

Abstract
Rosacea is a chronic inflammatory disease that affects the face skin. It is clinically classified into the following four subgroups depending on its location and severity: erythematotelangiectatic, papulopustular, phymatous, and ocular. Rosacea is a multifactorial disease triggered by favoring factors, the pathogenesis of which remains imperfectly understood. Recognized mechanisms include the innate immune system, with the implication of Toll-like receptors (TLRs) and cathelicidins; neurovascular deregulation involving vascular endothelial growth factor (VEGF), transient receptor potential (TRP) ion channels, and neuropeptides; and dysfunction of skin sebaceous glands and ocular meibomian glands. Microorganisms, genetic predisposition, corticosteroid treatment, and ultraviolet B (UVB) radiation are favoring factors. In this paper, we review the common and specific molecular mechanisms involved in the pathogenesis of cutaneous and ocular rosacea and discuss laboratory and clinical studies, as well as experimental models.
Introduction
Rosacea is a chronic inflammatory disease that mainly affects the central facial skin (cheeks, chin, nose, and central forehead); it is characterized by flushing, transient or persistent rash, inflammatory papules and pustules, telangiectasia, and ocular manifestations [1-3]. According to its clinical presentation, it has been classified by the National Committee of Experts into the four following subtypes: erythematotelangiectatic, papulopustular, phymatous, and ocular [4].
Rosacea affects about 10% of the population with a greater risk of sun-sensitive fair skin [5]; the disease can also develop in Asian and African populations [6]. The severity of the disease appears to depend on the patient’s gender and age, where rosacea is three times more frequent in women than in men but more severe in men and younger patients, suggesting that the more severe forms of the disease manifest sooner or that the disease improves over time [7]. The ocular variant of rosacea represents between 10% and 50% of the total rosacea population and is characterized by inflammation of the ocular surface tissues, including the eyelid edge (blepharitis) and eyes (tear film instability, eye irritation, red eyes, eye dryness, conjunctivitis, etc.) [8,9]. In the most severe cases, chronic corneal damage may lead to corneal neovascularization (CNV), corneal perforations, corneal ulcers, and corneal edemas, which compromise corneal transparency and lead to visual loss [3,10,11] (Figure 1).
Figure 1.
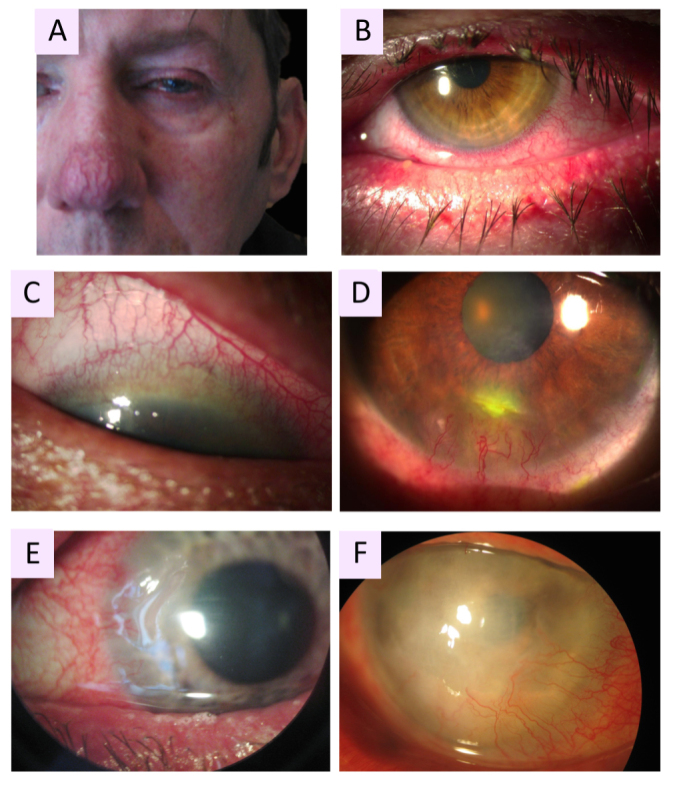
Clinical manifestations of oculocutaneous rosacea. A: Patient with phymatous rosacea–associated rhinophyma, blepharophyma, nasal and facial erythema with telangiectasia. B: Blepharophyma with thickened lid edges, lid margin telangiectasia, meibomian gland dysfunction (MGD). C: Corneal neovascularization (CNV) of ocular rosacea growing from the superior limbus with a crescent pattern forming a vascular pannus. D: Catarrhal corneal infiltrate caused by rosacea. E: Typical peripheral ulcerative keratitis (PUK) of rosacea, corresponding to sterile corneal melting of a crescentic area with newly formed stromal vessels. F: Advanced stage of ocular rosacea with white corneal infiltrates and whole corneal neovascularization, including the visual axis.
To date, the diagnosis of rosacea has been established clinically based on observation and interpretation of skin and ocular signs. While many patients show both ocular and skin signs, some may only show ocular signs, which can make diagnosis more difficult [12-14].
The mechanisms of rosacea are still unclear. In recent years, a multifactorial pathogenesis with genetic predisposition has been emphasized [15]. Many triggering factors, such as ultraviolet (UV) exposure, local inflammatory responses to skin microorganisms (associated with Demodex infestation), temperature changes, hot, cold, spicy foods, and stress have been considered to initiate or worsen the disease [16]. In addition, studies have demonstrated an implication of the skin and ocular immune system with changes in adaptative and innate cells, as well as neuroinflammatory changes [17] (Figure 2). Involvement of innate immunity has been advocated by high expression of the Toll-like receptor 2 (TLR-2) and the antimicrobial peptide of cathelicidin (LL-37) in the skin tissues of rosacea patients. In addition, the nucleotide-binding domain leucin-rich repeat and pyrin-containing receptor 3 (NLRP3) inflammasome appear to play a role in LL-37-mediated inflammation [18]. The infiltration of mast cells and the release of proinflammatory mediators, such as interleukin-1β (IL-1β), IL-6, IL-17, interferon-γ (IFN-γ), matrix metalloproteinase-2 (MMP-2), MMP-9, tumor necrosis factor-α (TNF-α), activated by nuclear factor-κB (NF-κB) [19-21], as well as the infiltration of CD4(+) T cells, contribute to the inflammatory signs [22,23]. Abnormal neural control of vascular flow contributes to enlargement of skin and ocular surface blood vessels. Such vascular content increases appear to relate to elevated levels of vascular endothelial growth factor (VEGF) and transient receptor potential (TRP) channel found in rosacea patients [24,25]. However, it remains unclear whether such vascular changes are a primary event or a consequence of inflammatory changes.
Figure 2.

Diagram showing the factors involved in rosacea pathophysiology.
Finally, the molecular steps involved in the pathogenesis of ocular rosacea are not fully known. Meibomian gland dysfunction has been recognized as a major component [13,26]. Like in the skin, activation of the innate and adaptive immune response and abnormal vascular regulations have been identified. In the tears and in the ocular surface tissues of ocular rosacea patients, high levels of TLR-4 and LL-37 were measured together with cell infiltration and the release of proinflammatory factors [13,27,28]. A link between Demodex infestation and ocular rosacea has also been advocated [29,30]. Overall, all these phenomena in the skin and eye act in synergy to maintain chronic inflammation at the cutaneous, epidermal, conjunctival, and perivascular interface, eventually leading to secondary fibrosis [31]. In this paper, we aim to review the common and specific pathogenic mechanisms of cutaneous and ocular rosacea and focus on the few models used to study this disease.
Deregulation of the immune system
Activation of immune-mediated inflammatory pathways appears to be at the center of the pathogenesis of rosacea and involves the coordinated activity of several cell types, such as mast cells and macrophages, and the release of proinflammatory mediators, such as IL-6, IL-1β, IL-18, or TNF-α [32,33]. Inhibition of these inflammatory pathways is correlated with clinical improvement.
Innate immune system
TLR-2/4 pathways
As part of the innate immune system, members of the TLRs, which recognize physical and chemical stimuli or microbial pathogens, are expressed on the surface of various skin cells, including keratinocytes, macrophages, and mast cells [34]. Induction of the innate immune response by TLR stimulation induces the controlled and limited activation of NF-κB and the subsequent production of cytokines, chemokines, and antimicrobial peptides [35]. However, uncontrolled activation of the innate immune system leads to deleterious consequences [36].
In the skin of rosacea patients, TLR-2 is overexpressed on the keratinocytes in the epidermis and on infiltrating cells in the dermis [19,21], enhancing skin sensitivity to external stimuli and the production of IL-8, IL-1β, and TNF-α by keratinocytes [20,37]. TLR-2 can also activate the NLRP3 inflammasome and induce cell death [18,38]. Components of the inflammasome, including caspase-1 and IL-1β, in the rosacea skin [39] worsen the inflammatory response by activating many other proinflammatory factors, such as IL-8, TNF-α and MMPs [20]. The transcription factor NF-κB, activated by TNF-α or directly stimulated by TLRs or LL-37 signaling, induces the expression of IL-1α, IL-1β, IL-18, and IL-33 [40,41].
Recently, erythroid differentiation regulator 1 (Erdr1) has been recognized as a cytokine suppressed by TLR-2 and the myeloid differentiation factor 88 (MyD88) pathways. Indeed, animals deficient in TLR-2 and MyD88 have elevated Erdr1 [42]. Erdr1, localized at the inner part of the cytoplasmic membrane and secreted through vesicles immediately under stress conditions, has been shown to be reduced in rosacea skin [43]. Erdr1 is negatively regulated by IL-18, a proinflammatory cytokine, and has a biphasic effect. It enhances cell survival at low concentrations and densities but increases cell death at high concentrations and densities. Recombinant Erdr1 significantly reduced the disease in a rosacea model induced by the injection of LL-37 in rodents [43].
TLRs are also present at the ocular surface, where TLR-2 and 4 are constitutively expressed in the corneal, limbic, and conjunctival epithelial cells [44,45]. Stimulation of TLRs by exogenous stimuli activates NF-κB and induces the production of cytokines and antimicrobial peptides, such as β-defensins [46]. In corneal inflammation in mice and humans, the roles of TLR-2 and 4 have been recognized [47]. In eyelid biopsies of patients with ocular rosacea, TLR-4 expression was significantly increased compared with normal subjects [28], and high levels of TLR-2 and 4 were detected in various ocular surface conditions, including dry eye syndrome, one of the hallmarks of ocular rosacea. Activated TLR-4 has been shown to induce the activation of TLR-2 and together enhance the transcription of inducible nitric oxide synthase (iNOS) [21]. In a rodent model of dry eye disease, the corneal expression of TLR-4 was significantly increased. TLR-4 inhibition reduced the severity of dryness and the expression of IL-1β, IL-6, and TNF-α, as well as the corneal infiltration of CD4(+) T cells [48]. These results suggest that TLR-4 participates in inflammatory responses of the ocular surface. Furthermore, TLR-2 expression was upregulated in the conjunctival and corneal cells and in the lacrimal glands in a rodent model of dry eye, as well as in conjunctival cells of patients with dry eye [49,50].
In the eyelid biopsies of patients with rosacea, Wladis et al. [51,52] showed that the NF-κB pathway was significantly increased in rosacea patients. Furthermore, patients showed increased levels of active mitogen-activated protein kinase (MAPK) protein 38 (p38) and extracellular signal-regulated kinase (Erk) compared with healthy subjects, acting in concert with NF-κB factor for the regulation of immune responses. In addition, TLRs, including TLR-2, are well known for promoting increased activation of p38 and Erk signaling.
In tears, rosacea patients have increased levels of IL-1α, IL-1β, IL-16, TNF-α, monocyte chemoattractant protein-1 (MCP-1), MMP-8, and MMP-9 [53-57]. IL-1 can increase the production and activation of MMPs, including MMP-9, contributing to eyelid and ocular surface irritation, epithelial defects, corneal ulcers, and CNV observed in severe rosacea patients [14]. Proinflammatory factors can also be released by resident cells, such as mast cells, or subsequently by infiltrating cells.
Cathelicidin-mediated inflammation/LL-37/KLK-5
Origin of cathelicidin in rosacea
Cathelicidins, classified as antimicrobial peptides (AMPs), play a major role in mammalian innate immune defense against bacteria. In humans, the Camp gene encodes the precursor peptide 18-kDa cationic antimicrobial protein (CAP-18), which is cleaved by kallikrein-related peptidase 5 (KLK-5) into the active forms LL-37 and FALL-39 [58-60]. LL-37, a-37-amino-acid amphipathic and helical peptide, is expressed in epithelial cells and in leukocytes, such as monocytes, neutrophils, T cells, natural killer (NK) cells, and B cells. Different cell types produce this peptide, such as keratinocytes, myeloid cells, neutrophils, and mast cells. Many studies have shown enhanced LL-37 expression in the skin biopsies of rosacea patients [61-63]. Elevated expression levels and activity of KLK-5 have also been described in the skin of rosacea patients [62], as well as in other skin diseases, such as atopic dermatitis, Netherton syndrome, or psoriasis [64]. Injection of KLK-5 into the skin of mice has been shown to cause dermatitis alleviated by inhibition of KLK-5 activity [65]. In human epidermal cells stimulated with calcitriol, the hormonally active form of vitamin D—which is known to induce expression of KLK-5 and LL-37—the inhibition of KLK-5 expression was correlated with a significant decrease in the expression levels of IL-6, IL-18, and MCP-1 [66]. Increasing the expression of TLR-2 increased the expression of KLK-5, and conversely, showed a link between TLR-2 and KLK-5 activation in rosacea-affected skin [19].
In a recent study, Sugimoto et al. [67] found that the expression of a Kazal-type lymphoepithelial inhibitor (LEKTI) in keratinocytes, known to control the activity of KLK-5, was upregulated in the biopsies of rosacea patients. Thus, this molecule could participate in the control of the aberrant activity of KLK-5 in the inflammatory reactions of rosacea. In addition to its activation by TLR-2, KLK-5 can also be activated by MMP-9 in rosacea [16,68]. Therefore, KLK-5 activity is tightly regulated by a balance of activators, such as TLR-2 and MMP-9, and repressors, such as LEKTI.
The increase in cathelicidin levels in rosacea patients could be amplified via the expression of the protease-activated receptor 2 (PAR-2), which is activated by KLK-5 and known to mediate inflammation in various tissues, including the skin [69]. Kim et al. [70] showed that PAR-2 levels were higher in skin biopsies from rosacea patients and that keratinocytes stimulation with PAR-2 in vitro increased the production of LL-37, IL-6, and IL-8.
Pathogenic role of cathelicidin in rosacea
The link between LL-37 and rosacea was demonstrated by Yamasaki et al. [61] in an inflammatory skin mouse model induced by the injection of LL-37, developing telangiectasia, erythema, and inflammation. Using the same model, Yuan et al. [71] showed that the skin expressed higher levels of TNF-α, IL-1β, IL-6, and TLR-2 compared with the control group. The Janus kinase/signal transducers and activators of transcription (JAK/STAT) signal transduction pathway also appeared to participate in the inflammatory mechanism in cutaneous rosacea mediated by LL-37. Using in vitro HaCaT cells, a spontaneously immortalized human keratinocyte line, treated with LL-37, Li et al. [72], further demonstrated the upregulation of JAK2 and STAT3, which upregulated the expression of proinflammatory cytokines, including TNF-α, IL-6, IL-8, and MCP-1. The inhibition of this pathway was followed by a decrease in the production of proinflammatory cytokines, suggesting that LL-37 cathelicidin may play a critical role in the inflammatory component of skin. In the LL-37-mediated skin inflammation model in mice, the number of neutrophils and macrophages was significantly increased in the skin together with the expression levels of chemokine (C-X-C motif) ligand (CXCL) 2 and chemokine (C-C motif) ligand (CCL) [71]. In addition, LL-37 and MMP-9 are expressed by neutrophils recruited from tissues in response to inflammation [73].
LL-37 promotes leukocyte chemotaxis and the release of proinflammatory mediators, such as IL-6 and MMP-9, from mast cells [74–78], where the density increases in skin rosacea and is correlated with the duration of disease [79]. In addition, the levels of chymase and tryptase released by mast cells were significantly elevated in the skin of mice injected with the LL-37 peptide and in skin biopsies of rosacea patients [73,80]. The role of mast cells was further confirmed when LL-37 injection in mice knocked out for mast cells did not develop any phenotype. Moreover, mast cell repopulation in these mice restored LL-37-induced skin inflammation and release of the proinflammatory factors IL-6 and MMP-9 [73].
Few of the conducted studies have been performed in the eye, and there is no clear evidence implicating LL-37 in ocular rosacea. LL-37 is naturally present in the conjunctiva, cornea, and Moll glands located between the eyelashes on the eyelid. Its expression increases in response to certain inflammatory processes, including corneal damage, corneal ulcers, or exposure to pathogenic microorganisms [81]. The α- and β-defensins expressed on the ocular surface are capable of inducing chemotaxis of monocytes and neutrophils, stimulating the production of cytokines and the release of histamine by mast cells [45,47], and they have been involved in corneal infections or dry eye [82]. More recently, Go et al. [27] demonstrated increased conjunctival LL-37 gene expression and increased tear levels of β-defensins in patients with ocular rosacea. However, whether LL-37 directly induces ocular rosacea has not been determined.
Mast cells
As described above, mast cells are essential in LL-37-induced skin inflammation, and their number increases in the rosacea skin, contributing to the inflammatory immune mechanism [73,80]. In addition, mast cells participate in fibrosis and vascular deregulation associated with rosacea. The histamine and tryptase released by these immune cells promote vasodilation, angiogenesis, and fibrosis by recruiting fibroblasts and MMPs. The fibroblasts stimulate mast cells and promote the release of MMP-1, which can also act on fibroblasts, facilitating fibrosis [78]. Clinically, patients with fibrosis have a phyma. Other factors are implicated in rosacea fibrosis, such as MMPs, serine proteases, growth factors, cytokines, and chemokines [17].
Mast cells play a role in the rosacea pathogenesis by enhancing vasodilation and angiogenesis via different interaction ways [83]. They can produce various proangiogenic molecules, such as VEGF and fibroblast growth factor (FGF), stimulating the migration and proliferation of vascular endothelial cells. FGF is a well-known proangiogenic molecule that, along with VEGF factor, appears to be pivotal in the regulation of angiogenesis [84]. Mast cells can also stimulate the release of proteinases and heparin, allowing the release of other proangiogenic factors and the recruitment of macrophages, among other cells [83]. In addition, during degranulation, mast cells release large amounts of histamine, which is a powerful vasodilator of the body that leads to inflammation, redness, and edema [85]. Finally, mast cells release chymase, a potent local angiotensin-converting enzyme that produces angiotensin II [86]. Angiotensin regulates vascular constriction and endothelial dysfunction, and angiotensin receptors are expressed not only in skin vessels but also in sebaceous glands and dermal fibroblasts [87]. Mast cell degranulation is activated by neuropeptides and sympathetic innervation contributing to the neurogenic inflammation pathway [88].
Adaptative immune system
T-cell-mediated responses
Little is known about the adaptive immune system in rosacea. The α- and β-defensins induce chemotaxis of T lymphocytes and stimulate the production of antibodies by B lymphocytes [45,47]. These molecules act at the interface between the innate immune system and the adaptive immune system. Extremely high levels of CD4(+) T cells have been observed around the hair follicles and in the perivascular region in rosacea skin [23], where the CD4(+) T cell response is dominated by T lymphocyte helper (TH)1/TH17 polarized immune cells, accompanied by a significant upregulation of IFN-γ and IL-17 [22,23]. In the mouse model of LL-37 peptide-induced inflammation, high levels of CD4(+) T cells were also found in the dermis [41]. TH17 cell-mediated immune responses are implicated in other dermatological pathologies, such as psoriatic dermatitis, in the recruitment of mast cells increases significantly with an upregulation of IL-17 [89].
B-cell-mediated responses
B lymphocytes and plasma cells seem to be also involved in the adaptive immune response in rosacea. This is shown by the significant increase in the expression level of a B cell marker, CD20, in skin biopsies [23].
Other actors involved in immune responses in rosacea
Moura et al. [21] suggested the implication of plasmacytoid dendritic cells (pDCs) in the inflammatory response in rosacea. These cells are present at the site of the lesion on the skin biopsies of patients with rosacea, as they are in psoriasis [90]. The cells stimulate CD4 helper T cells and B lymphocytes following certain viral infections and can secrete large quantities of IFN-α and β [91].
Recently, a new molecule has surfaced in the pathology of rosacea. Koebnerisin (S100A15) is an antimicrobial peptide involved in immune-mediated inflammatory responses in various dermatological pathologies, including psoriasis [92] and rosacea [93]. Regulated by TH1 and TH17 cells, koebnerisin acts as a chemoattractant for leukocytes [94,95]. Skin biopsies of patients with rosacea showed high levels of koebnerisin [93]. In vitro, human keratinocytes cultured with koebnerisin secrete MMP-9 and VEGF, suggesting that it could be proangiogenic [93]. Koebnerisin induced the production of TNF-α, IL-6, IL-8, and IL-1β in keratinocytes and leukocytes [92]. However, its role in ocular rosacea is unknown.
Endoplasmic reticulum (ER) stress protects cellular functions from internal disturbances, such as protein misfolding, and from external disturbances, such as exposure to UVB and microbes. It has been implicated in various skin diseases, such as melanoma and Darier’s disease [96]. In cutaneous rosacea, ER stress has been shown to increase the expression and reactivity of TLR-2 via activating transcription factor 4 (ATF4) signaling in epithelial cells [97]. In addition, it stimulates the production of the antimicrobial peptide LL-37 via the sphingosine-1-phosphate (S1P)-NK-κB/EBPα-dependent pathway [98]. Thus, the ER stress could participate in the inflammation in rosacea via LL-37.
Recently, Li et al. [99] highlighted the implication of a new molecule, decysin 1, coded by the ADAMDEC1 gene, in the pathophysiology of cutaneous rosacea. Decysin 1 is part of the ADAM family (a disintegrin and metalloproteinases), which are proteins linked to matrix metalloproteases and snake venom proteins. ADAMDEC1 has been associated with many inflammatory diseases, such as atherosclerosis and pulmonary sarcoidosis. ADAMDEC1 was strongly expressed in type M1 macrophages (proinflammatory) in skin biopsies of rosacea patients. Its inhibition prevented the polarization of the macrophages M1, and subsequent release of IL-6, iNOS, and TNF-α in the THP-1 cell line and in vivo in a murine model of rosacea induced by LL-37 [99]. Thus, ADAMDEC1 could be a new player in inflammation in the pathology of rosacea by regulating the polarization of M1 macrophages. In the context of the eye, there is no information in the literature linking this molecule to ocular rosacea and ocular surface pathologies.
In Table 1, the major molecular findings involved in the deregulation of the innate and adaptive immune systems and clinical effects are shown. In Figure 3, the possible mechanisms of involvement of the immunity system in the pathophysiology of rosacea is indicated.
Figure 3.

Possible mechanisms of involvement of both innate and adaptive immunity in rosacea pathophysiology. Activation of Toll-like receptor 2 and 4 (TLR-2, 4), which are expressed in different cell types in the skin and the eye, after stimulation by various external stimuli induces the production of the antimicrobial peptide human cathelicidin peptide (LL)-37 by activation of the serine protease kallikrein 5 (KLK-5), which cleaves the precursor peptide 18-kDa cationic antimicrobial protein (CAP-18) into the active form LL-37. KLK-5 is regulated by several other activators, including matrix metalloproteinase-9 (MMP-9), and repressors, such as the Kazal-type lymphoepithelial inhibitor (LEKTI). LL-37 activation by either KLK-5 or protease-activated receptor 2 (PAR-2) results in the activation and degranulation of mast cells, which release proinflammatory mediators, including MMPs, tumor necrosis factor- α (TNF-α), and interleukins (ILs), leading to the initiation or aggravation of inflammation at the cutaneous and ocular levels. Histamine release leads to fibrosis and the appearance of phyma. The activation of the nuclear factor kappa B (NF-κB) signaling pathway by TLRs or active MAPK protein 38 (p38) and extracellular signal-regulated kinase (Erk) can also activate the NF-κB pathway, leading to the release of several proinflammatory factors, including IL-33, IL-8, IL-18, and IL-1β. Their release can also be increased by the activation of the nucleotide-biding domain leucin-rich repeat and pyrin-containing receptor 3 (NLRP3) inflammasome. Endoplasmic reticulum (ER) stress participates in inflammatory mechanisms through the release of activating transcription factor 4 (ATF4), which promotes the activation of TLR-2. Sphingosine-1-phosphate (S1P) leads to the activation of enhancer-binding protein α (EBP-α), which also activates LL-37. Neutrophils release reactive oxygen species (ROS) and proteases. Macrophages, where M1 polarization appears to be favored by Adamdec1, also participate in inflammation by secreting other inflammatory factors, such as proteases or MMPs, TNF-α, ILs, and serine proteases. Plasmacytoid dendritic cells (pDCs) promote the activation of B and T cells. TH1 and TH17, polarized CD4(+) T cells of the adaptive immune system, secrete interferon-gamma (IFN-γ) and IL-17, respectively, leading to chemotaxis of other leukocytes by enhancing chemokine (C motif) ligand (CCL) and chemokine (C-X-C motif) ligand (CXCL) expression and increased activation of LL-37. TH17 cells are thought to stimulate the activity of koebnerisine (S100A15), an antimicrobial peptide that promotes the recruitment of inflammatory cells.
Deregulation of vascular control
The blood and lymphatic vessels are important for maintaining skin homeostasis. Increased skin blood flow, persistent vascular and lymphatic dilation, increased vascular permeability, and vascular hyperresponsiveness are signs of rosacea [100,101]. Cathelicidins and VEGF are involved in vasodilation and angiogenesis [102]. In skin biopsies of rosacea patients, VEGF expression is increased in the epidermis and in infiltrating cells, associated with enhanced expression of platelet/endothelial cell adhesion molecule-1 (CD31), a marker for the endothelium of blood vessels, and D240, a marker for lymphatic vessels [24,103,104]. Elevated levels of VEGF receptors, VEGF-R1 and VEGF-R2, were measured in the vascular endothelium, infiltrating mononuclear cells, and the serum of patients with rosacea [105].
LL-37 exerts proangiogenic effects through the activation of formyl peptide type 1 (FPR1) receptor, epidermal growth factor receptor (EGFR), and downstream signaling in epithelial cells [104]. LL-37 can also increase levels of VEGF in epidermal keratinocytes [36]. The injection of LL-37 into the skin of mice promoted erythema and vascular dilation [61], and LL-37 increased the density of microvessels and the expression of VEGF [41]. An important role of endothelial cells is their ability to express adhesion molecules, such as the vascular cell adhesion molecule-1 (VCAM-1) or the intracellular adhesion molecule-1 (ICAM-1) and selectins [106]. LL-37 has been shown to increase the expression of ICAM-1 on endothelial cells in patients with atherosclerosis [107]. In a recently released study, biopsies from rosacea patients showed high expression of the VCAM-1, ICAM-1, and E-selectin molecules, along with high expression of LL-37 [108].
It has been reported that increased ER stress, accompanied by an increase in LL-37, promotes angiogenesis. ER stress increases the expression of factor VEGF-A in rosacea [24], and ATF4 acts as a transcription factor for VEGF-A [109]. Thus, ER stress, via LL-37 and the control of the regulation of VEGF-A seems to be involved in vascular anomalies of rosacea.
Angiogenesis and inflammation can act as mutual enhancers [110]. VEGF has both an angiogenic and inflammatory regulatory role, and inhibition of vascular activation has been shown to have anti-inflammatory properties [17].
One of the severe complications of ocular rosacea is CNV and subsequent corneal remodeling and opacification. It is defined by the appearance of new vessels, which develop from preexisting vessels and invade the initially avascular cornea due to a disturbance in the balance between proangiogenic and antiangiogenic factors. Abnormal growth of blood vessels and the resulting corneal opacity affect corneal transparency [111]. CNV is associated with the production of TNF-α, MCP-1, IL-6, and proangiogenic factors, such as VEGF and FGF, by corneal endothelial cells, Langerhans cells, macrophages, and inflammatory cells [112]. VEGF deregulates the production of MMPs and stimulates the formation of blood vessels, and the angiogenic cascade is amplified by inflammation [112,113]. In the physiological state, corneal avascularity is maintained by soluble forms of the VEGF receptors, VEGF-R1 and 3, which act as endogenous blockers of the proangiogenic action of VEGF [111]. In contrast, VEGF stimulates corneal angiogenesis mainly by the activation of the membrane isoform VEGF-R2. Numerous studies have shown an upregulation of VEGF in the vascular corneas. Its expression is markedly increased in corneal epithelial cells with inflammation, in corneal endothelial cells, vascular endothelial cells, and in macrophage infiltrates [114]. IL-1β has also been shown to induce CNV [115]. In a model of CNV in rats by implantation of disks impregnated with an angiogenic stimulator (basic FGF or VEGF), the antagonism of IL-1β allowed a significant decrease in angiogenesis in vivo, with a decrease in corneal neovessels confirmed by a decrease in CD31 expression. Anti-angiogenic factors are also expressed in the corneal epithelium, such as thrombospondin-1 (TSP-1) and the pigment epithelium-derived factor (PEDF), which has been identified as a potent antiangiogenic factor contributing to the inhibition of the growth of new corneal vessels [116].
The cytokines IL-6 and IL-17 have been shown to induce CNV and stimulate the production of VEGF. Angiotensin II, a potent inducer of vascular inflammation, has been shown to combine with IL-6 and stimulate the induction of VEGF. Activation of STAT3 by IL-6 can also lead to the production of VEGF in corneal fibroblasts [117]. IL-17 promoted the production of VEGF in cultured fibroblasts, associating this molecule with angiogenesis and rosacea [118]. In addition, as seen in skin rosacea, the levels of IL-18, which is involved in the activation of the inflammasome, are increased. However, its role in the eye remains unclear. It has been shown that, in patients with macular edema because of retinal venous occlusion, IL-18 levels increase significantly after treatment, with high levels of IL-18 being correlated with good visual outcome [119]. An inverse relationship between IL-18 expression and VEGF has been shown. Indeed, in patients with ischemic retinopathy, the suppression of VEGF is accompanied by an increase in the expression of IL-18. In addition, injecting IL-18 intraocularly reduces VEGF-induced leakage and neovascularization. This shows that IL-18 and VEGF suppress the production and effects of the other on the vascular system, suggesting an antiangiogenic role for IL-18. To our knowledge, this relationship has not yet been explored in the case of ocular rosacea.
The expression and cellular distribution of leukocyte adhesion molecules have been studied in ocular rosacea [120,121]. Conjunctival biopsies from patients with ocular rosacea showed expression levels of LFA-1, its ligand ICAM-1, and VCAM-1, which showed perivascular cell distribution, compared with the control group. CD105, which is involved in angiogenesis and vascular remodeling, has shown low levels in the normal and healthy vascular endothelium and increased levels in patients with ocular rosacea with inflammatory activation [28]. Thus, the expression of these molecules allows the recruitment of leukocytes contributing to the immune response, as observed in cutaneous rosacea.
Deregulation of neuronal and neurovascular control
The neuronal control of inflammation and vascular dynamics could be a plausible link for the different signs of cutaneous and ocular rosacea. Patients with skin rosacea are sensitive to triggers, such as cold, heat, UV rays, stress, and many others, which indicate the relevance of the sensory and autonomic nervous systems in the pathogenesis of the disease. Sensory and autonomous nerves are important in controlling immunity and vessel dynamics. Close collocation between sensory nerves and blood vessels has been found in the skin of rosacea patients [122].
The TRP receptors hypothesis
Most of the environmental factors described above activate ion channels with TRP, a family of nonselective cation channels permeable to Ca2+ having sensory and signaling functions throughout the body and reactive to thermal, chemical, and mechanical stimuli. The TRP channels are divided into the seven following subfamilies: ankyrin (TRPA), cationic (TRPC), melastatin (TRPM), mucolipin (TRPML), mechanosensory (TRPN), polycystic (TRPP), and vanilloid TRP (TRPV). Different stimuli can activate TRP, including the following: pungent compounds, such as capsaicin (TRPV-1) and mustard oil (TRPA-1 and TRPV-1), heat (TRPV-1), cold (TRPA-1), UVB irradiation (TRPV-4), and cosmetics (TRPA-1) [123]. TRPs, which are located in sensory nerves and other nonneuronal cells, such as mast cells, dendritic cells, and endothelial cells and keratinocytes, are capable of triggering cell secretion events via the influx of cations, including Ca2+, and they are known to participate in nociceptive and neurogenic inflammatory processes. When the TRP channels are activated by external stimuli, such as heat, this leads to the release of vasoactive neuropeptides, such as substance P (SP), the peptide linked to the calcitonin gene (CGRP), the activator polypeptide of l pituitary adenylate cyclase (PACAP), the vasoactive intestinal peptide (VIP), or even bradykinin, associated with centrofacial vasodilation [20].
In rosacea skin, abnormally high expressions of certain TRPs have been found, including TRPV-1, which has proinflammatory properties and is involved in vasoregulation contributing to acute and chronic pain; TRPV-2, which is involved in vasodilation and immunomodulation; and TRPV-3, which is involved in inflammation and fibrosis [25]. Neurovascular deregulation of these receptors could contribute to the onset of flushing seen in rosacea skin. In particular, an increase in the density of nerve fibers, as well as an increase in the expression of TRPV-1 expression has been found in the skin of patients with erythematotelangiectatic rosacea compared with healthy skin [17]. Interestingly, channels TRPV-2 and 4 have been shown to be co-located with mast cells in the skin of rosacea patients [25]. In vivo, in the LL-37 mouse model, increased expression of TRPV-2 and TRPV-4 was shown in mouse skin, as was the colocalization of TRPV-2 with mast cells. In vitro, using primary murine mast cells, increased expression of TRPV-2 and TRPV-4 was observed in response to LL-37, and activation of TRPV-4 induced mast cell degranulation via Ca2+ flux [124]. LL-37 could increase the expression of TRPV-4 via the activation of a protein-coupled receptor, Mas-related gene X2 (MRGX2), leading to the activation of mast cells and their degranulation stimulated by the influx of Ca2+ [125]. This activation of mast cells could also involve the p38 and Erk MAPK signaling pathway. In fact, its inhibition showed a considerable decrease in the degranulation of mast cells induced by LL-37 [126]. Since TLR-2 signaling pathways are involved in neuroinflammation and neuronal damage [127], the association between neurons and immune deregulation could in part be explained by TLR-2.
The TRPC-1, TRPC-4, and TRPA-1 channels have also been shown to be involved in vascular permeability. In one study, Paria et al. [128] showed that TNF-α-induced expression of TRPC-1 led to increased Ca2+ influx and vascular permeability. Mice deficient in TRPC-4 showed reduced endothelial permeability [129]. TRPA-1, another subfamily of TRP channels, has been shown to be regulated in skin rosacea. Like TRPVs, TRPAs are expressed in sensory neurons, and in 50% of cases, coexpressed with TRPV-1 receptors, suggesting mutual control of transduction of stimuli in sensory neurons [130]. These channels are temperature sensitive and have been shown to be activated by spices, such as mustard oil and cinnamon, and thermal stimuli. In addition, they have been shown to mediate vasodilation [131]. In rat neurons, TRPA-1 channels have been shown to be collocated with PAR-2, which, as we have seen, contributes to inflammation and is activated by proteases, such as KLK-5. Therefore, PAR-2 appears to be involved in inflammation, as well as a neuronal process via TRPA-1, which further exacerbates the inflammatory response.
The eye is innervated by three types of nerves—sensory, sympathetic, and parasympathetic nerves. Several publications in the literature have studied the involvement of TRP channels and neurotransmitters in the eye. However, to our knowledge, there are no studies to date that directly link these molecules to ocular rosacea.
The cornea is densely innervated by sensory nerve fibers from the trigeminal nerve, which enters the corneal stroma through the limbus and innervates the corneal epithelium. Corneal innervation contributes to high sensitivity to physical and chemical stimuli [132]. Correct innervation is essential for maintaining the structure and function of the cornea. Different subtypes of TRP channels are expressed in cells and on human corneal sensory nerve endings, including TRPV-1–4, TRPA-1 and TRPM-8 and conjunctival including TRPV-1, 2, and 4 [133]. The degree of TRPV-1 immunoreactivity in the corneal neurons is also higher than that observed in the skin. In humans, TRPV-1 is expressed in all the corneal layers, in the basal layer of the conjunctiva, while TRPA-1 is only expressed in the corneal endothelium [134,135]. The TRPV-1 and TRPA-1 subtypes have been shown to be involved in the healing of corneal wounds by stimulating cell proliferation and migration, in transparency, in fibrosis, neovascularization, and inflammation with the release of proinflammatory cytokines including IL-6 and IL-8 via the p38 and Erk MAPK signaling pathway and the activation of NF-κB [136,137]. In a mouse model of CNV by chemical burn, the activation of TRPV-1 in all the corneal layers induced fibrosis, deregulated inflammation, neovascularization, opacification, and release of corneal resident cytokines leading to the activation of immune cells [138]. Corneal transparency was restored in mice deficient in TRPV-1, accompanied by a decrease in the release of proinflammatory cytokines. Improved healing has also been observed with repeated intraperitoneal injections of TRPV-1 antagonists [138]. In addition, TRPV-1 has been shown to be associated with transforming growth factor-beta 1 (TGF-β1) and to cause fibrosis [139]. A decrease in inflammation and fibrosis has also been shown in mice deficient for TRPA-1 [140]. In a recent study, the involvement of TRPA-1 in wound healing and neovascularization was studied using a model for the healing of corneal lesions in mice leading to CNV [141]. Removal of TRPA-1 significantly reduced expression of CD31, reduced infiltration of stromal macrophages, and expression levels of VEGF-A and TGF-β1 by 70%. Targeting the activation of TRPA-1 may be a therapeutic option to improve visual acuity by improving corneal transparency.
Studies have shown that TRPM-8 channels, which are sensitive to cold and expressed in the eyelid and cornea, are involved in neuropathic pain and dry eyes [142]. Mice deficient in TRPM-8 showed a reduction in basal secretion of tears, indicating a role for TRPM-8 in controlling basal lacrimal flow [143]. More recently, TRPV-1 and TRPA-1 channels have also been implicated in dry eye. In a tear-deficient dry eye model, stimulation with TRPA-1 agonist, mustard oil (agonist of TRPA-1), on the eyes showed a decrease in tear secretion by 50% and significantly increased spontaneous blinking [144].
The role of neurotransmitters
The TRPV receptors and neuropeptides produced are linked to the inflammatory immune mechanisms of rosacea through neurogenic inflammation, in which the sensory nerves contribute to inflammation. Elevated levels of neuropeptides, such as PACAP, SP, VIP, and CGRP, have also been found in the skin of rosacea patients, and they tend to increase the expression of TRPV channels [122]. Neuropeptides/neurotransmitters released from sympathetic and sensory nerve endings regulate the main immune functions, such as antigen presentation, antibody production, T cell activity, lymphocyte activity, and cytokine secretion (neuroinflammation). They can recruit and activate target immune cells, such as neutrophils, macrophages, and mast cells, which express the α- and β-adrenergic receptors, by the upregulation of vascular adhesion molecules [145-148]. The modulation of α-adrenergic receptors and the inhibition of β-adrenergic ones has been shown to be useful in some patients [122]. In turn, activated macrophages secrete proinflammatory factors, such as MMPs, IL-1, TNF-α, and IL-12, and activated neutrophils secrete reactive oxygen species (ROS) and proteases. Mast cells, stimulated by neuropeptides, cause the release of histamine, which is involved in vasodilation and inflammation; tryptase, a fibroblast chemoattracting agent, and MMPs [122,149,150]. The activation of fibroblasts and the production of MMPs can subsequently lead to cutaneous fibrosis observed in rosacea at an advanced stage.
SP has been shown to be involved in the vascular permeability and plasma extravasation seen in rosacea skin, while the other molecules are associated with vasodilation. The release of SP increases the expression of ICAM and VCAM in vascular endothelial cells and the release of VEGF from mast cells, promoting the proliferation and vascularization of endothelial cells [88]. The neuropeptide CGRP is a powerful vasodilator that contributes to neurogenic vasodilation and the recruitment of leukocytes. SP and CGRP have been shown to promote the polarization of Langerhans cells, which are epidermal dendritic cells involved in the presentation of the antigen, toward TH2-type immunity [151]. The neuropeptide PACAP also appears to be important in pathogenesis. The injection of PACAP into the skin of mice has been shown to upregulate the expression of proteases MMP-1 and MMP-9, as well as the expression of proinflammatory cytokines, such as TNF-α and CXCL2 [73,152]. The increased release of MMP-9 could further lead to inflammation via the LL-37 pathway because MMP-9 promotes the cleavage of cathelicidin into the LL-37 peptide [61]. PACAP can also stimulate the release of nitric oxide (NO) from endothelial cells, leading to vasodilation [153]. These observations suggest the involvement of neurogenic inflammation in the pathophysiological mechanism of cutaneous rosacea, although it is not yet clear whether neural activation precedes or follows the inflammatory infiltrate.
In the eye, the neurotransmitters SP and CGRP were detected in these corneal fibers. In addition to being known in neuroinflammatory processes and modulating the processing of the nociceptive signal, SP is involved in the healing of corneal wounds. It has been shown in mice that approximately 59% of the total epithelial innervation is occupied by nerves expressing SP [154]. In addition, co-expression of TRPV-1 with CGRP and with SP in the corneal neurons has been demonstrated [155]. Thus, TRPV-1 could increase the release of SP and CGRP in the cornea, as in the skin. When released from the corneal nerves, SP and CGRP contribute to the development of a local neurogenic inflammatory response [156].
In Table 2, the major molecular findings involved in the deregulation of both the vascular and neurovascular systems and clinical effects are shown. In Figure 4, the involvement of both neuronal and neurovascular systems in the pathophysiology of rosacea is indicated.
Figure 4.

Possible mechanisms of neuronal and neurovascular deregulation in the rosacea pathophysiology mediated by the transient receptor potential (TRP) channel family. Vanilloid TRP (TRPV) and ankyrin TRP (TRPA), among others, are expressed in neuronal and nonneuronal cells and activated by external stimuli, such as heat, cold, or UV B (UVB) irradiation, and they are significantly regulated upwards in rosacea. An influx of Ca2+ by the activation of these receptors induces the release of neuropeptides such as substance P (SP), the peptide related to the calcitonin gene (CGRP), the polypeptide activator of the pituitary adenylates (PACAP), the intestinal vasoactive peptide (VIP), and ATP. These neuropeptides can stimulate immune cells, such as mast cells, neutrophils, macrophages, and CD4+ T cells, that release proinflammatory mediators, such as matrix metalloproteinases (MMPs), tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), interleukins (ILs), proteases, and reactive oxygen species (ROS), leading to the initiation or aggravation of inflammation. Toll-like receptor 2 (TLR-2) activates kallikrein 5 (KLK-5), which activates the antimicrobial peptide of cathelicidin (LL-37). LL-37 is known to be an activating factor of mast cells. Furthermore, mast cells can also release VEGF, contributing to vasculogenesis. The action of neuropeptides on blood vessels leads to vasodilation, which is responsible for erythema and flushing, and to vascular permeability, which is responsible for plasma extravasation, leading to edema. The release of histamine from mast cells promotes vasodilation and the onset of fibrosis. The release of VEGF by mast cells promotes vasculogenesis. The sebaceous and meibomian glands exhibit sensory and autonomic nerve regulation with the expression of neurotransmitters. This regulation could contribute to the development of the disease.
Deregulation of the function of sebaceous and meibomian glands
The sebaceous glands
The surface of the skin is covered by an emulsion of water and grease, forming a hydrolipidic film constituting a protective barrier against microorganisms. The sebaceous glands are intradermal glands present all over the body that secrete sebum, a mixture of lipids and cellular debris, which is important to prevent drying of the skin and which enters the composition of the hydrolipidic film of the skin. Dysfunction of the sebaceous glands has been evidenced in various dermatological pathologies, such as acne vulgaris, atopic dermatitis, and psoriasis [157].
The link between the sebaceous glands and rosacea emerged when treatment with oral isotretinoin (retinoids), which inhibits sebum secretion by the sebaceous glands and was prescribed for acne, unexpectedly reduced erythema and papulopustules in patients with cutaneous rosacea [158]. The lipid analysis of skin hydrolipid film showed that rosacea is associated with a decrease in long-chain saturated fatty acids [159], resulting in reduced skin hydration, although the amounts and rates of sebum secretion are unchanged and do not correlate with the disease severity [157,160]. Alteration of the lipid film quality and the resulting skin dryness favor rosacea. Indeed, in 135 rosacea patients, the erythematotelangiectatic rosacea subtype was more common in dry skin than it was in seborrheic skin [161]. Sebocytes contribute to inflammatory response in rosacea, releasing many proinflammatory factors and adipokines, such as IL-6 [162]. When exposed to danger signals, human sebocytes rapidly acquire a competent immune status [163]. Recent data have shown that skin treated with α-bungarotoxin, an acetylcholine receptor antagonist, decreases sebum secretion and size, suggesting that autonomous innervation regulates the function of the sebaceous glands [164].
Several studies have shown a dysfunction of the sebaceous glands associated with a permeability barrier alteration in the skin of patients with rosacea. In a recent study, Medgyesi et al., have shown that all major components of the skin barrier are altered in papulopustular rosacea, including the altered expression of cornified envelope components, tight junction molecules, barriers alarmins, and AMPs [165]. The barrier disruption may contribute to disease pathophysiology.
The meibomian glands
The meibomian glands are sebaceous glands located in the epidermis of the lower and upper eyelids, which secrete meibum, a source of tear lipids. They play an essential role in the homeostasis of the ocular surface and in the stability of the tear film, which is the second line of defense of the ocular surface after the eyelids. Any meibomian gland dysfunction leads to poor lubrication of the ocular surface, which manifests by redness, swelling of the edge of the eyelids, and palpebral conjunctivitis around the orifices of the meibomian glands. The term “meibomian gland dysfunction” (MGD) was introduced in 1980 by Korb and Henriquez [166] to describe the obstruction in the meibomian gland by hyperkeratinization of the glandular and epithelium, accompanied by a change in the viscosity of the meibum leading to meibomitis. MGD can be secondary to medication intake such as antidepressants or antihistamines, to pollution, certain dermatological diseases such as rosacea, and infection with a microorganism, which is an important element in the development of MGD. The meibomitis represents a facet of the general dysfunction of the sebaceous glands in association with dermatological disorders of which cutaneous rosacea [167].
One of the main eye signs of ocular rosacea is MGD, as observed in several previous studies [13,26]. MGD has been reported in 50% to 93% of patients with ocular rosacea. MGD is the most common cause of dry eye, which is one of the symptoms of ocular rosacea [168]. Instability of the tear film damages the ocular surface epithelium, leading to the production of proinflammatory cytokines, which aggravate ocular surface inflammation and worsen the MGD [169]. The proinflammatory factors secondary to MGD cause inflammatory signs in eyelid edges [170].
A gene expression study was conducted on meibomian glands isolated from the eyelids of patients with MGD. Nearly 400 genes were differentially regulated, such as genes coding for small proline-rich proteins (SPRRs), calcium binding proteins S100 (S100A7, A8, and A9), or genes coding for keratins [171]. The SPRR proteins and A8 and A9 proteins have been shown to be involved in the development of the epidermis and keratinization [172,173]. Thus, they could be involved in the hyperkeratinization observed in patients with MGD. In addition, the proteins S100A8 and A9 are also involved in inflammatory processes, promoting the recruitment of leukocytes to the site of inflammation and the release of proinflammatory factors. Furthermore, these proteins form a heterodimer called calprotectin, known to be a TLR-4 agonist, which is overexpressed in patients with ocular rosacea [174]. Thus, these molecules could be partly responsible for the ocular inflammation observed in the eyes and the eyelid edges of patients with ocular rosacea.
Patients with ocular rosacea have been shown to have decreased tear production and a low clearance rate [13]. Barton et al. and Afonso et al. [53,54] have shown that, the lower the tear clearance rate in these patients, the higher the concentrations of IL-1α and MMP-9.
The meibomian glands present nervous regulation through their rich sensory, sympathetic, and parasympathetic innervation [175,176]. These nerve fibers express neurotransmitters, such as CGRP, SP, or VIP, released in the vicinity of the glands, which act on glandular receptors [177,178]. These compounds are known to stimulate lacrimal gland secretion [179] and could regulate meibomian gland secretion. However, this remains to be demonstrated.
Endogenous factors
Genetic predisposition
Rosacea patients are four times likelier to have a family member with rosacea [180]. Homozygous twins have clinical rosacea scores, as defined by the National Rosacea Society (NRS), that is higher than those observed for heterozygous twins; this demonstrates a contribution of genetic determinant in the disease [15].
A genome-wide association study (GWAS) showed that rosacea was associated with three alleles of human leukocyte antigen (HLA) class II, molecules that present antigens to immune cells [181]. Moreover, a single-nucleotide polymorphism (SNP) in the region of the HLA, the rs763035 polymorphism located between the Human Leukocyte Antigen – DR isotype (HLA-DRA) genes, an allele of the major histocompatibility complex of class I, and butyrophilin-like protein 2 (BTLN2)—which is also involved in the immune system in the activation of T lymphocytes [182]—have been associated with rosacea [181].
A null mutation polymorphism has been found in the gene that codes for glutathione S-transferase (GST), which is involved in cellular defense against oxidative stress [183]. The null genotypes GST mu 1 (GSTM1) and GST theta 1 (GSTT1) have been shown to be associated with a higher risk of developing the disease. The increase in ROS or the decrease in antioxidant potential, two characteristics observed in patients with rosacea, could be linked to the polymorphism of the GST gene.
SNP rs3733631, located in the tachykinin 3 receptor gene (TARC-3)—which comprises the endogenous ligand neurokinin B—was found in patients with rosacea, with a higher frequency than in the control group [184]. Tachykinins are a family of neuropeptides including substance P, neurokinin A, and neurokinin B, which bind to the TARC-1, TARC-2, and TARC-3 receptors, respectively. Neurons in the hypothalamus that express neurokinin B have been shown to promote skin vasodilation and estrogenic modulation of body temperature, leading to hot flashes in postmenopausal women, a characteristic common to rosacea [185]. The gene that codes for the TLR-2 receptor located on chromosome 4q32 has been shown to be adjacent to TARC-3 located at 4q25. Polymorphism in the TARC-3 receptor may contribute to the increased expression of TLR-2 seen in rosacea. This hypothesis remains to be confirmed.
The rs11168271 (ApaI), rs731336 (TaqI), and rs11568820 (Cdx2) polymorphisms in the vitamin D receptor (VDR) have been associated with rosacea. The authors demonstrated that polymorphisms of heterozygous and mutant ApaI alleles increased the risk of rosacea, while polymorphisms of mutant TaqI alleles decreased the risk of rosacea. Likewise, the heterozygous Cdx2 alleles increased the risk of rosacea, while the wild forms of the ApaI and mutant TaqI alleles reduced the risk. This demonstrates the involvement of vitamin D signaling in the disease [186,187].
Recently, Hayran et al. [105] investigated the association between polymorphisms in the gene that codes for the proangiogenic factor VEGF and rosacea. The study showed high expression of VEGF in the facial skin biopsies of patients with rosacea compared to the healthy group. Interestingly, they observed that the heterozygous and homozygous +405C/G polymorphism of the gene encoding VEGF increased the risk of rosacea by 1.7 times. A positive correlation between the severity of clinical signs and the +405C/G polymorphism has been found in patients with erythematotelangiectatic rosacea [105]. Although genetic predispositions favor the occurrence and severity of the disease, it seems to be one of the factors in a multifactorial and complex disease.
Corticosteroids
Corticosteroids are hormones secreted by the cortex of the adrenal glands. In this part of the gland, a distinction is made between mineralocorticoids (aldosterone) in the glomerular zone, glucocorticoids (GCs) in the fasciculate zone, and androgens in the reticulate zone.
Because of their anti-inflammatory and immunosuppressive properties, GCs are used in the treatment of many inflammatory ocular and skin conditions. However, GCs delay wound healing [188,189], and chronic treatment of dermatological diseases with steroids results in a clinical form similar to rosacea, called rosacea-like dermatitis induced by steroids or perioral dermatitis [190-192]. This includes erythema, pustules, and papules like those seen in rosacea skin. Thus, GCs could contribute to disease pathogenic mechanisms.
The skin is the target organ of systemic GCs. The GC receptor (GR) has been shown to be involved in the differentiation of keratinocytes and in the formation of the skin barrier. In addition, keratinocytes express several enzymes that are necessary in the GCs production pathway, such as cytochrome P450 11A1 (CYP11A1), cytochrome P450 17A1 (CYP17A1), cytochrome P450 21 (CYP21), or even the metastatic lymph node 64 (MLN64) protein [193]. The skin cells express human 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) and 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which transform cortisone into cortisol and vice versa, allowing activation and inactivation of GCs in the skin. In rosacea skin, the expression levels of CYP11A1, CYP21A2, and 11β-HSD1, as well as the expression of GR, increase significantly, suggesting de novo synthesis of GCs and their receptors simultaneously [194,195]. The sebaceous glands have been demonstrated to be a key target of GCs, acting as enzyme factories to produce GCs. The expression of CYP11A1 was increased in the sebaceous glands and in the epidermis of patients [194].
Surprisingly, GCs can enhance the expression of TLR-2 in human keratinocytes in culture through activation of the p38 MAPK signaling pathway, which is abolished by the GR antagonist RU 486 [196]. GCs induce TLR-2, which induces the recruitment of MyD88, a TLR-2 adaptor molecule [197]. However, a direct link between the expression of TLR-2 by GCs and the increase in inflammatory responses mediated by TLR-2 has not yet been demonstrated. Thus, GCs could paradoxically strengthen immunity by upregulating TLR-2. Different studies have shown that UV radiation, one of the aggravating factors of rosacea, upregulates 11β-HSD1 and downwardly adjusts 11β-HSD2, stimulating the production of GCs [198-200].
Although GCs represses the NF-κB pathway and the transcription of anti-inflammatory genes [201], hyperactivation of the mineralocorticoid receptor, which binds GCs and aldosterone with similar affinity, caused epidermal hyperplasia, impaired differentiation, and increased dermal infiltrates, correlating with increased NF-κB signaling and upregulation of TNF-α and IL-6 cytokines, showing that doses of GCs prone to saturate both receptors could contribute to GC-induced skin side effects [202,203]. GC-induced skin atrophy was also shown to result from inappropriate activation of the mineralocorticoid receptor, and it was prevented by mineralocorticoid receptor antagonism [204].
Synthetic GCs are widely used to treat several eye conditions. However, some patients receiving chronic treatment are likely to develop increased intraocular pressure, cataracts leading to vision loss, or sensitivity to infections [205]. In the cornea, GCs are known to inhibit the growth of vessels and decrease inflammation, but as in the skin, they are also associated with delayed healing of corneal wounds [206] through activation of the mineralocorticoid pathway [207-209].
The relationship between immunity and NF-κB and GCs in ocular rosacea has not yet been studied. It would be interesting to know whether there is a link between inflammatory immunity and corticosteroids in ocular rosacea. In Table 3, the major molecular findings of the aggravation of the disease by endogenous factors and clinical effects are shown.
Exogenous factors
Demodex
Demodex mites are other factors that may be related to the initiation or worsening of ocular and skin rosacea. Demodex folliculorum (D. folliculorum) and D. brevis are the two species described that can colonize human skin. D. folliculorum is found in hair follicles, while D. brevis is found mainly in the sebaceous and meibomian glands. Demodex mites colonize all areas of human skin, but a larger number is found in the facial skin [210]. These mites generally do not cause dermatological problems unless they reach a large number (≥5 mites/cm2) or enter the derma [211-213]. The association between Demodex and rosacea is strong, although the mechanisms remain unknown and controversial [214]. The mite population is five to seven times higher on skin biopsies of rosacea patients, and it correlates with the disease severity, being mostly found in papulopustular and infiltrating forms of the disease [39,211,212,215-219]. Quality and quantity of sebum are considered responsible for increased mite infestation [159,213,220,221], in that mite infestation is more frequent in drier or seborrheic skin [31] compared with normal skin [210]. Kubanov et al. [222] showed that Demodex colonization increases the duration of the disease (more than 5 years) and the likelihood of recurrence (from one to three relapses in 40% of patients), as well as that severe forms of rosacea are mostly associated with D. folliculorum.
Increased numbers of mites have been implicated in skin inflammation and the further aggravation of rosacea [39,215,223]. Indeed, Lacey et al. [214] have shown that live Demodex mites cocultivated with human sebocytes can modulate the immune response in vitro. In small numbers, mites tend to suppress the production of cytokines, as well as regulating downward expression of TLR-2, NF-κB, and CD180, a gene that encodes for a homologous TLR-4 receptor [37]. In greater numbers, they induce the release of proinflammatory factors, such as IL-8, and activate the TLR-2 pathway. The polysaccharide chitin contained in the mite exoskeleton is known to activate TLR-2 and facilitate the activation of the NLRP3 inflammasome [224]. Another pathogenic mechanism could be related to the mechanical blockade of hair follicles and sebaceous glands, causing disturbance of the skin barrier and tissue damage. Histologically, this phenomenon is supported by the presence of inflammatory infiltrations around enlarged hair follicles containing demodex mites in rosacea skin [225].
The inflammatory reaction could be aggravated by resident bacteria, such as Bacillus oleronius, released by dying mites, leading to the chemotaxis of more immune cells, such as neutrophils [226,227]. The secreted proinflammatory factors will attract more immune cells into the tissues, aggravating the inflammation reaction. In addition, activated neutrophils are known to release LL-37 and MMP-9, causing even more tissue damage [16]. More recently, Schaller et al. [228] showed that a dual anti-inflammatory and antiparasitic effect of ivermectin allows a significant reduction in Demodex mites and inflammatory genes.
In ophthalmology, Demodex is also known to play a role in chronic blepharitis, conjunctiva inflammation, corneal lesions, and dysfunction of the meibomian glands, all characteristics of ocular rosacea [30,229]. D. folliculorum is located in the follicles of eyelashes and causes anterior blepharitis associated with eyelash disorders. D. brevis is found in the sebaceous glands of the eyelashes and meibomian glands and is associated with posterior blepharitis with MGD and keratoconjunctivitis [230]. These phenomena can spread to the conjunctiva and cornea, causing keratitis and CNV in more severe cases, thereby compromising vision [231]. However, the spread of inflammation to the conjunctiva and cornea depends not only on the severity of the host’s inflammatory response but also on the distance from the Demodex infestation site. It has been shown that in patients with ocular rosacea with corneal complications, the number of D. brevis mites is significantly increased compared with patients who do not have corneal lesions. This observation is consistent with the fact that D. brevis is mainly found in the meibomian glands and can easily reach the conjunctiva and corneal tissues [213,232].
Eradication of Demodex is associated with a reduction in the tear cytokines IL-1β and IL-17, demonstrating the proinflammatory effect of the mites [233-235]. Bacillus oleronius may also be involved in the inflammatory eye process by enhancing the expression of inflammatory mediators MMP-3 and MMP-9 by corneal epithelial cells [236]. The reduction in the density of Demodex mites observed after treatment with topical ivermectin to 1%, an antihelminthic, was accompanied by a marked improvement in clinical skin and eye signs. In addition, a significant decrease in LL-37, IL-8, and TLR-2 was observed [237].
A better understanding of the mechanisms of action of these mites is needed to understand what triggers the transition from an initial immunosuppression to an immunostimulation state after Demodex colonization [238]. Yet, the presence of many mites may simply be an aggravating factor rather than a possible cause.
UV irradiation
UV and ROS
UV irradiation is a well-known trigger factor for cutaneous rosacea. Exposure to UV rays leads to changes in the extracellular matrix of the dermis and an inflammatory reaction with a predominance of neutrophils, an important source of ROS generated by both UVA and UVB rays. These free radicals can activate MMPs, which will cause breaks in dermal collagen, as well as inducing vascular damage and alterations in perivascular collagen [239].
Some studies have linked ROS to the etiology of cutaneous rosacea. In one study, Bakar et al. [240] showed that ROS levels were much higher in skin biopsies of rosacea patients compared with healthy controls. Oztas et al. [239] and Falay Gur et al. [220] studied the role of free radicals in the etiology of rosacea. They found that, in patients with mild rosacea, the antioxidant activity was higher to protect the skin, while in patients with severe rosacea, the activity of superoxide dismutase was low and levels of malondialdehyde high, demonstrating an antioxidant system defect in these patients. Tisma et al. [241] showed an increase in the ferritin content in the skin of rosacea patients following UV irradiation, as well as an increase in the resulting oxidative damage. It is known that iron overload can amplify the harmful effects of ROS, acting as a catalyst for reactions between ROS regulation and biomolecules. Excess iron has been shown in various dermatological pathologies, such as psoriasis or atopic eczema. More recently, in another study, Erdogan et al. [242] showed high oxidative stress in these patients, as well as an increased antioxidant status, probably as a compensatory mechanism. The level of advanced oxidation protein products (AOPPs), which reflect the oxidation of proteins derived from neutrophils and macrophages, was also increased. However, more studies are needed to determine whether the imbalance between the oxidant and antioxidant systems in the pathology of rosacea is a cause or consequence of this.
UV radiation is also associated with innate immunity in cutaneous rosacea with the production of cytokines and chemokines, such as IL-1β, IL-6, TNF-α, and IL-8, in a temporal and dose-dependent manner [243,244]. As mentioned above, to be active, IL-1β requires cleavage by caspase-1, a component of the NLRP3 inflammasome. Feldmeyer et al. [245] showed that keratinocytes exposed to UVB have the capacity to secrete IL-1β via the activation of the inflammasome, which shows that these cells can induce an inflammatory response following UVB irradiation. The release of ROS by cells, such as neutrophils, can also result in inflammation of the skin accompanied by activation of the inflammasome, upregulation of the NF-κB factor, release of proinflammatory cytokines, and other inflammatory mediators produced by keratinocytes and fibroblasts, leading to an exacerbation of inflammation [246].
UV-induced eye damage has been demonstrated in various human studies. The cornea is the most affected structure of the eye by UVB radiation, with an absorption of around 90% [132]. UV irradiation induces an inflammatory response, such as swelling of the stroma, and it is accompanied by significant leukocyte infiltration [247]. As in the case of the skin, at the ocular level, UVB can induce the production of ROS and proinflammatory cytokines [248]. UVB irradiation has been shown to increase the secretion levels of IL-6 and IL-8 by human corneal and conjunctival epithelial cells in vitro [249]. In one study, Chen et al. [250] showed that UVB irradiation in animals induces visible corneal inflammation and severe epithelialized exfoliation. High levels of TNF-α expression were observed in these animals compared to the control group. In addition, the corneal stroma has been strongly infiltrated by leukocytes. Korhonen et al. [251] have shown that exposure to UVB of human corneal epithelial cells (HCEs) in vitro induced an increase in the activity of NLRP3, as well as that of caspase-1, accompanied by an increase in the secretion of IL-1β. This shows that the inflammatory mechanism mediated by the activity of the inflammasome after exposure to UVB radiation also seems to be present in the eye. However, to date, no study has directly linked the effects of UV light and the immune inflammation mechanism in a specific case of ocular rosacea.
UV and LL-37
There seems to be a link between LL-37 and UVB in the pathophysiology of rosacea. Cutaneous rosacea patients exhibit an aggravated inflammatory response and production of ROS as a function of dose and time when they are exposed to UV radiation [252]. UVB-exposed keratinocytes cultured in the presence of LL-37 secrete higher levels of IL-1β and IL-18 than those that have not been cultured in the presence of the proinflammatory peptide, suggesting that the mechanism by which LL-37 causes the production of these cytokines could involve the activity of the inflammasome [253]. In another recent in vitro study, using HaCaT cells (transformed aneuploid immortal keratinocyte cell line from adult human skin) exposed to LL-37 and irradiated with UVB, Suhng et al. [254] showed a significant increase in the expression of IL-33, a member of the IL-1 family, compared with the LL-37 group alone or UVB alone. The levels of expression of other inflammatory cytokines, such as IL-1α and β, TNF-α, and IFN-γ, also increased. In particular, the expression of IL-1β was the most upregulated one. Therefore, the increased production of these proinflammatory cytokines in the presence of LL-37 may contribute to the susceptibility of rosacea patients to UV radiation.
Activation of LL-37 following UVB irradiation may partly result from ER stress induced by UV irradiation in the human epidermis [255], which may explain the increase in LL-37 levels in skin exposed to UVB. In one study, human skin fibroblasts (Hs68 cells) and mice exposed to UVB showed an increase in markers of ER stress proteins, such as CCAAT-enhancer-binding protein homologous protein (CHOP) and ATF4, in a dose-dependent manner. This effect was significantly reduced after treatment with trigonelline, a naturally occurring alkaloidal agent [256]. The synthesis of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] caused by UV radiation also induces the expression of cathelicidins in keratinocytes. Wang et al. [257] identified an element of response to vitamin D in the cathelicidin promoter. Serological levels of the active form of vitamin D3 have been shown to be higher in patients with rosacea [258]. In addition, it has been shown that 1,25(OH)2D3 can induce an increase in the expression of TLR-2 and KLK-5 [259]. In this sense, vitamin D seems to play a role in the immune system and in the proinflammatory cascades of rosacea by controlling the expression of certain molecules.
UV and vascular and neuronal systems
UVB radiation has been linked to angiogenesis by inducing epidermal thickening, dilatation, and hyperpermeability of blood vessels, leading to edema and erythema [260]. UVB has been shown to be able to modulate the angiogenic response by upregulating VEGF, FGF, and IL-8 and by downregulating TSP-1, an anti-angiogenic factor [261]. This is confirmed in skin biopsies of patients with rosacea, in which an inflammatory infiltrate is histologically present at the perivascular level [253]. Hirakawa et al. [262] showed that mice that overexpress VEGF-A are more sensitive to UVB irradiation. Conversely, blocking VEGF-A can reduce skin inflammation and vascular dilation after exposure to UVB. For mice that overexpress TSP-1 in epidermal keratinocytes, the damage induced by UVB is significantly reduced [263]. While the VEGF-A form is increased in the skin of mice after UVB irradiation, the VEGF-C form appears to be downregulated. In addition, inhibition of the VEGF-R3 receptor appears to worsen the phenotype induced by UVB exposure. Conversely, its activation attenuates clinical signs [264]. In addition, the release of IL-1β also contributes to the proangiogenic environment by modulating the activity of endothelial cells [265]. Overall, the inhibition of these pathways could be of interest in preventing photodamage in the pathophysiology of rosacea.
In addition, UVB radiation has been shown to affect the skin and cause sunburn by activating the TRPV-4 channels [266], related to the activity of LL-37, as seen before. After exposure to UVB, mice depleted for the Trpv-4 gene in keratinocytes showed much less sensitivity to thermal and mechanical stimuli, with a reduction in epidermal lesions demonstrating an important role for TRPV-4 in the damage to skin tissues induced by UVB. In addition, an increase in the expression of adhesion molecules like VCAM-1, ICAM-I, and E-selectin have been observed in vitro in human dermal microvascular endothelial cells (HDMECs) in cultures exposed to UVB. Their expression was even stronger than the exposure time and the dose of LL-37 [108]. Together, these observations may partially explain why exposure to UV rays promotes vascular inflammation in rosacea, which could go through LL-37 activity.
Chen et al. [250] have shown that eye irradiation with UVB in animals induces an increase in expression levels of VEGF compared with animals in the control group. Ocular exposure to UVB radiation leads to a significant reduction in innervation in corneal products and alters the density and morphology of sensory nerves, leading to nerve damage and disorganization of the corneal epithelium [132].
Previous studies have shown that neural activation leads to increased levels of the p38 MAPK signaling pathway, which is involved in cellular responses following inflammatory stimuli [267]. In a mouse model near UVB-induced photokeratitis, high levels of p38 and TRPV-1 were found in neurons and in neighboring satellite glial cells, which are known to release inflammatory and immune factors in response to inflammation and nerve damage. In Table 4, the major molecular findings of the aggravation of the disease by exogenous factors and clinical effects.
Study models
In vivo models of rosacea
Animal models have improved our understanding of the pathophysiology of various human diseases. These models, which mimic both diseases and conditions of the human skin and eye, are used to predict the effectiveness of a drug. Currently, there are few animal models mimicking cutaneous and ocular rosacea, which are mainly based on the development of clinical features observed in humans.
Inflammatory skin model induced by the injection of LL-37
Many studies have shown the central role of the antimicrobial peptide LL-37 in inflammatory and neurovascular processes at the cutaneous and ocular levels in patients with rosacea [61,62]. A model of inflammation induced by the injection of LL-37 into the skin of rodents has been developed, making it possible to best mimic the clinical characteristics and to study the inflammatory and neurological immune mechanisms of the disease [41]. It is an experimental animal model used systematically to study the pathophysiology of cutaneous rosacea. LL-37 injections into the skin of rodents were induced twice a day for 2 days. Animals sensitized to LL-37 exhibit scratching behaviors with the development of characteristics common to those observed in humans, namely, redness, erythema, and skin inflammation [41,61]. The clinical manifestation showed an infiltration of leukocytes at the site of the skin lesion, such as mast cells, neutrophils, macrophages, and CD4+ T lymphocytes, as well as an increased increase in inflammatory mediators, such as IL-1β, TNF-α, and IL-6. The increase in expression levels of the proangiogenic factor VEGF and the TRP involved in the detection of environmental stimuli (heat, cold, etc.) to which rosacea patients seem to be sensitive shows that this model of cutaneous rosacea induced by LL-37 is also relevant for studying the vascular and neurovascular systems [41,124,125,254].
The expression of LL-37 is also increased in patients with ocular rosacea [27]. The development of an ocular model by injection of LL-37 could be interesting to better understand its implications on the ocular level and the associated molecular mechanisms.
Skin and eye inflammation model induced by UVB light
UVB irradiation in murine models is often used to assess compounds tested on skin photoaging, photo damage, and skin inflammation accompanied by cytokine release and leukocyte infiltration [268,269]. The acute response to exposure to UVB radiation shares several characteristics common to those observed in humans, namely erythema and skin inflammation [16,262,270]. The mechanisms underlying the development of erythema in animals have not been approved as exactly mimicking those seen in humans with erythema associated with rosacea. However, this model makes it possible to study and assess different molecular targets for their ability to develop inflammation and vasoconstriction of blood vessels [271] present in rosacea patients in vivo.
At the ocular level, UVB radiation induces a corneal inflammatory response accompanied by the production of ROS and proinflammatory cytokines [248,250]. A model of photokeratitis by exposure of the eyes to UVB rays has been developed in rodents. This model causes corneal haze, inflammation, and fibrosis, leading to a decrease in visual acuity clinical features observed in patients with ocular rosacea [132,250]. This model does not directly mimic ocular rosacea, but it allows a better understanding of the molecular mechanisms induced by UVB irradiation, which could also be an aggravating factor of the disease.
In vitro models for rosacea
In the absence of relevant in vivo models, the development of in vitro models can prove to be an effective solution for a better understanding of molecular mechanisms. At both the skin and eye level, the use of organotypic skin and eye equivalents can be well-designed substitutes that mimic the skin and human eye. They are used as an alternative to animal models for drug permeability and toxicity tests.
NHEKs and HaCaTs
In vitro skin rosacea models further include the culture of normal human epidermal keratinocytes (NHEKs) and the immortalized cell line (HaCaT) of spontaneously transformed human keratinocytes from adult human skin. Epidermal keratinocytes in culture have a variety of applications in research on skin diseases and wound healing; they are also used for efficacy and toxicity testing of molecules.
In the study of the pathophysiology of cutaneous rosacea in vitro, these two models are the most used models to study inflammation during exposure to various factors, including the peptide LL-37 and UVB, which are two important players involved in the disease. When stimulated, these cells can express and release several inflammatory mediators, such as TNF-α and IL-1β, suggesting that these models of epidermal keratinocytes in culture are good models for studying inflammatory responses in vitro and evaluating the action of therapeutic molecules [254,261]. However, they do not explain the complete perplexity of the formation of skin lesions.
HCECs
The cells at the ocular surface of the corneal and conjunctiva are the cells most frequently exposed to external stimuli leading to the process of inflammation, corneal damage, and cell death. These cells constitute a component of the mucosal immune system and participate in defense mechanisms, notably by initiating an inflammatory response via the release of proinflammatory mediators [272]. They can respond to various stimuli, which is why these cells are interesting cell models. These are the two most widely used models for indirectly studying the pathophysiological molecular mechanisms of ocular rosacea, including the inflammatory mechanisms after exposure to UVB [251]. Corneal epithelial cells are also a good model for studying the interaction of pathogens with the ocular surface [273].
Conclusions and future directions
Despite major advances in understanding the pathophysiology of cutaneous and ocular rosacea, the exact mechanisms are still poorly understood. Current hypotheses (Figure 5) focus on the dysfunction of the immune, vascular, and neurovascular systems, which seem to be at the heart of pathogenesis, triggered by microbial or physical stimuli. Since meibomian glands, vessels, and immune cells, such as mast cells, are controlled by autonomous system innervation, it could be hypothesized that the manifestations of ocular rosacea could result from peripheral innervation deregulation, favored by hormonal, infectious, and physical factors. Since neurogenic inflammation is reaching a growing importance in the understanding of migraine [274] and skin inflammation [275], the molecular components of its deregulation in ocular rosacea should be analyzed. This analysis could help develop new study models for ocular rosacea.
Figure 5.

Major mechanisms of cutaneous and ocular rosacea. Excessive activation of innate immunity by various external and internal stimuli, including hormonal dysfunctions, microbes, reactive oxygen species (ROS) or ultraviolet (UV) radiation, leads to overexpression of Toll-like receptors (TLRs), including TLR-2 and 4. The release of proinflammatory cytokines, including interleukins (ILs) and tumor necrosis factor-α (TNF-α), and chemokines, including chemokine (C-X-C motif) ligand (CXCL) 1, CXCL2, CXCL5, and CXCL6, lead to inflammation and the development of papules, pustules, erythema, and edema. If the inflammation becomes persistent, patients develop a phyma, leading to fibrosis. At the same time, TRL-2 and 4 upregulate kallikrein 5 (KLK-5), which activates the cleavage of 18-kDa cationic antimicrobial protein (hCAP-18) into a biologically active peptide, LL-37. LL-37 facilitates the degranulation of mast cells, which leads to a greater release of matrix metalloproteinases (MMPs), IL-6, TNF-α, and the vascular endothelial growth factor (VEGF) participating in the cutaneous and ocular angiogenesis processes, which can lead to corneal neovascularization and appearance of telangiectasias. Microorganisms, including Demodex, can also directly disrupt the epidermal barrier, which can lead to worsening of inflammation processes. Heat and cold and environmental stimuli overactivate receptive potential channels transient receptor potential (TRP), including s), leading to a significant release of neuropeptides, including substance P (SP), the pituitary adenylate cyclase-activating polypeptide (PACAP), the peptide linked to the calcitonin gene (CGRP), and the vasoactive intestinal peptide (VIP), which cause neurovascular dysfunction, leading to the processes of inflammation, vasodilation, and angiogenesis with the appearance of redness and worsening of erythema and edema. Patients with ocular rosacea also have a dysfunction of the meibomian glands due to an alteration of the meibum, compared with the sebum in patients with cutaneous rosacea. Overexpression of the genes coding for the small proline-rich protein (SPRR) and calcium binding proteins (S100) A8 and A9 can lead to inflammation of the eyes and of the eyelid (blepharitis).
Table 1. Major molecular findings involved in the deregulation of the innate and adaptive immune system and clinical effects.
| Pathomechanism |
Major findings
Cutaneous rosacea Ocular rosacea |
Clinical features | |
|---|---|---|---|
|
Aberrant immune system
- Innate immune system |
• ↑ TLR-2
• Activation of NF-kB pathway
• ↑ Pro-inflammatory factors: IL-8, IL-1β, TNF-α, IL-18, IL-33
• ER stress promotes activation of TLR-2 |
• ↑ TLR-4
• Activation of NF-kB pathway
• ↑ Pro-inflammatory factors: IL-1α and β, IL-6, TNF-α, MMP-8, MMP-9 |
Cutaneous inflammation: • Papules • Pustules Ocular inflammation: • Irritation of the eyelid and ocular surface • Epithelial defects • Corneal ulcers • Corneal neovascularization |
| • ↑ Activation of the NLRP3 inflammasome: activation of caspase-1 and release of IL-1β |
• Activation of the MAPK pathway:
↑ p38 and Erk: release of pro-inflammatory factors |
||
| • ↑ KLK-5 and cathelicidin
• ↑ Release of pro-inflammatory LL-37: erythema, redness, inflammation,
release pro-inflammatory factors, leucocyte chemotaxis | |||
| • ↑ Mast cell density: release of MMP-9, IL-6 and histamine that promote inflammation and fibrosis
• ↑ Neutrophils and macrophages density |
• ↑ β-defensins in tears: release of pro-inflammatory factors; leucocytes chemotaxis and histamine release by mast cells |
||
| - Adaptative immune system | • ↑ CD4(+) T cells
• ↓ CD8(+) T cells |
||
| • ↑ B cells | |||
Table 2. Major molecular findings involved in the deregulation of both vascular and neurovascular system and clinical effects.
| Pathomechanism |
Major findings
Cutaneous rosacea Ocular rosacea |
Clinical features | |
|---|---|---|---|
|
Deregulation of vascular system |
• ↑ Blood flow, persistent vascular and lymphatic dilatation |
Cutaneous rosacea: • Angiogenesis • Vasodilatation • Erythema • Vascular permeability • Telangiectasia • Inflammation • Oedema • Fibrosis Ocular rosacea: • Angiogenesis (corneal neovascularization) • Vasodilatation • Erythema • Telangiectasia | |
| • ↑ CD31 and D240
• ↑ Expression levels of VCAM-1, ICAM-1, E-selectine and CD105 | |||
| • ↑ Expression levels of VEGF in epidermis and infiltrating cells, in corneal epithelial cells, vascular endothelial cells and macrophage infiltrates | |||
| • ↑ LL-37 promotes VEGF expression, erythema and vascular dilatation |
|
||
| • Mast cells participate in vascular mechanisms by releasing VEGF, bFGF and histamine | |||
| Deregulation of neurovascular system | • ↑ TRPV-1,2,3,4 and TRPA-1
• TRPV-2,4 stimulate mast cells degranulation
• LL-37 promotes TRPV-2,4 activation |
• TRPV-1 and TRPA-1 involved in corneal wound healing, corneal transparency, fibrosis, inflammation and neovascularization
• TRPM-8 involved in dry eye |
|
| • ↑ PACAP, SP, VIP and CGRP • SP involved in vascular permeability and plasma extravation • PACAP, VIP and CGRP involved in vasodilatation | • SP and CGRP involved in neuro-inflammatory processes and in corneal wound healing | ||
Table 3. Major molecular findings of the aggravation of the disease by endogenous factors and clinical effects.
| Pathomechanism | Major findings Cutaneous rosacea | Clinical features |
|---|---|---|
|
Endogenous factors
- Genetic predisposition |
• rs763035 polymorphism located between the HLA-DRA gene involved in immune system
• Null mutation polymorphism in the gene which codes for Glutathione S-transferase involved in oxidative stress
• rs3733631 polymorphism in the tachykinin 3 receptor gene involved in the neurovascular system
• rs11168271, rs731336 and rs11568820 polymorphisms in the vitamin D receptor
• Heterozygous and homozygous + 405C / G in the gene that codes for VEGF |
Cutaneous rosacea: • Activation or aggravation of inflammation |
| - Corticosteroids | • Cause a rosacea-like dermatitis induced by steroids • ↑ Expression levels of CYP11A1, CYP21A2, 11β-HSD2 and glucocorticoid receptor in skin rosacea • Glucocorticoids can enhance expression of TLR-2 and increased NF-kB signaling |
Table 4. Major molecular findings of the aggravation of the disease by exogenous factors and clinical effects.
| Pathomechanism |
Major findings
Cutaneous rosacea Ocular rosacea |
Clinical features | |
|---|---|---|---|
|
Exogenous factors
- Demodex |
• Mites population is 5 to 7 times higher on skin biopsies
• More frequent in drier or seborrheic skin
• Can activate TLR-2 and NLRP3 inflammasome and induce the release of pro-inflammatory factors |
• Mites population is higher in patients with ocular rosacea with corneal complications
• Can induce the release of pro-inflammatory factors |
Cutaneous and ocular rosacea: • Ocular angiogenesis • Activation or aggravation of cutaneous and ocular inflammation |
| - UV radiation | • ↑ ROS
• ↑ NLRP3 inflammasome activation and release of pro-inflammatory factors |
||
| • Promotes effects of LL-37: increased release of pro-inflammatory factors compared to LL-37 alone • ↑ Synthesis of vitamin D can promote the activation of TLR-2 and KLK-5 • ↑ Expression levels of VEGF-A, VCAM-1, ICAM-1 and E-selectine • ↑ TRPV-4 | |||
References
- 1.Steinhoff M, Schauber J, Leyden JJ. New insights into rosacea pathophysiology: A review of recent findings. J Am Acad Dermatol. 2013;69(Supplement 1):S15–26. doi: 10.1016/j.jaad.2013.04.045. [DOI] [PubMed] [Google Scholar]
- 2.Steinhoff M, Schmelz M, Schauber J. Facial Erythema of Rosacea - Aetiology, Different Pathophysiologies and Treatment Options. Acta Derm Venereol. 2016;96:579–86. doi: 10.2340/00015555-2335. [DOI] [PubMed] [Google Scholar]
- 3.Gallo RL, Granstein RD, Kang S, Mannis M, Steinhoff M, Tan J, Thiboutot D. Standard classification and pathophysiology of rosacea: The 2017 update by the National Rosacea Society Expert Committee. J Am Acad Dermatol. 2018;78:148–55. doi: 10.1016/j.jaad.2017.08.037. [DOI] [PubMed] [Google Scholar]
- 4.Wilkin J, Dahl M, Detmar M, Drake L, Feinstein A, Odom R, Powell F. Standard classification of rosacea: Report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584–7. doi: 10.1067/mjd.2002.120625. [DOI] [PubMed] [Google Scholar]
- 5.Gold LM, Draelos ZD. New and Emerging Treatments for Rosacea. Am J Clin Dermatol. 2015;16:457–61. doi: 10.1007/s40257-015-0156-2. [DOI] [PubMed] [Google Scholar]
- 6.Alexis AF, Callender VD, Baldwin HE, Desai SR, Rendon MI, Taylor SC. Global epidemiology and clinical spectrum of rosacea, highlighting skin of color: Review and clinical practice experience. J Am Acad Dermatol. 2019;80:1722–1729.e7. doi: 10.1016/j.jaad.2018.08.049. [DOI] [PubMed] [Google Scholar]
- 7.Alinia H, Tuchayi SM, James SM, Cardwell LA, Nanda S, Bahrami N, Awosika O, Richardson I, Huang KE, Feldman SR. Measurement of Disease Severity in a Population of Rosacea Patients. Dermatol Clin. 2018;36:97–102. doi: 10.1016/j.det.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Patiño-Ramírez BE, Rodríguez-García A, Díaz JC, Perfecto Ávalos Y. Alteraciones de la superficie ocular en pacientes con rosácea ocular. Rev Mex Oftalmol. 2012;86:86–96. [Google Scholar]
- 9.Vieira AC, Mannis MJ. Ocular rosacea: common and commonly missed. J Am Acad Dermatol. 2013;69(Suppl 1):S36–41. doi: 10.1016/j.jaad.2013.04.042. [DOI] [PubMed] [Google Scholar]
- 10.Rainer BM, Kang S, Chien AL. Rosacea: Epidemiology, pathogenesis, and treatment. Dermatoendocrinol. 2017;9:e1361574. doi: 10.1080/19381980.2017.1361574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.López-Valverde G, Garcia-Martin E, Larrosa-Povés JM, Polo-Llorens V, Pablo-Júlvez LE. Therapeutical Management for Ocular Rosacea. Case Rep Ophthalmol. 2016;7:237–42. doi: 10.1159/000446104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oltz M, Check J. Rosacea and its ocular manifestations. Optometry. 2011;82:92–103. doi: 10.1016/j.optm.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Alvarenga LS, Mannis MJ. Ocular Rosacea. Ocul Surf. 2005;3:41–58. doi: 10.1016/s1542-0124(12)70121-0. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Garcia A. Ocular Rosacea: Recent Advances in Pathogenesis and Therapy. In 2015. p. 175–199. [Google Scholar]
- 15.Aldrich N, Gerstenblith M, Fu P, Tuttle MS, Varma P, Gotow E, Cooper KD, Mann M, Popkin DL. Genetic vs Environmental Factors That Correlate With Rosacea: A Cohort-Based Survey of Twins. JAMA Dermatol. 2015;151:1213–9. doi: 10.1001/jamadermatol.2015.2230. [DOI] [PubMed] [Google Scholar]
- 16.Woo YR, Lim JH, Cho DH, Park HJ. Rosacea: Molecular Mechanisms and Management of a Chronic Cutaneous Inflammatory Condition. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17091562. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=27649161&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinhoff M, Buddenkotte J, Aubert J, Sulk M, Novak P, Schwab VD, Mess C, Cevikbas F, Rivier M, Carlavan I, Déret S, Rosignoli C, Metze D, Luger TA, Voegel JJ. Clinical, cellular, and molecular aspects in the pathophysiology of rosacea. J Investig Dermatol Symp Proc. 2011;15:2–11. doi: 10.1038/jidsymp.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–5. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 19.Yamasaki K, Kanada K, Macleod DT, Borkowski AW, Morizane S, Nakatsuji T, Cogen AL, Gallo RL. TLR2 Expression Is Increased in Rosacea and Stimulates Enhanced Serine Protease Production by Keratinocytes. J Invest Dermatol. 2011;131:688–97. doi: 10.1038/jid.2010.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmes AD, Steinhoff M. Integrative concepts of rosacea pathophysiology, clinical presentation and new therapeutics. Exp Dermatol. 2017;26:659–67. doi: 10.1111/exd.13143. [DOI] [PubMed] [Google Scholar]
- 21.Moura AKA, Guedes F, Rivitti-Machado MC, Sotto MN. Inate immunity in rosacea. Langerhans cells, plasmacytoid dentritic cells, Toll-like receptors and inducible oxide nitric synthase (iNOS) expression in skin specimens: case-control study. Arch Dermatol Res. 2018;310:139–46. doi: 10.1007/s00403-018-1806-z. [DOI] [PubMed] [Google Scholar]
- 22.Brown TT, Choi E-YK, Thomas DG, Hristov AC, Chan MP. Comparative analysis of rosacea and cutaneous lupus erythematosus: Histopathologic features, T-cell subsets, and plasmacytoid dendritic cells. J Am Acad Dermatol. 2014;71:100–7. doi: 10.1016/j.jaad.2014.01.892. [DOI] [PubMed] [Google Scholar]
- 23.Buhl T, Sulk M, Nowak P, Buddenkotte J, McDonald I, Aubert J, Carlavan I, Déret S, Reiniche P, Rivier M, Voegel JJ, Steinhoff M. Molecular and Morphological Characterization of Inflammatory Infiltrate in Rosacea Reveals Activation of Th1/Th17 Pathways. J Invest Dermatol. 2015;135:2198–208. doi: 10.1038/jid.2015.141. [DOI] [PubMed] [Google Scholar]
- 24.Smith JR, Lanier VB, Braziel RM, Falkenhagen KM, White C, Rosenbaum JT. Expression of vascular endothelial growth factor and its receptors in rosacea. Br J Ophthalmol. 2007;91:226–9. doi: 10.1136/bjo.2006.101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sulk M, Seeliger S, Aubert J, Schwab VD, Cevikbas F, Rivier M, Nowak P, Voegel JJ, Buddenkotte J, Steinhoff M. Distribution and expression of non-neuronal transient receptor potential (TRPV) ion channels in rosacea. J Invest Dermatol. 2012;132:1253–62. doi: 10.1038/jid.2011.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghanem VC, Mehra N, Wong S, Mannis MJ. The Prevalence of Ocular Signs in Acne Rosacea: Comparing Patients From Ophthalmology and Dermatology Clinics. Cornea. 2003;22:230–3. doi: 10.1097/00003226-200304000-00009. [DOI] [PubMed] [Google Scholar]
- 27.Gökçınar NB, Karabulut AA, Onaran Z, Yumuşak E, Budak Yıldıran FA. Elevated Tear Human Neutrophil Peptides 1–3, Human Beta Defensin-2 Levels and Conjunctival Cathelicidin LL-37 Gene Expression in Ocular Rosacea. Ocul Immunol Inflamm. 2019;27:1174–83. doi: 10.1080/09273948.2018.1504971. [DOI] [PubMed] [Google Scholar]
- 28.Wladis EJ, Carlson JA, Wang MS, Bhoiwala DP, Adam AP. Toll-like receptors and vascular markers in ocular rosacea. Ophthal Plast Reconstr Surg. 2013;29:290–3. doi: 10.1097/IOP.0b013e318293764c. [DOI] [PubMed] [Google Scholar]
- 29.Randon M, Liang H, El Hamdaoui M, Tahiri R, Batellier L, Denoyer A, Labbé A, Baudouin C. In vivo confocal microscopy as a novel and reliable tool for the diagnosis of Demodex eyelid infestation. Br J Ophthalmol. 2015;99:336–41. doi: 10.1136/bjophthalmol-2014-305671. [DOI] [PubMed] [Google Scholar]
- 30.Cheng AMS, Sheha H, Tseng SCG. Recent advances on ocular Demodex infestation. Curr Opin Ophthalmol. 2015;26:295–300. doi: 10.1097/ICU.0000000000000168. [DOI] [PubMed] [Google Scholar]
- 31.Addor FAS. Skin barrier in rosacea. An Bras Dermatol. 2016;91:59–63. doi: 10.1590/abd1806-4841.20163541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salamon M, Sysa-Jedrzejowska A, Lukamowicz J, Lukamowicz M, Swiatkowska E, Wozniacka A. Concentration of selected cytokines in serum of patients with acne rosacea. Przegl Lek. 2008;65:371–4. [PubMed] [Google Scholar]
- 33.Holmes AD, Spoendlin J, Chien AL, Baldwin H, Chang ALS. Evidence-based update on rosacea comorbidities and their common physiologic pathways. J Am Acad Dermatol. 2018;78:156–66. doi: 10.1016/j.jaad.2017.07.055. [DOI] [PubMed] [Google Scholar]
- 34.Miller LS. Toll-like receptors in skin. Adv Dermatol. 2008;24:71–87. doi: 10.1016/j.yadr.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 36.Yamasaki K, Gallo RL. Rosacea as a Disease of Cathelicidins and Skin Innate Immunity. J Investig Dermatol Symp Proc. 2011;15:12–5. doi: 10.1038/jidsymp.2011.4. [DOI] [PubMed] [Google Scholar]
- 37.Hari A, Flach TL, Shi Y, Mydlarski PR. Toll-like receptors: role in dermatological disease. Mediators Inflamm. 2010;2010:437246. doi: 10.1155/2010/437246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol. 2014;352:5. doi: 10.3389/fphys.2014.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casas C, Paul C, Lahfa M, Livideanu B, Lejeune O, Alvarez-Georges S, Saint-Martory C, Degouy A, Mengeaud V, Ginisty H, Durbise E, Schmitt AM, Redoulès D. Quantification of Demodex folliculorum by PCR in rosacea and its relationship to skin innate immune activation. Exp Dermatol. 2012;21:906–10. doi: 10.1111/exd.12030. [DOI] [PubMed] [Google Scholar]
- 40.Nasti TH, Timares L. Inflammasome activation of IL-1 family mediators in response to cutaneous photodamage. Photochem Photobiol. 2012;88:1111–25. doi: 10.1111/j.1751-1097.2012.01182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen M, Xie H, Chen Z, Xu S, Wang B, Peng Q, Sha K, Xiao W, Liu T, Zhang Y, Li J, Deng Z. Thalidomide ameliorates rosacea-like skin inflammation and suppresses NF-κB activation in keratinocytes. Biomed Pharmacother. 2019;116:109011. doi: 10.1016/j.biopha.2019.109011. [DOI] [PubMed] [Google Scholar]
- 42.Weis AM, Soto R, Round JL. Commensal regulation of T cell survival through Erdr1. Gut Microbes. 2018;9:458–64. doi: 10.1080/19490976.2018.1441662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim M, Kim K-E, Jung HY, Jo H, Jeong S-W, Lee J, Kim CH, Kim H, Cho D, Park HJ. Recombinant erythroid differentiation regulator 1 inhibits both inflammation and angiogenesis in a mouse model of rosacea. Exp Dermatol. 2015;24:680–5. doi: 10.1111/exd.12745. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Shen J, Beuerman RW. Expression of toll-like receptors in human limbal and conjunctival epithelial cells. Mol Vis. 2007;13:813–22. [PMC free article] [PubMed] [Google Scholar]
- 45.Redfern RL, Reins RY, McDermott AM. Toll-Like Receptor Activation Modulates Antimicrobial Peptide Expression by Ocular Surface Cells. Exp Eye Res. 2011;92:209–20. doi: 10.1016/j.exer.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hou A, Tin MQ, Tong L. Toll-like receptor 2-mediated NF-kappa B pathway activation in ocular surface epithelial cells. Eye Vis. 2017;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohammed I, Said DG, Dua HS. Human antimicrobial peptides in ocular surface defense. Prog Retin Eye Res. 2017;61:1–22. doi: 10.1016/j.preteyeres.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 48.Lee HS, Hattori T, Park EY, Stevenson W, Chauhan SK, Dana R. Expression of toll-like receptor 4 contributes to corneal inflammation in experimental dry eye disease. Invest Ophthalmol Vis Sci. 2012;53:5632–40. doi: 10.1167/iovs.12-9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Redfern RL, McDermott AM. Toll-Like Receptors in Ocular Surface Disease. Exp Eye Res. 2010;90:679–87. doi: 10.1016/j.exer.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Redfern RL, Patel N, Hanlon S, Farley W, Gondo M, Pflugfelder SC, McDermott AM. Toll-Like Receptor Expression and Activation in Mice with Experimental Dry Eye. Invest Ophthalmol Vis Sci. 2013;54:1554–63. doi: 10.1167/iovs.12-10739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wladis EJ, Swamy S, Herrmann A, Yang J, Carlson JA, Adam AP. Activation of p38 and Erk Mitogen-Activated Protein Kinases Signaling in Ocular Rosacea. Invest Ophthalmol Vis Sci. 2017;58:843–8. doi: 10.1167/iovs.16-20275. [DOI] [PubMed] [Google Scholar]
- 52.Wladis EJ, Lau KW, Adam AP. Nuclear Factor Kappa-B Is Enriched in Eyelid Specimens of Rosacea: Implications for Pathogenesis and Therapy. Am J Ophthalmol. 2019;201:72–81. doi: 10.1016/j.ajo.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 53.Barton K, Monroy DC, Nava A, Pflugfelder SC. Inflammatory cytokines in the tears of patients with ocular rosacea. Ophthalmology. 1997;104:1868–74. doi: 10.1016/s0161-6420(97)30014-1. [DOI] [PubMed] [Google Scholar]
- 54.Afonso AA, Sobrin L, Monroy DC, Selzer M, Lokeshwar B, Pflugfelder SC. Tear fluid gelatinase B activity correlates with IL-1alpha concentration and fluorescein clearance in ocular rosacea. Invest Ophthalmol Vis Sci. 1999;40:2506–12. [PubMed] [Google Scholar]
- 55.Määttä M, Kari O, Tervahartiala T, Peltonen S, Kari M, Saari M, Sorsa T. Tear fluid levels of MMP-8 are elevated in ocular rosacea–treatment effect of oral doxycycline. Graefes Arch Clin Exp Ophthalmol Albrecht Von Graefes Arch Klin Exp Ophthalmol. 2006;244:957–62. doi: 10.1007/s00417-005-0212-3. [DOI] [PubMed] [Google Scholar]
- 56.Wladis EJ, Iglesias BV, Adam AP, Gosselin EJ. Molecular Biologic Assessment of Cutaneous Specimens of Ocular Rosacea. Ophthal Plast Reconstr Surg. 2012;28:246–50. doi: 10.1097/IOP.0b013e31824dd9d4. [DOI] [PubMed] [Google Scholar]
- 57.Lam-Franco L, Perfecto-Avalos Y, Patiño-Ramírez BE, Rodríguez García A. IL-1α and MMP-9 Tear Levels of Patients with Active Ocular Rosacea before and after Treatment with Systemic Azithromycin or Doxycycline. Ophthalmic Res. 2018;60:109–14. doi: 10.1159/000489092. [DOI] [PubMed] [Google Scholar]
- 58.Larrick JW, Hirata M, Balint RF, Lee J, Zhong J, Wright SC. Human CAP18: a novel antimicrobial lipopolysaccharide-binding protein. Infect Immun. 1995;63:1291–7. doi: 10.1128/iai.63.4.1291-1297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agerberth B, Gunne H, Odeberg J, Kogner P, Boman HG, Gudmundsson GH. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci USA. 1995;92:195–9. doi: 10.1073/pnas.92.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sørensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951–9. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- 61.Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, Dorschner RA, Bonnart C, Descargues P, Hovnanian A, Morhenn VB, Gallo RL. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975–80. doi: 10.1038/nm1616. [DOI] [PubMed] [Google Scholar]
- 62.Coda AB, Hata T, Miller J, Audish D, Kotol P, Two A, Shafiq F, Yamasaki K, Harper JC, Del Rosso JQ, Gallo RL. Cathelicidin, kallikrein 5, and serine protease activity is inhibited during treatment of rosacea with azelaic acid 15% gel. J Am Acad Dermatol. 2013;69:570–7. doi: 10.1016/j.jaad.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park BW, Ha JM, Cho EB, Jin JK, Park EJ, Park HR, Kang HJ, Ko SH, Kim KH, Kim KJ. A Study on Vitamin D and Cathelicidin Status in Patients with Rosacea: Serum Level and Tissue Expression. Ann Dermatol. 2018;30:136–42. doi: 10.5021/ad.2018.30.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyer-Hoffert U. Reddish, scaly, and itchy: how proteases and their inhibitors contribute to inflammatory skin diseases. Arch Immunol Ther Exp (Warsz) 2009;57:345–54. doi: 10.1007/s00005-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 65.Two AM, Hata TR, Nakatsuji T, Coda AB, Kotol PF, Wu W, Shafiq F, Huang EY, Gallo RL. Reduction in serine protease activity correlates with improved rosacea severity in a small, randomized pilot study of a topical serine protease inhibitor. J Invest Dermatol. 2014;134:1143–5. doi: 10.1038/jid.2013.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thibaut de Ménonville S, Rosignoli C, Soares E, Roquet M, Bertino B, Chappuis J-P. Defoin-Platel/Chaussade C, Piwnica D. Topical Treatment of Rosacea with Ivermectin Inhibits Gene Expression of Cathelicidin Innate Immune Mediators, LL-37 and KLK5, in Reconstructed and Ex Vivo Skin Models. Dermatol Ther (Heidelb) 2017;7:213–25. doi: 10.1007/s13555-017-0176-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sugimoto S, Morizane S, Nomura H, Kobashi M, Sugihara S, Iwatsuki K. Toll-like receptor signaling induces the expression of lympho-epithelial Kazal-type inhibitor in epidermal keratinocytes. J Dermatol Sci. 2018;92:181–7. doi: 10.1016/j.jdermsci.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 68.Jang YH, Sim JH, Kang HY, Kim YC, Lee E-S. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol JEADV. 2011;25:544–8. doi: 10.1111/j.1468-3083.2010.03825.x. [DOI] [PubMed] [Google Scholar]
- 69.Rattenholl A, Steinhoff M. Proteinase-activated receptor-2 in the skin: receptor expression, activation and function during health and disease. Drug News Perspect. 2008;21:369–81. doi: 10.1358/dnp.2008.21.7.1255294. [DOI] [PubMed] [Google Scholar]
- 70.Kim JY, Kim YJ, Lim BJ, Sohn HJ, Shin D, Oh SH. Increased Expression of Cathelicidin by Direct Activation of Protease-Activated Receptor 2: Possible Implications on the Pathogenesis of Rosacea. Yonsei Med J. 2014;55:1648–55. doi: 10.3349/ymj.2014.55.6.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan X, Li J, Li Y, Deng Z, Zhou L, Long J, Tang Y, Zuo Z, Zhang Y, Xie H. Artemisinin, a potential option to inhibit inflammation and angiogenesis in rosacea. Biomed Pharmacother. 2019;117:109181. doi: 10.1016/j.biopha.2019.109181. [DOI] [PubMed] [Google Scholar]
- 72.Li T, Zeng Q, Chen X, Wang G, Zhang H, Yu A, Wang H, Hu Y. The therapeutic effect of artesunate on rosacea through the inhibition of the JAK/STAT signaling pathway. Mol Med Rep. 2018;17:8385–90. doi: 10.3892/mmr.2018.8887. [DOI] [PubMed] [Google Scholar]
- 73.Muto Y, Wang Z, Vanderberghe M, Two A, Gallo RL, Di Nardo A. Mast cells are key mediators of cathelicidin-initiated skin inflammation in rosacea. J Invest Dermatol. 2014;134:2728–36. doi: 10.1038/jid.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tchougounova E, Lundequist A, Fajardo I, Winberg J-O, Åbrink M, Pejler G. A Key Role for Mast Cell Chymase in the Activation of Pro-matrix Metalloprotease-9 and Pro-matrix Metalloprotease-2. J Biol Chem. 2005;280:9291–6. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 75.Bąbolewska E, Brzezińska-Błaszczyk E. Human-derived cathelicidin LL-37 directly activates mast cells to proinflammatory mediator synthesis and migratory response. Cell Immunol. 2015;293:67–73. doi: 10.1016/j.cellimm.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 76.Margalit A, Kowalczyk MJ, Żaba R, Kavanagh K. The role of altered cutaneous immune responses in the induction and persistence of rosacea. J Dermatol Sci. 2016;82:3–8. doi: 10.1016/j.jdermsci.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 77.Cardamone C, Parente R, Feo GD, Triggiani M. Mast cells as effector cells of innate immunity and regulators of adaptive immunity. Immunol Lett. 2016;178:10–4. doi: 10.1016/j.imlet.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 78.Wang L, Wang Y-J, Hao D, Wen X, Du D, He G, Jiang X. The Theranostics Role of Mast Cells in the Pathophysiology of Rosacea. Front Med. 2020;324:6. doi: 10.3389/fmed.2019.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aroni K, Tsagroni E, Kavantzas N, Patsouris E, Ioannidis E. A study of the pathogenesis of rosacea: how angiogenesis and mast cells may participate in a complex multifactorial process. Arch Dermatol Res. 2008;300:125–31. doi: 10.1007/s00403-007-0816-z. [DOI] [PubMed] [Google Scholar]
- 80.Kim M, Kim J, Jeong S, Jo H, Woo YR, Park HJ. Inhibition of mast cell infiltration in an LL-37-induced rosacea mouse model using topical brimonidine tartrate 0.33% gel. Exp Dermatol. 2017;26:1143–5. doi: 10.1111/exd.13381. [DOI] [PubMed] [Google Scholar]
- 81.Huang LC, Petkova TD, Reins RY, Proske RJ, McDermott AM. Multifunctional Roles of Human Cathelicidin (LL-37) at the Ocular Surface. Invest Ophthalmol Vis Sci. 2006;47:2369–80. doi: 10.1167/iovs.05-1649. [DOI] [PubMed] [Google Scholar]
- 82.Kolar SS, McDermott AM. Role of host-defence peptides in eye diseases. Cell Mol Life Sci. 2011;68:2201. doi: 10.1007/s00018-011-0713-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Norrby K. Mast cells and angiogenesis. APMIS Acta Pathol Microbiol Immunol Scand. 2002;110:355–71. doi: 10.1034/j.1600-0463.2002.100501.x. [DOI] [PubMed] [Google Scholar]
- 84.Adamis AP, Meklir B, Joyce NC. In situ injury-induced release of basic-fibroblast growth factor from corneal epithelial cells. Am J Pathol. 1991;139:961–7. [PMC free article] [PubMed] [Google Scholar]
- 85.Ebeigbe AB, Talabi OO. Vascular Effects of Histamine. Niger J Physiol Sci. 2014;29:7–10. [PubMed] [Google Scholar]
- 86.Ahmad S, Wright KN, Sun X, Groban L, Ferrario CM. Mast cell peptidases (carboxypeptidase A and chymase)-mediated hydrolysis of human angiotensin-(1–12) substrate. Biochem Biophys Res Commun. 2019;518:651–6. doi: 10.1016/j.bbrc.2019.08.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aleksiejczuk M, Gromotowicz-Poplawska A, Marcinczyk N, Przylipiak A, Chabielska E. The expression of the renin-angiotensin-aldosterone system in the skin and its effects on skin physiology and pathophysiology. J Physiol Pharmacol Off J Pol Physiol Soc. 2019;70 doi: 10.26402/jpp.2019.3.01. [DOI] [PubMed] [Google Scholar]
- 88.Choi JE, Di Nardo A. Skin Neurogenic inflammation. Semin Immunopathol. 2018;40:249–59. doi: 10.1007/s00281-018-0675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cho K-A, Park M, Kim Y-H, Woo S-Y. Th17 cell-mediated immune responses promote mast cell proliferation by triggering stem cell factor in keratinocytes. Biochem Biophys Res Commun. 2017;487:856–61. doi: 10.1016/j.bbrc.2017.04.141. [DOI] [PubMed] [Google Scholar]
- 90.Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, Burg G, Liu Y-J, Gilliet M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. 2005;202:135–43. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid Dendritic Cells: Recent Progress and Open Questions. Annu Rev Immunol. 2011;29:163–83. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Batycka-Baran A, Hattinger E, Zwicker S, Summer B, Zack Howard OM, Thomas P, Szepietowski JC, Ruzicka T, Prinz JC, Wolf R. Leukocyte-derived koebnerisin (S100A15) and psoriasin (S100A7) are systemic mediators of inflammation in psoriasis. J Dermatol Sci. 2015;79:214–21. doi: 10.1016/j.jdermsci.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 93.Batycka-Baran A, Hattinger E, Marchenkov A, Koziol M, Bieniek A, Szepietowski J, Ruzicka T, Wolf R. Koebnerisin (S100A15): A novel player in the pathogenesis of rosacea. J Am Acad Dermatol. 2019;80:1753–5. doi: 10.1016/j.jaad.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 94.Wolf R, Ruzicka T, Yuspa SH. Novel S100A7 (psoriasin)/S100A15 (koebnerisin) subfamily: highly homologous but distinct in regulation and function. Amino Acids. 2011;41:789–96. doi: 10.1007/s00726-010-0666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Borovaya A, Dombrowski Y, Zwicker S, Olisova O, Ruzicka T, Wolf R, Schauber J, Sárdy M. Isotretinoin therapy changes the expression of antimicrobial peptides in acne vulgaris. Arch Dermatol Res. 2014;306:689–700. doi: 10.1007/s00403-014-1477-3. [DOI] [PubMed] [Google Scholar]
- 96.Park K, Lee SE, Shin K-O, Uchida Y. Insights into the Role of ER Stress in Skin Function and Associated Diseases. FEBS J. 2019;286:413–25. doi: 10.1111/febs.14739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shimasaki S, Koga T, Shuto T, Suico MA, Sato T, Watanabe K, Morino-Koga S, Taura M, Okada S, Mori K, Kai H. Endoplasmic reticulum stress increases the expression and function of toll-like receptor-2 in epithelial cells. Biochem Biophys Res Commun. 2010;402:235–40. doi: 10.1016/j.bbrc.2010.09.132. [DOI] [PubMed] [Google Scholar]
- 98.Park K, Ikushiro H, Seo HS, Shin K-O, Kim YI, Kim JY, Lee Y-M, Yano T, Holleran WM, Elias P, Uchida Y. ER stress stimulates production of the key antimicrobial peptide, cathelicidin, by forming a previously unidentified intracellular S1P signaling complex. Proc Natl Acad Sci USA. 2016;113:E1334–42. doi: 10.1073/pnas.1504555113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li J, Yuan X, Tang Y, Wang B, Deng Z, Huang Y, Liu F, Zhao Z, Zhang Y. Hydroxychloroquine is a novel therapeutic approach for rosacea. Int Immunopharmacol. 2020;79:106178. doi: 10.1016/j.intimp.2019.106178. [DOI] [PubMed] [Google Scholar]
- 100.Cribier B. Rosacea under the microscope: characteristic histological findings. J Eur Acad Dermatol Venereol JEADV. 2013;27:1336–43. doi: 10.1111/jdv.12121. [DOI] [PubMed] [Google Scholar]
- 101.Perrigouard C, Peltre B, Cribier B. Étude histologique et immunohistochimique des anomalies vasculaires et inflammatoires de la rosacée. Ann Dermatol Venereol. 2013;140:21–9. doi: 10.1016/j.annder.2012.10.592. [DOI] [PubMed] [Google Scholar]
- 102.Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16–25. [PMC free article] [PubMed] [Google Scholar]
- 103.Gomaa AHA, Yaar M, Eyada MMK, Bhawan J. Lymphangiogenesis and angiogenesis in non-phymatous rosacea. J Cutan Pathol. 2007;34:748–53. doi: 10.1111/j.1600-0560.2006.00695.x. [DOI] [PubMed] [Google Scholar]
- 104.Kajiya K, Kajiya-Sawane M, Ono T, Sato K. Identification of an epidermal marker for reddened skin: Vascular endothelial growth factor A. J Dermatol. 2017;44:836–7. doi: 10.1111/1346-8138.13665. [DOI] [PubMed] [Google Scholar]
- 105.Hayran Y, Lay I, Mocan MC, Bozduman T, Ersoy-Evans S. Vascular endothelial growth factor gene polymorphisms in patients with rosacea: A case-control study. J Am Acad Dermatol. 2019;81:348–54. doi: 10.1016/j.jaad.2019.03.055. [DOI] [PubMed] [Google Scholar]
- 106.Muller WA. Mechanisms of Leukocyte Transendothelial Migration. Annu Rev Pathol. 2011;6:323–44. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Edfeldt K, Agerberth B, Rottenberg ME, Gudmundsson GH, Wang X-B, Mandal K, Xu Q, Yan Z. Involvement of the antimicrobial peptide LL-37 in human atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:1551–7. doi: 10.1161/01.ATV.0000223901.08459.57. [DOI] [PubMed] [Google Scholar]
- 108.Kulkarni NN, Takahashi T, Sanford JA, Tong Y, Gombart AF, Hinds B, Cheng JY, Gallo RL. Innate Immune Dysfunction in Rosacea Promotes Photosensitivity and Vascular Adhesion Molecule Expression. J Invest Dermatol. 2020;140:645–655.e6. doi: 10.1016/j.jid.2019.08.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pereira ER, Liao N, Neale GA, Hendershot LM. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mohr T, Haudek-Prinz V, Slany A, Grillari J, Micksche M, Gerner C. Proteome profiling in IL-1β and VEGF-activated human umbilical vein endothelial cells delineates the interlink between inflammation and angiogenesis. PLoS One. 2017;12 doi: 10.1371/journal.pone.0179065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Benayoun Y, Casse G, Forte R, Dallaudière B, Adenis J-P, Robert P-Y. Corneal neovascularization: epidemiological, physiopathological, and clinical features. J Fr Ophtalmol. 2013;36:627–39. doi: 10.1016/j.jfo.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 112.Sharif Z, Sharif W. Corneal neovascularization: updates on pathophysiology, investigations & management. Rom J Ophthalmol. 2019;63:15–22. [PMC free article] [PubMed] [Google Scholar]
- 113.Stevenson W, Cheng S-F, Dastjerdi MH, Ferrari G, Dana R. Corneal neovascularization and the utility of topical VEGF inhibition: ranibizumab (Lucentis) vs bevacizumab (Avastin). Ocul Surf. 2012;10:67–83. doi: 10.1016/j.jtos.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Azar DT. Corneal angiogenic privilege : angiogenic and antiangiogenic factors in corneal avascularity, vasculogenesis, and wound healing. Trans Am Ophthalmol Soc. 2006;104:264–302. [PMC free article] [PubMed] [Google Scholar]
- 115.Coxon A, Bolon B, Estrada J, Kaufman S, Scully S, Rattan A, Duryea D, Hu Y-L, Rex K, Pacheco E, Van G, Zack D, Feige U. Inhibition of interleukin-1 but not tumor necrosis factor suppresses neovascularization in rat models of corneal angiogenesis and adjuvant arthritis. Arthritis Rheum. 2002;46:2604–12. doi: 10.1002/art.10546. [DOI] [PubMed] [Google Scholar]
- 116.Zhang SX, Ma J. Ocular neovascularization: Implication of endogenous angiogenic inhibitors and potential therapy. Prog Retin Eye Res. 2007;26:1–37. doi: 10.1016/j.preteyeres.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 117.Zahir-Jouzdani F, Atyabi F, Mojtabavi N. Interleukin-6 participation in pathology of ocular diseases. Pathophysiology. 2017;24:123–31. doi: 10.1016/j.pathophys.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 118.Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, Robbins PD, Tahara H, Lotze MT. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–7. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 119.Shen J, Choy DF, Yoshida T, Iwase T, Hafiz G, Xie B, Hackett SF, Arron JR, Campochiaro PA. Interleukin-18 has Antipermeablity and Antiangiogenic Activities in the Eye; Reciprocal Suppression with VEGF. J Cell Physiol. 2014;229:974–83. doi: 10.1002/jcp.24575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zafirakis P, Voudouri A, Livir-Rallatos G, Livir-Rallatos C, Canakis C, Markomichelakis N, Baltatzis S. Leukocyte Adhesion Molecules in Ocular Rosacea. Invest Ophthalmol Vis Sci. 2002;43:2225–2225. [Google Scholar]
- 121.Pisella P-J, Brignole F, Debbasch C, Lozato P-A, Creuzot-Garcher C, Bara J, Saiag P, Warnet J-M, Baudouin C. Flow cytometric analysis of conjunctival epithelium in ocular rosacea and keratoconjunctivitis sicca. Ophthalmology. 2000;107:1841–9. doi: 10.1016/s0161-6420(00)00347-x. [DOI] [PubMed] [Google Scholar]
- 122.Schwab VD, Sulk M, Seeliger S, Nowak P, Aubert J, Mess C, Rivier M, Carlavan I, Rossio P, Metze D, Buddenkotte J, Cevikbas F, Voegel JJ, Steinhoff M. Neurovascular and Neuroimmune Aspects in the Pathophysiology of Rosacea. J Investig Dermatol Symp Proc Soc Investig Dermatol Inc Eur Soc Dermatol Res. 2011;15:53–62. doi: 10.1038/jidsymp.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Buddenkotte J, Steinhoff M. Recent advances in understanding and managing rosacea. F1000 Res. 2018;1885:7. doi: 10.12688/f1000research.16537.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mascarenhas NL, Wang Z, Chang Y-L, Di Nardo A. TRPV4 Mediates Mast Cell Activation in Cathelicidin-Induced Rosacea Inflammation. J Invest Dermatol. 2017;137:972–5. doi: 10.1016/j.jid.2016.10.046. [DOI] [PubMed] [Google Scholar]
- 125.Chen Y, Moore CD, Zhang JY, Hall RP, MacLeod AS, Liedtke W. TRPV4 Moves toward Center-Fold in Rosacea Pathogenesis. J Invest Dermatol. 2017;137:801–4. doi: 10.1016/j.jid.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen X, Niyonsaba F, Ushio H, Nagaoka I, Ikeda S, Okumura K, Ogawa H. Human cathelicidin LL-37 increases vascular permeability in the skin via mast cell activation, and phosphorylates MAP kinases p38 and ERK in mast cells. J Dermatol Sci. 2006;43:63–6. doi: 10.1016/j.jdermsci.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 127.Hoffmann O, Braun JS, Becker D, Halle A, Freyer D, Dagand E, Lehnardt S, Weber JR. TLR2 mediates neuroinflammation and neuronal damage. J Immunol. 2007;178:6476–81. doi: 10.4049/jimmunol.178.10.6476. [DOI] [PubMed] [Google Scholar]
- 128.Paria BC, Malik AB, Kwiatek AM, Rahman A, May MJ, Ghosh S, Tiruppathi C. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J Biol Chem. 2003;278:37195–203. doi: 10.1074/jbc.M304287200. [DOI] [PubMed] [Google Scholar]
- 129.Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91:70–6. doi: 10.1161/01.res.0000023391.40106.a8. [DOI] [PubMed] [Google Scholar]
- 130.Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J Biol Chem. 2010;285:15167–77. doi: 10.1074/jbc.M110.106153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pozsgai G, Bodkin JV, Graepel R, Bevan S, Andersson DA, Brain SD. Evidence for the pathophysiological relevance of TRPA1 receptors in the cardiovascular system in vivo. Cardiovasc Res. 2010;87:760–8. doi: 10.1093/cvr/cvq118. [DOI] [PubMed] [Google Scholar]
- 132.Chen S-J, Lee C-J, Lin T-B, Peng H-Y, Liu H-J, Chen Y-S, Tseng K-W. Protective Effects of Fucoxanthin on Ultraviolet B-Induced Corneal Denervation and Inflammatory Pain in a Rat Model. Mar Drugs. 2019;17 doi: 10.3390/md17030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Eguchi H, Hiura A, Nakagawa H, Kusaka S, Shimomura Y. Corneal Nerve Fiber Structure, Its Role in Corneal Function, and Its Changes in Corneal Diseases. BioMed Res Int. 2017;2017:3242649. doi: 10.1155/2017/3242649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Martínez-García MC, Martínez T, Pañeda C, Gallego P, Jimenez AI, Merayo J. Differential expression and localization of transient receptor potential vanilloid 1 in rabbit and human eyes. Histol Histopathol. 2013;28:1507–16. doi: 10.14670/HH-28.1507. [DOI] [PubMed] [Google Scholar]
- 135.Mergler S, Valtink M, Takayoshi S, Okada Y, Miyajima M, Saika S, Reinach PS. Temperature-Sensitive Transient Receptor Potential Channels in Corneal Tissue Layers and Cells. Ophthalmic Res. 2014;52:151–9. doi: 10.1159/000365334. [DOI] [PubMed] [Google Scholar]
- 136.Okada Y, Reinach PS, Shirai K, Kitano A, Kao WW-Y, Flanders KC, Miyajima M, Liu H, Zhang J, Saika S. TRPV1 involvement in inflammatory tissue fibrosis in mice. Am J Pathol. 2011;178:2654–64. doi: 10.1016/j.ajpath.2011.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang F, Yang H, Wang Z, Mergler S, Liu H, Kawakita T, Tachado SD, Pan Z, Capó-Aponte JE, Pleyer U, Koziel H, Kao WWY, Reinach PS. Transient receptor potential vanilloid 1 activation induces inflammatory cytokine release in corneal epithelium through MAPK signaling. J Cell Physiol. 2007;213:730–9. doi: 10.1002/jcp.21141. [DOI] [PubMed] [Google Scholar]
- 138.Okada Y, Reinach PS, Shirai K, Kitano-Izutani A, Miyajima M, Yamanaka O, Sumioka T, Saika S. Transient Receptor Potential Channels and Corneal Stromal Inflammation. Cornea. 2015;34(Suppl 11):S136–41. doi: 10.1097/ICO.0000000000000602. [DOI] [PubMed] [Google Scholar]
- 139.Yang Y, Wang Z, Yang H, Wang L, Gillespie SR, Wolosin JM, Bernstein AM, Reinach PS. TRPV1 potentiates TGFβ-induction of corneal myofibroblast development through an oxidative stress-mediated p38-SMAD2 signaling loop. PLoS One. 2013;8:e77300. doi: 10.1371/journal.pone.0077300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Okada Y, Shirai K, Reinach PS, Kitano-Izutani A, Miyajima M, Flanders KC, Jester JV, Tominaga M, Saika S. TRPA1 is required for TGF-β signaling and its loss blocks inflammatory fibrosis in mouse corneal stroma. Lab Investig J Tech Methods Pathol. 2014;94:1030–41. doi: 10.1038/labinvest.2014.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Usui-Kusumoto K, Iwanishi H, Ichikawa K, Okada Y, Sumioka T, Miyajima M, Liu C-Y, Reinach PS, Saika S. Suppression of neovascularization in corneal stroma in a TRPA1-null mouse. Exp Eye Res. 2019;181:90–7. doi: 10.1016/j.exer.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 142.Yang JM, Wei ET, Kim SJ, Yoon KC. TRPM8 Channels and Dry Eye. Pharmaceuticals. 2018;11 doi: 10.3390/ph11040125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Parra A, Madrid R, Echevarria D, del Olmo S, Morenilla-Palao C, Acosta MC, Gallar J, Dhaka A, Viana F, Belmonte C. Ocular surface wetness is regulated by TRPM8-dependent cold thermoreceptors of the cornea. Nat Med. 2010;16:1396–9. doi: 10.1038/nm.2264. [DOI] [PubMed] [Google Scholar]
- 144.Katagiri A, Thompson R, Rahman M, Okamoto K, Bereiter DA. Evidence for TRPA1 involvement in central neural mechanisms in a rat model of dry eye. Neuroscience. 2015;290:204–13. doi: 10.1016/j.neuroscience.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–8. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 146.Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- 147.Shi X, Wang L, Li X, Sahbaie P, Kingery WS, Clark JD. Neuropeptides contribute to peripheral nociceptive sensitization by regulating interleukin-1β production in keratinocytes. Anesth Analg. 2011;113:175–83. doi: 10.1213/ANE.0b013e31821a0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lindsey KQ, Caughman SW, Olerud JE, Bunnett NW, Armstrong CA, Ansel JC. Neural regulation of endothelial cell-mediated inflammation. J Investig Dermatol Symp Proc. 2000;5:74–8. doi: 10.1046/j.1087-0024.2000.00013.x. [DOI] [PubMed] [Google Scholar]
- 149.Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123:398–410. doi: 10.1111/j.1365-2567.2007.02705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Madva EN, Granstein RD. Nerve-derived transmitters including peptides influence cutaneous immunology. Brain Behav Immun. 2013;34:1–10. doi: 10.1016/j.bbi.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ding W, Stohl LL, Wagner JA, Granstein RD. Calcitonin gene-related peptide biases Langerhans cells toward Th2-type immunity. J Immunol. 2008;181:6020–6. doi: 10.4049/jimmunol.181.9.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Roosterman D, Goerge T, Schneider SW, Bunnett NW, Steinhoff M. Neuronal Control of Skin Function: The Skin as a Neuroimmunoendocrine Organ. Physiol Rev. 2006;86:1309–79. doi: 10.1152/physrev.00026.2005. [DOI] [PubMed] [Google Scholar]
- 153.Seeliger S, Buddenkotte J, Schmidt-Choudhury A, Rosignoli C, Shpacovitch V, von Arnim U, Metze D, Rukwied R, Schmelz M, Paus R, Voegel JJ, Schmidt WE, Steinhoff M. Pituitary adenylate cyclase activating polypeptide: an important vascular regulator in human skin in vivo. Am J Pathol. 2010;177:2563–75. doi: 10.2353/ajpath.2010.090941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.He J, Bazan HEP. Neuroanatomy and Neurochemistry of Mouse Cornea. Invest Ophthalmol Vis Sci. 2016;57:664–74. doi: 10.1167/iovs.15-18019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Murata Y, Masuko S. Peripheral and central distribution of TRPV1, substance P and CGRP of rat corneal neurons. Brain Res. 2006;1085:87–94. doi: 10.1016/j.brainres.2006.02.035. [DOI] [PubMed] [Google Scholar]
- 156.Sahbaie P, Shi X, Guo T-Z, Qiao Y, Yeomans DC, Kingery WS, Clark JD. Role of substance P signaling in enhanced nociceptive sensitization and local cytokine production after incision. Pain. 2009;145:341–9. doi: 10.1016/j.pain.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shi VY, Leo M, Hassoun L, Chahal DS, Maibach HI, Sivamani RK. Role of sebaceous glands in inflammatory dermatoses. J Am Acad Dermatol. 2015;73:856–63. doi: 10.1016/j.jaad.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 158.Hoting E, Paul E, Plewig G. Treatment of rosacea with isotretinoin. Int J Dermatol. 1986;25:660–3. doi: 10.1111/j.1365-4362.1986.tb04533.x. [DOI] [PubMed] [Google Scholar]
- 159.Ní Raghallaigh S, Bender K, Lacey N, Brennan L, Powell FC. The fatty acid profile of the skin surface lipid layer in papulopustular rosacea. Br J Dermatol. 2012;166:279–87. doi: 10.1111/j.1365-2133.2011.10662.x. [DOI] [PubMed] [Google Scholar]
- 160.Ní Raghallaigh S, Powell FC. Epidermal hydration levels in patients with rosacea improve after minocycline therapy. Br J Dermatol. 2014;171:259–66. doi: 10.1111/bjd.12770. [DOI] [PubMed] [Google Scholar]
- 161.Tan J, Blume-Peytavi U, Ortonne JP, Wilhelm K, Marticou L, Baltas E, Rivier M, Petit L, Martel P. An observational cross-sectional survey of rosacea: clinical associations and progression between subtypes. Br J Dermatol. 2013;169:555–62. doi: 10.1111/bjd.12385. [DOI] [PubMed] [Google Scholar]
- 162.Kovács D, Lovászi M, Póliska S, Oláh A, Bíró T, Veres I, Zouboulis CC, Ståhle M, Rühl R, Remenyik É, Törőcsik D. Sebocytes differentially express and secrete adipokines. Exp Dermatol. 2016;25:194–9. doi: 10.1111/exd.12879. [DOI] [PubMed] [Google Scholar]
- 163.Törőcsik D, Kovács D, Póliska S, Szentkereszty-Kovács Z, Lovászi M, Hegyi K, Szegedi A, Zouboulis CC, Ståhle M. Genome wide analysis of TLR1/2- and TLR4-activated SZ95 sebocytes reveals a complex immune-competence and identifies serum amyloid A as a marker for activated sebaceous glands. PLoS One. 2018;13 doi: 10.1371/journal.pone.0198323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li ZJ, Park SB, Sohn KC, Lee Y, Seo YJ, Kim CD, Kim YS, Lee JH, Im M. Regulation of lipid production by acetylcholine signalling in human sebaceous glands. J Dermatol Sci. 2013;72:116–22. doi: 10.1016/j.jdermsci.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 165.Medgyesi B, Dajnoki Z, Béke G, Gáspár K, Szabó IL, Janka EA, Póliska S, Hendrik Z, Méhes G, Törőcsik D, Bíró T, Kapitány A, Szegedi A. Rosacea Is Characterized by a Profoundly Diminished Skin Barrier. J Invest Dermatol. 2020;140:1938–1950. doi: 10.1016/j.jid.2020.02.025. [DOI] [PubMed] [Google Scholar]
- 166.Korb DR, Henriquez AS. Meibomian gland dysfunction and contact lens intolerance. J Am Optom Assoc. 1980;51:243–51. [PubMed] [Google Scholar]
- 167.Schaumberg DA, Nichols JJ, Papas EB, Tong L, Uchino M, Nichols KK. The International Workshop on Meibomian Gland Dysfunction: Report of the Subcommittee on the Epidemiology of, and Associated Risk Factors for, MGD. Invest Ophthalmol Vis Sci. 2011;52:1994–2005. doi: 10.1167/iovs.10-6997e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Suzuki T. Inflamed Obstructive Meibomian Gland Dysfunction Causes Ocular Surface Inflammation. Invest Ophthalmol Vis Sci. 2018;59:DES94–101. doi: 10.1167/iovs.17-23345. [DOI] [PubMed] [Google Scholar]
- 169.Mizoguchi S, Iwanishi H, Arita R, Shirai K, Sumioka T, Kokado M, Jester JV, Saika S. Ocular surface inflammation impairs structure and function of meibomian gland. Exp Eye Res. 2017;163:78–84. doi: 10.1016/j.exer.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Palamar M, Degirmenci C, Ertam I, Yagci A. Evaluation of dry eye and meibomian gland dysfunction with meibography in patients with rosacea. Cornea. 2015;34:497–9. doi: 10.1097/ICO.0000000000000393. [DOI] [PubMed] [Google Scholar]
- 171.Liu S, Richards SM, Lo K, Hatton M, Fay A, Sullivan DA. Changes in Gene Expression in Human Meibomian Gland Dysfunction. Invest Ophthalmol Vis Sci. 2011;52:2727–40. doi: 10.1167/iovs.10-6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hohl D, de Viragh PA, Amiguet-Barras F, Gibbs S, Backendorf C, Huber M. The small proline-rich proteins constitute a multigene family of differentially regulated cornified cell envelope precursor proteins. J Invest Dermatol. 1995;104:902–9. doi: 10.1111/1523-1747.ep12606176. [DOI] [PubMed] [Google Scholar]
- 173.Nukui T, Ehama R, Sakaguchi M, Sonegawa H, Katagiri C, Hibino T, Huh N-H. S100A8/A9, a key mediator for positive feedback growth stimulation of normal human keratinocytes. J Cell Biochem. 2008;104:453–64. doi: 10.1002/jcb.21639. [DOI] [PubMed] [Google Scholar]
- 174.Ehrchen JM, Sunderkötter C, Foell D, Vogl T, Roth J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol. 2009;86:557–66. doi: 10.1189/jlb.1008647. [DOI] [PubMed] [Google Scholar]
- 175.Cox SM, Nichols JJ. The Neurobiology of the Meibomian Glands. Ocul Surf. 2014;12:167–77. doi: 10.1016/j.jtos.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 176.Bründl M, Garreis F, Schicht M, Dietrich J, Paulsen F. Characterization of the innervation of the meibomian glands in humans, rats and mice. Ann Anat - Anat Anz. 2020;4:151609. doi: 10.1016/j.aanat.2020.151609. [DOI] [PubMed] [Google Scholar]
- 177.Kam WR, Sullivan DA. Neurotransmitter Influence on Human Meibomian Gland Epithelial Cells. Invest Ophthalmol Vis Sci. 2011;52:8543–8. doi: 10.1167/iovs.11-8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Knop E, Knop N, Millar T, Obata H, Sullivan DA. The International Workshop on Meibomian Gland Dysfunction: Report of the Subcommittee on Anatomy, Physiology, and Pathophysiology of the Meibomian Gland. Invest Ophthalmol Vis Sci. 2011;52:1938–78. doi: 10.1167/iovs.10-6997c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hodges RR, Dartt DA. Regulatory pathways in lacrimal gland epithelium. Int Rev Cytol. 2003;231:129–96. doi: 10.1016/s0074-7696(03)31004-6. [DOI] [PubMed] [Google Scholar]
- 180.Abram K, Silm H, Maaroos H-I, Oona M. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol JEADV. 2010;24:565–71. doi: 10.1111/j.1468-3083.2009.03472.x. [DOI] [PubMed] [Google Scholar]
- 181.Chang ALS, Raber I, Xu J, Li R, Spitale R, Chen J, Kiefer AK, Tian C, Eriksson NK, Hinds DA, Tung JY. Assessment of the Genetic Basis of Rosacea by Genome-Wide Association Study. J Invest Dermatol. 2015;135:1548–55. doi: 10.1038/jid.2015.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nguyen T, Liu XK, Zhang Y, Dong C. BTNL2, a Butyrophilin-Like Molecule That Functions to Inhibit T Cell Activation. J Immunol. 2006;176:7354–60. doi: 10.4049/jimmunol.176.12.7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yazici AC, Tamer L, Ikizoglu G, Kaya TI, Api H, Yildirim H, Adiguzel A. GSTM1 and GSTT1 null genotypes as possible heritable factors of rosacea. Photodermatol Photoimmunol Photomed. 2006;22:208–10. doi: 10.1111/j.1600-0781.2006.00220.x. [DOI] [PubMed] [Google Scholar]
- 184.Karpouzis A, Avgeridis P, Tripsianis G, Gatzidou E, Kourmouli N, Veletza S. Assessment of Tachykinin Receptor 3′ Gene Polymorphism rs3733631 in Rosacea. Int Sch Res Not. 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mittelman-Smith MA, Williams H, Krajewski-Hall SJ, McMullen NT, Rance NE. Role for kisspeptin/neurokinin B/dynorphin (KNDy) neurons in cutaneous vasodilatation and the estrogen modulation of body temperature. Proc Natl Acad Sci USA. 2012;109:19846–51. doi: 10.1073/pnas.1211517109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jansen T, Krug S, Kind P, Plewig G, Messer G. BsmI polymorphism of the vitamin D receptor gene in patients with the fulminant course of rosacea conglobata (rosacea fulminans). J Dermatol. 2004;31:244–6. doi: 10.1111/j.1346-8138.2004.tb00665.x. [DOI] [PubMed] [Google Scholar]
- 187.Akdogan N, Alli N, Incel Uysal P, Candar T. Role of serum 25-hydroxyvitamin D levels and vitamin D receptor gene polymorphisms in patients with rosacea: a case–control study. Clin Exp Dermatol. 2019;44:397–403. doi: 10.1111/ced.13769. [DOI] [PubMed] [Google Scholar]
- 188.Youm J-K, Park K, Uchida Y, Chan A, Mauro TM, Holleran WM, Elias PM. Local blockade of glucocorticoid activation reverses stress- and glucocorticoid-induced delays in cutaneous wound healing. Wound Repair Regen Off Publ Wound Heal Soc Eur Tissue Repair Soc. 2013;21:715–22. doi: 10.1111/wrr.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tiganescu A, Hupe M, Uchida Y, Mauro T, Elias PM, Holleran WM. Increased glucocorticoid activation during mouse skin wound healing. J Endocrinol. 2014;221:51–61. doi: 10.1530/JOE-13-0420. [DOI] [PubMed] [Google Scholar]
- 190.Rathi SK, Kumrah L. Topical corticosteroid-induced rosacea-like dermatitis: a clinical study of 110 cases. Indian J Dermatol Venereol Leprol. 2011;77:42–6. doi: 10.4103/0378-6323.74974. [DOI] [PubMed] [Google Scholar]
- 191.Tempark T, Shwayder TA. Perioral dermatitis: a review of the condition with special attention to treatment options. Am J Clin Dermatol. 2014;15:101–13. doi: 10.1007/s40257-014-0067-7. [DOI] [PubMed] [Google Scholar]
- 192.Tolaymat L, Hall MR. Perioral Dermatitis. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2020. [PubMed] [Google Scholar]
- 193.Nikolakis G, Zouboulis CC. Skin and glucocorticoids: effects of local skin glucocorticoid impairment on skin homeostasis. Exp Dermatol. 2014;23:807–8. doi: 10.1111/exd.12519. [DOI] [PubMed] [Google Scholar]
- 194.Hong JS, Han S, Lee JS, Lee C, Choi MH, Kim YK, Seo EY, Chung JH, Cho S. Abnormal Glucocorticoid Synthesis in the Lesional Skin of Erythematotelangiectatic Rosacea. J Invest Dermatol. 2019;139:2225–2228.e3. doi: 10.1016/j.jid.2019.02.036. [DOI] [PubMed] [Google Scholar]
- 195.Shimada-Omori R, Yamasaki K, Koike S, Yamauchi T, Aiba S.TLR3 augments glucocorticoid-synthetic enzymes expression in epidermal keratinocytes; Implications of glucocorticoid metabolism in rosacea epidermis. J Dermatol Sci 2020. 100-58-66 [DOI] [PubMed] [Google Scholar]
- 196.Shibata M, Katsuyama M, Onodera T, Ehama R, Hosoi J, Tagami H. Glucocorticoids enhance Toll-like receptor 2 expression in human keratinocytes stimulated with Propionibacterium acnes or proinflammatory cytokines. J Invest Dermatol. 2009;129:375–82. doi: 10.1038/jid.2008.237. [DOI] [PubMed] [Google Scholar]
- 197.Su Q, Pfalzgraff A, Weindl G. Cell type-specific regulatory effects of glucocorticoids on cutaneous TLR2 expression and signalling. J Steroid Biochem Mol Biol. 2017;171:201–8. doi: 10.1016/j.jsbmb.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 198.Skobowiat C, Sayre RM, Dowdy JC, Slominski AT. Ultraviolet radiation regulates cortisol activity in a waveband-dependent manner in human skin ex vivo. Br J Dermatol. 2013;168:595–601. doi: 10.1111/bjd.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Itoi S, Terao M, Murota H, Katayama I. 11β-Hydroxysteroid dehydrogenase 1 contributes to the pro-inflammatory response of keratinocytes. Biochem Biophys Res Commun. 2013;440:265–70. doi: 10.1016/j.bbrc.2013.09.065. [DOI] [PubMed] [Google Scholar]
- 200.Tiganescu A, Hupe M, Jiang YJ, Celli A, Uchida Y, Mauro TM, Bikle DD, Elias PM, Holleran WM. UVB induces epidermal 11β-hydroxysteroid dehydrogenase type 1 activity in vivo. Exp Dermatol. 2015;24:370–6. doi: 10.1111/exd.12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bekhbat M, Rowson SA, Neigh GN. Checks and balances: The glucocorticoid receptor and NFĸB in good times and bad. Front Neuroendocrinol. 2017;46:15–31. doi: 10.1016/j.yfrne.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Boix J, Bigas J, Sevilla LM, Iacobone M, Citton M, Torresan F, Caroccia B, Rossi GP, Pérez P. Primary aldosteronism patients show skin alterations and abnormal activation of glucocorticoid receptor in keratinocytes. Sci Rep. 2017;7:15806. doi: 10.1038/s41598-017-16216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nguyen VT, Farman N, Palacios-Ramirez R, Sbeih M, Behar-Cohen F, Aractingi S, Jaisser F. Cutaneous Wound Healing in Diabetic Mice Is Improved by Topical Mineralocorticoid Receptor Blockade. J Invest Dermatol. 2020;140:223–234. doi: 10.1016/j.jid.2019.04.030. [DOI] [PubMed] [Google Scholar]
- 204.Boix J, Nguyen VT, Farman N, Aractingi S, Pérez P. Mineralocorticoid receptor blockade improves glucocorticoid-induced skin atrophy but partially ameliorates anti-inflammatory actions in an irritative model in human skin explants. Exp Dermatol. 2018;27:185–7. doi: 10.1111/exd.13473. [DOI] [PubMed] [Google Scholar]
- 205.Sulaiman RS, Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in the eye. Steroids. 2018;133:60–6. doi: 10.1016/j.steroids.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kadmiel M, Janoshazi A, Xu X, Cidlowski JA. Glucocorticoid action in human corneal epithelial cells establishes roles for corticosteroids in wound healing and barrier function of the eye. Exp Eye Res. 2016;152:10–33. doi: 10.1016/j.exer.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Dahmana N, Mugnier T, Gabriel D, Kaltsatos V, Bertaim T, Behar-Cohen F, Gurny R, Kalia YN. Topical Administration of Spironolactone-Loaded Nanomicelles Prevents Glucocorticoid-Induced Delayed Corneal Wound Healing in Rabbits. Mol Pharm. 2018;15:1192–202. doi: 10.1021/acs.molpharmaceut.7b01028. [DOI] [PubMed] [Google Scholar]
- 208.Nguyen VT, Farman N, Maubec E, Nassar D, Desposito D, Waeckel L, Aractingi S, Jaisser F. Re-Epithelialization of Pathological Cutaneous Wounds Is Improved by Local Mineralocorticoid Receptor Antagonism. J Invest Dermatol. 2016;136:2080–9. doi: 10.1016/j.jid.2016.05.101. [DOI] [PubMed] [Google Scholar]
- 209.Zhao M, Mantel I, Gelize E, Li X, Xie X, Arboleda A, Seminel M, Levy-Boukris R, Dernigoghossian M, Prunotto A, Andrieu-Soler C, Rivolta C, Canonica J, Naud M-C, Lechner S, Farman N, Bravo-Osuna I, Herrero-Vanrell R, Jaisser F, Behar-Cohen F. Mineralocorticoid receptor antagonism limits experimental choroidal neovascularization and structural changes associated with neovascular age-related macular degeneration. Nat Commun. 2019;10:369. doi: 10.1038/s41467-018-08125-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhao Y, Peng Y, Wang X, Wu L, Wang M, Yan H, Xiao S. Facial dermatosis associated with Demodex: a case-control study. J Zhejiang Univ Sci B. 2011;12:1008–15. doi: 10.1631/jzus.B1100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gonzalez-Hinojosa D, Jaime-Villalonga A, Aguilar-Montes G, Lammoglia-Ordiales L. Demodex and rosacea: Is there a relationship? Indian J Ophthalmol. 2018;66:36–8. doi: 10.4103/ijo.IJO_514_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Erbağci Z, Ozgöztaşi O. The significance of Demodex folliculorum density in rosacea. Int J Dermatol. 1998;37:421–5. doi: 10.1046/j.1365-4362.1998.00218.x. [DOI] [PubMed] [Google Scholar]
- 213.Lacey N, Kavanagh K, Tseng SCG. Under the lash: Demodex mites in human diseases. Biochemist (Lond) 2009;31:2–6. [PMC free article] [PubMed] [Google Scholar]
- 214.Lacey N, Russell-Hallinan A, Powell FC. Study of Demodex mites: Challenges and Solutions. J Eur Acad Dermatol Venereol JEADV. 2016;30:764–75. doi: 10.1111/jdv.13517. [DOI] [PubMed] [Google Scholar]
- 215.Forton F, Seys B. Density of Demodex folliculorum in rosacea: a case-control study using standardized skin-surface biopsy. Br J Dermatol. 1993;128:650–9. doi: 10.1111/j.1365-2133.1993.tb00261.x. [DOI] [PubMed] [Google Scholar]
- 216.Bonnar E, Eustace P, Powell FC. The Demodex mite population in rosacea. J Am Acad Dermatol. 1993;28:443–8. doi: 10.1016/0190-9622(93)70065-2. [DOI] [PubMed] [Google Scholar]
- 217.Forton FMN, De Maertelaer V. Two Consecutive Standardized Skin Surface Biopsies: An Improved Sampling Method to Evaluate Demodex Density as a Diagnostic Tool for Rosacea and Demodicosis. Acta Derm Venereol. 2017;97:242–8. doi: 10.2340/00015555-2528. [DOI] [PubMed] [Google Scholar]
- 218.Turgut Erdemir A, Gurel MS, Koku Aksu AE, Falay T, Inan Yuksel E, Sarikaya E. Demodex mites in acne rosacea: reflectance confocal microscopic study. Australas J Dermatol. 2017;58:e26–30. doi: 10.1111/ajd.12452. [DOI] [PubMed] [Google Scholar]
- 219.Ahn CS, Huang WW. Rosacea Pathogenesis. Dermatol Clin. 2018;36:81–6. doi: 10.1016/j.det.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 220.Falay Gur T, Erdemir AV, Gurel MS, Kocyigit A, Guler EM, Erdil D. The investigation of the relationships of demodex density with inflammatory response and oxidative stress in rosacea. Arch Dermatol Res. 2018;310:759–67. doi: 10.1007/s00403-018-1857-1. [DOI] [PubMed] [Google Scholar]
- 221.Porta Guardia CA. Demodex folliculorum: its association with oily skin surface rather than rosacea lesions. Int J Dermatol. 2015;54:e14–7. doi: 10.1111/ijd.12398. [DOI] [PubMed] [Google Scholar]
- 222.Kubanov A, Gallyamova Y, Kravchenko A. Clinical picture, diagnosis and treatment of rosacea, complicated by Demodex mites. Dermatol Rep. 2019;11 doi: 10.4081/dr.2019.7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sattler EC, Hoffmann VS, Ruzicka T, Braunmühl TV, Berking C. Reflectance confocal microscopy for monitoring the density of Demodex mites in patients with rosacea before and after treatment. Br J Dermatol. 2015;173:69–75. doi: 10.1111/bjd.13783. [DOI] [PubMed] [Google Scholar]
- 224.Dai X, Sayama K, Tohyama M, Shirakata Y, Hanakawa Y, Tokumaru S, Yang L, Hirakawa S, Hashimoto K. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J Allergy Clin Immunol. 2011;127:806–14. doi: 10.1016/j.jaci.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 225.Moran EM, Foley R, Powell FC. Demodex and rosacea revisited. Clin Dermatol. 2017;35:195–200. doi: 10.1016/j.clindermatol.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 226.McMahon F, Banville N, Bergin DA, Smedman C, Paulie S, Reeves E, Kavanagh K. Activation of Neutrophils via IP3 Pathway Following Exposure to Demodex-Associated Bacterial Proteins. Inflammation. 2016;39:425–33. doi: 10.1007/s10753-015-0264-4. [DOI] [PubMed] [Google Scholar]
- 227.Lacey N, Delaney S, Kavanagh K, Powell FC. Mite-related bacterial antigens stimulate inflammatory cells in rosacea. Br J Dermatol. 2007;157:474–81. doi: 10.1111/j.1365-2133.2007.08028.x. [DOI] [PubMed] [Google Scholar]
- 228.Schaller M, Gonser L, Belge K, Braunsdorf C, Nordin R, Scheu A, Borelli C. Dual anti-inflammatory and anti-parasitic action of topical ivermectin 1% in papulopustular rosacea. J Eur Acad Dermatol Venereol JEADV. 2017;31:1907–11. doi: 10.1111/jdv.14437. [DOI] [PubMed] [Google Scholar]
- 229.Lee SH, Chun YS, Kim JH, Kim ES, Kim JC. The relationship between demodex and ocular discomfort. Invest Ophthalmol Vis Sci. 2010;51:2906–11. doi: 10.1167/iovs.09-4850. [DOI] [PubMed] [Google Scholar]
- 230.Liu J, Sheha H, Tseng SCG. Pathogenic role of Demodex mites in blepharitis. Curr Opin Allergy Clin Immunol. 2010;10:505–10. doi: 10.1097/ACI.0b013e32833df9f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kheirkhah A, Casas V, Li W, Raju VK, Tseng SCG. Corneal Manifestations of Ocular Demodex Infestation. Am J Ophthalmol. 2007;143:743–749.e1. doi: 10.1016/j.ajo.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 232.Zhang X-B, Ding Y-H, He W. The association between demodex infestation and ocular surface manifestations in meibomian gland dysfunction. Int J Ophthalmol. 2018;11:589–92. doi: 10.18240/ijo.2018.04.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Kim JH, Chun YS, Kim JC. Clinical and Immunological Responses in Ocular Demodecosis. J Korean Med Sci. 2011;26:1231–7. doi: 10.3346/jkms.2011.26.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kim JT, Lee SH, Chun YS, Kim JC. Tear cytokines and chemokines in patients with Demodex blepharitis. Cytokine. 2011;53:94–9. doi: 10.1016/j.cyto.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 235.Koo H, Kim TH, Kim KW, Wee SW, Chun YS, Kim JC. Ocular surface discomfort and Demodex: effect of tea tree oil eyelid scrub in Demodex blepharitis. J Korean Med Sci. 2012;27:1574–9. doi: 10.3346/jkms.2012.27.12.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.O’Reilly N, Gallagher C, Katikireddy KR, Clynes M, O’Sullivan F, Kavanagh K. Demodex-Associated Bacillus Proteins Induce an Aberrant Wound Healing Response in a Corneal Epithelial Cell Line: Possible Implications for Corneal Ulcer Formation in Ocular Rosacea. Invest Ophthalmol Vis Sci. 2012;53:3250–9. doi: 10.1167/iovs.11-9295. [DOI] [PubMed] [Google Scholar]
- 237.Schaller M, Pietschke K. Successful therapy of ocular rosacea with topical ivermectin. Br J Dermatol. 2018;179:520–1. doi: 10.1111/bjd.16534. [DOI] [PubMed] [Google Scholar]
- 238.Forton FMN. Elucidating the role of Demodex folliculorum in the pathogenesis of rosacea: exciting first steps…. Br J Dermatol. 2018;179:252–3. doi: 10.1111/bjd.16792. [DOI] [PubMed] [Google Scholar]
- 239.Oztas MO, Balk M, Ogüs E, Bozkurt M, Ogüs IH, Ozer N. The role of free oxygen radicals in the aetiopathogenesis of rosacea. Clin Exp Dermatol. 2003;28:188–92. doi: 10.1046/j.1365-2230.2003.01179.x. [DOI] [PubMed] [Google Scholar]
- 240.Bakar Ö, Demirçay Z, Yuksel M, Haklar G, Sanisoglu Y. The effect of azithromycin on reactive oxygen species in rosacea. Clin Exp Dermatol. 2007;32:197–200. doi: 10.1111/j.1365-2230.2006.02322.x. [DOI] [PubMed] [Google Scholar]
- 241.Tisma VS, Basta-Juzbasic A, Jaganjac M, Brcic L, Dobric I, Lipozencic J, Tatzber F, Zarkovic N, Poljak-Blazi M. Oxidative stress and ferritin expression in the skin of patients with rosacea. J Am Acad Dermatol. 2009;60:270–6. doi: 10.1016/j.jaad.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 242.Erdogan HK, Bulur I, Kocaturk E, Saracoglu ZN, Alatas O, Bilgin M. Advanced oxidation protein products and serum total oxidant/antioxidant status levels in rosacea. Adv Dermatol Allergol Dermatol Alergol. 2018;35:304–8. doi: 10.5114/ada.2018.76228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Brink N, Szamel M, Young AR, Wittern KP, Bergemann J. Comparative quantification of IL-1β, IL-10, IL-10r, TNFα and IL-7 mRNA levels in UV-irradiated human skin in vivo. Inflamm Res. 2000;49:290–6. doi: 10.1007/PL00000209. [DOI] [PubMed] [Google Scholar]
- 244.Schmutz J-L. Rosacée et ultraviolets. Ann Dermatol Venereol. 2006;133:467–9. doi: 10.1016/s0151-9638(06)70942-5. [DOI] [PubMed] [Google Scholar]
- 245.Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer H-D. The Inflammasome Mediates UVB-Induced Activation and Secretion of Interleukin-1β by Keratinocytes. Curr Biol. 2007;17:1140–5. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 246.Vemuri RC, Gundamaraju R, Sekaran SD, Manikam R. Major Pathophysiological Correlations of Rosacea: A Complete Clinical Appraisal. Int J Med Sci. 2015;12:387–96. doi: 10.7150/ijms.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mureşan S, Filip A, Mureşan A, Şimon V, Moldovan R, Gal AF, Miclăuş V. Histological findings in the Wistar rat cornea following UVB irradiation. Romanian J Morphol Embryol Rev Roum Morphol Embryol. 2013;54:247–52. [PubMed] [Google Scholar]
- 248.Pauloin T, Dutot M, Joly F, Warnet J-M, Rat P. High molecular weight hyaluronan decreases UVB-induced apoptosis and inflammation in human epithelial corneal cells. Mol Vis. 2009;15:577–83. [PMC free article] [PubMed] [Google Scholar]
- 249.Viiri J, Jauhonen HM, Kauppinen A, Ryhänen T, Paimela T, Hyttinen J, Sorri I, Laihia JK, Leino L, Kaarniranta K. Cis-urocanic acid suppresses UV-B-induced interleukin-6 and −8 secretion and cytotoxicity in human corneal and conjunctival epithelial cells in vitro. Mol Vis. 2009;15:1799–805. [PMC free article] [PubMed] [Google Scholar]
- 250.Chen S-J, Lee C-J, Lin T-B, Liu H-J, Huang S-Y, Chen J-Z, Tseng K-W. Inhibition of Ultraviolet B-Induced Expression of the Proinflammatory Cytokines TNF-α and VEGF in the Cornea by Fucoxanthin Treatment in a Rat Model. Mar Drugs. 2016;14 doi: 10.3390/md14010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Korhonen E, Piippo N, Hytti M, Kaarniranta K, Kauppinen A. NLRP3 inflammasome activation after ultraviolet B irradiation in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2016;57:327–327. doi: 10.1167/iovs.61.4.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Zheng Y, Niyonsaba F, Ushio H, Nagaoka I, Ikeda S, Okumura K, Ogawa H. Cathelicidin LL-37 induces the generation of reactive oxygen species and release of human alpha-defensins from neutrophils. Br J Dermatol. 2007;157:1124–31. doi: 10.1111/j.1365-2133.2007.08196.x. [DOI] [PubMed] [Google Scholar]
- 253.Salzer S, Kresse S, Hirai Y, Koglin S, Reinholz M, Ruzicka T, Schauber J. Cathelicidin peptide LL-37 increases UVB-triggered inflammasome activation: Possible implications for rosacea. J Dermatol Sci. 2014;76:173–9. doi: 10.1016/j.jdermsci.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 254.Suhng E, Kim BH, Choi YW, Choi HY, Cho H, Byun JY. Increased expression of IL-33 in rosacea skin and UVB-irradiated and LL-37-treated HaCaT cells. Exp Dermatol. 2018;27:1023–9. doi: 10.1111/exd.13702. [DOI] [PubMed] [Google Scholar]
- 255.Melnik BC. Endoplasmic reticulum stress: key promoter of rosacea pathogenesis. Exp Dermatol. 2014;23:868–73. doi: 10.1111/exd.12517. [DOI] [PubMed] [Google Scholar]
- 256.Lone AN, Malik AT, Naikoo HS, Raghu RSA, Tasduq S. Trigonelline, a naturally occurring alkaloidal agent protects ultraviolet-B (UV-B) irradiation induced apoptotic cell death in human skin fibroblasts via attenuation of oxidative stress, restoration of cellular calcium homeostasis and prevention of endoplasmic reticulum (ER) stress. J Photochem Photobiol B. 2020;202:111720. doi: 10.1016/j.jphotobiol.2019.111720. [DOI] [PubMed] [Google Scholar]
- 257.Wang T-T, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH. Cutting Edge: 1,25-Dihydroxyvitamin D3 Is a Direct Inducer of Antimicrobial Peptide Gene Expression. J Immunol. 2004;173:2909–12. doi: 10.4049/jimmunol.173.5.2909. [DOI] [PubMed] [Google Scholar]
- 258.Ekiz O, Balta I, Sen BB, Dikilitaş MC, Ozuğuz P, Rifaioğlu EN. Vitamin D status in patients with rosacea. Cutan Ocul Toxicol. 2014;33:60–2. doi: 10.3109/15569527.2013.797907. [DOI] [PubMed] [Google Scholar]
- 259.Morizane S, Yamasaki K, Kabigting FD, Gallo RL. Kallikrein expression and cathelicidin processing are independently controlled in keratinocytes by calcium, vitamin D(3), and retinoic acid. J Invest Dermatol. 2010;130:1297–306. doi: 10.1038/jid.2009.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Huggenberger R, Detmar M. The cutaneous vascular system in chronic skin inflammation. J Investig Dermatol Symp Proc Soc Investig Dermatol Inc Eur Soc Dermatol Res. 2011;15:24–32. doi: 10.1038/jidsymp.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Brauchle M, Funk JO, Kind P, Werner S. Ultraviolet B and H2O2 are potent inducers of vascular endothelial growth factor expression in cultured keratinocytes. J Biol Chem. 1996;271:21793–7. doi: 10.1074/jbc.271.36.21793. [DOI] [PubMed] [Google Scholar]
- 262.Hirakawa S, Fujii S, Kajiya K, Yano K, Detmar M. Vascular endothelial growth factor promotes sensitivity to ultraviolet B-induced cutaneous photodamage. Blood. 2005;105:2392–9. doi: 10.1182/blood-2004-06-2435. [DOI] [PubMed] [Google Scholar]
- 263.Yano K, Oura H, Detmar M. Targeted overexpression of the angiogenesis inhibitor thrombospondin-1 in the epidermis of transgenic mice prevents ultraviolet-B-induced angiogenesis and cutaneous photo-damage. J Invest Dermatol. 2002;118:800–5. doi: 10.1046/j.1523-1747.2002.01752.x. [DOI] [PubMed] [Google Scholar]
- 264.Kajiya K, Sawane M, Huggenberger R, Detmar M. Activation of the VEGFR-3 pathway by VEGF-C attenuates UVB-induced edema formation and skin inflammation by promoting lymphangiogenesis. J Invest Dermatol. 2009;129:1292–8. doi: 10.1038/jid.2008.351. [DOI] [PubMed] [Google Scholar]
- 265.Fan F, Stoeltzing O, Liu W, McCarty MF, Jung YD, Reinmuth N, Ellis LM. Interleukin-1β Regulates Angiopoietin-1 Expression in Human Endothelial Cells. Cancer Res. 2004;64:3186–90. doi: 10.1158/0008-5472.can-03-0407. [DOI] [PubMed] [Google Scholar]
- 266.Moore C, Cevikbas F, Pasolli HA, Chen Y, Kong W, Kempkes C, Parekh P, Lee SH, Kontchou N-A, Yeh I, Jokerst NM, Fuchs E, Steinhoff M, Liedtke WB. UVB radiation generates sunburn pain and affects skin by activating epidermal TRPV4 ion channels and triggering endothelin-1 signaling. Proc Natl Acad Sci USA. 2013;110:E3225–34. doi: 10.1073/pnas.1312933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Csáti A, Edvinsson L, Vécsei L, Toldi J, Fülöp F, Tajti J, Warfvinge K. Kynurenic acid modulates experimentally induced inflammation in the trigeminal ganglion. J Headache Pain. 2015;16:99. doi: 10.1186/s10194-015-0581-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Pyun H-B, Kim M, Park J, Sakai Y, Numata N, Shin J-Y, Shin H-J, Kim D-U, Hwang J-K. Effects of Collagen Tripeptide Supplement on Photoaging and Epidermal Skin Barrier in UVB-exposed Hairless Mice. Prev Nutr Food Sci. 2012;17:245–53. doi: 10.3746/pnf.2012.17.4.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Yogianti F, Kunisada M, Nakano E, Ono R, Sakumi K, Oka S, Nakabeppu Y, Nishigori C. Inhibitory effects of dietary Spirulina platensis on UVB-induced skin inflammatory responses and carcinogenesis. J Invest Dermatol. 2014;134:2610–9. doi: 10.1038/jid.2014.188. [DOI] [PubMed] [Google Scholar]
- 270.Rhodes LE, Belgi G, Parslew R, McLoughlin L, Clough GF, Friedmann PS. Ultraviolet-B-induced erythema is mediated by nitric oxide and prostaglandin E2 in combination. J Invest Dermatol. 2001;117:880–5. doi: 10.1046/j.0022-202x.2001.01514.x. [DOI] [PubMed] [Google Scholar]
- 271.Del Rosso JQ. Management of facial erythema of rosacea: what is the role of topical α-adrenergic receptor agonist therapy? J Am Acad Dermatol. 2013;69(Suppl 1):S44–56. doi: 10.1016/j.jaad.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 272.Chung S-H, Kweon M-N, Lee HK, Choi S-I, Yang J-Y, Kim EK. Toll-like receptor 4 initiates an innate immune response to lipopolysaccharide in human conjunctival epithelial cells. Exp Eye Res. 2009;88:49–56. doi: 10.1016/j.exer.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 273.Hozono Y, Ueta M, Hamuro J, Kojima K, Kawasaki S, Yamazaki K, Kinoshita S. Human corneal epithelial cells respond to ocular-pathogenic, but not to nonpathogenic-flagellin. Biochem Biophys Res Commun. 2006;347:238–47. doi: 10.1016/j.bbrc.2006.06.088. [DOI] [PubMed] [Google Scholar]
- 274.Edvinsson L, Haanes KA, Warfvinge K. Does inflammation have a role in migraine? Nat Rev Neurol. 2019;15:483–90. doi: 10.1038/s41582-019-0216-y. [DOI] [PubMed] [Google Scholar]
- 275.Siiskonen H, Harvima I. Mast Cells and Sensory Nerves Contribute to Neurogenic Inflammation and Pruritus in Chronic Skin Inflammation. Front Cell Neurosci. 2019;13:422. doi: 10.3389/fncel.2019.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]