerbrock NMJ, Lenhart PM, Fritz-Six KL, Kadmiel M, et al. 2013. Fetal-derived adrenomedullin mediates the innate immune milieu of the placenta. J Clin Invest. 123:2408–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W.. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. [DOI] [PubMed] [Google Scholar]
- Lifschytz E, Lindsley DL.. 1972. The role of X-chromosome inactivation during spermatogenesis. Proc Natl Acad Sci USA. 69:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loschiavo M, Nguyen QK, Duselis AR, Vrana PB.. 2007. Mapping and identification of candidate loci responsible for Peromyscus hybrid overgrowth. Mamm Genome 18:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF. 1961. Gene action in X-chromosome of mouse (Mus musculus L.). Nature 190:372–373. [DOI] [PubMed] [Google Scholar]
- Mack KL, Phifer-Rixey M, Harr B, Nachman MW.. 2019. Gene expression networks across multiple tissues are associated with rates of molecular evolution in wild house mice. Genes 10:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan MM, Blind RD.. 2016. Inositol polyphosphate multikinase (IPMK) in transcriptional regulation and nuclear inositide metabolism. Biochem Soc Trans. 44:279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeney M, Renard M, Schlecht-Louf G, Bouallaga I, Heidmann O, et al. 2007. Placental syncytins: genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc Natl Acad Sci USA. 104:20534–20539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manshouri R, Coyaud E, Kundu ST, Peng DH, Stratton SA, et al. 2019. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat Commun. 10:5125.[10.1038/s41467-019-12832-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10. [Google Scholar]
- Masly JP, Presgraves DC.. 2007. High-resolution genome-wide dissection of the two rules of speciation in Drosophila. PLoS Biol. 5:e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw S, Oakes CC, Martel J, Cirio MC, de Zeeuw P, et al. 2013. Loss of DNMT1o disrupts imprinted X chromosome inactivation and accentuates placental defects in females. PLoS Genet. 9:e1003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M.. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010:pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- Miljkovic-Licina M, Hammel P, Garrido-Urbani S, Lee BPL, Meguenani M, et al. 2012. Targeting Olfactomedin-like 3 inhibits tumor growth by impairing angiogenesis and pericyte coverage. Mol Cancer Ther. 11:2588–2599. [DOI] [PubMed] [Google Scholar]
- Miller SFC, Summerhurst K, Runker AE, Kerjan G, Friedel RH, et al. 2007. Expression of Plxdc2/TEM7R in the developing nervous system of the mouse. Gene Expr Patterns 7:635–644. [DOI] [PubMed] [Google Scholar]
- Moreira de Mello JC, Souza de Araujo ES, Stabellini R, Fraga AM, Santana de Souza JE, et al. 2010. Random X inactivation and extensive mosaicism in human placenta revealed by analysis of allele-specific gene expression along the X chromosome. PLoS ONE 5:e10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan K, Harr B, White MA, Payseur BA, Turner LM.. 2020. Disrupted gene networks in subfertile hybrid house mice. Mol Biol Evol. 37:1547–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morison IM, Ramsay JP, Spencer HG.. 2005. A census of mammalian imprinting. Trends Genet. 21:457–465. [DOI] [PubMed] [Google Scholar]
- Muller HJ. 1942. Isolating mechanisms, evolution, and temperature. Biol Symp. 6:71–125. [Google Scholar]
- Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E.. 2011. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev. 91:931–972. [DOI] [PubMed] [Google Scholar]
- O'Brien EK, Wolf JB.. 2017. The coadaptation theory for genomic imprinting. Evol Lett. 1:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, Patrat C, Thepot D, Peynot N, Fauque P, et al. 2011. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 472:370–374. [DOI] [PubMed] [Google Scholar]
- Orr HA, Turelli M.. 2001. The evolution of postzygotic isolation: accumulating Dobzhansky-Muller incompatibilities. Evolution 55:1085–1094. [DOI] [PubMed] [Google Scholar]
- Patten MM, Cowley M, Oakey RJ, Feil R.. 2016. Regulatory links between imprinted genes: evolutionary predictions and consequences. Proc Biol Soc. 283:20152760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicev M, Hiratsuka K, Swaggart KA, Dunn C, Muglia L.. 2015. Detecting endogenous retrovirus-driven tissue-specific gene transcription. Genome Biol Evol. 7:1082–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE.. 2012. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasschaert RN, Bartolomei MS.. 2014. Genomic imprinting in development, growth, behavior and stem cells. Development 141:1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Constancia M, Fowden A, Anderson N, Dean W, et al. 2003. Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J Physiol. 547:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribarska T, Ingenwerth M, Goering W, Engers R, Schulz WA.. 2010. Epigenetic inactivation of the placentally imprinted tumor suppressor gene TFPI2 in prostate carcinoma. Cancer Genomics Proteomics 7:51–60. [PubMed] [Google Scholar]
- Robinson MD, Oshlack A.. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanenko SA, Volobouev VT, Perelman PL, Lebedev VS, Serdukova NA, et al. 2007. Karyotype evolution and phylogenetic relationships of hamsters (Cricetidae, Muroidea, Rodentia) inferred from chromosomal painting and banding comparison. Chromosome Res. 15:283–297. [DOI] [PubMed] [Google Scholar]
- Safronova LD, Cherepanova EV, Vasil'eva NY.. 1999. Specific features of the first meiotic division in hamster hybrids obtained by backcrossing Phodopus sungorus and Phodopus campbelli. Russ J Genet. 35:184–188. [PubMed] [Google Scholar]
- Sanli I, Feil R.. 2015. Chromatin mechanisms in the developmental control of imprinted gene expression. Int J Biochem Cell Biol. 67:139–147. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Endo N, Rutledge SJ, Vogel R, Shinar D, et al. 1992. Identification of a new member of the steroid-hormone receptor superfamily that is activated by a peroxisome proliferator and fatty-acids. Mol Endocrinol. 6:1634–1641. [DOI] [PubMed] [Google Scholar]
- Scribner SJ, Wynne-Edwards KE.. 1994. Disruption of body temperature and behavior rhythms during reproduction in dwarf hamsters (Phodopus). Physiol Behav. 55:361–369. [DOI] [PubMed] [Google Scholar]
- Sears KE, Maier JA, Rivas-Astroza M, Poe R, Zhong S, et al. 2015. The relationship between gene network structure and expression variation among individuals and species. PLoS Genet. 11:e1005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Lefebvre L, Yu Y, Otto S, Krella A, et al. 2004. Loss-of-imprinting of Peg1 in mouse interspecies hybrids is correlated with altered growth. Genesis 39:65–72. [DOI] [PubMed] [Google Scholar]
- Sonderegger S, Pollheimer J, Knöfler M.. 2010. Wnt signalling in implantation, decidualisation and placental differentiation - review. Placenta 31:839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood R, Kalloway S, Mast AE, Hillard CJ, Weiler H.. 2006. Fetomaternal cross talk in the placental vascular bed: control of coagulation by trophoblast cells. Blood 107:3173–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic B, Manojlovic Z, Vied C, Badger CD, Stefanovic L.. 2019. Discovery and evaluation of inhibitor of LARP6 as specific antifibrotic compound. Sci Rep. 9:326. [10.1038/s41598-018-36841-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchová R, Gregorová S, Buckiová D, Kyselová V, Divina P, et al. 2004. Genetic analysis of X-linked hybrid sterility in the house mouse. Mamm Genome 15:515–524. [DOI] [PubMed] [Google Scholar]
- Storey JD, Bass AJ, Dabney A, Robinson D.. 2019. qvalue: Q-value estimation for false discovery rate control. R package version 2.18.0. http://github.com/jdstorey/qvalue
- Takagi N, Sasaki M.. 1975. Preferential inactivation of paternally derived X-chromosome in extraembryonic membranes of mouse. Nature 256:640–642. [DOI] [PubMed] [Google Scholar]
- Theiler K. 1972. The House Mouse: Development and Normal Stages from Fertilization to 4 Weeks of Age. New York, NY: Springer-Verlag. [Google Scholar]
- Turelli M, Moyle LC.. 2007. Asymmetric postmating isolation: darwin's corallary to Haldane's rule. Genetics 176:1059–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Orr HA.. 2000. Dominance, epistasis and the genetics of postzygotic isolation. Genetics 154:1663–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, et al. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11–33. [10.1002/0471250953.bi1110s43] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, et al. 2006. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell 11:711–722. [DOI] [PubMed] [Google Scholar]
- Vrana PB. 2007. Genomic imprinting as a mechanism of reproductive isolation in mammals. J Mammal. 88:5–23. [Google Scholar]
- Vrana PB, Fossella JA, Matteson P, del Rio T, O'Neill MJ, et al. 2000. Genetic and epigenetic incompatibilities underlie hybrid dysgenesis in Peromyscus. Nat Genet. 25:120–124. [DOI] [PubMed] [Google Scholar]
- Wake N, Takagi N, Sasaki M.. 1976. Non-random inactivation of the X chromosome in rat yolk sac. Nature 262:580–581. [DOI] [PubMed] [Google Scholar]
- Wang H, Morales-Levy M, Rose J, Mackey LC, Bodary P, et al. 2013. α(1,3)-Fucosyltransferases FUT4 and FUT7 control murine susceptibility to thrombosis. Am J Pathol. 182:2082–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Miller DC, Clark AG, Antczak DF.. 2012. Random X inactivation in the mule and horse placenta. Genome Res. 22:1855–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Soloway PD, Clark AG.. 2011. A survey for novel imprinted genes in the mouse placenta by mRNA-seq. Genetics 189:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M.. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder JA, Hewett EK, Gansner ME.. 2009. Molecular evolution of GYPC: evidence for recent structural innovation and positive selection in humans. Mol Biol Evol. 26:2679–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Ardiet DL, Saner C, Vilain N, Beermann F, et al. 2007. Normal hemopoiesis and lymphopoiesis in the combined absence of numb and numblike. J Immunol. 178:6746–6751. [DOI] [PubMed] [Google Scholar]
- Wolf JB. 2013. Evolution of genomic imprinting as a coordinator of coadapted gene expression. Proc Natl Acad Sci USA. 110:5085–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JB, Brandvain Y.. 2014. Gene interactions in the evolution of genomic imprinting. Heredity (Edinb). 113:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JB, Hager R.. 2006. A maternal–offspring coadaptation theory for the evolution of genomic imprinting. PLoS Biol. 4:e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff P, Jiang H, Wang G, Santos-González J, Köhler C.. 2015. Paternally expressed imprinted genes establish postzygotic hybridization barriers in Arabidopsis thaliana. eLife 4:e10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-I, Johnson NA, Palopoli MF.. 1996. Haldane's rule and its legacy: why are there so many sterile males? Trends Ecol Evol. 11:281–284. [DOI] [PubMed] [Google Scholar]
- Zakaria M, Ferent J, Hristovska I, Laouarem Y, Zahaf A, et al. 2019. The SHH receptor BOC is important for myelin formation and repair. Development 146:dev172502. [DOI] [PubMed] [Google Scholar]
- Zechner U, Reule M, Orth A, Bonhomme F, Strack B, et al. 1996. An X-chromosome linked locus contributes to abnormal placental development in mouse interspecific hybrids. Nat Genet. 12:398–403. [DOI] [PubMed] [Google Scholar]
- Zechner U, Shi W, Hemberger M, Himmelbauer H, Otto S, et al. 2004. Divergent genetic and epigenetic post-zygotic isolation mechanisms in Mus and Peromyscus. J Evol Biol. 17:453–460. [DOI] [PubMed] [Google Scholar]
- Zhang B, Horvath S.. 2005. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol. 4:17. [10.2202/1544-6115.1128] [DOI] [PubMed] [Google Scholar]
- Zou H, Yu D, Du X, Wang J, Chen L, et al. 2019. No imprinted XIST expression in pigs: biallelic XIST expression in early embryos and random X inactivation in placentas. Cell Mol Life Sci. 76:4525–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw sequencing reads are archived at NCBI under BioProject PRJNA306772. Accession numbers for individual libraries are provided in Supplementary Table S5. Supplementary material is available at figshare: https://doi.org/10.25386/genetics.14171075.