

Abstract
Almost two-thirds of patients with severe traumatic brain injury (TBI) develop some form of hemostatic disturbance, which contributes to poor outcome. While the initial head injury often leads to impaired clot formation, TBI is also associated with an increased risk of thrombosis. Most likely there is a progression from early bleeding to a later prothrombotic state. In this paper, we systematically review the literature on the time course of hemostatic disruptions following TBI. A MEDLINE search was performed for TBI studies reporting the trajectory of hemostatic assays over time. The search yielded 5,049 articles, of which 4,910 were excluded following duplicate removal as well as title and abstract review. Full-text assessment of the remaining articles yielded 33 studies that were included in the final review. We found that the first hours after TBI are characterized by coagulation cascade dysfunction and hyperfibrinolysis, both of which likely contribute to lesion progression. This is then followed by platelet dysfunction and decreased platelet count, the clinical implication of which remains unclear. Later, a poorly defined prothrombotic state emerges, partly due to fibrinolysis shutdown and hyperactive platelets. In the clinical setting, early administration of the antifibrinolytic agent tranexamic acid has proved effective in reducing head-injury-related mortality in a subgroup of TBI patients. Further studies evaluating the time course of hemostatic disruptions after TBI are warranted in order to identify windows of opportunity for potential treatment options.
Electronic supplementary material
The online version of this article (10.1007/s12028-020-01037-8) contains supplementary material, which is available to authorized users.
Background
Despite improvements in medical triage and tertiary care, traumatic brain injury (TBI) is associated with significant morbidity and mortality [1]. The initial injury is often followed by hemostatic disturbance, which is present in up to two-thirds of patients with severe TBI and associated with increased mortality [2, 3]. While it is still unclear exactly how TBI affects the coagulation system, the primary drivers appear to be platelet dysfunction, endothelial activation, disturbed fibrinolysis, endogenous anticoagulation, and inflammation [2, 4–6].
There is controversy regarding the exact nature of hemostatic disruption after TBI, and evidence exists to support the presence of both a hyper- and hypocoagulable state [2]. While the initial head injury often leads to impaired clot formation and exacerbation of hemorrhagic lesions [2, 7–9], TBI is also independently associated with an increased risk of venous thromboembolism [10–13] and ischemic stroke [14–18]. Most likely, there is a progression from early increased bleeding risk to a later prothrombotic state. However, an overlap and lack of distinction between the phases exists.
In this paper, we systematically review the literature on the time course of hemostatic disruptions following TBI, as reflected by temporal changes in selected laboratory assays, in order to highlight current evidence and potential future directions.
Methods
Search Strategy and Selection Criteria
A search strategy was decided upon (Supplementary file 1) and carried out using the MEDLINE database from its date of inception until December 2019. Titles and abstracts were independently screened to determine whether they met the inclusion criteria. Full texts of the chosen articles were assessed to confirm this. Reference lists of relevant articles were screened for additional studies.
Inclusion and Exclusion Criteria
Studies of adult and pediatric TBI patients that reported the time course (> 1 sample per patient) of one or several hemostatic assays were eligible for inclusion. The assays of primary interest were platelet count, prothrombin time (PT), partial thromboplastin time (PTT), activated partial thromboplastin time (APTT), D-dimer, thrombin antithrombin III complex (TAT), Prothrombin fragment 1 + 2 (F1 + 2), tissue plasminogen activator (t-PA), plasminogen activator inhibitor-1 (PAI-1), thromboelastography (TEG), thromboelastometry (TEM), Multiplate® (Roche Diagnostics, Basel, Switzerland) and VerifyNow® (Accumetrics, San Diego, CA, USA). There were no restrictions on methodological quality. Studies were excluded if they were non-English, and if it wasn’t possible to interpret data relating to patients with TBI (e.g. multitrauma studies without a subgroup analysis) or a specific laboratory assay (e.g. studies that grouped several assays together as one). For studies evaluating the effect of a hemostatic intervention, only data for the control group were extracted.
Data Abstraction
Using a customized form, we extracted data from the included studies and stored them in an electronic database. Where applicable, the following data were abstracted: study design, patient ages, cohort size, TBI severity, presence of extracranial injuries, pre-injury antithrombotic or anticoagulation medication, sampling times, time from injury to first sample, and the temporal trajectory for each assay. No meta-analysis was possible due to the heterogeneity of the data.
Results
Study Selection and Characteristics
The initial literature search generated 5,049 articles, of which 4,910 were excluded following duplicate removal as well as title and abstract review. Full-text assessment of the remaining 139 articles yielded 33 studies that were included in the final review (Fig. 1).
Fig. 1.
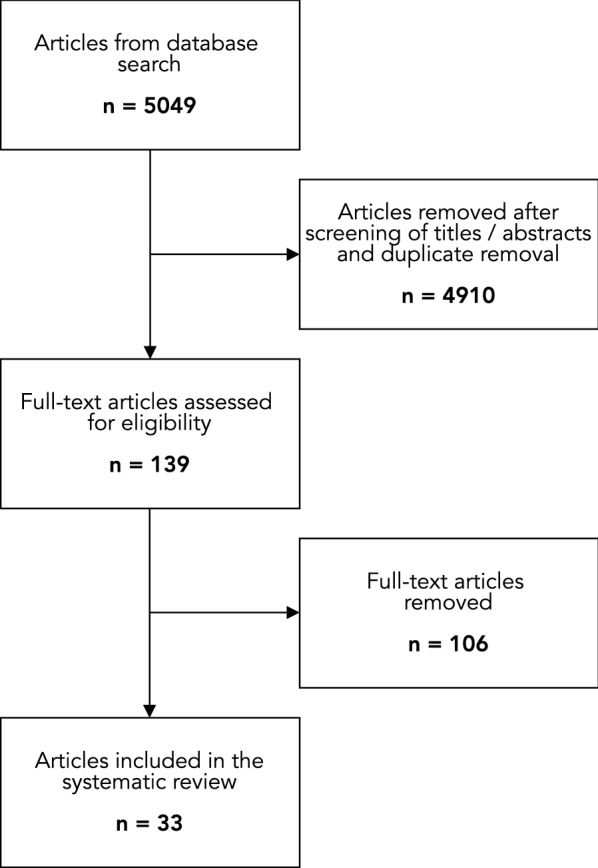
Schematic overview of the number of identified records for the systematic steps of the review process. Abbreviations: APTT = activated partial thromboplastin time; PT = prothrombin time; PTT = partial thromboplastin time
Platelets
Platelet Count
We included 17 studies with platelet count data, of which all but one found that platelet count decreased after injury [19–34]. In the largest study of 274 adults with severe TBI, mean platelet count was normal on admission and then decreased over time, with a steeper decline in more severe cases [20]. A similar pattern of posttraumatic decrease in platelet count was seen in the prospective studies [22, 23, 28] as well as in mild TBI [32]. Most often the platelet counts remained within reference ranges [19, 20, 24–26, 29, 30, 35], but some studies reported on the development of posttraumatic thrombocytopenia as well [21, 27, 28, 32]. The lowest platelet count levels were seen 1–5 days after injury [27–32], followed by a rebound to admission levels by day 5–9 [28, 30, 31]. One study found no change in platelet count over time, but only sampled blood tests on admission and 10–17 days after injury [35] (Table 1).
Table 1.
Platelet count
| Reference | Study design | TBI type | Age category | Patients (n) | Hemostatic comorbidities | Sampling times | First sample | Result |
|---|---|---|---|---|---|---|---|---|
|
Nakae |
Retrospective | Isolated severe TBI | Adults | 234 [18], 274 [19] | Excluded those with liver failure, hematological disease or malignancy | On admission and 3, 6, 12 h after injury | < 1 h after injury | Decrease in PLT count during time studied. Faster decrease in more severe cases. Median values stayed within reference range |
| Grenander [21] | RCT | Isolated TBI | Adults | 28 | Excluded those with known bleeding or coagulation disorders | On admission and 8, 16, 24, 36, 48, 72, 96, 120 h after injury | Mean 8.6 h after injury | Decrease in PLT count between admission and 48 h after injury, reaching an average level of 120 × 109/L, followed by an increase that did not reach normal values during time studied |
| Bredbacka [22] | Prospective observational | Isolated TBI | Adults | 20 | N/A | On admission and 1, 2, 3, 4, 5, 6 days after injury | < 24 h of injury | Decrease in PLT count that reached its lowest point 2 days after injury, with 50% of patients showing values < 150 × 109/L, followed by an increase that reached admission-values 5 days after injury |
| Kutcher [23] | Prospective observational | TBI | N/A | 62 | N/A | On admission and 6, 12, 24, 48, 72, 96, 120 h after injury | N/A | Decrease in PLT count between admission and 48 h after injury, followed by an increase that did not reach admission-values within time studied. Mean platelet count remained above 100 × 109/L |
| Vecht [24] | Prospective observational | Moderate to severe TBI | N/A | 34 | N/A | On admission and 1, 2, 3, 4, 5, 9, 16 days after injury | N/A | Decrease in PLT count between admission and 2–5 days after injury, followed by an increase that reached admission-values at 9 days after injury. Values within reference range |
| Auer [25] | Prospective observational | Severe isolated TBI | N/A | 30 | N/A | On admission and during the first 10 days after injury | N/A | Decrease in PLT count between admission and 3–5 days after injury, followed by an increase that reached admission-values by end of first week. Unclear if within reference range |
| Carrick [26] | Retrospective | Isolated moderate to severe TBI | All ages | 184 | N/A | On admission and 72 h after injury | N/A | Thrombocytopenia in 14% on admission that increased to 46% after 72 h |
| Hulka [27] | Retrospective | TBI | Adults | 91 | N/A | N/A | 3.1 ± 0.4 h | Thrombocytopenia in 0% on admission and in 30% 2–6 h after injury, followed by a decrease to 20% after 18–24 h |
| Engström [28] | Retrospective | Severe TBI | N/A | 27 | N/A | On admission and 24 h after injury | N/A | Decrease in PLT count over time. Some patients were below reference range but mean value unclear |
| Lustenberger [29] | Retrospective | Isolated severe TBI | N/A | 278 | N/A | On admission and every 6 h until clinically stable | N/A | Decrease in PLT count between admission and 24 h after injury, followed by an increase that did not reach admission values during time studied. Mean value below reference range at lowest point |
| Nekludov [30] | Prospective observational | Isolated severe TBI | Adults | 20 | Alcoholism in 50%. Excluded those with other known coagulation disorders | On admission and 3 days after injury | 17 ± 12 h after injury | Decrease in PLT count over time. Unclear if within reference range |
| Fair [31] | Prospective observational | Isolated TBI | Adults | 141 | Excluded those with known coagulation disorders | On admission and 6, 12, 24, 48 h after injury | N/A | Decrease in PLT count over time. Unclear if within reference range |
| Briggs [32] | Interventional | Isolated TBI | Adults | 5 | Excluded those with history of thrombocytopenia, platelet disorder, or other preexisting coagulopathy | On admission and 4–12 h after injury | N/A | Decrease in PLT count over time. Values within reference range |
| Wang [33] | Interventional | Isolated moderate TBI | Adults | 83 | Excluded those with known coagulation disorders | 12, 24, 48, 72 h after injury | N/A | Decrease in PLT count over time. Values within reference range |
| Scherer [34] | Prospective observational | Isolated severe TBI | Adults | 20 | N/A | On admission and 3 h after admission | N/A | Decrease in PLT count over time. Values within reference range |
| Touho [35] | Prospective observational | Isolated TBI | N/A | 12 | N/A | On admission and 10–17 days after injury | < 24 h of injury | No change in PLT count over time. Values within reference range |
h hours; N/A not available; RCT randomized control trial; TBI traumatic brain injury
Platelet Function
We included 6 studies with platelet function data. The methods used were TEG, Multiplate® (Roche Diagnostics, Basel, Switzerland) and spectrophotometry. All studies found that platelet function decreased after injury [22, 24, 29, 36–38]. This appeared to be independent of platelet transfusion and pre-injury antithrombic therapy [36, 37]. In the largest prospective study of 153 adults with TBI, mean adenosine diphosphate (ADP)-induced aggregation was below-normal on admission and continued to decrease during the first 24 h after injury [36]. Five other studies reported a similar pattern of normal to below-normal platelet function on admission that then decreased over time in stimulation to collagen (COL) [24], ADP, thrombin receptor activating peptide (TRAP) and arachidonic acid (AA or ASPI) [22, 37]. The lowest platelet function levels were observed 6–48 h after injury [24, 29, 37, 38] and then returned to normal after 2–16 days [22, 29, 37, 38]. In the two studies that observed patients for more than one week, platelet function increased to above-normal levels (i.e. hyperactive platelets) following their initial decline [29, 37] (Table 2).
Table 2.
Platelet function
| Reference | Study design | TBI type | Age category | Patients (n) | Antithrombotic treatment (n, %) | Hemostatic comorbidities | Sampling times | First sample | Result |
|---|---|---|---|---|---|---|---|---|---|
| Nekludov [30] | Prospective observational | Isolated severe TBI | Adults | 20 | 0 (0%) | Alcoholism in 50%. Excluded those with other known coagulation disorders | On admission and 3 days after injury | 17 ± 12 h after injury | Below-normal platelet function on admission (Multiplate: AA/ASPI and ADP), that returned to normal by day 3 in most cases although some patients normalized after 2–3 weeks |
| Vecht [38] | Prospective observational | Moderate to severe TBI | N/A | 34 | N/A | N/A | On admission and 1, 2, 3, 4, 5, 9 days and 2, 3, 4, 5, 6 weeks after injury | < 2 h of injury | Below-normal platelet function on admission (Vitatron spectrophotometer: ADP), with the lowest value on day 2, that returned to normal after 9–16 days |
| Kutcher [23] | Prospective observational | TBI | N/A | 62 | 5 (8%) | N/A | On admission and 6, 12, 24, 48, 72, 96, 120 h after injury | N/A | Low-to-normal platelet function on admission (Multiplate: ADP, TRAP, AA/ASPI, COL), that decreased to below-normal levels 6 h after injury, before returning to normal after 24 h (TRAP, COL), 96 h (ADP) or 120 h (AA/ASPI) |
| Briggs [32] | Prospective interventional | Isolated TBI | Adults | 5 | 0 (0%) | Excluded those with history of thrombocytopenia, platelet disorder, or other preexisting coagulopathy | On admission and 4–12 h after injury | N/A | Below-normal platelet function on admission (Multiplate: COL), with the lowest value after 4–12 h. Same trend for AA/ASPI but within reference ranges |
| Lindblad [37] | Retrospective | TBI, admitted to ICU | Adults | 178 | 47 (26%) | N/A | N/A | N/A | Below-normal and decreasing platelet function (Multiplate®: AA/ASPI) during first 10 h after injury, that returned to normal after 4–5 days. Results independent of COX-inhibition and platelet transfusion |
| Guillotte [36] | Prospective observational | TBI | Adults | 153 | 62 (41%) | N/A | N/A | N/A | Below-normal and decreasing platelet function in moderate-to-severe TBI (TEG: ADP) during first 24 h after injury. Results independent of antithrombotic medication |
AA/ASPI acetylsalicylic acid test; ADP adenosine diphosphate test; COL collage test; h hours; N/A not available; TBI traumatic brain injury; TEG thromboelastography; TRAP thrombin receptor activating peptide-6 test
Coagulation Cascade
PT, PTT and APTT
We included 12 studies with PT, PTT and/or APTT data. Most of these studies noted that TBI was followed by a decrease in markers of coagulation cascade function [19, 21, 32, 33, 39, 40]. In the largest study of 441 patients with isolated TBI, mean APTT was increased during the first 12 h after injury, with a mean value of 16 s (reference range: 10.3–13.4 s) [39]. A similar pattern, although within reference ranges, was seen in another study of 234 adult severe TBI patients, where APTT and INR significantly increased during the first 3 h after injury, reaching median values of 29.2 s (reference range: 24–36 s) and 1.07 (reference range: 0.8–1.2), respectively [19]. These observations were further supported by three smaller studies that all identified posttraumatic abnormally increased PT, INR and PTT that peaked at 2–6 h after injury [21, 33, 40]. In addition, one study found that Glasgow Coma Scale (GCS) was inversely proportional to PT, indicating that severer TBI cases induce a more pronounced coagulation cascade dysfunction [40]. Following the initial phase, values then seemed to return to normal after 12 h to several days following injury [32, 33, 40]. Two studies saw no significant change in these coagulation cascade assays over time [23, 25] (Table 3).
Table 3.
APTT, PT, INR
| Reference | Study design | TBI type | Age category | Patients (n) | Antithrombotic treatment (n, %) | Hemostatic comorbidities | Sampling times | First sample | Results |
|---|---|---|---|---|---|---|---|---|---|
| Halpern [39] | Retrospective | Isolated TBI | N/A | 441 | N/A | N/A | On admission and 6, 12, 18, 24, 48, 72 h after injury | < 1 h of injury | Abnormally elevated APTT during first 12 h after injury, with values then returning to normal |
| Nakae [19] | Retrospective | Isolated severe TBI | Adults | 234 | 0 (0%) | Excluded those with liver failure, hematological disease or malignancy | On admission and 3, 6, 12 h after injury | < 1 h of injury | Increase in INR and APTT from admission to 3 h after injury. Median values within reference range, with some outliers |
| Hulka [27] | Retrospective observational | TBI | Adults | 91 | N/A | N/A | N/A | 3.1 ± 0.4 h | Abnormally elevated PT (> 15 s) in 25% on admission that increased to 60% 2–6 h after injury, and then returned to 25% 12–18 h after injury. Also abnormally elevated PTT (> 35 s) in 20% on admission that increased to 60% 2–6 h after injury and then returned to 20% 6–12 h after injury |
| Touho [35] | Prospective observational | Isolated TBI | N/A | 12 | N/A | N/A | On admission and 10–17 days after injury | < 24 h of injury | No change in PT or PTT during time studied |
| Fair [31] | Prospective observational | Isolated TBI | Adults | 141 | 20 (14%) ASA | Excluded those with known coagulation disorders | On admission and 6, 12, 24, 48 h after injury | N/A | INR and APTT showed a trend toward prolongation over time, but stayed within reference ranges |
| Vecht [24] | Prospective observational | Moderate to severe TBI | N/A | 34 | N/A | N/A | On admission and 1, 2, 3, 4, 5, 9, 16 days after injury | N/A | PPT decreased on admission and then increased on days 1, 2 and 3 after injury |
| Carrick [26] | Retrospective observational | Isolated moderate to severe TBI | All ages | 184 | N/A | N/A | On admission and 72 h after injury | N/A | Increased bleeding (PT > 14.2 s or PTT > 38.4 s) in 21% of patients on admission that increased to 41% 72 h after injury |
| Pahatouridis [40] | Prospective observational | Isolated moderate TBI | Adults | 61 | 0 (0%) | Excluded those with known coagulopathy | On admission and 4, 6, 24, 48, 72 h after injury | N/A | Abnormally elevated PT in 78% on admission that increased to 100% 3–6 h after injury, before returning to 77% and 36% after 48 and 72 h, respectively. Lower GCS associated with increased PT |
| Wang [33] | Interventional | Isolated moderate TBI | Adults | 83 | 0 (0%) | Excluded those with known coagulation disorders | 12, 24, 48, 72 h after injury | N/A | No change in PT or APTT during time studied |
| Scherer [34] | Prospective observational | Isolated severe TBI | Adults | 20 | N/A | N/A | On admission and after 3 h | N/A | Slight increase in APTT over time (within reference ranges), but PT unchanged |
| Lustenberger [29] | Retrospective | Isolated severe TBI | N/A | 278 | N/A | N/A | On admission and every 6 h until clinically stable | N/A | Increase in INR from admission to 6 h after injury, which at that point was abnormally elevated. Returned to normal after 60 h |
APTT activated partial thromboplastin time; ASA acetylsalicylic acid; h hours; INR international normalized ratio; N/A not available; PT prothrombin time; PPT partial thromboplastin time; TBI traumatic brain injury
TAT and F1 + 2
We included 5 studies with TAT and/or F1 + 2 data. All of these studies found that TAT and F1 + 2 were elevated during the first day of injury [26, 27, 41–43]. In the largest study, which included 28 patients with isolated TBI, mean TAT was elevated on admission (mean 8.6 h after injury) and then decreased to reference levels after 5 days [27]. The other studies reported a similar pattern of increased TAT and/or F1 + 2 levels on admission that then decreased, reaching reference ranges from 24 h up to one week after injury [26, 41–43] (Table 4).
Table 4.
Thrombin generation: TAT and F1 + 2
| Reference | Study design | TBI type | Age category | Patients (n) | Antithrombotic treatment (n, %) | Hemostatic comorbidities | Sampling times | First sample | Results |
|---|---|---|---|---|---|---|---|---|---|
| Grenander [21] | RCT | Isolated TBI | Adults | 28 | 0 (0%) | Excluded those with known bleeding or coagulation disorders | On admission and 8, 16, 24, 36, 48, 72, 96, 120 h after injury | Mean 8.6 h after injury | Elevated TAT on admission that then decreased and reached reference levels 5 days after injury |
| Nekludov [41] | Prospective observational | Isolated severe TBI | Adults | 11 | 0 (0%) | Excluded those with known coagulation disorders or alcohol abuse | On admission and 3 days after injury | 15.4 ± 2.6 h after injury | Elevated TAT and F1 + 2 on admission that decreased and reached reference levels 24 h after injury |
| Sørensen [42] | Prospective observational | Isolated severe TBI | All ages | 14 | N/A | N/A | On admission and 1, 2, 3, 7 days after injury | N/A | Elevated TAT and F1 + 2 on admission that then decreased but remained elevated during time studied |
| Gando [43] | Prospective observational | Isolated TBI | Adults | 5 | 0 (0%) | Excluded those with preexisting coagulation disorders | On admission and 1, 2, 3, 4 days after injury | N/A | Elevated TAT and F1 + 2 on admission that then decreased but remained elevated during time studied |
| Scherer [34] | Prospective observational | Isolated severe TBI | Adults | 20 | N/A | N/A | A and 3 h after admission | N/A | Elevated TAT and F1 + 2 on admission that then decreased but remained elevated during time studied |
F1 + 2 = prothrombin fragment F1 + 2; h hours; TAT thrombin–antithrombin complex; TBI traumatic brain injury
Fibrinolysis
D-dimer
We included 11 studies with D-dimer data, all of which reported increased levels after injury [19, 23, 26, 27, 33, 35, 40, 41, 43–47]. In the largest prospective study of 141 patients with isolated TBI, mean D-dimer was increased on admission with mean peak levels of 8 µg/mL (normal range: 0.0–1.0 µg/mL) observed 6 h after trauma. This was then followed by a decrease over time [23]. Another study of 234 TBI patients identified a D-dimer peak within 3 h of injury in patients with head abbreviated injury scale (AIS) 5, and within 3–6 h in those with head AIS 4 [19], where median levels reached as high as 45.3 µg/mL. In these studies, D-dimer levels were also correlated with injury severity [19, 40] and hemorrhage progression [23]. A similar pattern of posttraumatic increase in D-dimer was seen in several other smaller studies as well [26, 27, 33, 35, 40, 41, 43–47]. In respect of time to normalization, most studies were limited by short follow-up time [19, 23, 26, 33, 40, 41, 46, 47], but one reported that 55% of patients normalized their D-dimer levels within 3 days of injury [40] (Table 5).
Table 5.
D-dimer
| Reference | Study design | TBI type | Age category | Patients (n) | Antithrombotic treatment (n, %) | Hemostatic comorbidities | Sampling times | First sample | Results |
|---|---|---|---|---|---|---|---|---|---|
| Fair [31] | Prospective observational | Isolated TBI | Adults | 141 | 20 (14%) ASA | Excluded those with known coagulation disorders | On admission and 6, 12, 24, 48 h after injury | N/A | Increased D-dimer on admission with a peak 6 h after injury, followed by a decrease during the remainder of the study period. D-dimer levels also correlated with hemorrhage progression |
| Nakae [19] | Retrospective | Isolated severe TBI | Adults | 234 | 0 (0%) | Excluded those with liver failure, hematological disease or malignancy | On admission and 3, 6, 12 h after injury | < 1 h of injury | Increased D-dimer on admission with a peak 3–6 h after injury, followed by a decrease during the remainder of the study period. More severe injury was associated with earlier peak levels |
| Touho [35] | Prospective observational | Isolated TBI | N/A | 12 | N/A | N/A | On admission and 10–17 days after injury | < 24 h of injury | Increased FDP on admission as compared to 10–17 days after injury |
| Grenander [21] | RCT | Isolated TBI | Adults | 28 | 0 (0%) | Excluded those with known bleeding or coagulation disorders | On admission and 8, 16, 24, 36, 48, 72, 96, 120 h after injury | Mean 8.6 h after injury | Increased D-dimer on admission, followed by a decrease during the remainder of the study period |
| Nekludov [41] | Prospective observational | Isolated severe TBI | Adults | 11 | 0 (0%) | Excluded those with known coagulation disorders or alcohol abuse | On admission and 3 days after injury | 15.4 ± 2.6 h after injury | Increased D-dimer on admission, followed by a decrease to a moderately elevated plateau during the following days |
| Gando [43] | Prospective observational | Isolated TBI | Adults | 5 | 0 (0%) | Excluded those with preexisting coagulation disorders | On admission and 1, 2, 3, 4 days after injury | N/A | Increased D-dimer on admission, followed by a decrease during the remainder of the study period |
| Pahatouridis [40] | Prospective observational | Isolated moderate TBI | Adults | 61 | 0 (0%) | Excluded those with known coagulopathy | On admission and after 4, 6, 24, 48, 72 h after injury | N/A | Increased D-dimer on admission that remained during day 1, followed by a decrease during the remainder of the study period. Lower GCS corresponded to increased D-dimer levels |
| DeFazio [44] | Retrospective | Isolated severe TBI | Adults | 44 | N/A | N/A | On admission and 24, 48, 72 h after injury | < 3 h of injury | Increased D-dimer on admission, followed by a decrease during the remainder of the study period. Median values at 24 h greater in patient with poorer clinical status |
| Foaud [45] | Prospective case control | Isolated TBI, admitted to ICU | Pediatric | 46 | N/A | N/A | On admission and day 3, 14 after injury | N/A | Increased D-dimer on admission, followed by a decrease during the remainder of the study period |
| Suehiro [46] | Retrospective | Moderate to severe TBI | All ages | 9 | N/A | N/A | On admission and 1, 3, 5, 7 days after injury | N/A | Increased D-dimer on admission, followed by a decrease during the remainder of the study period |
| Karri [47] | Retrospective | Severe TBI | Adults | 25 | N/A | N/A | On admission and 2, 4, 6 h after admission | N/A | Increased D-dimer on admission, followed by a decrease during the remainder of the study period |
| Scherer [34] | Prospective observational | Isolated severe TBI | Adults | 20 | N/A | N/A | A and 3 h after admission | N/A | Increased D-dimer highest on admission that had decreased three hours later |
| Hulka [27] | Retrospective | TBI | Adults | 49 | N/A | N/A | N/A | N/A | Immediate and persistent elevation of D-dimer concentration in 70% of the population on admission, that decreased to 40% 18 h after injury |
FDP fibrin and fibrinogen degradation product; h hours; TBI traumatic brain injury
PAI-1 and t-PA
We included 4 studies with PAI-1 and/or t-PA data. In the study of 141 TBI patients by Fair et al., PAI-1 and t-PA were elevated on admission and then decreased over time [23]. A similar pattern of posttraumatic increase in PAI-1 and t-PA, with the highest levels seen on admission, was observed in the other three smaller studies as well [43, 46, 48] (Table 6).
Table 6.
Plasminogen activators and inhibitors
| Reference | Study design | TBI type | Age category | Patients (n) | Antithrombotic treatment (n, %) | Hemostatic comorbidities | Sampling times | First sample | Results |
|---|---|---|---|---|---|---|---|---|---|
| Fair [31] | Prospective observational | Isolated TBI | Adults | 141 | 20 (14%) ASA | Excluded those with known coagulation disorders | On admission and 6, 12, 24, 48 h after injury | N/A | Increased PAI-1 and t-PA on admission that then decreased over time studied |
| Gando [43] | Prospective observational | Isolated TBI | Adults | 5 | 0 (0%) | Excluded those with preexisting coagulation disorders | On admission and 1, 2, 3, 4 days after injury | N/A | Increased PAI-1 on admission that then decreased to normal levels 5 days after injury |
| Becker [48] | Prospective observational | Severe TBI | Pediatric | 27 | 0 (0%) | N/A | On admission and 1, 3–5, 7–9, 21 and 35 days later | < 2 h of injury | Increased PAI-1 and t-PA on admission that then decreased over time studied |
| Karri [47] | Retrospective | Severe TBI | Adults | 25 | N/A | N/A | On admission and 2, 4, 6 h after admission | N/A | Increased t-PA on admission that then decreased over time studied. Increased PAI-1 with peak levels 4 h after admission. uPA increase over time studied |
H hours; PAI-1 plasminogen activator inhibitor 1; TBI traumatic brain injury; t-PA tissue plasminogen activator; uPA urokinase plasminogen activator
Viscoelastic Assays
We included 5 studies with viscoelastic assay data. In a study of 91 pediatric patients with severe TBI, elevated TEG clot lysis (LY30) was noted within the first hour and decreased over time [49]. In another study by the same group, where early sampling was not performed, no significant change in TEG LY30 was detected. Similarly, in samples taken regularly during the first two days after trauma, Fair et al. found no significant change in TEG LY30, although it is unclear when the first sample was taken [23]. Another ROTEM study of 83 adults with isolated mild TBI, where blood sampling was performed 12–72 h after injury, found no significant change in clotting time (CT), clot formation time (CFT) or maximum clot firmness (MCF). Following the initial phase, two pediatric studies from the same center observed that TEG LY30 then decreased below reference ranges, indicative of fibrinolysis shutdown. In the first study, where sampling was performed on admission and once daily, fibrinolysis shutdown was the most common phenotype between postinjury day 1 and continuing through postinjury day 3 [50]. In the follow-up study, in which early sampling was performed with a higher frequency, fibrinolysis shutdown was the most dominant phenotype already 3 h after trauma [49] (Table 7).
Table 7.
Viscoelastic assays
| Reference | Study design | TBI type | Age category | Patients (n) | Antithrombotic treatment (n, %) | Hemostatic comorbidities | Sampling times | First sample | Results |
|---|---|---|---|---|---|---|---|---|---|
| Fair [31] | Prospective observational | Isolated TBI | Adults | 141 | 20 (14%) ASA | Excluded those with known coagulation disorders | On admission and 6, 12, 24, 48 h after injury | N/A | Normal TEG LY30 during time studied |
| Wang [33] | Interventional | Isolated moderate TBI | Adults | 83 | 0 (0%) | Excluded those with known coagulation disorders | NO admission, 12, 24, 48, 72 h after injury | N/A | Normal ROTEM CT, CFT or MCF during time studied |
| Leeper [50] | Prospective observational | Severe TBI | Pediatric | 39 | N/A | Excluded those with preexisting coagulation disorders | On admission and 1, 2, 3, 4 days after injury | N/A | Normal LY30 on admission that decreased to fibrinolysis shutdown ranges starting on postinjury day 1 and continuing to postinjury day 3 |
| Leeper [49] | Prospective observational | Severe TBI | Pediatric | 91 | N/A | Excluded those with preexisting coagulation disorders | N/A | N/A | Elevated TEG LY30 within the first hour after injury, followed by a decrease toward fibrinolysis shutdown which had become the predominant phenotype already 3 h after trauma |
| Massaro [57] | Prospective observational | Moderate to severe TBI | Adults | 25 | 0 (0%) | Alcohol use in 68% | On admission and 1–2, 2–3, 3–4, 4–5 days after injury | Mean 18 h after injury | TEG MA, TG and G higher at late stage (> 48 h) compared to early stage (0–48 h) after injury |
CFT clot formation time; CT clotting time; h hours; MA maximum amplitude; MCF maximum clot firmness; G G-value; TBI traumatic brain injury; TEG thromboelastography; TG thrombin generation; ROTEM rotational thromboelastometry
Discussion
TBI is often followed by hemostatic disturbance [2, 3], which has been associated with worse clinical outcome [51]. It is important to note that hemorrhagic lesion progression primarily occurs during the first hours after injury, with one study of mild TBI reporting that lesion progression had stopped in 75% of patients within 2 h, and in 97% within 24 h, of injury [52]. This highlights the window of opportunity for the treatment of deranged hemostasis. The aim of this study was to review the literature on the time course of hemostatic disruptions following TBI. Thirty-three studies were included, contributing data on temporal changes in primary hemostasis, coagulation cascade function and fibrinolysis. To the best of our knowledge, this is the first review of its kind and contributes findings that are important for patient management and future study design.
Primary Hemostasis
Clinically, primary hemostasis is assessed by platelet count and platelet function [53]. In this review, we observed that TBI was followed by a decrease in platelet count and function that appeared to be independent of platelet transfusions and antithrombic therapy [36, 37]. Platelet count typically stayed within reference ranges [19, 20, 24–26, 29, 30, 35] and returned to admission levels within two weeks [28, 30, 31], while platelet function decreased below reference levels about 6–48 h after injury [24, 29, 37, 38]. The reason behind this is likely multifactorial. Consumption of platelet–fibrin clot formation probably decreases platelet count early on, a decrease which is aggravated by dilution due to extensive fluid resuscitation therapy. Additional decreases in platelet count and function may be the result of consumptive depletion and exhaustion [2]. Platelet dysfunction could also be driven by endothelial injury [54], with endothelial disruption catalyzing systemic activation of platelets that renders them inert to subsequent stimulation [55], as well as factors within soluble plasma that render platelets inactive after trauma [56]. Platelets also undergo structural change following trauma, which could decrease their function, but it remains unclear whether this process occurs in isolated TBI [55].
Two studies also found that platelets became hyperactive following their initial dysfunction [29, 37]. This was supported by a TEG study that noted increasing maximum amplitude (MA) levels 7 days after TBI [57] (MA represents clot strength that is mainly dependent on platelet function). Platelet hyperactivity could explain the increased risk of thrombosis seen in TBI patients [10–18, 58], but future studies are warranted to assess its clinical significance.
In summary, platelet dysfunction and decreased platelet count were evident during the acute postinjury phase. In patients surviving the initial insult, platelets became hyperactive, although the exact time frame remains to be determined (Fig. 2).
Fig. 2.

Conceptual figure of the time course of hemostatic disruptions after traumatic brain injury
Clinical Implications
Platelet dysfunction was seen between six hours to several days after injury. Considering the fact that lesion progression is most prevalent within the first hours of trauma [52], this might imply that platelet alterations are not a main cause behind exacerbation of hemorrhagic lesions in TBI. This is supported by the fact that posttraumatic platelet dysfunction has not been associated with intracranial lesion progression [37]. However, while hemorrhagic expansion of the primary brain contusions is a chief concern, delayed noncontiguous hemorrhagic lesions at remote locations may also appear in the evolution of a patient’s clinical course [59]. This could be caused by platelet dysfunction. Further adding to the uncertainty regarding the clinical significance of platelet dysfunction in TBI, platelet function assays were originally designed to detect antiplatelet medication efficacy, and have secondarily been extrapolated to trauma, making interpretation of cutoff values unclear. The assays are also limited by the absence of endothelium and flow conditions, as well as variability related to platelet count and hematocrit level [54]. Highlighting this, studies of platelet function-targeted therapy have failed to demonstrate clinical efficacy. While there is some evidence that platelet transfusion can correct platelet dysfunction in TBI [60], others studies suggest that transfusions are more likely to improve aspirin-induced, rather than trauma-induced, dysfunction [24]. Desmopressin could be an alternative to platelet transfusions, though there exist only mixed results from low-quality studies [61, 62]. Thus, it is unclear whether posttraumatic platelet dysfunction contributes to lesion progression in TBI, and there is also no proven treatment for TBI-induced platelet dysfunction.
Coagulation Cascade
In the clinical setting, coagulation cascade function is assessed by PT, PTT and APTT, as well as surrogate markers of thrombin generation such as TAT and F1 + 2. We observed that TBI was followed by a temporary increase in APTT, PTT and PT [19, 21, 33, 39, 40], indicating decreased coagulation cascade function. This seemed to be apparent on admission and was most pronounced during the first hours after injury [19, 21, 33, 40]. TAT and F1 + 2 were also elevated during this time [26, 27, 41–43], indicating that the coagulation cascade dysfunction was secondary to increased thrombin generation. Therefore, the changes are probably the result of consumption, where excess clotting leads to depletion and/or dysfunction of coagulation factors in plasma [39]. Many believe that this is due to trauma-induced unregulated tissue factor release [58, 63], which would stimulate thrombin generation and consumption of clotting factors in the ways observed in this review, but evidence remains elusive. Thus, the first few hours following TBI are characterized by coagulation cascade dysfunction that is likely secondary to unregulated thrombin production (Fig. 2).
Clinical Implications
Coagulation cascade dysfunction appears to occur when the risk of intracranial lesion progression is high [52], supporting a theory that it contributes to hemorrhagic expansion in TBI. This presents a possible window of opportunity for interventions. In general trauma, there has been much interest in the use of fresh frozen plasma (FFP), which contains both fibrinogen and clotting factors [64], to limit blood loss [2]. In isolated TBI, retrospective evidence has found that early plasma transfusion is associated with improved survival in patients with multifocal intracranial hemorrhage [65], but no randomized controlled trials have been performed to validate this. Prothrombin complex concentrate (PCC) provides another theoretical treatment strategy for coagulation cascade dysfunction secondary to consumption of clotting factors, and is currently recommended for the emergency reversal of vitamin K anticoagulant therapy in TBI [2]. One retrospective study of general trauma patients found that PCC decreased mean INR in patients without warfarin treatment [66], but there is currently no recommendation for its use in patients with isolated TBI without anticoagulation therapy [67]. Administration of recombinant FVIIa has also been associated with reduced hemorrhage progression in TBI, albeit with a higher rate of thrombosis, but the effect size was small and the benefit of routine recombinant FVIIa remains unclear [68]. Thus, there is a sound theoretical basis for the belief that consumption of clotting factors contributes to lesion progression in TBI, but there are yet no evidence-based treatment options to prevent this.
Fibrinolysis
Fibrinolysis is usually assessed using D-dimer and, to a certain extent, viscoelastic assays. Some studies also use t-PA, which stimulates conversion of plasminogen to plasmin, as well as PAI-1, an inhibitor of t-PA [69]. We observed an increase in D-dimer following TBI [19, 23, 26, 27, 33, 35, 40, 41, 43–47], with peak levels seen during the first 6 h after injury [19, 23, 40]. This shows that TBI is followed by an early increase in fibrinolytic activity, which also seems to be proportional to injury severity [19]. This is supported by the observation that PAI-1 and t-PA are increased during the same time period [23, 43, 46, 48]. However, only one study could verify this using viscoelastic assays during the first hour after injury [49]. This could mean that hyperfibrinolysis is only present within this short time interval, or be due to an arbitrary threshold for hyperfibrinolysis [70] and LY30′s relative lack of sensitivity to changes in fibrinolysis [23]. Highlighting this, a study of general trauma found that 90% of patients with hyperfibrinolysis on D-dimer, t-PA and plasmin‐α2AP did not meet criteria based on TEM, leading the authors to conclude that TEM may be an unreliable measurement of endogenous fibrinolytic activity [71]. Thus, hyperfibrinolysis is evident in the acute phase following TBI, but it remains unclear exactly for how long it continues (Fig. 2).
Clinical Implications
It has been suggested that hyperfibrinolysis is a main driving force behind trauma-induced bleeding disorders in TBI, and can cause hemorrhage expansion via the degradation of coagulation factors, breakdown of the formed fibrin clot, and impairment of clot formation as a result of excessive generation of fibrin degradation products. Moreover, from a time-based standpoint, hyperfibrinolysis appears to occur when the risk of intracranial lesion progression is high [52]. A subgroup analysis from the CRASH-3 randomized trial found that administration of tranexamic acid (TXA) was associated with a reduction in 28-day head injury-related mortality if administered within 3 h to patients with mild-to-moderate isolated TBI [72]. This indicates that antifibrinolytic drugs can provide effective treatment. However, the CRASH-3 study protocol was modified, reducing the time from injury to randomization from 8 to 3 h, and no treatment effect was seen in patients with severe TBI. Limiting the time window after TBI probably increased the chance that TXA was administered when hyperfibrinolysis actually took place. Based on the observations in this review, one would expect prehospital administration of TXA to be even more effective, a treatment option which has been assessed in a completed but not yet published trial (ClinicalTrials.gov Identifier: NCT01990768). Other antifibrinolytic drugs, such as aprotinin, epsilon-aminocaproic acid and aminomethylbenzoic acid, are all also theoretically sound options but have not been assessed in TBI. Thus, early hyperfibrinolysis appears to contribute to lesion progression, and there is evidence in support of antifibrinolytic treatment in a subgroup of TBI patients if administered early.
Prothrombotic State
TBI is also independently associated with an increased risk of thromboembolic complications that persist after index hospitalization [10–18, 73]. Most likely, initial hypocoagulability transitions to a subacute hypercoagulability phase. Recent evidence suggests that posttraumatic fibrinolytic shutdown is associated with a higher risk of thromboembolic events in TBI [74]. In the pediatric study by Leeper et al., fibrinolysis shutdown (as indicated by low TEG LY30 levels) was the predominant phenotype already 3 h after injury [49]. Similarly, in their study using TEM in adult patients with moderate-severe TBI, Massaro et al. identified a progressive and delayed hypercoagulable state that was more common > 48 h, compared to < 48 h, after injury [57]. With regard to platelet function, two studies found that multiplate levels increased above reference ranges 1–2 weeks following initial trauma [29, 37]. Collectively, these results indicate a delayed prothrombotic state characterized by initial fibrinolysis shutdown, which appears within hours to days of injury, and later possible platelet hyperactivity. However, the time point at which a patient becomes phenotypically prothrombotic remains unclear, and will most likely be a combination of these factors countered by any residual coagulation cascade dysfunction and thrombocytopenia. These findings warrant particular consideration with regard to several unresolved clinical dilemmas in order to decrease the rates of VTEs and capillary microthrombi seen in vulnerable TBI patients, such as the timing of postinjury pharmacological thromboprophylaxis and resumption of oral anticoagulation therapy. However, more studies on long-term hemostatic disruptions after TBI are needed before any further conclusions can be drawn.
Age Differences
The three studies that included patients of all ages did not perform any analysis to differentiate between adult and pediatric populations [32, 42, 45]. In the four studies that focused exclusively on pediatric populations, posttraumatic elevations in D-dimer [44], PAI-1, t-PA [48] and TEG LY30 [49, 50] were noted. Thus, pediatric patients seem to experience posttraumatic hyperfibrinolysis, but a direct comparison to adults is lacking. Moreover, to date, no studies have assessed whether pediatric patients undergo posttraumatic changes in platelet count, platelet function and coagulation cascade function as well.
Systemic Versus Localized Hemostasis
The primary contusion in TBI is the result of ruptured microvessels. In the penumbra and surrounding regions, molecular processes can then be activated leading to hemorrhagic progression or new lesions [75]. The addition of hemostatic disturbance contributes to lesion progression [76]. For all studies included in this review, the assumption has been made that hemostatic assays sampled from the systemic circulation are applicable to the brain vasculature as well. However, the complexity of the cerebral microvessel walls, and their differences in structure and function compared to extracranial vessels, might suggest that hemostatic response differs regionally [77]. In severe sepsis for example, certain organs have been found to be at a greater risk of thrombosis than others, indicating that hemostatic disruption might partly be a localized phenomenon [78]. With this said, there is still very little evidence that blood or plasma is altered in its passage through the brain, or that the brain environment alters blood components under normal conditions [79].
Study Limitations
There are some limitations to this study that should be mentioned. For one, TBI is heterogeneous and multifactorial, consisting of a mix of pathologic processes that can be active in different patients and in different regions of the brain. In addition, the methodological quality of the included studies reviews was variable, with many being observational cohort studies without control groups and variable numbers of participants. Studies also varied in terms of sampling frequency and follow-up time. Different cutoff levels and parameters were also used for the hemostatic assays, which makes standardization and interpretation difficult. We also only made use of a single search database to perform our systematic review, which could exclude potentially valuable articles [80]. However, we also screened the reference lists of the included studies and thus consider this risk to be minor. Despite its limitations, we believe that clinically helpful and valid conclusions can be drawn from this review, and will be useful in designing future studies and clinical practice guidelines.
Conclusions and Future Directions
Altered hemostasis, with early hemorrhagic progression and later thrombotic complications, is a substantial and ongoing challenge in the management of TBI. There is currently a lack of well-designed prospective trials reviewing the temporal trend in hemostatic disruption, making it difficult to identify windows of opportunity for potential treatment options. Moreover, most data underlying the initiation and progression of hemostatic disruption in TBI are speculative and not linked to causative data. Based on the results from this review, it appears that TBI is followed by both early and delayed hemostatic disturbance. The first hours after TBI are characterized by a derangement of the coagulation system, as a consequence of massive and systemic activation, and hyperfibrinolysis, both of which likely contribute to lesion progression. This is then followed by decreased platelet function and count, the clinical significance of which remains unclear. Thereafter, a poorly defined prothrombotic state emerges, which might be characterized by fibrinolysis shutdown and hyperactive platelets. In the clinical setting, only early administration of TXA has proved effective in reducing the risk of head-injury related mortality in a subgroup of TBI patients. Further studies aimed at analyzing the progress of hemostatic disruption following TBI are warranted. In particular, studies with high-frequency early sampling are warranted in order to characterize the cause behind intracranial lesion progression, as well as those with longer follow-up times in order to understand the delayed prothrombotic state. Studies using point-of-care viscoelastic assays are also of particular interest, as they have the potential of providing real-time information in the clinical setting.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Open access funding provided by Karolinska Institute.
Abbreviations
- AA
Arachidonic acid
- ADP
Adenosine diphosphate
- AIS
Abbreviated injury scale
- APTT
Activated partial thromboplastin time
- ASPI
Aspirin
- CFT
Clotting formation time
- COL
Collagen
- CT
Clotting time
- F1 + 2
Prothrombin fragment 1 + 2
- FFP
Fresh-frozen plasma
- GCS
Glasgow coma scale
- LY30
Lysis index 30
- MA
Maximum amplitude
- MCF
Maximum clot firmness
- PAI-1
Plasminogen activator inhibitor-1
- PCC
Prothrombin complex concentrate
- PT
Prothrombin time
- PTT
Partial thromboplastin time
- t-PA
Tissue plasminogen activator
- TAT
Thrombin antithrombin III complex
- TBI
Traumatic brain injury
- TEG
Thromboelastography
- TEM
Thromboelastometry
- TRAP
Thrombin receptor-activating peptide
- TXA
Tranexamic acid
Funding
No funding was received for this research project.
Conflict of interest
Marc Maegele has received honoraria for speaker´s bureaus, participation in advisory board meetings and congress travel support from IL-Werfen/TEM International and Haemonetics. BMB has received speaker’s fees from CSL Behring. All other authors declare no competing interests.
Ethical Approval
We confirm that authorship requirements have been met and the final manuscript was approved by all authors.
Consent for Publication
We confirm that this manuscript has not been published elsewhere and is not under consideration by another journal.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Stocchetti N, Zanier ER. Chronic impact of traumatic brain injury on outcome and quality of life: a narrative review. Crit Care. 2016;20:1–10. doi: 10.1186/s13054-016-1318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maegele M, Schöchl H, Menovsky T, Maréchal H, Marklund N, Buki A, et al. Coagulopathy and haemorrhagic progression in traumatic brain injury: advances in mechanisms, diagnosis, and management. Lancet Neurol. 2017;16:630–677. doi: 10.1016/S1474-4422(17)30197-7. [DOI] [PubMed] [Google Scholar]
- 3.Harhangi BS, Kompanje EJO, Leebeek FWG, Maas AIR. Coagulation disorders after traumatic brain injury. Acta Neurochir (Wien) 2008;150:165–175. doi: 10.1007/s00701-007-1475-8. [DOI] [PubMed] [Google Scholar]
- 4.Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood. 2016;128:1043–1049. doi: 10.1182/blood-2016-01-636423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herbert JP, Guillotte AR, Hammer RD, Scott LN. Coagulopathy in the setting of mild traumatic brain injury: Truths and consequences. Brain Sci. 2017;7:92. doi: 10.3390/brainsci7070092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fletcher-Sandersjöö A, Maegele M, Bellander B-M. Does complement-mediated hemostatic disturbance occur in traumatic brain injury? A literature review and observational study protocol. Int J Mol Sci. 2020;21:1596. doi: 10.3390/ijms21051596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Folkerson LE, Sloan D, Cotton BA, Holcomb JB, Tomasek JS, Wade CE. Predicting progressive hemorrhagic injury from isolated traumatic brain injury and coagulation. Surgery. 2015;158:655–661. doi: 10.1016/j.surg.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Zhang F, Dong JF. Coagulopathy induced by traumatic brain injury: systemic manifestation of a localized injury. Blood. 2018;131:2001–2006. doi: 10.1182/blood-2017-11-784108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan Q, Sun YR, Wu XHX, Yu J, Li ZQ, Du ZY, et al. Coagulopathy in traumatic brain injury and its correlation with progressive hemorrhagic injury: a systematic review and meta-analysis. J Neurotrauma. 2016;33:1279–91. doi: 10.1089/neu.2015.4205. [DOI] [PubMed] [Google Scholar]
- 10.Skrifvars MB, Bailey M, Presneill J, French C, Nichol A, Little L, et al. Venous thromboembolic events in critically ill traumatic brain injury patients. Intensive Care Med. 2017;43:419–428. doi: 10.1007/s00134-016-4655-2. [DOI] [PubMed] [Google Scholar]
- 11.Strollo BP, Bennett GJ, Chopko MS, Guo WA. Timing of venous thromboembolism chemoprophylaxis after traumatic brain injury. J Crit Care. 2018;43:75–80. doi: 10.1016/j.jcrc.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Glass NE, Vadlamani A, Hwang F, Sifri ZC, Kunac A, Bonne S, et al. Bleeding and thromboembolism after TBI in the elderly: a real conundrum. J Surg Res. 2019;235:615–20. doi: 10.1016/j.jss.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 13.Hachem LD, Mansouri A, Scales DC, Geerts W, Pirouzmand F. Anticoagulant prophylaxis against venous thromboembolism following severe traumatic brain injury: a prospective observational study and systematic review of the literature. Clin Neurol Neurosurg. 2018;175:68–73. doi: 10.1016/j.clineuro.2018.09.032. [DOI] [PubMed] [Google Scholar]
- 14.Albrecht JS, Liu X, Smith GS, Baumgarten M, Rattinger GB, Gambert SR, et al. Stroke incidence following traumatic brain injury in older adults HHS public access. J Head Trauma Rehabil. 2015;30:62–67. doi: 10.1097/HTR.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowalski RG, Haarbauer-Krupa JK, Bell JM, Corrigan JD, Hammond FM, Torbey MT, et al. Acute ischemic stroke after moderate to severe traumatic brain injury: incidence and impact on outcome. Stroke. 2017;48:1802–1809. doi: 10.1161/STROKEAHA.117.017327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu SW, Huang LC, Chung WF, Chang HK, Wu JC, Chen LF, et al. Increased risk of stroke in patients of concussion: A nationwide cohort study. Int J Environ Res Public Health. 2017;14:230. doi: 10.3390/ijerph14030230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eric Nyam TT, Ho CH, Chio CC, Lim SW, Wang JJ, Chang CH, et al. Traumatic brain injury increases the risk of major adverse cardiovascular and cerebrovascular events: a 13-year, population-based study. World Neurosurg. 2019;122:e740–e53. doi: 10.1016/j.wneu.2018.10.130. [DOI] [PubMed] [Google Scholar]
- 18.McFarlane TD, Love J, Hanley S, Dixon BE, Hammond FM. Increased risk of stroke among young adults with serious traumatic brain injury. J Head Trauma Rehabil. 2019;1. [DOI] [PubMed]
- 19.Nakae R, Takayama Y, Kuwamoto K, Naoe Y, Sato H, Yokota H. Time course of coagulation and fibrinolytic parameters in patients with traumatic brain injury. J Neurotrauma. 2016;33:688–95. doi: 10.1089/neu.2015.4039. [DOI] [PubMed] [Google Scholar]
- 20.Nakae R, Yokobori S, Takayama Y, Kuwamoto K, Naoe Y, Yokota H. Age-related differences in fibrinolytic parameters in patients with acute traumatic brain injury. Surg Neurol Int 2017;8. [DOI] [PMC free article] [PubMed]
- 21.Lustenberger T, Talving P, Kobayashi L, Inaba K, Lam L, Plurad D, et al. Time course of coagulopathy in isolated severe traumatic brain injury. Injury. 2010;41:924–928. doi: 10.1016/j.injury.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 22.Nekludov M, Bellander B-MM, Blombäck M, Wallen HN, Blomback M, Wallen HN. Platelet dysfunction in patients with severe traumatic brain injury. J Neurotrauma. 2007;24:1699–706. doi: 10.1089/neu.2007.0322. [DOI] [PubMed] [Google Scholar]
- 23.Fair K, Farrell D, McCully B, Rick E, Dewey EN, Hilliard C, et al. Fibrinolytic activation in patients with progressive intracranial hemorrhage after traumatic brain injury. New York: Mary Ann Liebert Inc.; 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briggs A, Gates JD, Kaufman RM, Calahan C, Gormley WB, Havens JM. Platelet dysfunction and platelet transfusion in traumatic brain injury. J Surg Res. 2015;193:802–806. doi: 10.1016/j.jss.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Cao H, Zhang X, Ge L, Bie L. The effect of hypertonic saline and mannitol on coagulation in moderate traumatic brain injury patients. Am J Emerg Med. 2017;35:1404–7. doi: 10.1016/j.ajem.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 26.Scherer RU, Spangenberg P. Procoagulant activity in patients with isolated severe head trauma. Crit Care Med. 1998;26:149–156. doi: 10.1097/00003246-199801000-00031. [DOI] [PubMed] [Google Scholar]
- 27.Grenander B, Bredbacka S, Rydvall A, Ároch R, Edner G, Koskinen LOD, et al. Antithrombin treatment in patients with traumatic brain injury: a pilot study. J Neurosurg Anesthesiol. 2001;13:49–56. doi: 10.1097/00008506-200101000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Bredbacka S, Edner G. Soluble fibrin and D-dimer as detectors of hypercoagulability in patients with isolated brain trauma. J Neurosurg Anesthesiol. 1994;6:75–82. doi: 10.1097/00008506-199404000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, et al. Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg. 2012;73:13–19. doi: 10.1097/TA.0b013e318256deab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vecht CJ, Sibinga CT, Minderhoud JM. Disseminated intravascular coagulation and head injury. J Neurol Neurosurg Psychiatry. 1975;38:567–571. doi: 10.1136/jnnp.38.6.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auer L. Disturbances of the coagulatory system in patients with severe cerebral trauma. I. Acta Neurochir. 1978;43:51–9. doi: 10.1007/BF01809225. [DOI] [PubMed] [Google Scholar]
- 32.Carrick M, Tyroch A, Youens C, Handley T. Subsequent development of thrombocytopenia and coagulopathy in moderate and severe head injury: support for serial laboratory examination. J Trauma. 2005;58:725–729. doi: 10.1097/01.ta.0000159249.68363.78. [DOI] [PubMed] [Google Scholar]
- 33.Hulka F, Mullins RJ, Frank EH. Blunt brain injury activates the coagulation process. Arch Surg. 1996;131:923–927. doi: 10.1001/archsurg.1996.01430210021004. [DOI] [PubMed] [Google Scholar]
- 34.Engström M, Romner B, Schalén W, Reinstrup P. Thrombocytopenia predicts progressive hemorrhage after head trauma. J Neurotrauma. 2005;22:291–296. doi: 10.1089/neu.2005.22.291. [DOI] [PubMed] [Google Scholar]
- 35.Touho H, Hirakawa K, Hino A, Karasawa J, Ohno Y. Relationship between abnormalities of coagulation and fibrinolysis and postoperative intracranial hemorrhage in head injury. Neurosurgery. 1986;19:523–531. doi: 10.1227/00006123-198610000-00005. [DOI] [PubMed] [Google Scholar]
- 36.Guillotte AR, Herbert JP, Madsen R, Hammer RD, Litofsky NS. Effects of platelet dysfunction and platelet transfusion on outcomes in traumatic brain injury patients. Brain Inj. 2018;32:1849–1857. doi: 10.1080/02699052.2018.1536805. [DOI] [PubMed] [Google Scholar]
- 37.Lindblad C, Thelin EP, Nekludov M, Frostell A, Nelson DW, Svensson M, et al. Assessment of platelet function in traumatic brain injury-A retrospective observational study in the neuro-critical care setting. Front Neurol. 2018;9:15. doi: 10.3389/fneur.2018.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vecht CJ, Minderhoud JM, Sibinga CT. Platelet aggregability in relation to impaired consciousness after head injury. J Clin Pathol. 1975;28:814–820. doi: 10.1136/jcp.28.10.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halpern CH, Reilly PM, Turtz AR, Stein SC. Traumatic coagulopathy: the effect of brain injury. J Neurotrauma. 2008;25:997–1001. doi: 10.1089/neu.2008.0548. [DOI] [PubMed] [Google Scholar]
- 40.Pahatouridis D, Alexiou GA, Zigouris A, Mihos E, Drosos D, Voulgaris S. Brain injury coagulopathy in moderate head injury. The role of early administration of low molecular weight heparin. 2010; [DOI] [PubMed]
- 41.Nekludov M, Antovic J, Bredbacka S, Blombäck M. Coagulation abnormalities associated with severe isolated traumatic brain injury: cerebral arterio-venous differences in coagulation and inflammatory markers. J Neurotrauma. 2007;24:174–180. doi: 10.1089/neu.2006.0173. [DOI] [PubMed] [Google Scholar]
- 42.Sørensen JV, Jensen HP, Rahr HB, Borris LC, Lassen MR, Fedders O, et al. Haemostatic activation in patients with head injury with and without simultaneous multiple trauma. Scand J Clin Lab Invest. 2009;53:659–665. doi: 10.3109/00365519309092568. [DOI] [PubMed] [Google Scholar]
- 43.Gando S, Nanzaki S, Kemmotsu O. Coagulofibrinolytic changes after isolated head injury are not different from those in trauma patients without head injury. J Trauma. 1999;46:1070–6. doi: 10.1097/00005373-199906000-00018. [DOI] [PubMed] [Google Scholar]
- 44.Foaud HMA, Labib JR, Metwally HG, El-Twab KMA. Plasma D-dimer as a prognostic marker in ICU admitted egyptian children with traumatic brain injury. J Clin Diagn Res. 2014;8:PC01–6. doi: 10.7860/JCDR/2014/9489.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suehiro E, Fujiyama Y, Kiyohira M, Motoki Y, Nojima J, Suzuki M. Probability of soluble tissue factor release lead to the elevation of d-dimer as a biomarker for traumatic brain injury. Neurol Med Chir (Tokyo) 2019;59:63–67. doi: 10.2176/nmc.oa.2018-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karri J, Cardenas JC, Matijevic N, Wang YW, Choi S, Zhu L, et al. Early fibrinolysis associated with hemorrhagic progression following traumatic brain injury. Shock. 2017;48:644–650. doi: 10.1097/SHK.0000000000000912. [DOI] [PubMed] [Google Scholar]
- 47.Defazio M V., Rammo RA, Robles JR, Bramlett HM, Dietrich WD, Bullock MR. The potential utility of blood-derived biochemical markers as indicators of early clinical trends following severe traumatic brain injury. World Neurosurg. 2014. p. 151–8. [DOI] [PMC free article] [PubMed]
- 48.Becker S, Schneider W, Kreuz W, Jacobi G, Scharrer I, Nowak-Göttl U. Post-trauma coagulation and fibrinolysis in children suffering from severe cerebro-cranial trauma. Eur J Pediatr. 1999;158(Suppl 3):S197–202. doi: 10.1007/pl00014355. [DOI] [PubMed] [Google Scholar]
- 49.Leeper CM, Strotmeyer SJ, Neal MD, Gaines BA. Window of opportunity to mitigate trauma-induced coagulopathy: fibrinolysis shutdown not prevalent until 1 hour post-injury. Ann Surg. 2019;270:528–534. doi: 10.1097/SLA.0000000000003464. [DOI] [PubMed] [Google Scholar]
- 50.Leeper CM, Neal MD, McKenna CJ, Gaines BA. Trending fibrinolytic dysregulation: fibrinolysis shutdown in the days after injury is associated with poor outcome in severely injured children. Ann Surg. 2017;266:508–515. doi: 10.1097/SLA.0000000000002355. [DOI] [PubMed] [Google Scholar]
- 51.Greuters S, van den Berg A, Franschman G, Viersen VA, Beishuizen A, Peerdeman SM, et al. Acute and delayed mild coagulopathy are related to outcome in patients with isolated traumatic brain injury. Crit Care. 2011;15:R2. doi: 10.1186/cc9399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Homnick A, Sifri Z, Yonclas P, Mohr A, Livingston D. The temporal course of intracranial haemorrhage progression: How long is observation necessary? Injury. 2012;43:2122–2125. doi: 10.1016/j.injury.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 53.Tynngård N, Lindahl TL, Ramström S. Assays of different aspects of haemostasis - what do they measure? Thromb J 2015. [DOI] [PMC free article] [PubMed]
- 54.Vulliamy P, Kornblith LZ, Kutcher ME, Cohen MJ, Brohi K, Neal MD. Alterations in platelet behavior after major trauma: adaptive or maladaptive? Platelets 2020. p. 1–10. [DOI] [PMC free article] [PubMed]
- 55.Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG. Platelet activation and function after trauma. J Trauma. 2001;51:639–647. doi: 10.1097/00005373-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 56.Verni CC, Davila A, Balian S, Sims CA, Diamond SL. Platelet dysfunction during trauma involves diverse signaling pathways and an inhibitory activity in patient-derived plasma. J Trauma Acute Care Surg. 2019;86:250–9. doi: 10.1097/TA.0000000000002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Massaro AM, Doerfler S, Nawalinski K, Michel B, Driscoll N, Ju C, et al. Thromboelastography defines late hypercoagulability after TBI: a pilot study. Neurocrit Care. 2015;22:45–51. doi: 10.1007/s12028-014-0051-3. [DOI] [PubMed] [Google Scholar]
- 58.Stein SC, Graham DI, Chen X-H, Smith DH. Association between intravascular microthrombosis and cerebral ischemia in traumatic brain injury. Neurosurgery. 2004;54:687–91. doi: 10.1227/01.neu.0000108641.98845.88. [DOI] [PubMed] [Google Scholar]
- 59.Kurland D, Hong C, Aarabi B, Gerzanich V, Simard JM. Hemorrhagic progression of a contusion after traumatic brain injury: a review. New York: Mary Ann Liebert, Inc.; 2012. pp. 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Furay E, Daley M, Teixeira PG, Coopwood TB, Aydelotte JD, Malesa N, et al. Goal-directed platelet transfusions correct platelet dysfunction and may improve survival in patients with severe traumatic brain injury. J Trauma Acute Care Surg. 2018;85:881–7. doi: 10.1097/TA.0000000000002047. [DOI] [PubMed] [Google Scholar]
- 61.Kim DY, O’Leary M, Nguyen A, Kaji A, Bricker S, Neville A, et al. The effect of platelet and desmopressin administration on early radiographic progression of traumatic intracranial hemorrhage. J Neurotrauma. 2015;32:1815–21. doi: 10.1089/neu.2014.3728. [DOI] [PubMed] [Google Scholar]
- 62.Furay EJ, Daley MJ, Satarasinghe P, Lara S, Aydelotte JD, Teixeira PG, et al. Desmopressin is a transfusion sparing option to reverse platelet dysfunction in patients with severe traumatic brain injury. J Trauma Acute Care Surg. 2020;88:80–6. doi: 10.1097/TA.0000000000002521. [DOI] [PubMed] [Google Scholar]
- 63.Wada T, Gando S, Maekaw K, Katabami K, Sageshima H, Hayakawa M, et al. Disseminated intravascular coagulation with increased fibrinolysis during the early phase of isolated traumatic brain injury. Crit Care. 2017;21:219. doi: 10.1186/s13054-017-1808-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duguid J, O’Shaughnessy DF, Atterbury C, Maggs PB, Murphy M, Thomas D, et al. Guidelines for the use of fresh-frozen plasma, cryoprecipitate and cryosupernatant. Br. J. Haematol. 2004. p. 11–28. [DOI] [PubMed]
- 65.Chang R, Folkerson LE, Sloan D, Tomasek JS, Kitagawa RS, Choi HA, et al. Early plasma transfusion is associated with improved survival after isolated traumatic brain injury in patients with multifocal intracranial hemorrhage. Surgery. 2017;161:538–45. doi: 10.1016/j.surg.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joseph B, Amini A, Friese RS, Houdek M, Hays D, Kulvatunyou N, et al. Factor IX complex for the correction of traumatic coagulopathy. J Trauma Acute Care Surg. 2012. p. 828–34. [DOI] [PubMed]
- 67.Ferreira J, Delossantos M. The clinical use of prothrombin complex concentrate. J Emerg Med. 2013;44:1201–1210. doi: 10.1016/j.jemermed.2012.12.022. [DOI] [PubMed] [Google Scholar]
- 68.Narayan RK, Maas AIR, Marshall LF, Servadei F, Skolnick BE, Tillinger MN. Recombinant factor viia in traumatic intracerebral hemorrhage: Results of a dose-escalation clinical trial. Neurosurgery. 2008;62:776–786. doi: 10.1227/01.neu.0000316898.78371.74. [DOI] [PubMed] [Google Scholar]
- 69.Longstaff C. Measuring fibrinolysis: from research to routine diagnostic assays. J Thromb Haemost 2018. p. 652–62. [DOI] [PMC free article] [PubMed]
- 70.Ramos CR, Moore EE, Manco-Johnson ML, Silliman CC, Chapman MC, Banerjee A. The incidence and magnitude of fibrinolytic activation in trauma patients: a rebuttal. J Thromb Haemost 2013; p. 1435–7. [DOI] [PMC free article] [PubMed]
- 71.Raza I, Davenport R, Rourke C, Platton S, Manson J, Spoors C, et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost. 2013;11:307–314. doi: 10.1111/jth.12078. [DOI] [PubMed] [Google Scholar]
- 72.CRASH-3 trial collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet 2019;394:1713–23. [DOI] [PMC free article] [PubMed]
- 73.Olufajo OA, Yorkgitis BK, Cooper Z, Rios-Diaz A, Metcalfe D, Havens JM, et al. How long should we fear? Long-term risk of venous thromboembolism in patients with traumatic brain injury. J Trauma Acute Care Surg. Lippincott Williams and Wilkins; 2016. p. 71–7. [DOI] [PubMed]
- 74.Meizoso JP, Karcutskie CA, Ray JJ, Namias N, Schulman CI, Proctor KG. Persistent fibrinolysis shutdown is associated with increased mortality in severely injured trauma patients. J Am Coll Surg. 2017;224:575–582. doi: 10.1016/j.jamcollsurg.2016.12.018. [DOI] [PubMed] [Google Scholar]
- 75.Hay JR, Johnson VE, Young AMH, Smith DH, Stewart W. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol Oxford Academic. 2015;74:1147–1157. doi: 10.1097/NEN.0000000000000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang D, Gong S, Jin H, Wang J, Sheng P, Zou W, et al. Coagulation parameters and risk of progressive hemorrhagic injury after traumatic brain injury: a systematic review and meta-analysis. Biomed Res Int. 2015;2015:261825. doi: 10.1155/2015/261825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spatz M, Micic D, Mrsulja BB, Klatzo I. Cerebral microvessels as mediators of cerebral transport. Adv Neurol. 1978;20:189–196. [PubMed] [Google Scholar]
- 78.Levi M, Van Der Poll T, Schultz M. Systemic versus localized coagulation activation contributing to organ failure in critically ill patients. Semin Immunopathol Springer; 2012. p. 167–79. [DOI] [PMC free article] [PubMed]
- 79.Del Zoppo GJ, Izawa Y, Hawkins BT. Hemostasis and alterations of the central nervous system. Semin Thromb Hemost. 2013;39:856–875. doi: 10.1055/s-0033-1357490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rollin L, Darmoni S, Caillard JF, Gehanno JF. Searching for high-quality articles about intervention studies in occupational health—What is really missed when using only the Medline database? Scand J Work Environ Heal. 2010;36:484–487. doi: 10.5271/sjweh.3082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.