

A scaffold-mediated system was developed to enhance the local delivery of Cas9 complexes for targeting leukemia stem cells in AML.
Abstract
Leukemia stem cells (LSCs) sustain the disease and contribute to relapse in acute myeloid leukemia (AML). Therapies that ablate LSCs may increase the chance of eliminating this cancer in patients. To this end, we used a bioreducible lipidoid-encapsulated Cas9/single guide RNA (sgRNA) ribonucleoprotein [lipidoid nanoparticle (LNP)–Cas9 RNP] to target the critical gene interleukin-1 receptor accessory protein (IL1RAP) in human LSCs. To enhance LSC targeting, we loaded LNP-Cas9 RNP and the chemokine CXCL12α onto mesenchymal stem cell membrane–coated nanofibril (MSCM-NF) scaffolds mimicking the bone marrow microenvironment. In vitro, CXCL12α release induced migration of LSCs to the scaffolds, and LNP-Cas9 RNP induced efficient gene editing. IL1RAP knockout reduced LSC colony-forming capacity and leukemic burden. Scaffold-based delivery increased the retention time of LNP-Cas9 in the bone marrow cavity. Overall, sustained local delivery of Cas9/IL1RAP sgRNA via CXCL12α-loaded LNP/MSCM-NF scaffolds provides an effective strategy for attenuating LSC growth to improve AML therapy.
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy characterized by aberrant proliferation and impaired differentiation and is the leading type of acute leukemia in adults, accounting for the most deaths of all leukemia subtypes in the United States (1–3). With a 28% 5-year survival rate, AML is associated with a poor prognosis and a high relapse rate, although most patients achieve remission following upfront chemotherapy (3, 4). Recent studies have demonstrated the importance of preexisting leukemia stem cells (LSCs) that have an inherent resistance to chemotherapy (5, 6). These LSCs have self-renewal and are quiescent in most cases—characteristics that enable them to drive AML initiation, progression, and relapse (7, 8). LSC gene expression signatures have been shown to predict clinical outcomes of AML, establishing a crucial need to address LSC populations to improve AML outcomes (9, 10).
Chronic inflammation of the bone marrow has been associated with progression of myeloid malignancies (11). Interleukin-1 receptor accessory protein (IL1RAP) is a necessary co-receptor in the interleukin-1 (IL-1) proinflammatory signaling pathway (12). IL1RAP is consistently overexpressed in multiple genetic subtypes of AML stem cells and progenitor cells (13–15) while being minimally expressed in normal hematopoietic stem and progenitor cells (HSPCs) (13–15). Overexpression of IL1RAP has also been found in stem and progenitor cells in other disease types including chronic myeloid leukemia (16, 17) and high-risk myelodysplastic syndromes (13), suggesting its critical role in myeloid malignancies. The importance of IL1RAP extends beyond IL-1 signaling as IL1RAP also interacts with the receptor tyrosine kinases FLT3 (fms-like tyrosine kinase 3) and c-KIT (mast/stem cell growth factor receptor kit) to promote AML progression (18). Targeting IL1RAP is therefore a promising approach to treat AML as it could disrupt multiple downstream oncogenic pathways.
The CRISPR system has demonstrated unprecedented potential for treating cancer and genetic diseases through gene editing (19). Cas9 endonuclease is currently the most widely used CRISPR-associated nuclease (20). A safe and efficient CRISPR-Cas9 delivery platform is needed to exploit the potential of gene editing therapy to treat AML. Numerous approaches have been developed to deliver CRISPR-Cas9 components (DNA, mRNA, and protein), but these approaches suffer from several limitations (21). Viral delivery is the most commonly used methodology for delivering CRISPR-Cas9 vectors in vivo. Although viral plasmid delivery using adeno-associated viruses is effective and does not result in genomic integration, its use is limited by off-target effects and the need for multiple viruses to deliver all of the components, which can reduce gene editing efficiency (22–24). Nonviral delivery systems have been developed to circumvent these limitations (21). Nanoparticle-based delivery of the CRISPR-Cas9 ribonucleoprotein (RNP) complex achieves high delivery efficiency, no risk of genomic integration, and transient gene regulation with low off-target effects (25). Of the various nanoparticle-based delivery carriers, lipidoid nanoparticles (LNPs) are attractive due to their proven effectiveness for gene and protein delivery in vitro and in vivo (26–28). Recently, Wang et al. (29) demonstrated the use of bioreducible LNPs to deliver CRISPR-Cas9 RNPs for gene recombination in vivo. These LNPs accelerated the endosomal release of encapsulated Cas9 RNP complexes, as the nanoparticles rapidly collapse in the reducing environment, allowing more efficient gene editing with low off-target effects (21). LNPs have shown high-efficiency Cas9 RNP delivery and genome editing in several cell types, but their application to gene editing to treat acute leukemia has not been examined.
A limitation of in vivo CRISPR-Cas9 gene editing is the low efficiency of targeted gene editing following systemic administration (30, 31). To improve the tissue specificity of systemic CRISPR-Cas9 delivery, local activation of genome editing has been achieved by applying external stimuli such as magnetic fields (32), optical radiation (33), and chemical cues (34). However, these systems often require multiple dosing due to their inability to generate therapeutic levels of genome editing at the target site. Furthermore, the difficulty of controlling systemic dissemination can lead to high genotoxicity and undesirable off-target effects. Therefore, a local CRISPR-Cas9 delivery system is desirable for genome editing within a specific target tissue or organ. To address these issues, we propose an innovative scaffold-mediated CRISPR-Cas9 delivery system by immobilizing CRISPR-Cas9 complexes on the surface of an injectable scaffold for local administration. The surface-immobilized CRISPR-Cas9 complexes would facilitate interaction of host cells and improve the delivery to target cells. Furthermore, the complexes would stay longer at the injection site due to attachment to the scaffold as compared to free complexes, leading to a more sustained delivery of CRISPR-Cas9 complexes.
Here, we develop a local CRISPR-Cas9 delivery system that targets LSCs in bone marrow and achieves sustained delivery of Cas9/IL1RAP single guide RNA (sgRNA) to improve gene editing efficiency and therapeutic efficacy (Fig. 1). The system consists of injectable, bioreducible lipidoid-encapsulated Cas9/sgRNA RNP loaded together with the chemoattractant CXCL12α onto mesenchymal stem cell membrane–coated nanofibril (MSCM-NF) that mimics the bone marrow environment. We characterize and optimize the delivery system in terms of CXCL12α release, LSC migration, and gene editing efficiency in vitro and apply the system to evaluate the therapeutic efficacy of IL1RAP gene editing in LSCs in a xenotransplantation assay in vivo.
Fig. 1. Schematic of the Cas9 RNP delivery system.
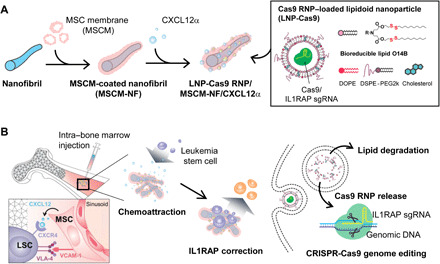
(A) Lipidoid nanoparticle (LNP)–encapsulated Cas9 RNP delivery system. PCL nanofibril (NF) that mimic the bone tissue environment were coated with mesenchymal stem cell membrane (MSCM) and were loaded with CXCL12α cytokine and LNP-coated Cas9 RNP. (B) The LNP-Cas9 RNP/MSCM-NF/CXCL12α complex can be injected into the bone marrow cavity to induce chemoattraction of leukemia blasts or LSCs, to more effectively deliver the gene editing cargo to these cells.
RESULTS
Preparation and characterization of LNPs
Lipidoid nanoparticles have been shown to be efficient carrier systems for delivering Cas9/sgRNA complexes into cells (29, 35, 36). To optimize an LNP system that targets LSCs, we synthesized and tested eight bioreducible lipids. We synthesized a single 14-carbon hydrophobic tail (O-14B) containing disulfide bonds and then reacted this bioreducible acrylate tail with eight different head groups (lipid library numbers: 76, 77, 80, 87, 123, 306, 400, and 401) (Fig. 2A and fig. S1, A and B). LNPs were formulated by combining each lipid with cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2k) at a 16:4:1:1 weight ratio as described previously (29, 35, 36). LNP size, polydispersity index (PDI), and zeta potential were measured by dynamic light scattering (fig. S1C). The LNPs had a positive surface charge (42.1 to 58.1 mV) and had diameters of 73.3 to 162.2 nm. We previously confirmed the structural integrity and long-term storage stability of blank and Cas9/sgRNA RNP complex-loaded LNPs (35, 36). Next, we tested the gene editing efficiency of the LNPs in leukemia cells. Cas9/IL1RAP sgRNA complexes (Cas9 RNP) were encapsulated by LNPs by self-assembly and were used to treat THP-1 cells, a human leukemic cell line that expresses IL1RAP. After 48 hours of treatment, Cas9 RNP complexed with the cationic LNPs 76-O14B (L76) and 77-O14B (L77) showed the highest gene editing efficiency in leukemia cells (fig. S1D) and were selected for further studies.
Fig. 2. Synthesis and characterization of Cas9-loaded lipidoid nanoparticles (LNP-Cas9) and MSCM-NF.
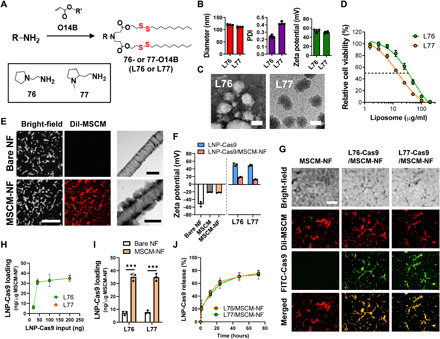
(A) Synthesis of bioreducible lipids 76-O14B and 77-O14B. (B) Size, PDI, and zeta potential of blank LNPs, measured by dynamic light scattering. (C) TEM images of LNP-Cas9 nanoparticles L76-Cas9 and L77-Cas9. Scale bars, 100 nm. (D) Cytotoxicity of blank LNPs L76 and L77. Cell viability was measured using CellTiter-Glo after 24 hours of treatment. (E) (Left) CLSM images of bare NF and MSCM-NF. MSCM was stained with DiI dye (red) to show colocalization of NF and MSCM. Scale bar, 50 μm. (Right) TEM images of bare NF and MSCM-NF. Scale bars, 500 nm. (F) Zeta potential of free LNP-Cas9 or LNP-Cas9 on MSCM-NF. (G) CLSM images showing colocalization of FITC-LNP-Cas9 and DiI-MSCM-NF. Scale bar, 50 μm. (H) Loading efficiency profile of LNP-Cas9 on MSCM-NF as a function of the amount of LNP-Cas9 added. (I) LNP-Cas9 loading of bare NF or MSCM-NF as 200 ng of LNP-Cas9 was added. (J) Release profile and efficiency of LNP-Cas9 on MSCM-NF. Data are means ± SD. ***P < 0.0001; Student’s t test.
L76 has a 2-(pyrrolidin-1-yl)ethan-1-amine head group, and L77 has a 2-(1-methylpyrrolidin-2-yl)ethan-1-amine head group. Blank L76 and L77 have a similar size (120.2 and 111.2 nm, with PDIs of 0.2 and 0.4, respectively) and surface charge (53.8 and 50.7 mV) (Fig. 2B). L76 and L77 complexed with Cas9 proteins (L76-Cas9 and L77-Cas9) exhibited spherical morphologies (Fig. 2C), consistent with our previous studies (29, 35, 36). To test the cytotoxicity of unloaded L76 and L77 to leukemia cells, THP-1 cells were treated with blank LNPs and cell viability was analyzed after 24 hours of treatment. The median inhibitory concentration (IC50) of L76 was 35.56 μg/ml (R2 = 0.9938) and the IC50 of L77 was 15.83 μg/ml (R2 = 0.9965) (Fig. 2D).
Optimization of LNPs L76 and L77 for Cas9/sgRNA delivery in leukemia cells
To optimize the Cas9-loaded LNP system, we prepared LNP-Cas9 RNP complexes using different LNP-to-Cas9 weight ratios (4:1, 6:1, 8:1, and 10:1) and evaluated their gene editing efficiency and cytotoxicity to leukemia cells. We found that an 8:1 LNP-to-Cas9 ratio (32 μg/ml of LNPs and 4 μg/ml of Cas9 proteins) achieved the best gene editing efficiency (18.2% for L76 and 18.1% for L77), comparable to that of Lipofectamine CRISPRMAX for L76 (18.6%) (fig. S2A). THP-1 cells treated with L76- or L77-Cas9 RNP remained ~50% viable after 24 hours of treatment at the 8:1 weight ratio (fig. S2B). Therefore, an 8:1 ratio of LNP to Cas9 (at an LNP concentration of 32 μg/ml) was selected for use in subsequent studies.
Preparation of MSCM-NF
Nanofibril (NF) was prepared by fragmenting electrospun poly(ε-caprolactone) (PCL) nanofibers as we described previously (37). Briefly, PCL nanofibers were torn apart in a blender and were subsequently hydrolyzed in alkaline solution. The NF was rod-shaped with a diameter of 1.0 ± 0.2 μm and a length of 22.0 ± 10.1 μm (fig. S3). After fragmentation, the nanofiber surface was rough with grooves due to the harsh hydrolysis of the PCL backbone.
Cell membrane–coated biomaterials mimic the physicochemical properties of the cell type used for coating and can improve the targeting efficiency of nanoparticles and reduce immunogenicity in vivo (38). To increase the affinity of LSCs for the NF, we cloaked the NF in MSCM. Bone marrow stromal cells (BMSCs) including MSCs provide pivotal microenvironmental components that support LSC survival (39). BMSCs secrete C-X-C motif chemokine 12 (CXCL12), which is a ligand of C-X-C chemokine receptor 4 (CXCR4). Secretion of CXCL12 contributes to LSC migration and homing to the bone marrow niche (39–41). Leukemia cells also highly express very late antigen-4 (VLA-4), a cell surface ligand for vascular cell adhesion molecule-1 (VCAM-1) on BMSCs. The VLA-4/VCAM interaction facilitates the adhesion of AML cells to their niche (42). We performed flow cytometry analysis to determine the expression levels of these microenvironmental components in leukemia THP-1 cells and bone marrow–derived MSCs (fig. S4). We confirmed that VLA-4 was highly expressed on THP-1 cells and stromal ligand VCAM-1 was present on MSCs (fig. S4A). In addition, we demonstrated that surface CXCR4 was expressed on THP-1 cells (fig. S4B).
Bone marrow–derived MSCM for coating NF was prepared as described previously (43–45). After hypotonic lysis of cultured MSCs, the cell membranes were collected by ultracentrifugation and were sonicated to form cell membrane–derived vesicles. The MSCM was labeled with the lipophilic carbocyanine dye DiI, and the NF was mixed with the vesicles after sonication, resulting in coating of NF with membrane. Fluorescence signal from the membranes was distributed evenly on the NF (Fig. 2E) and was retained on NF for 2 weeks (fig. S5), indicating a stable cell membrane coating. Transmission electron microscopy (TEM) imaging showed a thin (173.8 ± 46.4 nm) membrane layer on the NF surface. The surface zeta potential of the MSCM-NF was similar to that of MSCM (Fig. 2F). Together, these results confirmed coating of the NF with MSCM.
LNP-Cas9 loading of MSCM-NF
To achieve localized and sustained Cas9 delivery in the bone marrow niche, Cas9-encapsulated LNPs were loaded onto MSCM-NF scaffolds via electrostatic interactions. We visualized the LNP-Cas9 loading of MSCM-NF by using DiI-labeled MSCM and fluorescein isothiocyanate (FITC)–labeled Cas9 (Fig. 2G). Green fluorescence signals of FITC-Cas9–encapsulated LNP were detected along with red fluorescence signals of DiI–MSCM-NF, confirming LNP-Cas9 loading onto the MSCM-NF scaffolds. LNP-Cas9 adsorbed onto MSCM-NF within 30 min (fig. S6A), and the amount of LNP loaded was LNP concentration dependent (Fig. 2H and fig. S6B). Both L76 and L77 showed similar LNP-Cas9 loading efficiencies and profiles. Before loading, the surface charges of the MSCM-NF, L76-Cas9, and L77-Cas9 were −22.2, 53.8, and 50.7 mV, respectively (Fig. 2F). Following LNP-Cas9 loading, the surface charge of the MSCM-NF increased drastically, from −22.2 to 19.4 mV for L76-Cas9 and 13.3 mV for L77-Cas9, indicating adsorption of the positively charged LNPs onto the MSCM-NF surface. MSCM-NF not only improved the LNP-Cas9 loading (Fig. 2I) but also showed the attenuated LNP-Cas9 release as compared to bare NF (Fig. 2J and fig. S6C), suggesting that the cell membrane could function as a drug reservoir.
Leukemia cell uptake of LNP-Cas9/MSCM-NF and gene editing
To assess leukemia cell uptake of MSCM-NF, THP-1 cells were incubated with MSCM-NF or bare NF. THP-1 cells demonstrated greater clustering with MSCM-NF than with bare NF (fig. S7A). We compared the uptake of LNP-Cas9/MSCM-NF with that of free LNP-Cas9 following incubation with THP-1 cells for 24 hours (fig. S7B). Most of the cells treated with free LNP-Cas9 were FITC positive after 4 hours (Fig. 3A and fig. S7B). LNP-Cas9/MSCM-NF also demonstrated substantial internalization, with over 85% FITC-positive cells. On the basis of microscopy, the THP-1 cells did not take up the lengthy NF, indicating that the LNP-Cas9 complexes were released from MSCM-NF (Fig. 3B). After 24 hours, released LNP-Cas9 localized to the nucleus of THP-1 cells (Fig. 3B). LNP-Cas9/MSCM-NF showed low cytotoxicity (Fig. 3C), with a gene editing efficiency similar to that of the bolus Cas9 delivery system (Fig. 3D). After a week of gene editing, the gene editing efficiency of LNP-Cas9 RNP/MSCM-NF increased 1.9-fold, while that of free LNP-Cas9 RNP increased only 1.2-fold (fig. S8, A and B). These results suggest that nanofibril-mediated LNP-Cas9 RNP delivery achieves more stable gene editing than bolus delivery by providing localized and sustained delivery to AML cells. Together, the MSCM-NF Cas9 delivery system achieves high leukemia cell uptake efficiency, low cytotoxicity, and efficient gene editing.
Fig. 3. LNP-Cas9 loaded onto MSCM-NF showed low leukemia cell toxicity, high uptake efficiency, and comparable gene editing efficiency to bolus delivery.
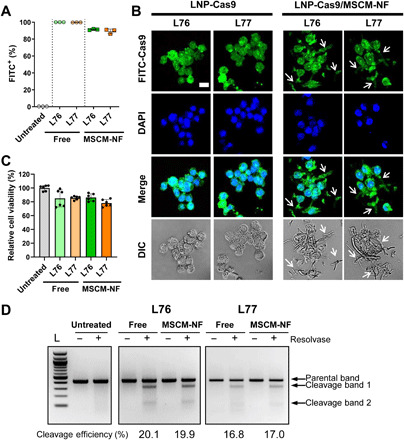
(A) Cell uptake rate of FITC-labeled free LNP-Cas9 or LNP-Cas9 on MSCM-NF after 4 hours of treatment. The FITC-positive population was measured by flow cytometry. Data are means ± SD. P < 0.0001 was observed between free and MSCM-NF groups. One-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. (B) CLSM images showing intracellular uptake of FITC-LNP-Cas9 in leukemia THP-1 cells after 24 hours of treatment. Nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI). THP-1 cells did not take up the lengthy NF (arrow). Scale bar, 25 μm. (C) Cytotoxicity of free LNP-Cas9 RNP or LNP-Cas9 RNP on MSCM-NF. Cell viability was measured after 24 hours of treatment and normalized to the untreated group. n = 2 independent studies. Data are means ± SD. No significant difference was observed between free and MSCM-NF groups. One-way ANOVA with Tukey’s multiple comparison test. (D) Gene editing efficiency of free LNP-Cas9 RNP and LNP-Cas9 RNP/MSCM-NF in leukemia cells. THP-1 cells were incubated with free LNP-Cas9 RNP or LNP-Cas9 RNP/MSCM-NF for 2 days, and cells were harvested for a gene editing detection assay. Free, Free LNP-Cas9 RNP. MSCM-NF, LNP-Cas9 RNP/MSCM-NF. L, 100-bp DNA ladder.
Optimization of leukemia cell-to-nanofibril ratio
To determine the optimal leukemia cell-to-NF ratio, different numbers of THP-1 cells (5 × 104 to 1 × 106 cells) were incubated with FITC-labeled L76-Cas9/MSCM-NF (2 μg of FITC-Cas9/50 μg of MSCM-NF), and the cell uptake efficiency was evaluated by flow cytometry. All groups showed over 95% FITC-positive cells after 24 hours of treatment; however, the median fluorescence intensity of internalized FITC-Cas9 decreased with increased cell number (fig. S9A). Gene-cleavage analysis showed that L76-Cas9/IL1RAP sgRNA/MSCM-NF (2 μg of Cas9/50 μg of MSCM-NF) achieved a similar gene editing efficiency (~16%) in the range of 5 × 104 to 5 × 105 leukemia cells but decreased to ~12% at ≥1 × 106 cells (fig. S9B). These results suggest that the ratio of leukemia cells to NF in vitro (and the NF dose in vivo) is a critical parameter for effective gene editing because the delivery efficiency of scaffold system depends on the available surface area of the scaffold.
CXCL12α-loaded LNP-Cas9/MSCM-NF induces leukemia cell chemoattraction and gene editing
We next investigated whether loading MSCM-NF with CXCL12α enhanced recruitment of leukemia cells. CXCL12α was loaded onto the MSCM-NF via electrostatic interactions [CXCL12α (pI 9.92) is positively charged at neutral pH]. The amount of CXCL12α loaded on MSCM-NF was controlled by adjusting the amount of CXCL12α added (Fig. 4A). CXCL12α dissociated from MSCM-NF within an hour, consistent with weak electrostatic interactions between CXCL12α and MSCM-NF (Fig. 4B). The amount of CXCL12α released was proportional to the amount loaded (Fig. 4B).
Fig. 4. LNP-Cas9 RNP/MSCM-NF/CXCL12α induces chemoattraction of leukemia cells and delivers Cas9 RNP to induce gene editing.
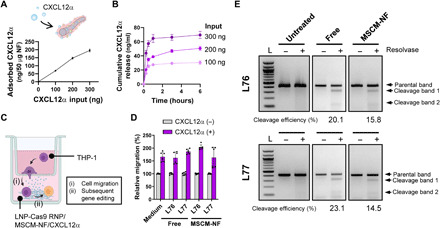
(A) Loading profiles of CXCL12α on MSCM-NF. (B) CXCL12α release profiles from CXCL12α-loaded MSCM-NF for different loading amounts of CXCL12α (100, 200, or 300 ng of CXCL12α for 50 μg of MSCM-NF). The amount of CXCL12α released was proportional to the amount loaded. (C) Schematic of migration assay evaluating (i) cell migration by CXCL12α and (ii) the subsequent gene editing by LNP-Cas9 RNP delivery. (D) The percentage of THP-1 cells migrating to the lower chamber was measured after 4.5 hours of incubation. n = 2 to 3 independent studies. (E) Gene editing efficiency of LNP-Cas9 RNP/MSCM-NF/CXCL12α in migrated THP-1 cells. THP-1 cells that migrated to the lower chamber were collected, and a gene editing detection assay was performed after 2 days of treatment. L, 100-bp DNA ladder. Data are presented as means ± SD.
To evaluate the chemoattraction of leukemia cells by CXCL12α-loaded MSCM-NF, cell migration assays were performed. An insert with a permeable membrane (pore size, 5 μm) separated two chambers. THP-1 cells were loaded into the upper chamber, and free LNP-Cas9 RNP or LNP-Cas9 RNP/MSCM-NF were placed in the lower chamber (Fig. 4C). For the free LNP-Cas9 RNP groups, CXCL12α-containing medium was added to the lower chamber; for the CXCL12α-loaded MSCM-NF groups, no CXCL12α was added (Fig. 4C). After 4.5 hours of incubation, cell migration from the upper chamber into the lower chamber was quantified. In the presence of CXCL12α in the lower chamber, the cell migration rate increased by 1.5- to 2.0-fold in all groups (Fig. 4D). The migration rate of the CXCL12α-loaded MSCM-NF group was comparable to that of the CXCL12α-containing medium group (Fig. 4D). Together, these results indicate that CXCL12α-loaded MSCM-NF can recruit leukemia cells to a site of interest.
We further assessed IL1RAP gene editing in the THP-1 cells that migrated to LNP-Cas9/IL1RAP sgRNA/MSCM-NF in the lower chamber. After 48 hours of incubation, editing of IL1RAP in THP-1 cells was generated by free L76- and L77-Cas9 RNP, and by L76- and L77-Cas9 RNP/MSCM-NF (Fig. 4E). Nanofibril-mediated delivery of LNP-Cas9 RNPs showed a slightly lower gene editing efficiency (15.8% for L76 and 14.5% for L77) than bolus delivery (20.1 and 23.1%) at 48 hours. Overall, these results suggest that CXCL12α-loaded LNP-Cas9/MSCM-NF can induce leukemia cell chemoattraction and achieve efficient gene editing by providing localized delivery to AML cells. For further in vivo studies, L76 was selected over L77 because L76 showed a slightly lower cytotoxicity at the same concentration compared to L77 (fig. S2B) and higher gene editing efficiency when loaded on MSCM-NF (Figs. 3D and 4E).
In vivo biodistribution of LNP-Cas9/MSCM-NF
Previous work has shown that free LNP-Cas9 RNP nanoparticles spread rapidly from the injection site through the circulatory system (36). To examine whether loading LNP-Cas9 onto MSCM-NF increases the retention of LNP-Cas9 in vivo, we monitored the biodistribution of LNP-Cas9/MSCM-NF in mice using an in vivo imaging system. Free L76-Cas9 or L76-Cas9/MSCM-NF were injected into the bone marrow cavity in the right tibia of BALB/c mice via an intra–bone marrow route. Images were acquired at 0, 30, 60, 90, and 120 min after injection (Fig. 5A). For mice receiving free L76-Cas9, the fluorescence signal of L76-Cas9 rapidly spread around the injection site immediately after injection, vanished in the bone marrow cavity within 30 min, and was detected in the bladder at 1 hour after injection. However, in mice receiving L76-Cas9/MSCM-NF, the fluorescence signal of LNP-Cas9 remained localized in the injection site for 2 hour (Fig. 5B). Overall, these results indicate that the retention time of LNP-Cas9 in the bone marrow cavity is increased by loading LNP-Cas9 onto MSCM-NF.
Fig. 5. Targeting IL1RAP via the LNP-Cas9 RNP/MSCM-NF system affects LSC colony-forming ability and reduces long-term leukemic burden.
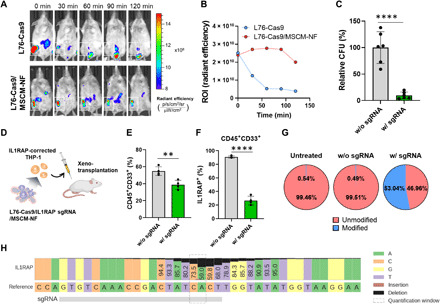
(A) In vivo imaging system (IVIS) images of in vivo biodistribution. Alexa Fluor 647–labeled free L76-Cas9 or L76-Cas9/MSCM-NF were injected into the right tibia of BALB/c mice (white square). The NP clearance rate was monitored using an IVIS. (B) The ROI (region of interest) (radiant efficiency), as described in (A), showing that free L76-Cas9 NPs were cleared from the bone marrow injection site faster than L76-Cas9/MSCM-NF. (C) CFU, colony-forming unit assay. THP-1 cells were incubated with L76-Cas9 RNP/MSCM-NF for 48 hours before plating in methylcellulose-based medium with recombinant human cytokines. The colony-forming ability was measured after 14 days of culture. n = 2 independent studies. (D) Schematic of mouse xenograft model with ex vivo IL1RAP gene editing treatment. THP-1 cells were treated with L76-Cas9 RNP/MSCM-NF for 2 days, and an equal number of THP-1 cells were transplanted into sublethally irradiated NSG mice. Human leukemic burden in recipient mice was measured by flow cytometry after 8 weeks or when the mice appeared moribund. (E) Percentage of human CD45+CD33+ population in the bone marrow harvested from recipient mice, as described in (D). (F) Percentage of human IL1RAP expression in CD45+CD33+ population in bone marrow harvested from recipient mice. (G) Identification of unmodified and modified IL1RAP alleles by amplicon sequencing analysis. THP-1 cells were treated with L76-Cas9 RNP/MSCM-NF as described in (D). DNA was harvested from cells before xenotransplantation for sequencing. (H) Nucleotide distribution near the IL1RAP sgRNA targeting site. The percentage of each base in the amplicon is shown based on the sequencing reads. w/o sgRNA, L76-Cas9/MSCM-NF without IL1RAP sgRNA. w/ sgRNA, L76-Cas9/MSCM-NF with IL1RAP sgRNA. Data are means ± SD. **P < 0.01 and ****P < 0.0001; Student’s t test.
Gene editing of IL1RAP affects LSC function
To determine whether IL1RAP gene editing via the LNP-Cas9 RNP/MSCM-NF system affects LSC function, we measured the colony-forming capacity of THP-1 cells following Cas9/IL1RAP sgRNA delivery (fig. S10A). THP-1 cells were plated in methylcellulose-based medium with recombinant human cytokines, and the growth, proliferation, and differentiation ability of the cells were assessed. We observed a 0.80-fold reduction of the IL1RAP-positive population in the knockout group after 5 days of transfection, indicating that IL1RAP knockout may affect cell surface IL1RAP expression or antibody binding of IL1RAP (fig. S10B). The L76-Cas9 RNP/MSCM-NF group had significantly fewer colonies formed, only ~10% of the colonies formed in the control group lacking sgRNA (Fig. 5C), indicating that LSC function was significantly impaired following IL1RAP knockout.
We next investigated the long-term effects of IL1RAP gene editing on LSCs by performing a xenotransplantation assay. THP-1 cells were treated ex vivo with L76-Cas9 RNP/MSCM-NF containing IL1RAP sgRNA (Fig. 5D). Two days later, an equal number of THP-1 cells were transplanted into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. At 8 weeks after transplantation, the leukemic burden of the recipient mice was assessed by determining the frequency of human CD45+CD33+ cells in the bone marrow of mouse femurs using flow cytometry. Unlike the control group that lacked IL1RAP sgRNA, L76-Cas9 RNP/MSCM-NF with IL1RAP sgRNA successfully induced IL1RAP gene editing (fig. S10C), reduced IL1RAP expression in THP-1 cells (fig. S10D), and significantly reduced the human leukemic burden in recipient mice (Fig. 5E). These results are consistent with the loss of LSC function following IL1RAP gene editing. IL1RAP protein expression in engrafted human CD45+CD33+ cells from bone marrow was significantly lower in the L76-Cas9 RNP/MSCM-NF–treated group than in the control group (Fig. 5F). We also assessed the on-target gene editing activity of L76-Cas9 RNP/MSCM-NF by amplicon next-generation sequencing. The L76-Cas9 RNP/MSCM-NF–treated cells showed a high editing efficiency (53%) at the Cas9 cleavage site within the IL1RAP locus, with no editing of the IL1RAP locus in control groups (untreated or without IL1RAP sgRNA) (Fig. 5, G and H). The nucleotide distribution across the entire amplicon also demonstrated that the Cas9 RNP delivery system mostly induced deletions around the sgRNA targeting site (Fig. 5H and fig. S10, E to G). Collectively, these results indicate that the LNP-Cas9 RNP/MSCN-NF system is able to successfully deliver Cas9/sgRNA RNP complexes into LSCs and induce editing of the IL1RAP gene. The defective function of IL1RAP caused by gene knockout leads to impaired colony formation and engraftment by the LSCs.
DISCUSSION
Preventing relapse after initial treatment is a pressing limitation of current AML therapies including previously unidentified therapies such as chimeric antigen receptor cellular therapy (46, 47). LSCs are thought to be responsible for the high incidence of AML relapse owing to their capacity for self-renewal and their quiescence, which promote resistance to chemotherapy (15, 48). Therefore, elimination of LSCs is crucial for improving the prognoses of patients with AML. Targeting IL1RAP for AML treatment is attractive for several reasons. IL1RAP is selectively overexpressed on leukemia stem and progenitor cells across different AML subtypes but is minimally expressed on normal HSPCs (13, 14, 18, 49). No significant hematopoietic dysfunction is observed in Il1rap−/− mice (17). IL1RAP promotes AML pathogenesis through multiple oncogenic signaling pathways, including IL-1 signaling, FLT3, and c-KIT (15, 18). High expression of IL1RAP has been associated with poor overall survival in AML (13). Therefore, targeting IL1RAP in LSCs is a promising strategy for treating AML.
In this study, we applied the CRISPR-Cas9 gene editing system to achieve longer and more complete elimination of IL1RAP function than with previous methods targeting IL1RAP (14, 17, 49, 50). Recent approaches have relied on antibodies against IL1RAP to recruit an immune response to eliminate AML cells or to suppress the proliferation of AML cells via an IL-1 signaling blockade (14, 49). Although antibody-dependent cellular cytotoxicity can kill LSCs, therapeutic effects may be limited because the immune systems of most patients with AML are impaired. Here, we show that knocking out IL1RAP via LNP delivery of Cas9/sgRNA RNP reduced IL1RAP protein expression in leukemia cells and reduced leukemia cell clonogenicity in vitro and resulted in decreased leukemic burden in vivo. Our future work will improve the lipidoid-based nanoparticle design for Cas9 RNP delivery for in vitro and in vivo applications in normal HSPCs and in other blood cancer treatments.
Local delivery of Cas9 RNP offers several advantages over systemic administration for targeting LSCs or HSPCs in the bone marrow. Local injection of Cas9 RNP enables direct access to the bone marrow cavity for highly localized delivery and lower systemic genotoxicity (51). However, rapid clearance of nanometer-size particles carrying Cas9 RNP from the injection site remains a hurdle for in vivo gene editing (31, 52, 53). To enhance the efficiency of gene editing in LSCs in bone marrow, we developed a scaffold-mediated Cas9 RNP delivery system. By loading Cas9 RNP onto the surface of NF, the in vivo retention time of Cas9 RNP at the injection site was notably increased compared to that of free Cas9 RNP. Scaffold-mediated drug delivery provides excellent intracellular uptake due to a high local drug concentration and direct contact between cells and drug (54–59), resulting in an in vitro genome editing efficiency similar to that of bolus delivery (60). Furthermore, loading Cas9 RNP/nanoparticle complexes onto a scaffold may protect complexes from enzymatic degradation, maintaining an elevated concentration of Cas9 RNP for a longer time period (56, 61). Likewise, Cas9 RNP-loaded NF exhibited comparable intracellular uptake and in vitro gene editing efficiency to free Cas9 RNP. In addition, its longer in vivo retention time and reduced cytotoxicity would be beneficial for stable local delivery of Cas9 RNP. Future investigations will focus on in vivo gene editing in a leukemic mouse model; we expect that the in vivo gene editing performance of Cas9 RNP delivered via NF will be superior to that of free Cas9 RNA delivered via local injection.
To further improve the LSC targeting efficiency of the scaffold in bone marrow, we prepared a mesenchymal stem cell–like scaffold. NF can be used as the scaffold due to their extracellular matrix–like fibrous structure, large surface area for drug loading and cell adhesion, and ability to create three-dimensional cell-cell and cell-matrix interactions (37, 62, 63). Cell membrane coating replicates the complex cell surface properties and critical functions of the cell type used (43, 44, 64). For example, platelet membrane–cloaked nanoparticles display selective adhesion to damaged vasculatures and enhanced binding to platelet-adhering pathogens (44), cancer cell membrane–coated nanoparticles exhibited homologous targeting and immune-invasion characteristics (65), and pancreatic beta cell membrane–coated nanofiber scaffolds improve beta cell proliferation rate and functions such as glucose-dependent insulin secretion versus the same beta cells cultured in unmodified nanofiber scaffolds (45). In the current study, we showed that the MSCM layer coating the NF was slightly thicker and rougher than that of membranes on spherical nanoparticles. Cell attachment to NF was notably accelerated by coating the NF with MSCM. Emptied cell membrane vesicles are prone to fuse with solid substrates to reduce their high surface energy (44, 66). Although fusion of MSCM vesicles with NF and nanoparticles is driven by a similar mechanism, the different dimensional and mechanophysical characteristics of the core materials may affect the coating thickness and surface roughness (45). We assume that the smaller the surface area of the core material, the more cell membrane vesicles aggregate during fusion. However, NF were completely covered with an MSCM that proved stable for 2 weeks, allowing attachment of leukemia cells via VCAM-1/VLA-4 interactions.
The cell membrane coating of the NF also served as a drug reservoir. The drug loading and release profiles of the MSCM-NF likely depend on the surface charge density, size, and hydrophobicity of the drug payloads (37). Coating the NF with MSCM significantly improved the payloads of LNP-Cas9 and CXCL12α; LNP-Cas9 loading efficiency of MSCM-NF was 4.5-fold higher than that of bare NF (Fig. 2I). Positively charged LNP-Cas9 adsorbed to bare NF but was released rapidly (within 24 hours; fig. S6C), whereas LNP-Cas9 remained attached to MSCM-NF for 3 days. CXCL12α was also loaded onto MSCM-NF. We observed a burst release of CXCL12α within 3 hours (~40% of CXCL12α loaded on the NF). The MSCM-NF thus recapitulated the characteristics of interactions between MSCs and HSPCs in bone marrow and improved the gene editing of leukemia cells via scaffold-mediated Cas9 RNP delivery. From a clinical translation viewpoint, a potential limitation is the drug loading capacity of the scaffold, which is dependent on the interaction between the cell membrane coating of the scaffold and the drugs (i.e., CXCL12α and Cas9 RNP). Because the surface area of the scaffold is limited, it may be difficult to achieve a high loading level. Reducing the fiber size to increase the surface area is one solution. Another approach is to focus only on highly potent therapeutics such as the ones applied in this study.
This NF scaffold–mediated CRISPR-Cas9 delivery system provides a safe and efficient Cas9 delivery platform for targeting LSCs to improve AML therapy. Further studies will focus on controlling the degradation rate of the NF to balance gene editing and clearance of the scaffold from the body. Nanofibril size will also be optimized to improve Cas9 payload and increase the dose for intra–bone marrow injection to achieve higher in vivo gene editing efficiency. Combination therapy of local delivery of Cas9 RNP with systemic delivery of anticancer drugs and sensitizers might be required for successful AML therapy in the future.
MATERIALS AND METHODS
Materials
A gRNA sequence against the IL1RAP gene (GTGTCAAACCGACTATCACT) was selected using CRISPOR (http://crispor.tefor.net/). Synthetic sgRNAs with chemical modifications were purchased from Synthego. Primers were purchased from Integrated DNA Technologies (IDT). IL1RAP forward primer: CCCATGAAACTCCCAGTGCA; IL1RAP reverse primer: GGAACTAGAAGTGGGCAGCA. Cas9 proteins were purchased from Thermo Fisher Scientific (TrueCut Cas9 Protein v2) or IDT (Alt-R S.p. HiFi Cas9 Nuclease V3).
Mice
All experiments were conducted using protocols approved by the Institutional Animal Care and Use Committees (biodistribution assay) or the University Committee on Animal Resources (xenotransplantation assay). All mice in this study (BALB/cJ, 000651; NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, NSG, 005557) were purchased from the Jackson Laboratory.
Human cell culture
The THP-1 human monocytic leukemia cell line was acquired from the American Type Culture Collection (TIB-202), and human bone marrow–derived mesenchymal stem cells were purchased from Texas A & M College of Medicine. All cells were maintained in a humidified incubator at 37°C/5% CO2. THP-1 cells were cultured in RPMI medium (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 1% penicillin-streptomycin (P/S; Gibco), and 0.05 mM 2-mercaptoethanol (Gibco). MSCs were maintained in minimum essential medium α (Gibco) supplemented with 20% FBS and 1% P/S.
Synthesis of LNPs
Synthesis of LNPs was performed as described previously (29, 35, 36) with slight modifications. Bioreducible lipids were acquired from Tufts University. All lipids have an O-14B containing a disulfide bond, combined with different head groups (lipid library numbers: 76, 77, 80, 87, 123, 306, 400, and 401). To formulate the LNPs, the bioreducible lipids were mixed with cholesterol (Avanti Polar Lipids), DOPE (Avanti Polar Lipids), and DSPE-PEG2k (Avanti Polar Lipids) in a chloroform solution (Sigma-Aldrich). The organic solvent was evaporated under vacuum overnight and the resulting lipid films were dissolved in ethanol (99%) (Fisher Scientific). The resulting solution was mixed with sodium acetate buffer (25 mM, pH 5.2) and added dropwise into an aqueous solution with the other half of the DSPE-PEG2k to produce the final mixture at a 16:4:1:1 weight ratio of bioreducible lipid:cholesterol:DOPE:DSPE-PEG2k. The mixture was then dialyzed against distilled water overnight with 1000-Da molecular weight cutoff dialysis tubing (VWR). LNP size, PDI, and zeta potential were measured by dynamic light scattering analysis with a Zetasizer (Nano ZS90, Malvern Panalytical). To test LNP cytotoxicity, THP-1 cells were seeded in a 96-well white plate at 1 × 104 cells per well. LNPs or vehicle was added to each well, and the plate was incubated at 37°C for 24 hours. Cell viability was measured with a CellTiter-Glo 2.0 Assay (Promega).
Fabrication of PCL NF
NF was fabricated by fragmenting electrospun PCL nanofibers as described previously (37). Briefly, PCL (molecular weight 50,000; Polysciences) was dissolved in a 3:1 (v/v) mixture of chloroform:methanol at a PCL concentration of 20% (w/v). The PCL solution was electrospun at 15 kV at a flow rate of 0.5 ml/hour through a 27-gauge needle. Electrospun nanofibers were deposited onto an aluminum foil ground at a ground-to-needle distance of 15 cm. Electrospun nanofibers were hydrolyzed in 1 M sodium hydroxide at 37°C for 6 hours. Nanofibril fragments were collected by centrifugation at 12,000 rpm for 5 min and were washed three times with distilled water. Morphological changes of nanofiber and NF scaffolds before and after fragmentation were observed by scanning electron microscopy (Zeiss Sigma VP, Carl Zeiss Microscopy).
Extraction and preparation of MSCM
Human MSCs were harvested and resuspended in hypotonic lysing buffer [20 mM tris-HCl (pH 7.5), 10 mM KCl, 2 mM MgCl2, and 1× protease inhibitor cocktail (Sigma-Aldrich)] and were incubated for 15 min at 4°C. Cell membranes were mechanically fragmented with an ultrasonic cell disruptor (QSonica Sonicators). Samples were centrifuged at 3200g for 5 min to collect the supernatant. Cell pellets were resuspended in hypotonic lysing buffer to collect additional supernatant. The combined supernatant was centrifuged at 10,000g for 5 min to remove cell debris. The supernatant was then ultracentrifuged at 100,000g for 30 min to pellet cell membrane. Pelleted cell membrane was resuspended in phosphate-buffered saline (PBS) supplemented with 1× protease inhibitors, and samples were ultrasonicated and stored at 4°C until use. All the above steps were performed using cold buffer or at 4°C. For labeling cell membranes, cells were resuspended in PBS and were stained with Vybrant DiI Cell-Labeling Solution (5 μl/106 cells/ml; Thermo Fisher Scientific) for 10 min at 37°C. Cells were washed three times with PBS to remove extra dye before cell membrane extraction.
MSCM coating of NF
MSCM (0.1 mg/ml) was sonicated for 5 min and then mixed with NF (1 mg) in PBS at pH 7.4. The mixture was incubated at room temperature for 30 min and then at 4°C for 12 hours with shaking at 200 rpm. Cell membrane–coated NF was collected by centrifugation at 8000 rpm for 5 min and were washed three times with PBS to remove free cell membrane. The cell membrane coating of NF was confirmed by TEM (Talos F200X, Thermo Fisher Scientific). TEM samples were stained with 2% (w/v) phosphotungstic acid for 1 min before observation. DiI-labeled cell membrane (DiI-MSCM) was used for fluorescence microscopy (Eclipse TS100, Nikon). The stability of the DiI-MSCM coating on NF was evaluated by monitoring fluorescence from fibrils in PBS at 37°C for 14 days.
LNP-Cas9 loading and release of MSCM-NF
To load LNP-Cas9 onto MSCM-NF, Cas9 protein (2 μg) encapsulated in LNP was mixed with MSCM-NF (50 μg) in PBS (pH 7.4, 100 μl) for 30 min at 37°C with gentle shaking. LNP-Cas9/MSCM-NF was collected by centrifugation at 12,000 rpm for 10 min. The surface charge of the LNP-Cas9/MSCM-NF was measured with a Zetasizer. To evaluate the LNP-Cas9 loading, FITC-Cas9 was encapsulated in LNPs and FITC-Cas9 LNP was incubated with MSCM-NF in PBS. The fluorescence intensity of FITC-Cas9 in the supernatant was measured with a microplate reader (FLUOstar OPTIMA, BMG Labtech), and the loading efficiency was evaluated based on the decrease in fluorescence intensity of FITC-Cas9 over time. FITC-Cas9 LNP/MSCM-NF was also observed by fluorescence microscopy (Eclipse TS100, Nikon). To evaluate the release rate and profile of LNP-Cas9, FITC-Cas9 LNP/MSCM-NF (50 μg) was incubated in PBS (pH 7.4, 500 μl) at 37°C for 72 hours. The FITC-Cas9 LNP release was measured with a microplate reader, and the release amount was calculated based on a standard curve of FITC-Cas9 LNP.
CXCL12α loading and release from MSCM-NF
CXCL12α (100, 200, and 300 ng) (PeproTech) in PBS was mixed with LNP-Cas9/MSCM-NF (50 μg) and incubated for 1 hour at 37°C. The amount of unbound CXCL12α was quantified by enzyme-linked immunosorbent assay (ELISA) (RayBiotech) to evaluate CXCL12α loading. CXCL12α-loaded MSCM-NF (50 μg) was incubated in 500 μl of RPMI with 10% FBS at 37°C. The supernatant (100 μl) was collected and supplemented with fresh RPMI medium (100 μl) at each time point. The amount of CXCL12α released was quantified by ELISA. For other in vitro and in vivo studies, LNP-Cas9 RNP was first loaded onto MSCM-NF and then CXCL12α was incorporated.
Intracellular delivery of LNP-encapsulated Cas9/sgRNA nanoparticles
For cell transfection, cells were seeded in a 24-well plate or 96-well U-bottom plate at 5 × 104 cells per well unless otherwise indicated. Briefly, Cas9 protein (2 μg) and IL1RAP sgRNA (0.4 μg) were encapsulated within LNPs in Opti-MEM I Reduced Serum Medium (Gibco) with gentle shaking for 15 min at room temperature. LNP-Cas9 RNP was loaded onto MSCM-NF (50 μg) for 30 min at 37°C with gentle shaking. LNP-Cas9 RNP/MSCM-NF was collected by centrifugation at 12,000 rpm for 10 min. LNP complexes were added dropwise to wells for transfection. Cell viability was measured with a CellTiter-Glo 2.0 Assay (Promega) after 24 hours of treatment unless otherwise specified. Gene editing efficiency was measured using the Guide-it Mutation Detection Kit (Takara Bio) after 48 hours of treatment unless otherwise indicated. Lipofectamine CRISPRMAX Cas9 Transfection Reagent (Thermo Fisher Scientific) was used for comparison.
Gene editing detection assay
Gene editing efficiency was measured using a Guide-it Mutation Detection Kit (Takara Bio). Cells were harvested, washed with 1× PBS, and lysed after 48 hours of treatment unless otherwise specified. The DNA sequence surrounding the CRISPR targeting site was amplified from each lysate sample by polymerase chain reaction (PCR) with IL1RAP gene–specific primers. After amplification, DNA hybridization was performed using the PCR product, and Guide-it Resolvase was added to each sample followed by a 15-min incubation at 37°C. The resulting amplicon samples were run on a 2% agarose gel at 100 V for 55 min and were imaged using a MultiImage Light Cabinet (Alpha Innotech) or ChemiDoc Imaging System (Bio-Rad). Cleavage efficiency was calculated using the following equation [Nature Materials method (67) and GeneArt Kit protocol]: Cleavage efficiency (%) = 1 − [(1 − fraction cleaved)½] × 100. Fraction cleaved = sum of intensities of cleaved bands/sum of intensities of cleaved and parental bands.
Cell viability assay
Cell viability assays were performed using a CellTiter-Glo 2.0 Assay. At the indicated time points, cells (150 μl) in a 96-well white plate were mixed with CellTiter-Glo 2.0 Reagent (50 μl). The plate was incubated at room temperature for 30 min with gentle rocking to induce cell lysis and stabilize luminescent signal. Luminescence in each well was measured with a FLUOstar OPTIMA Microplate Reader.
Flow cytometry analysis
Cell surface protein expression was measured by flow cytometry. For immunophenotyping, THP-1 cells or MSCs were washed with 1× PBS with 0.5% FBS and were stained with antibodies against human IL1RAP (PE, clone 89412, R&D), VLA-4 (CD49d; PE, clone 9F10, eBioscience), VCAM-1 (CD106; PE-Cyanine7, clone STA, eBioscience), or CXCR4 (CD184; APC, clone 12G5, BD Pharmingen). DAPI (4′,6-diamidino-2-phenylindole; Invitrogen) was used for live/dead cell staining. Flow cytometry analysis was performed on an LSR II or LSRFortessa instrument (BD Biosciences). Data analysis was conducted using FlowJo software (BD).
Colony-forming unit assay
THP-1 cells (5 × 104) were transfected with LNP-Cas9 RNP/MSCM-NF. After 48 hours of incubation, cells were mixed with MethoCult H4434 Classic medium (STEMCELL Technologies) supplemented with GCSF (10 ng/ml) (PeproTech) and 1% P/S, and were dispensed into 35-mm dishes at 1 × 104 cells per plate. Plates were incubated at 37°C and colonies were counted after 14 days of culture.
Migration assay
A migration assay was performed to assess the ability of CXCL12α to recruit THP-1 cells to the LNP-Cas9/MSCM-NF scaffolds. Briefly, 2 × 105 THP-1 cells were loaded into the upper chamber of a Millicell hanging cell culture with a 5-μm pore-size insert (Millipore) in a 24-well plate. LNP-Cas9 RNP/MSCM-NF/CXCL12α (4 μg of Cas9, 100 μg of MSCM-NF, and 300 ng of CXCL12α) was placed in the lower chamber. For the free LNP-Cas9 RNP group, free LNP-Cas9 RNP was mixed with CXCL12α-containing medium (100 ng/ml) and was added to the lower chamber. After 4.5 hours of incubation at 37°C, cells migrating to the lower chamber were quantified using a CellTiter-Glo 2.0 Assay (Promega).
In vivo imaging system
Alexa Fluor 647–labeled Cas9 was used for in vivo fluorescence imaging. Free L76-Cas9 or L76-Cas9–loaded MSCM-NF were injected into the tibia of BALB/c mice and were imaged with an IVIS Spectrum system (PerkinElmer) with excitation at 640 nm and emission at 650 to 670 nm.
Ex vivo treatment and xenotransplantation
THP-1 cells were seeded in a 96-well U-bottom plate at 2.5 × 105 cells per well and were treated with L76-Cas9 RNP/MSCM-NF (2 μg of Cas9 and 50 μg of MSCM-NF) at 37°C for 2 to 3 days. NSG mice were sublethally irradiated (2.5 gray) on the day before transplantation. On the day of transplantation, live cells were harvested using Ficoll Paque Plus (GE Healthcare) to remove the NF. Cells were washed and resuspended in 1× PBS with 2% FBS at 1 × 107 cells/ml. Each mouse was injected with 1 × 106 cells via the tail vein. Mice were euthanized at 8 weeks after transplantation or when they appeared moribund. To evaluate the engrafted cells in recipient mice, bone marrow cells from mice were harvested and stained with antibodies against human CD33 (FITC, clone HIM3-4, BD), CD45 (PE-Cy7, clone HI30, BD), and IL1RAP (PE, clone 89412, R & D), and were analyzed by flow cytometry. Some THP-1 cells treated with L76-Cas9 RNP/MSCM-NF were set aside and harvested after 2 days of treatment for a gene editing detection assay and amplicon sequencing analysis.
Amplicon sequencing analysis
Amplicon PCR samples (~400 bp) were amplified using reagents from a Guide-it Mutation Detection Kit (Takara Bio) as described above. Amplicon PCR products were purified with a QIAquick PCR Purification Kit (Qiagen). Amplicon-based next-generation sequencing analyses were performed by Genewiz (Amplicon-EZ). Sequencing data were analyzed by the University of Rochester Genomics Research Center.
Statistical analysis
Statistical analysis was performed with GraphPad Prism software (GraphPad Software) using a Student’s t test or one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test where applicable. P < 0.05 was considered statistically significant.
Acknowledgments
We would like to thank the Flow Cytometry Core of the Columbia Center, the University of Rochester Medical Center Flow Cytometry Core, and the University of Rochester Genomics Research Center for technical support. Funding: This study was supported by the Columbia University startup fund, the Leukemia and Lymphoma Society (LLS grant ID: 7020-19), and the National Research Foundation of Korea (GRL 2015K1A1A2032163). Author contributions: T.-C.H., H.S.K., M.W.B., and K.W.L. designed the experiments. T.-C.H., H.S.K., Y.C., Y.L., M.W.L., C.C., H.W., and J.G. performed the experiments. T.-C.H., H.S.K., Y.C., Y.L., C.D.P., and J.M.A. analyzed the data. H.K. provided support and critical advice. Q.X. provided critical materials and discussion. T.-C.H., H.S.K., M.W.B., and K.W.L. wrote the manuscript. M.W.B. and K.W.L. managed and supported the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabg3217/DC1
REFERENCES AND NOTES
- 1.Dohner H., Weisdorf D. J., Bloomfield C. D., Acute myeloid leukemia. N. Engl. J. Med. 373, 1136–1152 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Shallis R. M., Wang R., Davidoff A., Ma X., Zeidan A. M., Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 36, 70–87 (2019). [DOI] [PubMed] [Google Scholar]
- 3.N. Howlader, A. M. Noone, M. Krapcho, D. Miller, A. Brest, M. Yu, J. Ruhl, Z. Tatalovich, A. Mariotto, D. R. Lewis, H. S. Chen, E. J. Feuer, K. A. Cronin, SEER Cancer Statistics Review, 1975–2016 (National Cancer Institute, Bethesda, MD, April 2019); https://seer.cancer.gov/csr/1975_2016/.
- 4.Estey E., Dohner H., Acute myeloid leukaemia. Lancet 368, 1894–1907 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Shlush L. I., Mitchell A., Heisler L., Abelson S., Ng S. W. K., Trotman-Grant A., Medeiros J. J. F., Rao-Bhatia A., Jaciw-Zurakowsky I., Marke R., McLeod J. L., Doedens M., Bader G., Voisin V., Xu C., McPherson J. D., Hudson T. J., Wang J. C. Y., Minden M. D., Dick J. E., Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 547, 104–108 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Ding L., Ley T. J., Larson D. E., Miller C. A., Koboldt D. C., Welch J. S., Ritchey J. K., Young M. A., Lamprecht T., McLellan M. D., McMichael J. F., Wallis J. W., Lu C., Shen D., Harris C. C., Dooling D. J., Fulton R. S., Fulton L. L., Chen K., Schmidt H., Kalicki-Veizer J., Magrini V. J., Cook L., McGrath S. D., Vickery T. L., Wendl M. C., Heath S., Watson M. A., Link D. C., Tomasson M. H., Shannon W. D., Payton J. E., Kulkarni S., Westervelt P., Walter M. J., Graubert T. A., Mardis E. R., Wilson R. K., DiPersio J. F., Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas D., Majeti R., Biology and relevance of human acute myeloid leukemia stem cells. Blood 129, 1577–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishikawa F., Yoshida S., Saito Y., Hijikata A., Kitamura H., Tanaka S., Nakamura R., Tanaka T., Tomiyama H., Saito N., Fukata M., Miyamoto T., Lyons B., Ohshima K., Uchida N., Taniguchi S., Ohara O., Akashi K., Harada M., Shultz L. D., Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 25, 1315–1321 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Eppert K., Takenaka K., Lechman E. R., Waldron L., Nilsson B., van Galen P., Metzeler K. H., Poeppl A., Ling V., Beyene J., Canty A. J., Danska J. S., Bohlander S. K., Buske C., Minden M. D., Golub T. R., Jurisica I., Ebert B. L., Dick J. E., Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 17, 1086–1093 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Ng S. W., Mitchell A., Kennedy J. A., Chen W. C., McLeod J., Ibrahimova N., Arruda A., Popescu A., Gupta V., Schimmer A. D., Schuh A. C., Yee K. W., Bullinger L., Herold T., Görlich D., Büchner T., Hiddemann W., Berdel W. E., Wörmann B., Cheok M., Preudhomme C., Dombret H., Metzeler K., Buske C., Löwenberg B., Valk P. J., Zandstra P. W., Minden M. D., Dick J. E., Wang J. C., A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 540, 433–437 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Pietras E. M., Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 130, 1693–1698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinarello C. A., Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barreyro L., Will B., Bartholdy B., Zhou L., Todorova T. I., Stanley R. F., Ben-Neriah S., Montagna C., Parekh S., Pellagatti A., Boultwood J., Paietta E., Ketterling R. P., Cripe L., Fernandez H. F., Greenberg P. L., Tallman M. S., Steidl C., Mitsiades C. S., Verma A., Steidl U., Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood 120, 1290–1298 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Askmyr M., Agerstam H., Hansen N., Gordon S., Arvanitakis A., Rissler M., Juliusson G., Richter J., Jaras M., Fioretos T., Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood 121, 3709–3713 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Ho T.-C., LaMere M., Stevens B. M., Ashton J. M., Myers J. R., O’Dwyer K. M., Liesveld J. L., Mendler J. H., Guzman M., Morrissette J. D., Zhao J., Wang E. S., Wetzler M., Jordan C. T., Becker M. W., Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 128, 1671–1678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaras M., Johnels P., Hansen N., Agerstam H., Tsapogas P., Rissler M., Lassen C., Olofsson T., Bjerrum O. W., Richter J., Fioretos T., Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc. Natl. Acad. Sci. U.S.A. 107, 16280–16285 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agerstam H., Hansen N., von Palffy S., Sanden C., Reckzeh K., Karlsson C., Lilljebjorn H., Landberg N., Askmyr M., Hogberg C., Rissler M., Porkka K., Wadenvik H., Mustjoki S., Richter J., Jaras M., Fioretos T., IL1RAP antibodies block IL-1-induced expansion of candidate CML stem cells and mediate cell killing in xenograft models. Blood 128, 2683–2693 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Mitchell K., Barreyro L., Todorova T. I., Taylor S. J., Antony-Debre I., Narayanagari S. R., Carvajal L. A., Leite J., Piperdi Z., Pendurti G., Mantzaris I., Paietta E., Verma A., Gritsman K., Steidl U., IL1RAP potentiates multiple oncogenic signaling pathways in AML. J. Exp. Med. 215, 1709–1727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright A. V., Nunez J. K., Doudna J. A., Biology and applications of CRISPR Systems: Harnessing nature’s toolbox for genome engineering. Cell 164, 29–44 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Stella S., Alcon P., Montoya G., Class 2 CRISPR-Cas RNA-guided endonucleases: Swiss Army knives of genome editing. Nat. Struct. Mol. Biol. 24, 882–892 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Liu C., Zhang L., Liu H., Cheng K., Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 266, 17–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kay M. A., State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 12, 316–328 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Ran F. A., Cong L., Yan W. X., Scott D. A., Gootenberg J. S., Kriz A. J., Zetsche B., Shalem O., Wu X., Makarova K. S., Koonin E. V., Sharp P. A., Zhang F., In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin H., Song C. Q., Dorkin J. R., Zhu L. J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A., Gupta A., Bolukbasi M. F., Walsh S., Bogorad R. L., Gao G., Weng Z., Dong Y., Koteliansky V., Wolfe S. A., Langer R., Xue W., Anderson D. G., Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leader B., Baca Q. J., Golan D. E., Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 7, 21–39 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Staahl B. T., Benekareddy M., Coulon-Bainier C., Banfal A. A., Floor S. N., Sabo J. K., Urnes C., Munares G. A., Ghosh A., Doudna J. A., Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 35, 431–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zuris J. A., Thompson D. B., Shu Y., Guilinger J. P., Bessen J. L., Hu J. H., Maeder M. L., Joung J. K., Chen Z. Y., Liu D. R., Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang M., Zuris J. A., Meng F., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q., Georgakoudi I., Liu D. R., Xu Q., Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 113, 2868–2873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin H., Xue W., Chen S., Bogorad R. L., Benedetti E., Grompe M., Koteliansky V., Sharp P. A., Jacks T., Anderson D. G., Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 32, 551–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun W., Ji W., Hall J. M., Hu Q., Wang C., Beisel C. L., Gu Z., Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew. Chem. Int. Ed. Engl. 54, 12029–12033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu H., Zhang L., Tong S., Lee C. M., Deshmukh H., Bao G., Spatial control of in vivo CRISPR-Cas9 genome editing via nanomagnets. Nat. Biomed. Eng. 3, 126–136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nihongaki Y., Kawano F., Nakajima T., Sato M., Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 33, 755–760 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Dow L. E., Fisher J., O’Rourke K. P., Muley A., Kastenhuber E. R., Livshits G., Tschaharganeh D. F., Socci N. D., Lowe S. W., Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 33, 390–394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y., Yang T., Yu Y., Shi N., Yang L., Glass Z., Bolinger J., Finkel I. J., Li W., Xu Q., Combinatorial library of chalcogen-containing lipidoids for intracellular delivery of genome-editing proteins. Biomaterials 178, 652–662 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Li Y., Bolinger J., Yu Y., Glass Z., Shi N., Yang L., Wang M., Xu Q., Intracellular delivery and biodistribution study of CRISPR/Cas9 ribonucleoprotein loaded bioreducible lipidoid nanoparticles. Biomater. Sci. 7, 596–606 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Kim H. S., Yoo H. S., Surface-polymerized biomimetic nanofibrils for the cell-directed association of 3-D scaffolds. Chem. Commun. 51, 306–309 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Fang R. H., Kroll A. V., Gao W., Zhang L., Cell membrane coating nanotechnology. Adv. Mater. 30, e1706759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Konopleva M. Y., Jordan C. T., Leukemia stem cells and microenvironment: Biology and therapeutic targeting. J. Clin. Oncol. 29, 591–599 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tavor S., Petit I., Porozov S., Avigdor A., Dar A., Leider-Trejo L., Shemtov N., Deutsch V., Naparstek E., Nagler A., Lapidot T., CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 64, 2817–2824 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Rombouts E. J., Pavic B., Löwenberg B., Ploemacher R. E., Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood 104, 550–557 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Gruszka A. M., Valli D., Restelli C., Alcalay M., Adhesion deregulation in acute myeloid leukaemia. Cell 8, 66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang R. H., Hu C.-M., Luk B. T., Gao W., Copp J. A., Tai Y., O’Connor D. E., Zhang L., Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 14, 2181–2188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu C.-M., Fang R. H., Wang K.-C., Luk B. T., Thamphiwatana S., Dehaini D., Nguyen P., Angsantikul P., Wen C. H., Kroll A. V., Carpenter C., Ramesh M., Qu V., Patel S. H., Zhu J., Shi W., Hofman F. M., Chen T. C., Gao W., Zhang K., Chien S., Zhang L., Nanoparticle biointerfacing by platelet membrane cloaking. Nature 526, 118–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen W., Zhang Q., Luk B. T., Fang R. H., Liu Y., Gao W., Zhang L., Coating nanofiber scaffolds with beta cell membrane to promote cell proliferation and function. Nanoscale 8, 10364–10370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansrivijit P., Gale R. P., Barrett J., Ciurea S. O., Cellular therapy for acute myeloid leukemia—Current status and future prospects. Blood Rev. 37, 100578 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Gerstung M., Papaemmanuil E., Martincorena I., Bullinger L., Gaidzik V. I., Paschka P., Heuser M., Thol F., Bolli N., Ganly P., Ganser A., McDermott U., Dohner K., Schlenk R. F., Dohner H., Campbell P. J., Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat. Genet. 49, 332–340 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pollyea D. A., Jordan C. T., Therapeutic targeting of acute myeloid leukemia stem cells. Blood 129, 1627–1635 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Agerstam H., Karlsson C., Hansen N., Sanden C., Askmyr M., von Palffy S., Hogberg C., Rissler M., Wunderlich M., Juliusson G., Richter J., Sjostrom K., Bhatia R., Mulloy J. C., Jaras M., Fioretos T., Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc. Natl. Acad. Sci. U.S.A. 112, 10786–10791 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warda W., Larosa F., Neto Da Rocha M., Trad R., Deconinck E., Fajloun Z., Faure C., Caillot D., Moldovan M., Valmary-Degano S., Biichle S., Daguindau E., Garnache-Ottou F., Tabruyn S., Adotevi O., Deschamps M., Ferrand C., CML hematopoietic stem cells expressing IL1RAP can be targeted by chimeric antigen receptor-engineered T cells. Cancer Res. 79, 663–675 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Zhang F., Wen Y., Guo X., CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 23, R40–R46 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Moghimi S. M., Exploiting bone marrow microvascular structure for drug delivery and future therapies. Adv. Drug Deliv. Rev. 17, 61–73 (1995). [Google Scholar]
- 53.Awasthi V. D., Garcia D., Goins B. A., Phillips W. T., Circulation and biodistribution profiles of long-circulating PEG-liposomes of various sizes in rabbits. Int. J. Pharm. 253, 121–132 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Jang J. H., Bengali Z., Houchin T. L., Shea L. D., Surface adsorption of DNA to tissue engineering scaffolds for efficient gene delivery. J. Biomed. Mater. Res. A 77, 50–58 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu K., Xu J., Liu M., Song W., Yan J., Gao S., Zhao L., Zhang Y., Induction of osteogenic differentiation of stem cells via a lyophilized microRNA reverse transfection formulation on a tissue culture plate. Int. J. Nanomedicine 8, 1595–1607 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen R., Zhang H., Yan J., Bryers J. D., Scaffold-mediated delivery for non-viral mRNA vaccines. Gene Ther. 25, 556–567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Low W. C., Rujitanaroj P. O., Lee D. K., Messersmith P. B., Stanton L. W., Goh E., Chew S. Y., Nanofibrous scaffold-mediated REST knockdown to enhance neuronal differentiation of stem cells. Biomaterials 34, 3581–3590 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Shin J., Cho J. H., Jin Y., Yang K., Lee J. S., Park H. J., Han H. S., Lee J., Jeon H., Shin H., Cho S. W., Mussel adhesion-inspired reverse transfection platform enhances osteogenic differentiation and bone formation of human adipose-derived stem cells. Small 12, 6266–6278 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Chooi W. H., Ong W., Murray A., Lin J., Nizetic D., Chew S. Y., Scaffold mediated gene knockdown for neuronal differentiation of human neural progenitor cells. Biomater. Sci. 6, 3019–3029 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Chin J. S., Chooi W. H., Wang H., Ong W., Leong K. W., Chew S. Y., Scaffold-mediated non-viral delivery platform for CRISPR/Cas9-based genome editing. Acta Biomater. 90, 60–70 (2019). [DOI] [PubMed] [Google Scholar]
- 61.A. K. Pannier, T. Segura, Surface-and hydrogel-mediated delivery of nucleic acid nanoparticles, in Nanotechnology for Nucleic Acid Delivery (Springer, 2013), pp. 149–169. [DOI] [PubMed] [Google Scholar]
- 62.Mao W., Kang M. K., Shin J. U., Son Y. J., Kim H. S., Yoo H. S., Coaxial hydro-nanofibrils for self-assembly of cell sheets producing skin bilayers. ACS Appl. Mater. Interfaces 10, 43503–43511 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Lee S., Kim H. S., Yoo H. S., Electrospun nanofibrils embedded hydrogel composites for cell cultivation in a biomimetic environment. RSC Adv. 7, 54246–54253 (2017). [Google Scholar]
- 64.Parodi A., Quattrocchi N., van de Ven A. L., Chiappini C., Evangelopoulos M., Martinez J. O., Brown B. S., Khaled S. Z., Yazdi I. K., Enzo M. V., Isenhart L., Ferrari M., Tasciotti E., Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 8, 61–68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shao D., Li M., Wang Z., Zheng X., Lao Y.-H., Chang Z., Zhang F., Lu M., Yue J., Hu H., Yan H., Chen L., Dong W.-f., Leong K. W., Bioinspired diselenide-bridged mesoporous silica nanoparticles for dual-responsive protein delivery. Adv. Mater., e1801198 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Hu C.-M., Zhang L., Aryal S., Cheung C., Fang R. H., Zhang L., Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. U.S.A. 108, 10980–10985 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shahbazi R., Sghia-Hughes G., Reid J. L., Kubek S., Haworth K. G., Humbert O., Kiem H. P., Adair J. E., Targeted homology-directed repair in blood stem and progenitor cells with CRISPR nanoformulations. Nat. Mater. 18, 1124–1132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabg3217/DC1