

Graphical abstract

Keywords: Taiwan Biobank, Extensive whole-genome sequencing, De novo mutations, Cardiovascular diseases
Abstract
Introduction
A population-specific genomic reference is important for research and clinical practice, yet it remains unavailable for Han Chinese (HC) in Taiwan.
Objectives
We report the first whole genome sequencing (WGS) database of HC (1000 Taiwanese genome (1KTW-WGS)) and demonstrate several applications to cardiovascular medicine.
Methods
Whole genomes of 997 HC were sequenced to at least 30X depth. A total of 20,117 relatively healthy HC individuals were genotyped using a customized Axiom GWAS array. We performed a genome-wide genotype imputation technique using IMPUTE2.
Results
We identified 26.7 million single-nucleotide variants (SNVs) and 4.2 million insertions-deletions. Of the SNVs, 16.1% were novel relative to dbSNP (build 152), and 34.2% were novel relative to gnomAD. A total of 18,450 healthy HC individuals were genotyped using a customized Genome-Wide Association Study (GWAS) array. We identified hypertension-associated variants and developed a hypertension prediction model based on the correlation between the WGS data and GWAS data (combined clinical and genetic models, AUC 0.887), and also identified 3 novel hyperlipidemia-associated variants. Each individual carried an average of 16.42 (SD = 3.72) disease-causing variants. Additionally, we established an online SCN5A (an important cardiac gene) database that can be used to explore racial differences. Finally, pharmacogenetics studies identified HC population-specific SNVs in genes (CYP2C9 and VKORC1) involved in drug metabolism and blood clotting.
Conclusion
This research demonstrates the benefits of constructing a population-specific genomic reference database for precision medicine.
Introduction
The human genome contains an enormous amount of information about human evolution, development, and medicine. To discover the underlying genetic causes of diseases, comprehensive whole-genome sequencing (WGS) is needed to interrogate all types of genetic variation, including single-nucleotide variants (SNVs) and structural and de novo variants. High-throughput genotyping and sequencing have shown how diversity in the sequence of the human genome affects human diversity. A huge number of variants identified by WGS presents new opportunities and challenges for researchers and clinicians.
Several large-scale WGS projects have been completed. Foremost among them are the 1000 Genomes (1000G) Project; the National Heart, Lung and Blood Institute’s GO Exome Sequencing Project for European Americans and African Americans; the integrative Japanese Genome Variation Database (iJGVD); and gnomAD. All of them have provided valuable information about human genome diversity across different ethnic populations worldwide. In the past few decades, studies based on WGS and whole-exome sequencing have identified rare variants associated with diseases. Therefore, it is essential to study large samples to build a complete database of genetic variations for each population.
Taiwan is a small but densely populated island in East Asia (~23.5 million inhabitants in 36,543 km2, a population larger than that of the Netherlands but with a smaller geographic area). The majority of the population (>95%) in Taiwan is of Han Chinese (HC) ancestry [1]. In the Taiwan Biobank (TWB), a total of 20,117 relatively healthy HC individuals were genotyped using the customized Axiom Genome-Wide Association Study (GWAS) array. Although the GWAS chip was specifically designed for Taiwanese people, it only contains around 600,000 SNV loci. This number is dramatically lower than the number of bases in a whole human genome (3 billion) and thus we performed this WGS study in order to provide better variant identification. Here, we describe the insights gained from sequencing the whole genomes of 997 HC individuals in Taiwan. Lastly, we performed population genetic analyses and applied the WGS data to the improvement of patient care in the cardiovascular field. We also investigated the correlation between the WGS data and the GWAS data and developed a hypertension prediction model based on GWAS data.
Materials and methods
The Han Chinese study population. Taiwan is an island with a population of approximately 23.5 million people. The majority (>95%) of Taiwanese are of HC ancestry and mostly immigrated from southeast China over the past 4 centuries, whereas ~2% are of aboriginal ancestry (Austronesian) [2]. This study was based on WGS data from the white blood cells of 997 unrelated, relatively healthy HC individuals randomly selected from among the 20,117 HC participating in the TWB. Since the resulting sequence database has almost 1000 genomes, it was called 1KTW-WGS. The detailed design of the TWB and individual enrollment are described in the Supplementary Note. Supplementary Fig. 1 shows the locations of the recruitment centers. No ethnicity outliers were included in the 1KTW-WGS project. All participants provided written informed consent, and all DNA samples and personal information were analyzed anonymously. This study was approved by National Taiwan University Hospital (201305043RINB by Research Ethics Committee B) and by the Ethics and Governance Council of the TWB (TWBR10507-03 by IRB-Biomedical Science Research, Academia Sinica). The TWB is governed by the Ethics and Governance Council and the National Ministry of Health and Welfare.
Data generation and processing. We sequenced the whole genomes of 997 HC using either Illumina Hi-Seq 2500 or Ion Torrent-Proton technology (499 genomes using Illumina, 498 genomes using Ion Torrent-Proton). The overall analysis workflows for the two platforms are illustrated in Supplementary Fig. 2 and Supplementary Fig. 3, respectively. In general, the two pipelines followed the standard protocols provided by the two companies, and all analysis parameters were set at their default values. The details of procedures are described in the Supplementary Note.
Customized whole-genome genotyping and quality control. The TWB used a customized Axiom GWAS array that includes 653,291 SNVs specific for the HC population in Taiwan and constructed a population-specific reference for the HC population in Taiwan, which was used in our previous study [3]. A total of 20,117 relatively healthy HC individuals were genotyped using the customized array in the current study. Quality control of the customized GWAS array with PLINK software [4] includes a call rate greater than95%, Hardy-Weinberg equilibrium > 10−4, and ATP calling. Detection of uncertain kinships and ethnicity outliers was also performed as described in our previous study [3].
Imputation performance and application of 1KTW-WGS. We performed a genome-wide genotype imputation technique using IMPUTE2 [5] to estimate untyped genotypes from known haplotype information in order to compare 1KTW-WGS with 1000G Phase 3 East Asian (EAS) sequences. We randomly selected 406 of the 499 individuals sequenced by Illumina to construct a phased reference panel and used the remaining 93 individuals to validate the imputed genotypes.
Applying the 1KTW-WGS data to discover novel genetic variants associated with hyperlipidemia. A SNV, rs7115242, was identified with a P value of 5.72*10−8 in our previous GWAS, which was conducted to identify important and predictive SNVs related to hyperlipidemia. However, novel variants associated with hyperlipidemia were not detectable due to the limitations of using an SNV microarray. Therefore, to demonstrate the application of the 1KTW-WGS database, we explored novel variants associated with rs7115242. Initially, a linkage disequilibrium (LD) analysis was performed for rs7115242 using the single nucleotide polymorphism annotation and proxy search (SNAP) web site [6]. To focus on the East Asian population, we selected the Chinese Han Beijing (CHB) and Japanese in Tokyo (JPT) populations as reference groups. The genotyping data for the reference panel were obtained from the 1000G Pilot 1 project [7], and confidence interval testing (used D prime as the parameter) in Haploview 4.2 software was used to partition the chromosome into blocks. The physical coordinates for the two SNPs closest to the boundary of the LD block were determined according to the R2 value (R2≧0.8) [8], [9], [10]. Subsequently, the chromosomal locations for the LD block were utilized to select nonsynonymous mutations identified from the next-generation sequencing (NGS) data in the TWB. Three bioinformatics algorithms—Sorting Tolerant From Intolerant (SIFT) [11], Protein Variation Effect Analyzer (PROVEAN) [12], and Polymorphism Phenotyping v2 (PolyPhen-2) [13]—were used to assess the functional significance of the nonsynonymous mutations.
Development of a prediction model for hypertension. Hypertension was defined as (i) systolic blood pressure ≥ 140 mmHg, or (ii) diastolic blood pressure ≥ 90 mmHg, (iii) or self-reported hypertension patients on medications; all other subjects were classified as non-hypertensive. We genotyped 18,450 samples with the TWB genotyping array. We excluded samples with (i) sample call rate > 95%, (ii) heterozygosity rate > 5 standard deviations from the population mean, (iii) closely related individuals (identity by descent rating > 0.1875), and (iv) non–East Asian outliers identified by principal component analysis (PCA) of the studied samples and the three major reference populations (Africans, Europeans, and East Asians) in the International HapMap Project. We then applied standard quality-control criteria with PLINK software [4] for variants, excluding those with (i) SNV call rate > 95%, (ii) minor allele frequency < 5%, and (iii) Hardy–Weinberg equilibrium P ≤ 1.0 X10−4, and (iv) different missing rate between cases and controls (P < 0.00001). Subsequently, for each of the remaining SNVs, a logistic regression model was used to evaluate the association of the SNV with hypertension after adjustment for age, gender, and body mass index (BMI). PCA was used for the assessment of population stratification. The first 8 principal components were used to adjust the GWAS analysis. The genome-wide P value significance threshold (P < 10−8) was defined to select SNVs significantly associated with hypertension. Lastly, a prediction model for hypertension was developed based on the significant SNVs using a logistic regression model. We selected candidate SNVs to construct a polygenic risk score after LD clumping of the variants with P < 5 × 10-5. The score was created using the equation , where k is the number of selected SNVs and is the regression coefficient for each that was derived from logistic regression.
Development of an online database for identifying SCN5A variants in sudden arrhythmia death syndrome. Because sudden arrhythmia death syndrome is inheritable and is one of the primary causes of sudden death in children and young adults, databases of disease-relevant mutations (e.g., Human Gene Mutation Database (HGMD), ClinVar) are often used to identify potentially pathogenic variants for clinical genetic testing or basic research. We annotated variants in the HC WGS data that were listed as disease-causing in the HGMD and ClinVar [14], [15], and established an online gene database to easily explore racial differences and the potential functional impact of identified variants. The design of the database is summarized in Supplementary Fig. 4. Briefly, we collected all nucleotide variants of SCN5A from the TWB [16] and 3 online databases, including the 1000G database (phase 3) [7], the iJGVD [17], and gnomAD [18]. The detailed characteristics of the four datasets and procedures are summarized in Supplementary Table 1 and the Supplementary Note, and each population was classified into Asian or non-Asian groups according to their ethnic background. The proportion test embedded in the R language was performed to assess whether DNA variants in SCN5A show different allele frequencies between the two groups (proportion test: P < 0.001). The online database for SCN5A variants was developed using two programming languages, Python Flask and MySQL.
Statistical analysis
For the comparison of minor allele frequency (MAF) across different groups, a proportion test was conducted, and P values less than0.05 were reported as significant. For SNVs analyzed for hypertension and hyperlipidemia, a logistic regression model was utilized, and confounding factors including age, gender, BMI, and principal components were adjusted. The LD associations were evaluated to identify tag SNVs within a LD block.
Data availability
The URL for the online database for SCN5A variants is http://140.112.136.14:8000. The 1KTW-WGS database is open to the public (the Taiwan Biobank website is https://taiwanview.twbiobank.org.tw/browse38).
Results
We randomly selected 997 unrelated HC individuals from the TWB to construct 1KTW-WGS, identified novel variants in their genomes, and applied the WGS data to clinical practice and patient care in the cardiovascular field.
Sequencing and variant discovery
After considering the quality and abundance of DNA samples, uncertain kinship, and ethnicity outliers (Supplementary Table 2), we sequenced the whole genomes of 997 HC and identified a total of 26,051,907 SNVs and 3,592,314 indels with at least 30X coverage. Of the SNVs, 13,624,601 (32.7%) were novel compared to gnomAD, while 2,266,663 of the indels (43.7%) were novel. Because HC in both Taiwan and Japan are East Asian, we compared our WGS data with that of 1,070 Japanese individuals (1KJPN) from iJGVD [17]. Numbers of SNVs, novel SNVs, indels, and novel indels were similar to those in 1KJPN (Supplementary Table 3). At first, two different cutoffs (20X or 30X) of the read depth were utilized to identify DNA variants in this study (Supplementary Table 3). Since the average depth of the NGS data in this study was around 30X and the number of identified variants was comparable to that from the 1KJPN Biobank, we decided to use 30X as the cutoff in this study. Per individual, the mean total number of SNVs with at least 30X coverage was 1,894,528.7 (range 585,773–2,740,034), and the mean number of novel SNVs was 52,394.1 (16,037–91,629), whereas the mean total number of indels was 133,869 (31,182–306,432) and the mean number of novel indels was 8,746.7 (3,403–51233) (Fig. 1). The two platforms detected similar numbers of SNVs relative to dbSNP build 152 and the combined datasets of dbSNP and 1000G, whereas more novel SNVs were detected by the Ion Torrent-Proton platform than the Illumina platform (Supplementary Table 4).
Fig. 1.

The distribution of the number of variants and novel variants per individual in 1KTW-WGS.
Comparing the number of indels between protein-coding regions and non-coding regions using the dbSNP 152 and 1000G phase 3 public databases as references, we found that there were fewer indels in protein-coding regions than in non-coding regions, but the percentage of novel indels was higher in protein-coding regions than in non-coding regions (Fig. 2).
Fig. 2.

Distribution and number of indels in protein-coding regions (a) and non-coding regions (b) in 1KTW-WGS using the dbSNP (build 152) and 1000 Genomes (1 KG) phase 3 public databases as references. Novel indels are colored blue. Indels in dbSNP 152 alone are colored orange, whereas indels in dbSNP 152 and 1000 Genomes are colored gray.
We inspected the length and number of indels by genomic location. The overall shape of the distribution for size frequency showed that larger indels were less frequent than smaller indels (Fig. 2), irrespective of their location in a coding or non-coding region. Comparison of the length distribution of indels between protein-coding regions and non-coding regions showed that indel lengths were similar, from −30 to 30 base pairs. However, the number of large indels (>20 bp) was higher in non-coding regions than in protein-coding regions (Supplementary Fig. 5). Compared with the distribution of indel lengths inside and outside protein-coding regions of the Icelandic population (the only population that provided this data) [18], our data also showed more deletions were called than insertions. However, Gudbjartsson et al. observed a deficit of deletions and insertions that were not multiples of three, which was different from our finding.
In protein-coding regions, we found that the percentage of indels that were multiples of three was higher compared to that in non-coding regions, regardless of the MAF (Supplementary Fig. 6). The MAF of most large indels was relatively low (<0.5%) compared to the MAF of small indels in coding regions, but this difference in MAF distribution between large indels and small indels was not found in non-coding regions (Supplementary Fig. 6).
Comparison of 1KTW-WGS with existing databases to reveal novel variants
As expected, the majority of SNVs (65.8%) in the 1KTW-WGS database were present in the Asian subset of gnomAD-genome (EAS) or gnomAD-genome-all, which implied that common alleles segregated across Asian populations. Of these variants, 16.1% of SNVs in the 1KTW-WGS database were novel relative to dbSNP build 152, 53.4% were novel relative to the combined dataset of dbSNP and 1000G phase 3, and 34.2% were novel relative to gnomAD-genome-all (Fig. 3, Supplementary Table 5). This demonstrates the value of performing WGS in individual populations in greater depth. On the other hand, we also compared the results generated by two different platforms (Illumina vs. Ion Torrent-Proton), which showed similar results.
Fig. 3.
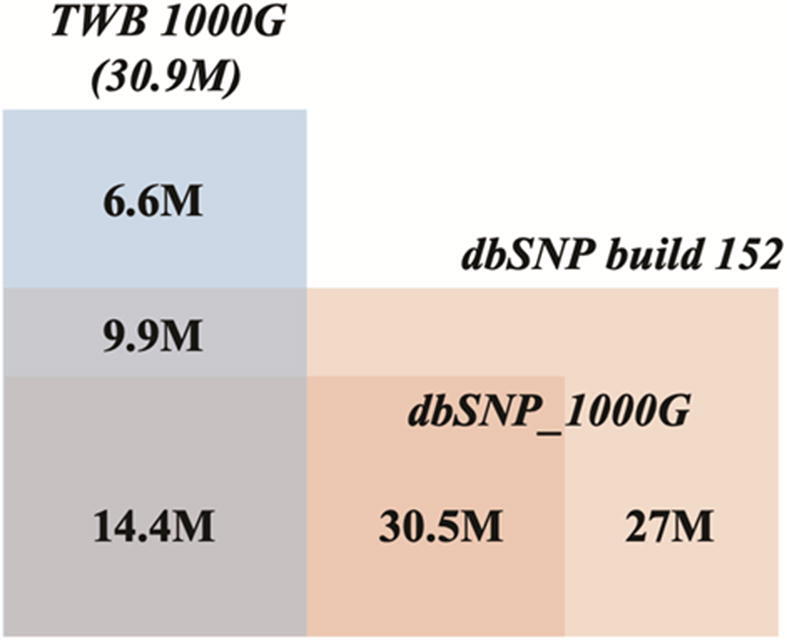
Venn diagram of all SNVs and indels discovered in 1KTW-WGS relative to dbSNP (Build 152), and the 1000 Genomes Project (1000G) Phase 3. M, million.
Variants categorized by the burden of loss of function
In a clinical setting, predicting the biological consequences of variants is an important application of sequencing data. To particularly evaluate and compare the burden of loss-of-function (LOF) variants, we classified these variants into four categories as described in previous studies: (1) LOF, including stop-gain or -loss variants, frameshift indels, splice donor or acceptor variants, and initiator codon variants; (2) moderate impact, including missense variants, in-frame indels and splice-region variants; (3) low impact, including synonymous variants and 3′- and 5′-UTR variants; and (4) others, including deep intronic and intergenic variants. Table 1 shows the frequency distribution of variants based on functional annotation. We found that the percentage of variants with a MAF below 0.5% was 76.3%, 75.5%, 62.3% and 63.2% in the categories for the LOF, moderate impact, low impact, and other categories, respectively. In each category, the proportion of novel variants with an MAF below 0.5% was similar to the proportion of total variants with an MAF below 0.5%. We also found that the proportion of total variants with a MAF below 0.5% was similar in the categories of LOF, moderate impact, low impact and other, between the two NGS platforms. The exception was indels, which were higher in the Ion Torrent-Proton platform than the Illumina platform (Supplementary Table 6). Notably, in Supplementary Table 6, we only included indels with MAF<0.5% to do the comparisons, i.e., rare indels. We note that the indels identified in the Ion Torrent-Proton system had higher MAFs and thus under such criteria (MAF < 0.5%), the number of novel indels from the Ion Torrent-Proton system was less than that from the Illumina system (888,033 versus 1045,081). However, if we calculate the proportion of novel indels versus total indels, the Ion Torrent-Proton system still has a slightly higher number (57.57% versus 56.63%). This might be explained by the presence of false positive indels, as reported by previous studies [19], [20].
Table 1.
The frequency distribution of variants based on functional annotations*
| Type | SNV |
Indel |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| MAF | > 5% | 0.5–5% | < 0.5% | All | > 5% | 0.5–5% | < 0.5% | All | |
| 3′ or 5′ UTR, upstream, downstream, Synonymous | Total | 217,916 | 143,562 | 618,151 | 979,629 | 33,692 | 30,607 | 86,387 | 150,686 |
| Novela | 2674 | 4045 | 137,780 | 144,499 | 7867 | 12,585 | 48,616 | 69,068 | |
| % of total variants | 3.703 | 3.925 | 3.61 | 3.674 | 3.492 | 3.624 | 3.558 | 3.556 | |
| % of novel variants | 3.298 | 3.77 | 3.367 | 3.376 | 3.244 | 3.422 | 3.375 | 3.368 | |
| Stop gain or loss, frameshift variant, splicing variant | Total | 647 | 605 | 4943 | 6195 | 937 | 1696 | 7561 | 10,194 |
| Novela | 4 | 16 | 1171 | 1191 | 441 | 1163 | 5463 | 7067 | |
| % of total variants | 0.011 | 0.017 | 0.029 | 0.023 | 0.097 | 0.201 | 0.312 | 0.241 | |
| % of novel variants | 0.005 | 0.015 | 0.029 | 0.028 | 0.182 | 0.316 | 0.379 | 0.345 | |
| Missense, in-frame variant | Total | 18,893 | 18,097 | 114,202 | 151,192 | 470 | 461 | 2824 | 3755 |
| Novela | 32 | 330 | 17,752 | 18,114 | 64 | 134 | 1668 | 1866 | |
| % of total variants | 0.321 | 0.495 | 0.667 | 0.567 | 0.049 | 0.055 | 0.116 | 0.089 | |
| % of novel variants | 0.039 | 0.3 | 0.43 | 0.42 | 0.026 | 0.03 | 0.11 | 0.09 | |
| Intronic, intergenic | Total | 5,647,133 | 3,494,935 | 16,385,854 | 25,527,922 | 929,818 | 811,862 | 2,331,119 | 4,072,799 |
| Novela | 78,362 | 102,894 | 3,935,234 | 4,116,490 | 234,153 | 353,881 | 1,384,933 | 1,972,967 | |
| % of total variants | 95.965 | 95.563 | 95.694 | 95.736 | 96.363 | 96.121 | 96.014 | 96.115 | |
| % of novel variants | 96.657 | 95.907 | 96.17 | 96.173 | 96.548 | 96.226 | 96.131 | 96.197 | |
| Overall | Total | 5,884,589 | 3,657,199 | 17,123,150 | 26,664,938 | 964,917 | 844,626 | 2,427,891 | 4,237,434 |
| Novela | 81,072 | 107,285 | 4,091,937 | 4,280,294 | 242,525 | 367,763 | 1,440,680 | 2,050,968 | |
Not observed in dbSNP build 152 and 1000 Genomes Project Phase 3; MAF: minor allele frequency; SNV: single nucleotide variant; Indel: insertion or deletion.
Identifying a causal variant in patients with an inherited Mendelian disease is crucial in clinical genetic testing. Online Mendelian Inheritance in Man (OMIM) is a public and popular database of reported genes and mutations of rare diseases and mutations with high penetrance [21]. For Mendelian diseases, the causal variants in the OMIM database are most often LOF or moderate-impact mutations [21]. Reliable estimates of genotype frequency in a population play an extremely important role when filtering candidate variants in a patient with an inherited Mendelian disease. The 1KTW-WGS database provides the distribution of mean genotype counts per individual by frequency and variant impact (Table 2, Supplementary Fig. 7). Our sequenced individuals carried on average 149 LOF variants, of which 1.4 were only seen in 1 or 2 of the 997 sequenced individuals (MAF < 0.04%) and are thus likely candidates for dominant determinants of rare traits. In the context of rare recessive traits, we found that 3.3 individuals were homozygous for a LOF variant with MAF < 3.2%, a threshold that would correspond to 1 in 997 individuals being homozygous under Hardy-Weinberg equilibrium (Supplementary Table 7).
Table 2.
Variant load of coding mutations and disease-associated variations per individual in 1KTW-WGS.
| Rare variant (<0.5%) |
Low frequency variant (0.5–5%) |
Common variant (>5%) |
||||
|---|---|---|---|---|---|---|
| Mean | (s.d.) | Mean | (s.d.) | Mean | (s.d.) | |
| Total loss of functiona | 19.76 | 13.79 | 27.26 | 7.88 | 171.05 | 26.31 |
| Non-synonymous | 135.67 | 27.57 | 245.81 | 54.11 | 2032.78 | 415.2 |
| Probably damaging | 0.29 | 0.55 | 0.36 | 0.62 | 1.47 | 0.97 |
| Splicing varianta | 10.88 | 3.71 | 3.5 | 1.32 | 10.39 | 3.51 |
| Stop gaina | 5.67 | 2.34 | 6.55 | 2.57 | 70.42 | 18.71 |
| Synonymous | 51.37 | 9.79 | 114.13 | 25.97 | 1123.95 | 242.7 |
| HGMD (only disease-causing mutations) | 0.03 | 0.17 | 0.99 | 0.74 | 11.25 | 3.62 |
| OMIMC | 0.23 | 0.47 | 0.25 | 0.5 | 1.25 | 0.88 |
| Loss of function (>20BP deletion)a | 0.03 | 0.17 | 0.11 | 0.32 | 0.53 | 0.75 |
| Indel frameshift (<20 BP)a | 11.34 | 13.05 | 15.44 | 5.82 | 72.04 | 15.32 |
| Indel non-frameshift (<20 BP)a | 3.47 | 16.87 | 4.91 | 3.15 | 68.2 | 13.48 |
| All SNVsa | 599.8 | 126.1 | 1008.38 | 239.15 | 9982.2 | 2230 |
| Novela,b | 26.42 | 12.4 | 5.58 | 3.98 | 14.01 | 5.97 |
| Total conserved | 191.49 | 36.61 | 365.23 | 79.53 | 3221.63 | 671.2 |
| Total bases deleted | 7,007,314 bases | |||||
Only SNV sites at which ancestral state can be assigned with high confidence and that are highly conserved (GERP > 2.0) are reported. OMIM, Online Mendelian Inheritance in Man; HGMD, The Human Genetic Mutation Database professional version.
No conservation filter applied but used SNPAncestralAllele; bNot observed in dbSNP build 152 and 1000 Genomes Project Phase 3; cOnly counts damaging and possible damaging variants.
Functional variants
We found that the overall patterns and per-individual distributions of LOF SNVs (variants introducing premature stop codons or variants interrupting splice sites) and missense variants in 1KTW-WGS were consistent with those found in the 1000G Project. Regarding the individual variant load of coding mutations, each individual carried an average of 218.07 LOF SNVs, 99.49 LOF indels, and 0.67 LOF large deletions. Interestingly, 78.4% of the LOF SNVs for each individual are common variants. In contrast, considering rare LOF SNVs (MAF < 0.5%) alone, each individual carried an average of 5.67 nonsense variants, 10.88 variants interrupting a splice site, 11.34 frameshift indels, and 0.03 larger deletions (Table 2).
Because databases of disease-relevant mutations are often employed to identify potential variants of interest, we annotated variants in 1KTW-WGS that were listed as disease-causing mutations (DMs) in the HGMD [14]. We identified 1139 variants with at least 30X coverage as DMs, and each individual carried an average of 16.42 (SD = 3.72) DM variants whose GERP score was greater than 2.0 (Table 2, Supplementary Fig. 8). ClinVar is another well-known and popular disease-relevant database (https://www.ncbi.nlm.nih.gov/clinvar/). In addition to annotating disease-causing variants in 1KTW-WGS based on the data in the HGMD database, we listed some variants in 1KTW-WGS that were classified as pathogenic in ClinVar (Table 3).
Table 3.
HGMD or ClinVar reported disease-causing or pathogenic mutations in sudden arrhythmic death syndrome or cardiomyopathy in 1KTW-WGS individuals.
| Gene | Transcript | Reference/Mutation allele | rsID | Disease | Disease prevalence | Phenotypic manifestation | Inheritance pattern | Database | 1000G |
gnomAD |
2KJPN |
ChinaMap |
Mutation allele frequency in 1KTW- WGS |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EU | AFR | EAS | SAS | EU (non-F) | AFR | EA | SA | ||||||||||||
| SCN3B | NM_018400.3 | c.328G > A | rs147205617 | Brugada syndrome | 0.0012* | Syncope, SCD | AD | HGMD | 0 | 0 | 0.002* | 0 | 0.00003 | 0.0006* | 0.0024* | 0.0005* | 0.002* | 0.0029 | 0.003 |
| KCNH2 | NM_000238.3 | c.2771G > A | rs199473009 | Long QT syndrome | 0.0005 | Syncope, SCD | AD or AR | HGMD | NA | NA | NA | NA | 0 | 0 | 0.0008 | 0 | NA | NA | 0.0012 |
| KCNJ5 | NM_000890.3 | c.1159G > C | rs199830292 | Long QT syndrome | 0.0005 | Syncope, SCD | AD or AR | HGMD/ClinVar | 0 | 0 | 0.001 | 0 | 0 | 0 | 0.0025 | 0 | 0.0015 | 0.0015 | 0.0029 |
| MYBPC3 | NM_000256.3 | c.1000G > A | rs573916965 | HCM | 0.002 | Syncope, SCD | AD | HGMD | 0.001 | 0 | 0.004 | 0 | 0.000008 | 0 | 0.0035 | 0 | 0.0043 | 0.0047 | 0.0026 |
| MYBPC3 | NM_000256.3 | c.2761C > G | rs367729718 | HCM | 0.002 | Syncope, SCD, heart failure | AD | HGMD | 0 | 0 | 0.006 | 0 | 0 | 0 | 0.0021 | 0.0007 | NA | 0.0011 | 0.0029 |
| MYH7 | NM_000257.3 | c.77C > T | rs186964570 | HCM | 0.002 | Syncope, SCD, heart failure | AD | HGMD | 0 | 0 | 0.0079 | 0 | 0.00004 | 0.00004 | 0.0068 | 0.0001 | 0.0039 | 0.0107 | 0.0046 |
| DSP | NM_004415.3 | c.7964C > G | rs193922671 | CM | 0.0002–0.0004 | Syncope, SCD, heart failure | AD or AR | ClinVar | NA | NA | NA | NA | 0 | 0 | 0.0008 | 0.0002 | NA | 0.0009 | 0.0019 |
| DMD | NM_004006.2 | c.748G > T | rs128626239 | Duchenne muscular dystrophy | 0.00015–0.0002 | Muscular dystrophy, scoliosis, intellectual disability | X-link | ClinVar | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 0.0003 |
AD = Autosomal dominant; AFR = African; AR = Autosomal recessive; CM: cardiomyopathy; EA = EAS = East Asian; EA = European American; EU = European; F = Finnish; HCM: Hypertrophic cardiomyopathy; HGMD = The Human Gene Mutation database; NA = not available; SA = South Asian; SAS = South Asian; SCD = sudden cardiac death.
As the strength of negative selection increases, we expect a greater fraction of very rare variants (FVRVs), defined the same as in previous work [17], which are not usually included on GWAS chips. In FVRV analyses, we only considered positions with a coverage of at least 30X. A detailed breakdown of FVRVs by variant annotation category is shown in Fig. 4. More than 40% of the total identified missense variants were FVRVs, which was the highest among all mutation types and genomic regions (Fig. 4a). In the Japanese population [17], the FVRV was highest in intergenic regions. This finding suggested that although Japanese and HC in Taiwan are both East Asian, the effect of negative selection is different in the two ethnic populations. In addition, we were surprised to find that variants with a greater impact on gene-encoded proteins tended to have a higher FVRV. Approximately 40–50% of the total identified variants predicted to be probably/possibly damaging by PolyPhen-2 or LOF variants by the Variant Effect Predictor were very rare variants (Fig. 4b).
Fig. 4.

(a) Fractions of very rare variants in different genomic locations in 1KTW-WGS. (b) Fractions of very rare variants with different predicted functions using in silico analysis in 1KTW-WGS. VEP, Variant Effect Predictor.
Genotyping consistency between 1KTW-WGS and a customized GWAS array and imputation performance
Since we used two platforms of NGS, we tested the genotyping consistency of each platform separately. Four hundred ninety-nine individuals underwent both WGS by Illumina and the customized GWAS array, whereas 498 individuals underwent both WGS by Ion Torrent-Proton and the customized GWAS array. There were 610,225 and 585,535 overlapping SNVs, and the consistency rates were 98.7% and 95.6%, respectively. We also compared 1KTW-WGS and the samples in 1000G (phase 3: EAS) for genotyping imputation. The highest R2 value (the measure of imputation accuracy) was achieved with 1KTW-WGS plus 1000G (phase 3: EAS), irrespective of a high or low MAF; the mean R2 values were 0.7 for rare SNVs (MAF 0.01–1%), 0.7–0.85 for low-frequency SNVs (MAF 1–5%), and 0.88–0.94 for common SNVs (MAF > 5%) (Supplementary Fig. 9). In addition, we compared the number of imputed SNVs using 1KTW-WGS and 1000G (phase 3: EAS) separately versus 1KTW-WGS plus 1000G (phase 3: EAS). In general, the combination of 1KTW-WGS plus 1000G (phase 3: EAS) could impute more variants, especially rare variants, than the use of 1KTW-WGS or 1000G (phase 3: EAS) alone (Supplementary Fig. 10). The significant improvement in imputation accuracy and the number of imputed SNVs using the 1KTW-WGS data suggest the importance of constructing and examining a population-specific reference panel.
Applying 1KTW-WGS sequencing data to the discovery of novel genetic variants associated with hyperlipidemia
The results of a LD analysis using the SNAP web site indicated that the nucleotide variants located within chr11:116264865–116432950 (GRCh 37/hg19) had R2 values higher than 0.5. Based on their chromosomal coordinates, 43 non-synonymous variants were reported from the WGS data in the TWB (Supplementary Table 8). To assess whether these variants have functional effects on their downstream proteins, three bioinformatics algorithms—SIFT, PROVEAN, and PolyPhen-2—were utilized. As shown in Supplementary Table 8, 5 of the 43 variants were predicted to be damaging by all three algorithms. Among them, three (chr11: 116633749; chr11: 116661392; chr11: 117128401) were novel, and two of them were reported as SNVs: rs200949753 in BUD13 and rs2075291 in APOA5. These variants were identified based on rs7115242, which was significantly associated with the dysregulation of lipids and lipoproteins in the HC population in our prior GWAS (P = 5.72*10−8, unpublished). In addition, we used the proportion test to examine the allele frequencies of the 5 damaging variants in the Asian and non-Asian groups. The results showed that one of the 5 variants, rs2075291, located in APOA5 which regulates triglyceride levels, was present at a significantly higher proportion in the Asian group (P < 0.001).
Identification of hypertension-related SNVs and development of a prediction model for hypertension
Because of the high genotyping consistency between 1KTW-WGS and the customized GWAS array and the large sample size of the customized GWAS array with high quality controls, we used the data from the customized GWAS array to identify hypertension-related SNVs and develop a prediction model for hypertension in the HC population. After quality control of genotyping data and excluding pre-hypertensive individuals, the GWAS analysis was performed on 10,678 unrelated individuals (2,936 hypertension patients and 7,742 non-hypertensive individuals) from the TWB, and 577,656 autosomal SNVs passed stringent quality controls. The Manhattan plot showed 1 region in chromosome 4 with genome-wide significance (P < 5 × 10−8, Supplementary Fig. 11). In this region, rs16998073 in the FGF5 gene was the most significant SNV (P = 2.16 × 10−10). Next, we utilized a genome-wide P value threshold (P < 5x10−5) to select significant SNVs (N = 48) for development of the prediction model (Supplementary Table 9). To avoid the issue of collinearity among the predictors, we used an LD pruning approach to filter out independent SNVs. The baseline prediction model (clinical model) was developed based on gender, alcohol consumption, family history, hyperlipidemia, BMI, glucose, triglyceride, and microalbumin. Besides using clinical variables to develop the clinical model, we further incorporated a polygenic risk score to develop an advanced model (combined clinical and genetic models). We selected 26 SNVs to construct a polygenic risk score after LD clumping of the 48 variants with P value < 5 × 10−5 (Supplementary Table 10). Receiver operating characteristic curve analysis of the clinical model and the advanced model are shown in Supplementary Fig. 12. The predictive performance of the advanced model was slightly better than the clinical model (area under curve (AUC) 0.87 vs. 0.85). In addition, the advanced model had slightly better predictive performance for hypertension in females than in males (AUC 0.887 vs. 0.858, Supplementary Table 11).
Comparison of allele frequency of pharmacogenetics-related SNVs identified in WGS databases across multiple populations
To demonstrate the clinical applications of 1KTW-WGS data, we have listed examples of clinical scenarios relevant to genetic variants in Table 4. Warfarin is a widely used oral anticoagulant for preventing or treating thromboembolism in patients with atrial fibrillation or a mechanical valve. It is a well-known clinical issue that warfarin displays large inter-individual and inter-ethnic differences in the dose required to achieve its anticoagulation effects [22], [23]. SNVs in the CYP2C9 gene (rs1057910, rs1799853) and the VKORC1 gene (rs9923231) have been reported to be associated with the dose requirement for warfarin [24], [25], [26]. Table 4 shows that the MAFs of these three SNVs in 1KTW-WGS were significantly higher or lower than those in Caucasians and African Americans. This genetic difference may be one of the reasons explaining the different dosage requirements for warfarin across populations (Supplementary Fig. 13).
Table 4.
Examples of pharmacogenetics in clinical scenarios and comparisons of minor allele frequency across different populations.
| Drug name | Gene | Genetic variants | Minor Allele Frequency |
Clinical application or impact | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | Alt. | 1KTWWGS | 1000GCHB | 1000GCHS | 1000G JPT |
gnomADEAS | gnomAD SAS |
1000GCEU | gnomAD non-Finish |
gnomAD AFR |
ChinaMap | ||||
| Warfarin | VKORC1 | rs9923231 | C | T | 0.865 | 0.956* | 0.890 | 0.903 | 0.901* | NA | 0.429* | 0.368* | 0.102* | NA | Weekly warfarin dosage |
| CYP2C9 | rs1057910 | A | C | 0.034 | 0.038 | 0.047 | 0.019 | 0.033 | 0.109* | 0.066 | 0.068* | 0.012* | 0.0459 | ||
| rs1057910 | A | G | NA | NA | NA | NA | NA | NA | NA | <0.000 | 0.00007 | NA | |||
| rs1799853 | C | T | 0.001 | <0.000 | 0.004 | <0.000 | 0.0004 | 0.047* | 0.152* | 0.127* | 0.021* | 0.0012 | |||
| Affects the metabolism of clopidogrel and the risk of coronary stenting thrombosis or cardiovascular events | |||||||||||||||
| Clopidogrel | CYP2C19*2 | rs4244285 | G | A | 0.348 | 0.335 | 0.352 | 0.3220 | 0.308* | 0.325 | 0.131* | 0.147* | 0.178* | 0.3117 | |
| rs3758580 | C | T | 0.305 | 0.335 | 0.352 | 0.3220 | 0.310 | 0.327 | 0.131 | 0.147* | 0.179* | 0.3121 | |||
| rs181297724 | G | C | 0.0048 | <0.000 | 0.004 | 0.009 | 0.004 | 0.00007* | <0.000 | 0.0012* | 0.00004* | 0.0053 | |||
| rs181297724 | G | A | NA | NA | NA | NA | NA | NA | NA | <0.000 | 0.00007 | NA | |||
| rs17878459 | G | C | <0.000 | <0.000 | <0.000 | <0.000 | <0.000 | 0.0078* | 0.0250 | 0.033 | 0.008 | 0.00024 | |||
| rs778258371 | G | A | <0.000 | NA | NA | NA | NA | NA | NA | <0.000 | <0.000 | 0.00005 | |||
| rs144036596 | G | A | NA | <0.000 | <0.000 | <0.000 | 0.0001 | 0.0001 | <0.000 | 0.0003 | 0.0002 | 0.00014 | |||
| rs144036596 | G | C | NA | NA | NA | NA | NA | NA | NA | 0.00005 | <0.000 | NA | |||
| rs144036596 | G | T | NA | NA | NA | NA | NA | NA | NA | 0.00001 | <0.000 | NA | |||
| rs550527959 | A | T | <0.000 | <0.000 | <0.000 | <0.000 | <0.000 | <0.000 | <0.000 | <0.000 | <0.000 | NA | |||
| CYP2C19*3 | rs4986893 | G | A | 0.055 | 0.044 | 0.048 | 0.072 | 0.063 | 0.004* | 0* | 0.00025* | 0.00038* | 0.0485 | ||
| rs763625282 | T | A | <0.000 | NA | NA | NA | NA | NA | NA | <0.000 | <0.000 | 0.0005 | |||
| rs144036596 | G | A | <0.000 | <0.000 | <0.000 | <0.000 | 0.00011 | 0.0001 | <0.000 | 0.00029 | 0.00021 | 0.00014 | |||
| rs144036596 | G | C | NA | NA | NA | NA | NA | NA | NA | 0.00005 | <0.000 | NA | |||
| rs144036596 | G | T | NA | NA | NA | NA | NA | NA | NA | 0.00001 | <0.000 | NA | |||
* Indicates P value < 0.01 compared with 1KTW-WGS; AFR: African/African American; CEU: European; CHB: Chinese-Bejing; CHS: Chinese-South; JPT: Japanese; EAS: East Asian; NA: not available; SAS: South Asian; Ref: reference; Alt: alternate; CHC: chronic hepatitis C; <0.000 means the actual number is not provided in the databases, but it should be less than < 0.000001.
Clopidogrel is the standard of care for patients receiving coronary stenting [27], [28], [29]. It becomes an active metabolite after processing by cytochrome P-450 (CYP) enzymes to achieve its antiplatelet effect. LOF alleles (*2 or *3) of the CYP2C19 gene have been reported to be associated with clopidogrel response and cardiac ischemic events in patients with coronary stenting or acute coronary syndrome [30], [31]. It is reported that the prevalence of the CYP2C19 LOF variants is 35% to 45% among blacks, and 25% to 35% in whites, whereas it is 55% to 70% among Asians. The prevalence of CYP2C19 poor metabolizers, which means individuals carrying 2 LOF alleles, is 5% among blacks and whites, whereas it is higher among Asians (10–20%). In addition to the CYP2C19*2 allele, 10% to 20% of Asians also carry another defective allele, CYP2C19*3 [32], [33], [34]. The differences in the MAFs of SNVs in CYP2C19*2 and CYP2C19*3 across different populations are shown in Table 4, showing substantial ethnic differences in the distribution and type of CYP2C19 LOF alleles.
A very recent project entitled 'ChinaMap' reported the genomes of 10,588 individuals (http://www.mbiobank.com/) [35]. We compared ChinaMap with the TWB, and found that in general, the results between them are very similar, and only a few loci showed minor differences. For example, rs199473009, associated with long QT syndrome, was not reported in ChinaMap but showed a MAF of 0.002 in the TWB (Table 3). Another SNV, rs4244285 in Table 4, had 35% frequency in the TWB but around 31% in ChinaMap. Thus, minor differences do exist between these two datasets.
Applying 1KTW-WGS to the investigation of the impact of ethnic genetic differences on the SCN5A gene and development of an online database
It is well known that SCN5A (cardiac voltage-gated sodium channel α-subunit) is an important cardiac gene that can cause several inheritable life-threatening arrhythmic diseases, including long QT syndrome, Brugada syndrome, and dilated cardiomyopathy [36], [37], [38], [39]. Mutations in SCN5A can in sudden cardiac death, especially in children or young adults. For physicians and patients in clinical practice, genetic testing for patients with inheritable arrhythmic diseases and family members is strongly recommended to obtain an early diagnosis, a treatment plan, and risk stratification [40]. The SCN5A gene is an example of the usefulness of 1KTW-WGS in clinical genetic testing. When a SCN5A variant is identified in a patient, a physician could use our online database to easily explore the potential functional impact of the identified SCN5A variant in a race-specific way. An online tool was developed to demonstrate this application of the 1KTW-WGS database.
A total of 1877 DNA variants in SCN5A were identified in 1KTW-WGS. A proportional test revealed that 349 variants showed significant differences in allele frequencies between the Asian and non-Asian groups (proportion test: P < 0.001), and among them, 191 variants had higher allele frequencies in the Asian group (Supplementary Table 12). Intriguingly, none of the 349 significant variants were reported in the HGMD database, suggesting that these significant SCN5A variants identified in healthy volunteers are not causal mutations in inheritable arrhythmic diseases or cardiomyopathy.
As shown in Supplementary Fig. 14, we developed an online database for SCN5A variants that accesses five datasets derived from different populations (Supplementary Table 1). Briefly, users can input the chromosomal coordinates of interest to query SCN5A variants (Supplementary Fig. 14a), and the database is able to output a spreadsheet including the genetic structure, allele frequency in different populations, and the pathogenic level in previous studies, if available (Supplementary Fig. 14b). A tutorial and an example of the use of the database are described in the Supplementary Note.
With advancements in high-throughput technology, such as microarrays and NGS, more SCN5A variants in Brugada syndrome are expected to be identified, and thus this SCN5A database can serve as an important reference system. Users can easily explore racial differences and the potential functional impact of identified SCN5A variants. Importantly, the results indicate that the development of a reference database in healthy general populations can facilitate the screening of important and pathogenic DNA variants associated with inheritable arrhythmic diseases or cardiomyopathy, since these variants seldom overlap between the 1KTW-WGS and HGMD databases.
Discussion
We have deeply sequenced the whole genomes of 997 HC individuals in Taiwan and constructed 1KTW-WGS, the first large reference database for the HC population. The results presented here reflect the wealth of knowledge that can be gleaned from WGS data, providing a comprehensive understanding of the genetic structure of the HC population and a basis to uncover associations between DNA variants and phenotypes and develop clinical applications. The observed proportion of novel variants that were not identified in the 1000G and gnomAD databases demonstrated the value of in-depth population-specific WGS studies in HC.
Large-scale investigation of human genetic variations suggested that recently increased human population growth has caused an excess of rare genetic variants, most of which possibly arose in the past 5,000–10,000 years [41]. It is reasonable to hypothesize that these rare variants are population-specific. As a result, it would be difficult to impute these variants from a reference database generated from diverse genetic backgrounds. Here, we showed that the number of imputed variants increased with the combined use of both a population-specific reference (1KTW-WGS data) and a non-specific reference (1000G phase 3). These results demonstrated that combining sequencing datasets within and across populations can maximize sensitivity and resolution for the discovery of DNA variants.
The density and frequency of variants in genes and genomic regions afford important information about the strength of natural selection acting on them [42], [43]. Because deleterious mutations generally disappear from populations faster than neutral mutations via natural selection, SNVs observed at lower frequencies in a population are indicative of natural selection. Furthermore, the strength of natural selection (purifying selection) differs among various functional genomic categories. We evaluated the relative influence of natural selection on SNVs of each functional category in the same way as previous large sequencing projects [41], [44], [45]. In 1KTW-WGS, we found that the non-synonymous FVRVs was higher in all categories than in the 1000G dataset [17]. Furthermore, the non-synonymous FVRVs in both 1KTW-WGS and 1000G was higher than the FVRVs of intergenic regions in both datasets, implying this region is under weak natural selection. However, the FVRVs in 3′-UTRs and introns in 1KTW-WGS was significantly higher than the FVRVs in intergenic regions, whereas the FVRVs in 3′-UTRs and introns in the 1000G dataset was lower than the FVRVs in intergenic regions [17]. This might be due to low coverage in these regions in the 1000G dataset rather than a signature of weak purifying selection in UTRs and introns. With regard to structural variants, the overall shape of the distribution for size-frequency showed that larger structural events are less frequent than smaller ones, presumably reflecting the relatively deleterious nature of larger structural changes. The difference between the length distributions of coding and noncoding indels is possibly the result of negative selection against frameshift indels [46], [47]. Overall, our results are consistent with previous reports [42] showing that variants with a greater impact on gene products tended to have a higher FVRVs, illustrating that these variants can be associated with diseases and could be useful in capturing causal variants in future studies.
In the 1KTW-WGS database, we observed that a relatively healthy individual carries an average of 16.42 disease-causing variants reported in HGMD. This raises the question of how any person remains disease-free. One possible explanation is that the existence of modifier alleles induces incomplete penetrance or variable expression of DMs, which is dependent on the carrier’s genetic background [48]. An alternative explanation is that HGMD contains a large number of false-positive DMs [49]. Of the 1139 DMs identified in 1KTW-WGS, 30% had a MAF greater than 1%, which is higher than the prevalence of many of the diseases described in HGMD. The majority of these mutations were common in 1KTW-WGS, suggesting that these variants are not subject to strong selective pressure and are likely phenotypically benign. In other words, given the inheritance patterns of the diseases conferred by these variants, many individuals in 1KTW-WGS should have been affected by diseases with profound physical or structural cardiac abnormalities or lethal arrhythmias (Table 3). For example, one of these variants (MYH7 c.77C > T, rs186964570; T variant for hypertrophic cardiomyopathy) was reported as a DM in HGMD. The prevalence of hypertrophic cardiomyopathy (MIM 192600), an autosomal dominant disease, is estimated to be 0.2%. However, some unrelated 1KTW-WGS individuals were heterozygous carriers of this variant (prevalence = 0.46%, ~2.3-fold higher than the disease prevalence). Thus, establishing population-specific WGS reference datasets (null expectation) is crucial in defining guidelines for investigating the causality of variants [50]. This observation also emphasizes the need for caution in assigning pathogenicity to variants purely based on in silico predicted impact on protein structure, which is widely used in clinical genetic testing for patients with inherited diseases.
Of the hypertension-associated regions with genome-wide significance in this study, rs16998073 in FGF5 was the most significant SNV in Taiwan’s HC population. In a previous meta-analysis focusing on 4 SNVs, rs16998073 was significantly associated with hypertension risk in East Asians [51], consistent with our finding. This suggests that rs16998073 may be used as a predictive marker for hypertension in clinical practice. On the other hand, we found that adding a weighted genetic risk score into the prediction model generated by clinical variables only slightly improved the prediction of hypertension, implying that clinical risk factors such as age and BMI have a stronger predictive power for hypertension than genetic variants. This is consistent with previous studies [52], [53]. We also identified rs2075291, which had a significantly higher frequency in the Asian population. This variant is located in APOA5, which regulates triglyceride levels, suggesting that it may play a role in the regulation of lipid biosynthesis. Furthermore, previous studies have demonstrated that abnormal lipid expression levels are an important risk factor in early-onset breast cancer patients [54], [55]. Our previous study also showed that deletion of the APOA1/C3/A4/A5 gene cluster occurred in approximately 30% of breast cancer patients in Taiwan (data not shown). Taken together, the WGS data from the TWB further revealed three novel SNVs that might be potential genetic regulators of lipid biosynthesis.
There were limitations in this study. First, we used two NGS platforms (Illumina and Ion Torrent-Proton) in the TWB, which might cause batch effects in the data. To address this issue, we performed joint calling and selected the variants identified in common by both Illumina and Ion Torrent-Proton platforms to do the comparisons. The difference in MAF was calculated by subtracting the MAF in the Illumina platform from the MAF in the Ion Torrent-Proton platform. The results are illustrated in Supplementary Fig. 15. Notably, no major differences were observed, suggesting the reproducibility of the two platforms to do the variant analysis. Similar to the MAF difference analysis, we selected the variants identified in common to both platforms for the PCA plot. The first principal component explained about 20% of the variance and adequately distinguished the two platforms (Supplementary Fig. 16). Additionally, we did PCA of the samples analyzed in the TWB and the 1000G CHB population. The results are illustrated in Supplementary Fig. 17. Notably, the plot showed a triangle shape, which suggested that some batch effects exist among the 3 different platforms. However, the values of the first and second principal components are only 8.2% and 5.08%. Therefore, the differences are not very large and the results across the 3 different platforms could be reliably compared each other. Second, although 1KTW-WGS was established, population-wide sequencing with a larger sample size (e.g., > 100 K) is warranted to discover the full spectrum of SNVs and structural variants. Third, all participants who were enrolled in TWB were volunteers. They were not randomly sampled from the HC population. Lastly, in our study, Torrent Variant Caller (TVC) was utilized for Ion Torrent-Proton data, whereas iSAAC Variant Caller was used for Illumina data. No study has directly compared the callers of the two types of the data, but a previous study [56] suggested that the consistency of different callers used with Illumina is up to 91.7%, whereas the variations of the different callers used with Ion Torrent-Proton data ranged from 1.3% to 34.6%. Furthermore, some previous reports indicated that the false discovery rate of SNVs and indels was relatively higher in the Ion Torrent-Proton system than in the Illumina system [57], [58]. In addition, several studies suggested that higher false positive rates were observed when using the Ion Torrent-Proton system versus the Illumina system [59], [60], [61]. One plausible explanation for this is that the data from the Illumina system have better sequencing quality and stable read length. These advantages give the Illumina system better performance in identification of rare variants because of better alignment quality as well as more uniform and consistent reads.
Conclusions
In this study, we used the 1KTW-WGS reference database to discover novel HC population-specific variants, identified novel hyperlipidemia-related variants, developed a hypertension prediction model, and established an online SCN5A tool for genetic testing. Taken together, the results demonstrate the necessity of constructing a population-specific genomic reference, which can pave the way for precision medicine and population health initiatives.
The cost of WGS is gradually decreasing. Thus, we expect that a more comprehensive interrogation of genetic variants will advance basic research and support development of diagnostic tools, healthcare customization, and preventive therapeutics for human diseases.
Acknowledgments
Acknowledgments
We sincerely thank all staff related to or working in the TWB, Academia Sinica, Taipei, Taiwan, for establishing the biobank. Financial support for this research was partially provided through grants from the Ministry of Science and Technology, Taiwan (grant numbers: MOST 104-2314-B-002-193-MY3, MOST 106-2314-B-002-047-MY3, MOST 106-2314-B-002-134-MY2, MOST 106-2314-B-002-206, MOST 107-2314-B-002-009, MOST 108-2314-B-002-007 and MOST 107-2314-B-002-261-MY3), Taiwan Health foundation, National Taiwan University Hospital (grant numbers: NTUH 105-S3077, NTUH-105-S2995, NTUH-UN105-012, NTUH 106-S3469, NTUH 106-S3458, and NTUH 106-018), and National Taiwan University (grant number: GTZ300). We are also grateful to the staff of the Sixth Core Lab, Department of Medical Research, National Taiwan University Hospital, for technical support.
Competing financial interests
The authors declare that they have no competing interests.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2020.12.003.
Contributor Information
Eric Y. Chuang, Email: jjmjuang@ntuh.gov.tw.
Chen-Yang Shen, Email: bmcys@ibms.sinica.edu.tw.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Executive Yuan tRoCT. The Republic of China Yearbook; 2016.
- 2.Executive Yuan tRoCT. The Republic of China Yearbook; 2013.
- 3.Chen C.H., Yang J.H., Chiang C.W.K., Hsiung C.N., Wu P.E., Chang L.C. Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project. Hum Mol Genet. 2016;25(24):5321–5331. doi: 10.1093/hmg/ddw346. PubMed PMID: 27798100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559-75. Epub 2007/08/19. doi: 10.1086/519795. PubMed PMID: 17701901; PubMed Central PMCID: PMC1950838. [DOI] [PMC free article] [PubMed]
- 5.Browning B.L., Browning S.R. Genotype imputation with millions of reference samples. Am J Hum Genet. 2016;98(1):116–126. doi: 10.1016/j.ajhg.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O'Donnell C.J., de Bakker P.I. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24(24):2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68-74. doi: 10.1038/nature15393. PubMed PMID: 26432245; PubMed Central PMCID: PMCPMC4750478. [DOI] [PMC free article] [PubMed]
- 8.Kawakami T., Backstrom N., Burri R., Husby A., Olason P., Rice A.M. Estimation of linkage disequilibrium and interspecific gene flow in Ficedula flycatchers by a newly developed 50k single-nucleotide polymorphism array. Mol Ecol Resour. 2014;14(6):1248–1260. doi: 10.1111/1755-0998.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Bakker P.I., Yelensky R., Pe'er I., Gabriel S.B., Daly M.J., Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37(11):1217–1223. doi: 10.1038/ng1669. PubMed PMID: 16244653. [DOI] [PubMed] [Google Scholar]
- 10.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. PubMed PMID: 15297300. [DOI] [PubMed] [Google Scholar]
- 11.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. PubMed PMID: 19561590. [DOI] [PubMed] [Google Scholar]
- 12.Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7(10) doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics. 2013;Chapter 7:Unit7 20. doi: 10.1002/0471142905.hg0720s76. PubMed PMID: 23315928; PubMed Central PMCID: PMC4480630. [DOI] [PMC free article] [PubMed]
- 14.Stenson P.D., Mort M., Ball E.V., Evans K., Hayden M., Heywood S. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862-8. Epub 2015/11/20. doi: 10.1093/nar/gkv1222. PubMed PMID: 26582918; PubMed Central PMCID: PMCPMC4702865. [DOI] [PMC free article] [PubMed]
- 16.Rcggp R. Aritmie cardiache rare in eta pediatrica. Clin Pediatr. 1963;45:656–683. [PubMed] [Google Scholar]
- 17.Nagasaki M., Yasuda J., Katsuoka F., Nariai N., Kojima K., Kawai Y. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun. 2015;6:8018. doi: 10.1038/ncomms9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-scale whole-genome sequencing of the Icelandic population. Nature genetics. 2015;47(5):435-44. Epub 2015/03/26. doi: 10.1038/ng.3247. PubMed PMID: 25807286. [DOI] [PubMed]
- 19.Yeo ZX, Wong JC, Rozen SG, Lee AS. Evaluation and optimisation of indel detection workflows for ion torrent sequencing of the BRCA1 and BRCA2 genes. BMC genomics. 2014;15:516. Epub 2014/06/26. doi: 10.1186/1471-2164-15-516. PubMed PMID: 24962530; PubMed Central PMCID: PMCPMC4079958. [DOI] [PMC free article] [PubMed]
- 20.Yeo ZX, Chan M, Yap YS, Ang P, Rozen S, Lee AS. Improving indel detection specificity of the Ion Torrent PGM benchtop sequencer. PLoS One. 2012;7(9):e45798. Epub 2012/10/03. doi: 10.1371/journal.pone.0045798. PubMed PMID: 23029247; PubMed Central PMCID: PMCPMC3446914. [DOI] [PMC free article] [PubMed]
- 21.McKusick V.A. Mendelian Inheritance in Man and its online version. OMIM. Am J Human Genet. 2007;80(4):588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi H., Wilkinson G.R., Caraco Y., Muszkat M., Kim R.B., Kashima T. Population differences in S-warfarin metabolism between CYP2C9 genotype-matched Caucasian and Japanese patients. Clin Pharmacol Ther. 2003;73(3):253–263. doi: 10.1067/mcp.2003.26a. PubMed PMID: 12621390. [DOI] [PubMed] [Google Scholar]
- 23.Xie H.G., Kim R.B., Wood A.J., Stein C.M. Molecular basis of ethnic differences in drug disposition and response. Annu Rev Pharmacol Toxicol. 2001;41:815–850. doi: 10.1146/annurev.pharmtox.41.1.815. PubMed PMID: 11264478. [DOI] [PubMed] [Google Scholar]
- 24.Schwarz U.I., Ritchie M.D., Bradford Y., Li C., Dudek S.M., Frye-Anderson A. Genetic determinants of response to warfarin during initial anticoagulation. New Engl J Med. 2008;358(10):999–1008. doi: 10.1056/NEJMoa0708078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nunnelee JD. Review of an Article: The international Warfarin Pharmacogenetics Consortium (2009). Estimation of the warfarin dose with clinical and pharmacogenetic data. NEJM 360 (8): 753-64. J Vasc Nurs. 2009;27(4):109. doi: 10.1016/j.jvn.2009.09.001. PubMed PMID: 19914573. [DOI] [PubMed]
- 26.Rieder M.J., Reiner A.P., Gage B.F., Nickerson D.A., Eby C.S., McLeod H.L. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. New Engl J Med. 2005;352(22):2285–2293. doi: 10.1056/NEJMoa044503. PubMed PMID: 15930419. [DOI] [PubMed] [Google Scholar]
- 27.Yusuf S., Zhao F., Mehta S.R., Chrolavicius S., Tognoni G., Fox K.K. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. New Engl J Med. 2001;345(7):494–502. doi: 10.1056/NEJMoa010746. PubMed PMID: 11519503. [DOI] [PubMed] [Google Scholar]
- 28.Levine G.N., Bates E.R., Blankenship J.C., Bailey S.R., Bittl J.A., Cercek B. 2015 ACC/AHA/SCAI Focused Update on Primary Percutaneous Coronary Intervention for Patients With ST-Elevation Myocardial Infarction: An Update of the 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention and the 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction. J Am Coll Cardiol. 2016;67(10):1235–1250. doi: 10.1016/j.jacc.2015.10.005. PubMed PMID: 26498666. [DOI] [PubMed] [Google Scholar]
- 29.Levine GN, Bates ER, Bittl JA, Brindis RG, Fihn SD, Fleisher LA, et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines: An Update of the 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention, 2011 ACCF/AHA Guideline for Coronary Artery Bypass Graft Surgery, 2012 ACC/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the Diagnosis and Management of Patients With Stable Ischemic Heart Disease, 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction, 2014 AHA/ACC Guideline for the Management of Patients With Non-ST-Elevation Acute Coronary Syndromes, and 2014 ACC/AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery. Circulation. 2016;134(10):e123-55. Epub 2016/03/31. doi: 10.1161/cir.0000000000000404. PubMed PMID: 27026020. [DOI] [PubMed]
- 30.Shuldiner A.R., O'Connell J.R., Bliden K.P., Gandhi A., Ryan K., Horenstein R.B. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA. 2009;302(8):849–857. doi: 10.1001/jama.2009.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mega J.L., Simon T., Collet J.P., Anderson J.L., Antman E.M., Bliden K. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA. 2010;304(16):1821–1830. doi: 10.1001/jama.2010.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Man M., Farmen M., Dumaual C., Teng C.H., Moser B., Irie S. Genetic variation in metabolizing enzyme and transporter genes: comprehensive assessment in 3 major East Asian subpopulations with comparison to Caucasians and Africans. J Clin Pharmacol. 2010;50(8):929–940. doi: 10.1177/0091270009355161. PubMed PMID: 20173083. [DOI] [PubMed] [Google Scholar]
- 33.Lee J.M., Park S., Shin D.J., Choi D., Shim C.Y., Ko Y.G. Relation of genetic polymorphisms in the cytochrome P450 gene with clopidogrel resistance after drug-eluting stent implantation in Koreans. The Am J Cardiol. 2009;104(1):46–51. doi: 10.1016/j.amjcard.2009.02.045. PubMed PMID: 19576320. [DOI] [PubMed] [Google Scholar]
- 34.Hwang S.J., Jeong Y.H., Kim I.S., Koh J.S., Kang M.K., Park Y. The cytochrome 2C19*2 and *3 alleles attenuate response to clopidogrel similarly in East Asian patients undergoing elective percutaneous coronary intervention. Thromb Res. 2011;127(1):23–28. doi: 10.1016/j.thromres.2010.10.021. PubMed PMID: 21075428. [DOI] [PubMed] [Google Scholar]
- 35.Cao Y., Li L., Xu M., Feng Z., Sun X., Lu J. The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals. Cell Res. 2020 doi: 10.1038/s41422-020-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q., Shen J., Splawski I., Atkinson D., Li Z., Robinson J.L. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80(5):805–811. doi: 10.1016/0092-8674(95)90359-3. PubMed PMID: 7889574. [DOI] [PubMed] [Google Scholar]
- 37.Baruteau AE, Kyndt F, Behr ER, Vink AS, Lachaud M, Joong A, et al. SCN5A mutations in 442 neonates and children: genotype-phenotype correlation and identification of higher-risk subgroups. Eur Heart J. 2018;39(31):2879-87. Epub 2018/07/31. doi: 10.1093/eurheartj/ehy412. PubMed PMID: 30059973. [DOI] [PubMed]
- 38.Juang J.J., Horie M. Genetics of Brugada syndrome. J Arrhythm. 2016;32(5):418–425. doi: 10.1016/j.joa.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Remme CA, Verkerk AO, Nuyens D, van Ginneken AC, van Brunschot S, Belterman CN, et al. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation. 2006;114(24):2584-94. Epub 2006/12/06. doi: 10.1161/CIRCULATIONAHA.106.653949. PubMed PMID: 17145985. [DOI] [PubMed]
- 40.Priori S.G., Wilde A.A., Horie M., Cho Y., Behr E.R., Berul C. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace. 2013;15(10):1389–1406. doi: 10.1093/europace/eut272. PubMed PMID: 23994779. [DOI] [PubMed] [Google Scholar]
- 41.Fu W., O'Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493(7431):216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khurana E., Fu Y., Colonna V., Mu X.J., Kang H.M., Lappalainen T. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science. 2013;342(6154):1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrovski S., Wang Q., Heinzen E.L., Allen A.S., Goldstein D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9(8) doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buchanan C.C., Torstenson E.S., Bush W.S., Ritchie M.D. A comparison of cataloged variation between International HapMap Consortium and 1000 Genomes Project data. J Am Med Inform Assoc. 2012;19(2):289–294. doi: 10.1136/amiajnl-2011-000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knetsch CW, Connor TR, Mutreja A, van Dorp SM, Sanders IM, Browne HP, et al. Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the Netherlands, 2002 to 2011. Euro Surveill. 2014;19(45):20954. PubMed PMID: 25411691; PubMed Central PMCID: PMCPMC4518193. [DOI] [PMC free article] [PubMed]
- 46.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eilbeck K., Lewis S.E., Mungall C.J., Yandell M., Stein L., Durbin R. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6(5):R44. doi: 10.1186/gb-2005-6-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper D.N., Krawczak M., Polychronakos C., Tyler-Smith C., Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132(10):1077–1130. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cassa C.A., Tong M.Y., Jordan D.M. Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum Mutat. 2013;34(9):1216–1220. doi: 10.1002/humu.22375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508(7497):469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xi B, Shen Y, Reilly KH, Wang X, Mi J. Recapitulation of four hypertension susceptibility genes (CSK, CYP17A1, MTHFR, and FGF5) in East Asians. Metabolism: clinical and experimental. 2013;62(2):196-203. Epub 2012/09/11. doi: 10.1016/j.metabol.2012.07.008. PubMed PMID: 22959498. [DOI] [PubMed]
- 52.Sun D, Liu J, Xiao L, Liu Y, Wang Z, Li C, et al. Recent development of risk-prediction models for incident hypertension: An updated systematic review. PloS one. 2017;12(10):e0187240. Epub 2017/10/31. doi: 10.1371/journal.pone.0187240. PubMed PMID: 29084293; PubMed Central PMCID: PMCPMC5662179. [DOI] [PMC free article] [PubMed]
- 53.Izawa H, Yamada Y, Okada T, Tanaka M, Hirayama H, Yokota M. Prediction of Genetic Risk for Hypertension. Hypertension. 2003;41(5):1035-40. doi: doi:10.1161/01.HYP.0000065618.56368.24. [DOI] [PubMed]
- 54.Laisupasin P., Thompat W., Sukarayodhin S., Sornprom A., Sudjaroen Y. Comparison of serum lipid profiles between normal controls and breast cancer patients. J Lab Phys. 2013;5(1):38–41. doi: 10.4103/0974-2727.115934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang S.J., Hou M.F., Tsai S.M., Wu S.H., Hou L.A., Ma H. The association between lipid profiles and breast cancer among Taiwanese women. Clin Chem Lab Med. 2007;45(9):1219–1223. doi: 10.1515/CCLM.2007.263. PubMed PMID: 17663634. [DOI] [PubMed] [Google Scholar]
- 56.Hwang S., Kim E., Lee I., Marcotte E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep. 2015;5:17875. doi: 10.1038/srep17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cooper G.M., Goode D.L., Ng S.B., Sidow A., Bamshad M.J., Shendure J. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods. 2010;7(4):250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bragg L.M., Stone G., Butler M.K., Hugenholtz P., Tyson G.W. Shining a light on dark sequencing: characterising errors in Ion Torrent PGM data. PLoS Comput Biol. 2013;9(4) doi: 10.1371/journal.pcbi.1003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quail M.A., Smith M., Coupland P., Otto T.D., Harris S.R., Connor T.R. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kleftogiannis D., Punta M., Jayaram A., Sandhu S., Wong S.Q., Gasi Tandefelt D. Identification of single nucleotide variants using position-specific error estimation in deep sequencing data. BMC Med Genomics. 2019;12(1):115. doi: 10.1186/s12920-019-0557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The URL for the online database for SCN5A variants is http://140.112.136.14:8000. The 1KTW-WGS database is open to the public (the Taiwan Biobank website is https://taiwanview.twbiobank.org.tw/browse38).