

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes acute, highly transmissible respiratory infection in humans and a wide range of animal species. Its rapid global spread has resulted in a major public health emergency, necessitating commensurately rapid research to improve control strategies. In particular, the ability to effectively retrace transmission chains in outbreaks remains a major challenge, partly due to our limited understanding of the virus’ underlying evolutionary dynamics within and between hosts. We used high-throughput sequencing whole-genome data coupled with bottleneck analysis to retrace the pathways of viral transmission in two nosocomial outbreaks that were previously characterised by epidemiological and phylogenetic methods. Additionally, we assessed the mutational landscape, selection pressures, and diversity at the within-host level for both outbreaks. Our findings show evidence of within-host selection and transmission of variants between samples. Both bottleneck and diversity analyses highlight within-host and consensus-level variants shared by putative source-recipient pairs in both outbreaks, suggesting that certain within-host variants in these outbreaks may have been transmitted upon infection rather than arising de novo independently within multiple hosts. Overall, our findings demonstrate the utility of combining within-host diversity and bottleneck estimations for elucidating transmission events in SARS-CoV-2 outbreaks, provide insight into the maintenance of viral genetic diversity, provide a list of candidate targets of positive selection for further investigation, and demonstrate that within-host variants can be transferred between patients. Together these results will help in developing strategies to understand the nature of transmission events and curtail the spread of SARS-CoV-2.
Keywords: SARS-CoV-2, transmission dynamics, bottleneck, within-host variants, selection, nonsynonymous, South Africa, NGS whole-genome sequencing
1. Introduction
The emergence and spread of a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in Wuhan, China, resulted in a public health emergency of international concern in just under two months (Zhu et al. 2020; WHO 2020). In South Africa, the first officially diagnosed case of SARS-CoV-2 was reported on 5 March 2020. Strict public health mitigation strategies and non-pharmaceutical interventions played a critical role in controlling the COVID-19 pandemic in South Africa, lowering the new cases reported each day to approximately 1,770 by 31 October 2020. Unfortunately, this brief lull was followed by the emergence of the 501Y.V2 variant and a resurgence in the pandemic (NICD 2020). Understanding the patterns of transmission and the selection pressures acting on viral populations leading up to that point in time could be critical to preventing a recurrence of the surge in infections and excess deaths such as that South Africa experienced between November 2020 and February 2021 (Tegally et al. 2020), which ultimately killed between 513 and 4027 (SAMRC 2020).
Phylogenetic inference can be used together with epidemiological investigations to elucidate transmission events and retrace transmission chains (He et al. 2020; Lauring 2020; Bajaj and Purohit 2020; Guo et al., 2020); however, phylogenetic reports based on consensus sequences (i.e. one sequence per case) only represent the dominant viral lineage in a host and thus provide limited resolution in transmission analyses (Mavian et al. 2020). Whole-genome analyses integrating within-host diversity have been proposed as a better alternative for capturing viral genetic diversity, including low-frequency variants in viral populations present within a given host (Sanjuan et al. 2004). Indeed, several studies have already revealed the existence of substantial genetic variation in within-host viral populations of SARS-CoV-2 (Wolfel et al. 2020; Shen et al. 2020; Lythgoe et al. 2020; Butler et al. 2020; Nelson et al. 2020a). For example, within-host analyses have revealed large numbers of within-host (intrahost) single nucleotide variants (iSNVs), including nonsynonymous (amino acid changing) iSNVs in SARS-CoV-2-positive nasopharyngeal and oropharyngeal swabs (Zhou et al. 2020; Siqueira et al. 2020) and bronchoalveolar lavage fluid samples (Shen et al. 2020). Importantly, Wang et al. (2020) have shown that different samples with matching consensus sequences can exhibit different iSNVs. Nevertheless, Shen et al. (2020) could not confirm the transmission of any iSNVs across two confirmed source-recipient pairs in the Wuhan area, suggesting either strong purifying selection or the stochastic occurrence and disappearence of within-host variants upon or following the transmission bottleneck (Shen et al. 2020). It therefore remains unclear whether iSNVs can be used to improve tracing of viral transmission and enhance our understanding of the evolutionary dynamics of SARS-CoV-2.
Although many (i)SNVs are unlikely to affect viral fitness, others can potentially result in viral genotypes with altered pathogenicity, improved host-specific adaptations (such as immune evasion phenotypes) or generally improved replication and/or transmission kinetics (Gojobori et al. 1990; Lucas et al. 2001). Examples of SNVs with such properties are those found in the recently emerged N501Y lineages, which putatively increases both the transmissibility of these viruses (Tegally et al. 2020; Rambaut et al. 2020; Faria et al. 2021; du Plessis et al. 2021) and their capacity to evade population-level immunity (Fontanet et al. 2021). The emergence of the 501Y lineages re-emphasizes the need to better understand how SARS-CoV-2 genomic diversity arises, the fitness costs and benefits of individual arising mutations, and the evolutionary pressures that ultimately drive some mutations to high frequencies in global populations.
Leveraging genomic and epidemiological data (Giandhari et al. 2020; Pillay et al. 2020) from two well-characterized nosocomial SARS-CoV-2 outbreaks with whole genome diversity analysis, we performed an in-depth analysis of multiple inferred SARS-CoV-2 transmission chains. Further, we analysed the frequency distribution of (i)SNVs within- and between-hosts to show how combining within-host diversity and bottleneck estimation can yield improved power to retrace transmission chains during SARS-CoV-2 outbreaks.
2. Methods
2.1 Two SARS-CoV-2 nosocomial outbreaks
This study analysed 109 SARS-CoV-2 positive cases from two nosocomial outbreaks in the Kwazulu-Natal province of South Africa. The first outbreak (CH1; thirty-five cases analysed) lasted four weeks, while the second outbreak (CH3; seventy-four cases analysed) lasted six weeks. Both outbreaks occurred at a time of relatively limited community transmission. Timelines for the two outbreaks are shown in Fig. 1.
Figure 1.

Phylogenetic analysis of the two outbreaks showing the clustering of sequences across hospital departments and associated Pangolin lineages to which the sequences belong. A) Pyhlogeny of samples from CH1 outbreak. B) Phylogeny of samples from the CH3 outbreak. Inset of each phylogeny is the TempEst plot showing showing the clocklike signal. Sample clustering was not consistent with the epidemiological settings in CH3.
2.2 Epidemiological investigation and identification of transmission chains
The investigation of transmission chains and clusters during the outbreaks was conducted by researchers at the Kwazulu Natal Research and Innovation Sequencing Platform (KRISP) and the University of Kwazulu Natal. Investigation methods included medical record reviews, ward visits, and interviews with healthcare workers and hospital management. Detailed timelines of patient cases were constructed to generate hypotheses on the spread of infection among patients and healthcare workers within the two hospitals and inference of putative transmission pairs (KRISP 2020).
2.3 Real time-polymerase chain reaction
All cases were confirmed using comparative real time (RT)-polymerase chain reaction (PCR). We used the TaqPath COVID-19 CE-IVD RT-PCR Kit (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. The assays target genomic regions (ORF1ab, S protein and N protein) of the SARS-CoV-2 genome. RT-PCR was performed on a QuantStudio 7 RT-PCR instrument (Life Technologies, Carlsbad, CA).
2.4 Whole-genome sequencing and assembly
We performed cDNA synthesis from RNA using random primers followed by gene-specific multiplex PCR using the ARTIC protocol. Briefly, extracted RNA was converted to cDNA using the Superscript IV First Strand synthesis system (Life Technologies, Carlsbad, CA) and random hexamer primers. SARS-CoV-2 whole genome amplification by multiplex PCR was carried out using primers designed on Primal Scheme (http://primal.zibraproject.org/) to generate 400-bp amplicons with an overlap of seventy base pairs covering the thirty-base pair SARS-CoV-2 genome. PCR products were cleaned up using AmpureXP purification beads (Beckman Coulter, High Wycombe, UK) and quantified using the Qubit dsDNA High Sensitivity assay on the Qubit 4.0 instrument (Life Technologies, Carlsbad, CA).
The Illumina Nextera Flex DNA Library Prep kit was used according to the manufacturer’s protocol to prepare uniquely indexed paired-end libraries of genomic DNA. Sequencing libraries were normalized to 4 nM, pooled and denatured with 0.2 N sodium acetate. Sample (12 pM) library was spiked with one per cent PhiX (PhiX Control v3 adapter-ligated library used as a control). Libraries were loaded onto a 500-cycle v2 MiSeq Reagent Kit and run on the Illumina MiSeq instrument (Illumina, San Diego, CA). Raw reads coming from Illumina sequencing were assembled using Genome Detective 1.126 (https://www.genomedetective.com/) and the Coronavirus Typing Tool (Vilsker et al. 2019; Cleemput et al. 2020a). The initial assembly obtained from Genome Detective was polished by aligning mapped reads to the references and filtering out low-quality variants using the bcftools 1.7-2 mpileup method. All variants were confirmed visually with bam files using Geneious (Biomatters Ltd, New Zealand). Indels resulting in mid-gene stop codons and frameshifts were reverted to wild type (Cleemput et al. 2020b). All of the sequences were deposited in GISAID (https://www.gisaid.org/) (Shu and Mccauley 2017).
2.5 Phylogenetic analysis
Phylogenetic analysis was performed to verify the epidemiologically inferred transmission chains (Fig. 1). Sequences from CH1 and CH3 outbreaks were aligned in MAFFT (Nakamura et al. 2018) and the alignments were manually edited in Geneious to fix codon misalignments and remove insertions and frameshifts. Maximum likelihood tree topologies were then inferred from the subsequent alignments in IQTREE (Nguyen et al. 2015) using the generalized time-reversible substitution model (Tavare 1986). These trees were used to check the temporal molecular clock signal of each outbreak cluster in TempEst (Rambaut et al. 2016). After good molecular clock signals were established, samples of 9,000 similarly likely phylogenetic trees were inferred using a Bayesian Markov Chain Monte Carlo approach implemented in BEAST v 1.1.10 (Suchard et al. 2018). Runs were executed under a strict molecular clock assumption with a strong mutation rate prior (8 × 10−4 subsitution/site/year; SD: 0.5 × 10−3) with a chain length of 100 million steps in the chain (sampling every 10,000 steps). Markov chain Monte Carlo runs were assessed in Tracer v 1.6.0 (Rambaut et al. 2018) for good convergence and proper mixing, that is for effective sample size values >200 for each estimated parameter. The estimated root of each cluster was recorded and Maximum Clade Credibility (MCC) trees were constructed in TreeAnnotator, discarding the first ten per cent of sampled trees as burn-in (i.e. each MCC tree represented a sample of 9,000 similarly likely trees). Resulting trees were visualized using the R package, ggtree (Yu 2020).
2.6 Within-host variants identification
We used LoFreq v.2.1.5 (Wilm et al. 2012) to call iSNVs (intrahost SNVs), including low-frequency variants. Initial variants were called relative to the Wuhan-Hu-1 NC_045512.2 reference at sites with a minimum sequencing depth of ×100 and employing a false discovery rate (FDR) cutoff of one per cent, as proposed by Costello et al. (2018) and Pightling et al. (2019) after thorough empirical evaluation of Illumina-based library preparation and sequencing errors. LoFreq automatically eliminates all variants that have a P-value below the FDR threshold and have ≥85 per cent of reads mapping to just one strand to avoid strand bias. Additional filtering to further eliminate strand biases was performed using customized scripts to retain only minor alleles that were present at a frequency ≥5 per cent, supported by at least two per cent of the total reads, and supported by a minimum of five reads on each strand, following Shen et al. (2020). Positions with more than one minor allele were filtered out to minimize false discovery, that is only biallelic sites were considered. Only variants in protein-coding regions were analyzed. Variants passing these criteria were then annotated using snpEff v. 4.5 (Cingolani et al. 2012b) and SnpSift v.4.3t (Cingolani et al. 2012a).
2.7 Mutational spectra
To characterize the mutational spectra, we considered only variants for which the reference allele matched the reference (Wuhan-Hu-1). For SNVs, the alternative allele was required to be the dominant allele (i.e. frequency >50%) and for iSNVs (within-host variants), only alleles with alternative allele frequency less than fifty per cent were considered. Reference minor alleles (i.e. alleles where the reference base was the minor allele) were not considered in this study.
2.8 Technical controls
To evaluate the efficiency and consistency of our sequencing process and variant identification protocols, we thoroughly assessed two representative biological replicates. The first replicate, CC00686518T6, was sequenced twice in separate runs to assess potential biases introduced by the sequencing step. The second replicate, EG00465499T6, was taken from a new aliquot and sequenced to assess the possibility of contamination. Both replicates yielded similar results that were highly concordant in terms of variants identified and their associated frequencies (Fig. 2C, Supplementary Table S5). Supplementary Fig. S6 shows the correlation after positions that were not reproducible or occurred at an allele frequency less than five per cent in the second replicate had been filtered out. Furthermore, we sequenced two samples that had previously tested negative for SARS-CoV-2 and, for these, we did not obtain any viral reads on assembly, confirming the suitability of our DNA preprocessing, extraction, sequencing and analysis pipeline for the study.
Figure 2.

Overview of general diversity of SARS-CoV-2 genomes from South African patients. (A) Nucleotide changes in SARS-CoV-2 genomes. (B) Distribution of variant frequencies across nucleotide changes. (C) Regression plot showing the correlation between frequencies of mutations in the two replicates. Outliers colored in red show variants that only occurred in a single replicate or at very low frequencies (<5%) in the second replicate and as such were filtered out. (D) The upset plot shows the distribution of iSNVs and SNVs across the outbreaks. The vertical bar chart shows the size of the intersection and the black dots and lines show the combination of iSNVs and SNVs. The horizontal bars show the unconditional frequency count of variants within each group. (E) Sequence variability detected in SARS-CoV-2 overlaid with major protein coding regions in the genome. Variants that only occurred as SNVs in more than ten samples are labelled in black while those that also occurred as iSNVs and in more than ten samples as SNVs are marked in red.
2.9 Nucleotide diversity and selection inference
To quantify within-host genetic diversity and infer selection pressures acting on SARS-CoV-2 protein-coding genes, we estimated nonsynonymous (πN; amino acid changing) and synonymous (πS; not amino acid changing) nucleotide diversity using the software SNPGenie (Nelson et al. 2015; https://github.com/chasewnelson/SNPGenie), which implements the method of Nei and Gojobori (1986). The null hypothesis of neutrality (πN = πS) was evaluated with Z-tests using a bootstrap method (codon unit, 10,000 replicates for genes, 1,000 replicates for sliding windows), where πN > πS is consistent with positive selection favoring nonsynonymous variants and πN < πS with purifying (negative) selection favouring the elimination of nonsynonymous variants. Sites overlapping more than one protein-coding gene, including the entirety of ORF9b and ORF9c (both located in the +1 reading frame of N), were excluded from the analysis. Sliding windows of thirty codons were chosen based on the suggestion of (Harrison et al. 2014) and because this did not exceed the length of ORF10 (thirty-nine codons).
2.10 Transmission analyses
We hypothesized that direct or closely linked transmission pairs are likely to share a significant number of iSNVs. To determine the iSNVs shared between putatively related cases and the possibility of these iSNVs having been transmitted, we compared variant calling results of candidate source-recipient pairs of samples. For the CH1 outbreak, we leveraged the putative transmission source-recipient pairs that had previously been inferred by epidemiological investigation and supported by phylogenetic analysis.
Unlike CH1, no transmission pairs had been previously identified for the CH3 outbreak. To infer candidate pairs, we thus treated each sample both as a potential source and recipient for all possible pairs. We permutated the samples to generate source-recipient pairs as below;
where n is the number of samples. This resulted in 5,402 pairs for seventy-four samples, that is each pair was considered twice, once with each member treated as the source or recipient. Pairs with negative sample date differences were eliminated (n = 2,578, 48%), that is a possible source was required to predate a possible recipient, while both pairs were retained if they were collected on the same day. The remaining 2,824 (52%) pairs were further analysed for transmission events.
We examined consensus-level SNP distances between putative pairs using the snp-dists package. An SNP distance of up to two was allowed between putative transmission pairs (Tables 2 and 3; Supplementary Table S4). We also computed the L1-Norm distance between pairs first by determining the absolute difference between the frequencies of the variants shared by putative pairs and then adding to the result the frequencies of the variants that were present in only one of the samples in the pair.
Table 2.
Common consensus mutations shared between putative source-recipient pairs in the CH1 outbreak.
| Source | Recipient |
|---|---|
| P3 (C241T, C3037T, C14408T, A23403G) | P7 (C241T, C3037T, C14408T, A23403G) |
| P10 (C241T, C3037T, C14408T, A23403G) | |
| HW4 (C241T, C3037T, C14408T, A23403G) | |
| P22 (C241T, C3037T, C14408T, C16376T, A23403G) | |
| P5 (C241T, C3037T, C14408T, C16376T, A23403G) | |
| P20 (C241T, C3037T, C14408T, C16376T, A23403G) | |
| P27 (C241T, C3037T, C14408T, C16376T, A23403G) | |
| P29 (C241T, C3037T, C14408T, C16376T, A23403G) | |
| HW4 (C241T, C3037T, C14408T, A23403G) | P11 (C241T, C3037T, C14408T, A23403G) |
| P15 (C241T, C3037T, C14408T, A23403G) | |
| P7 (C241T, C3037T, C14408T, A23403G) | P23 (C241T, C3037T, C14408T, C16376T, A23403G) |
| X1 (C241T, C2997T, C3037T, C14408T, A23403G) | |
| P26 (C241T, C3037T, C14408T, C16376T, A16561C, A23403G) |
Mutations in bold were present in the recipient but not in the source. SNP distances between the genomes were confirmed using snp-dists package. Mutations were called relative to the Wuhan-Hu-1 reference (NC044512.2).
Table 3.
Shared iSNVs, days between samples, SNP distance and bottleneck estimates of CH1 outbreak putative source–recipient pairs.
| Source_ Recipient outbreak ID | Days between samples | SNP distance | Shared iSNVs | Shared iSNV count | Bottle Neck Estimate | lower CI | upper CI | L1_ norm |
|---|---|---|---|---|---|---|---|---|
| P3_P10 | 5 | 0 | U11288G|A12240G|A13003G|A20465G | 4 | 4 | 2 | 8 | 13 |
| P3_HW4 | 7 | 0 | G11241A|G11243C|U11288G|A11556U|A12240G|A13003G| G14707A|A20465G|U22507A|C29187U|A29188G | 11 | 11 | 6 | 19 | 13 |
| P3_P13 | 12 | 1 | U11288G|A11556U|A12240G|A13003G|A13587U| G14707A|G18181U|A20465G|U22507A|U22514A | 10 | 17 | 9 | 34 | 8 |
| P3_P21 | 14 | 1 | U11288G|A11556U|A12240G|A13003G|G14707A|G18181U| A20465G|U22507A|G22763A|C29187U|A29188G | 11 | 10 | 6 | 17 | 13 |
| P3_P22 | 16 | 1 | U11288G|A11556U|A12240G|A13003G|G14707A|G18181U| A20465G|U22507A|G22763A|C29187U|A29188G | 11 | 13 | 8 | 23 | 7 |
| P3_P5 | 5 | 1 | A11556U|A12240G|G22763A|G23302A|C23306G| C29187U|A29188G | 7 | 1000 | 311 | 1,000 | 11 |
| P3_P7 | 11 | 0 | A12240G|A13003G|A13587U|G18181U|G23302A|C23306G | 6 | 32 | 12 | 74 | 6 |
| P3_P29 | 15 | 1 | A12240G|A13003G|G14707A|G18181U|A20465G| U22507A|G22763A | 7 | 5 | 3 | 8 | 12 |
| P3_P20 | 14 | 1 | A11556U|A12240G|A13003G|G14707A|G18181U| A20465G|C29187U|A29188G | 8 | 7 | 4 | 13 | 11 |
| P3_P27 | 14 | 1 | 0 | 942 | 1 | 1,000 | 5 | |
| HW4_P11 | 5 | 0 | U11288G|A11556U|A12240G|A13003G|G14707A|C17933G| U20135A|U22507A | 8 | 8 | 5 | 16 | 15 |
| HW4_P15 | 6 | 0 | 0 | 704 | 1 | 1,000 | 14 | |
| P7_P23 | 3 | 1 | A12240G|A13003G|U17928G|A17929C|C17933G|G18181U| C18904U|U20135A|A20387G | 9 | 38 | 15 | 83 | 12 |
| P7_X1 | 0 | 1 | C12053G|A12240G|A13003G|A13587U|G17252U|A17256G| U17928G|A17929C|C17933G|G18181U|C18904U| U24552C|C25132A|A25136G | 14 | 8 | 6 | 12 | 21 |
| P7_P26 | 3 | 2 | 0 | 725 | 1 | 1,000 | 6 |
Three pairs shared no iSNVs even though other recipients sharing the same source had iSNVs present in the source.
2.11 Bottleneck estimation
We applied the beta-binomial model implemented in the BB Bottleneck software in the exact mode with an upper bound (NbMax) of 1,000 (Sobel Leonard et al. 2017), to estimate the size of the founding population (i.e. the total number of virions transmitted) of the virus transmitted from the source to the recipient. The beta binomial model is superior to the mutation counting method in its ability to account for variant calling thresholds and stochastic viral replication dynamics within the recipient (Sobel Leonard et al. 2017). The method assumes that for a given variant present in the source, the number of transmitted virions carrying the variant is binomially distributed with the bottleneck size refering to the number of trials and transmission probability (success) to the variant frequency in the source (Popa et al. 2020).
3. Results
This study focused on the analysis of 109 SARS-CoV-2 cases from two different nosocomial outbreaks. Clinical characteristics of infected individuals are reported in Supplementary Table S1. The 109 cases were further categorized by outbreak as CH1 (35/109, 32%) and CH3 (74/109, 68%). Of the thirty-five samples collected from CH1 that were available for our analysis, 16 (45.7%) had putative transmission linkages that were supported by phylogenetic inference (Fig. 1A, Table 3). Samples from the beginning of the CH3 outbreak (24/74, 32.4%) were grouped by hospital department and reported social networks. Time-scaled Bayesian phylogenies were inferred for samples that yielded high quality genomes (coverage >90, n = 21/24). Phylogenetic analyses suggested multiple introductions of the virus, that is patients in the same epidemiological group such as the recovery room had different viral profiles (Fig. 1B), and therefore no putative transmission pairs were confirmed.
3.1 Allele frequencies and the mutational landscapes of SARS-CoV-2 genes
All 109 whole-genome sequences analyzed yielded near full-length genomes with coverage greater than ninety per cent and the average read depth ranging from 158.71 to 5046.56 (Supplementary Table S2). Sequencing depth was not detectably associated with the number of iSNVs recorded (R = -0.13, P = 0.18) (Supplementary Fig. S2). In total, 1,841 (1,232 unique) iSNVs were identified across coding regions of the 109 CH1 and CH3 samples at minor allele frequencies (MAFs) between five per cent and fifty per cent (Fig. 2A and B). Higher numbers of individual types of the nucleotide substitutions were observed in CH1 than CH3 samples, with the exception of C→A (45 vs 56) and C→U (147 vs 160). We also observed 820 SNVs, with different mutational patterns to iSNVs. These include C→U (n = 284), A→G (n = 130), G→U (n = 63), and G→A (n = 62) as the most prevalent. Overall, the frequency of the SNVs was higher in CH3 samples than in CH1 samples.
In terms of location in the SARS-CoV-2 genome, a large proportion of iSNVs (0.84) and SNVs (0.79) were found within the S and ORF1ab genes, with a high concentrations specifically in the nsp3 protein of the ORF1ab gene (Supplementary Fig. S3F, Table S3), however, to objectively compare the accumulation of variants in different genes/ORFs, we normalized the variant counts to gene lengths. This revealed higher mutation loads in the N and S genes as well as ORF3a (Table 1). The SNVs identified were spread across 1,337 (4.5%) positions of the genomes of the 109 samples. Three positions (14,707, 1,637, 20,465) demonstrated fixation of the alternative allele in more than ten per cent of the samples in CH1 compared to seven positions (7,064, 16,376, 24,034, 28,628, and 28,881/2/3; Fig. 2E) in CH3.
Table 1.
Summary of iSNVs present at frequencies between 5% and 50% in the 109 SARS-CoV-2 genomes classified according to import on the genes and ORFs in which they occur.
| Gene | Length | High | Moderate | Low | Total, N (v/kbgl)a |
|---|---|---|---|---|---|
| (nonsense) | (non-synonymous) | (synonymous) | |||
| ORF1ab | 21,393 | 41 | 1234 | 466 | 1,741 (81.38) |
| S | 3,822 | 32 | 287 | 141 | 460 (120.36) |
| ORF3a | 828 | 3 | 62 | 32 | 97 (117.15) |
| E | 228 | 2 | 15 | 5 | 22 (96.49) |
| M | 669 | 3 | 42 | 13 | 58 (86.7) |
| ORF6 | 186 | 0 | 2 | 14 | 16 (86.02) |
| ORF7a | 366 | 0 | 11 | 16 | 27 (73.77) |
| ORF7b | 132 | 1 | 1 | 0 | 2 (15.15) |
| ORF8 | 366 | 1 | 16 | 9 | 26 (71.04) |
| N | 1260 | 5 | 139 | 60 | 204 (161.9) |
| ORF10 | 117 | 0 | 5 | 3 | 8 (68.38) |
| Total, N | 88 | 1,814 | 759 |
In the last column, total mutation counts are normalized to number of mutations per kilobase for easy comparison. Majority of the iSNVs detected were nonsynonymous.
N = (total variants in gene/gene length) × 1,000.
The mutations could be further divided into 1,814 nonsynonymous, followed by 759 synonymous, and 88 nonsense (stop lost/gained) mutation categories (Table 1, Supplementary Table S3). Of the observed iSNVs, the majority of nonsynonymous (757 vs 524), synonymous (282 vs 196) and nonsense (54 vs 28) were found in the CH1 samples compared to CH3 samples. In contrast, a larger fraction of nonsynonymous (380 vs 153) and synonymous (236 vs 45) SNVs were found in CH3 than in CH1 samples, unlike nonsense SNVs (4 vs 2).
The iSNVs were distributed in eleven protein-coding viral genes with variable frequencies. Individual iSNVs that were found in multiple different patients samples were most commonly found in genes encoding non-structural proteins, that is nsp8 (A12240G in 24/109 samples), nsp14 (G18181T in 22/109 samples), nsp6 (A11556T in 20/109 samples), nsp9 (A13003G in 20/109 samples), nsp13 (A17929C and T17928G in 17/109 samples), S (T25312A in 17/109 samples), nsp2 (T1483C in 15/109 samples) and nsp15 (T20135A in 15/109 samples).
We also observed high-frequency SNVs A23403G (109/109 in S gene), C14408T (108/109 in nsp12), C3037T (95/109 in nsp3) in the analyzed samples (Fig. 2E). Other genes (E and M) and proteins (nsp5, nsp7, snp9, nsp10, nsp11, ORF6, ORF7a, ORF7b, ORF8, and ORF10) were well conserved, with iSNVs and SNVs frequencies consistently less than ten per cent (Supplementary Fig. S3A and F).
3.2 Transmission of consensus mutations between source-recipient pairs in the CH1 outbreak
Here, we used consensus mutations (SNVs) to explore the transmission dynamics of SARS-CoV-2 within and across samples of sixteen in-hospital patients (P) and healthcare workers (HW). In our report into a nosocomial outbreak of SARS-CoV-2 infections at one of the private hospitals in Durban, South Africa, phylogenetic inferences showed that inpatient-3 (P3, source) infected by the index patient sustained the chains of transmission generating secondary clusters of recipients including HW4 and P7. Table 2 shows common consensus mutations (C241T, C3037T, C14408T, and A23403G) found in the viral consensus sequences of (Cluster1) and its putative recipients (P5, P7, P20, P27, and P29). In addition, P5, P20, P27, and P29 also developed an additional mutation C16376T. Similar mutations were found in secondary clusters 2 (HW4) and 3 (P7), with additional mutations found in consensus sequences of patient P26 (A16561C) and X1 (C2997T) in cluster 3. Development of the additional mutation could be attributed to multiple transmitted strains or selection pressure within the different hosts.
3.3 Transmission dynamics of shared within-host variants between samples
We investigated whether minor alleles with frequencies between five per cent and fifty per cent observed were shared between the epidemiologically inferred source and recipient pairs, and whether these could have been indicative of specific transmission events within the CH1 and CH3 hospital outbreaks.
From the sixteen CH1 samples analyzed, we observed 720 iSNVs. The CH1 iSNVs consisted of 412 nonsynonymous, 154 synonymous, and 29 nonsense variants (Supplementary Table S3). We assessed the evidence for the transmission of shared iSNVs between samples. We found that HW4 (source) was most likely to have transmitted eight iSNVs to the recipient P11 (Fig. 3F, Table 3). In addition, P7 potentially passed on nine iSNVs to P23 (Fig. 3B) and fourteen to X1. Of the fourteen, one (A20465G) later became established as an SNV (Fig. 3L). Furthermore, P3 shared seven iSNVs with P5 (Fig. 3A) and six with P7 (Fig. 3B). P3 was likely to have transmitted four iSNVs to P10, one of which later established as an SNV (Fig. 3G). There were eight shared iSNVs between P3 and P20, with two later established as SNVs (Fig. 3J). P3 shared seven iSNVs with P29, four of which fixed as SNVs (Fig. 3K). In contrast, there was no evidence of shared iSNVs between P3 (source) and recipient P27 (Table 3). Similarly, source HW4 did not share any iSNVs with the recipient P15, nor did source P7with recipient P26. Twenty-nine iSNVs were shared by two or more samples (Supplementary Fig. S1). These findings are consistent with the results from the bottleneck analysis (Table 3, Supplementary Table S4). Based on these findings, we see potentially linked transmission pairs are likely to share a number of iSNVs. Figure 4 shows a reconstruction of the transmission links from the epidemiological investigation incorporating within-host diversity.
Figure 3.

Transmission dynamics of shared within-host variants between CH1 outbreak samples. Each plot shows shared iSNVs between putative donor (red)/recipient (blue) pairs as evidence for transmission. (A) The presence of shared iSNVs between P3 and P5, (B) between P3 and P7. P7 further infected X1 (L) and P23 (C). P3 also sustained a transmission chain through HW4 (I) who infected P11 (F). Finally, P3 infected and transmitted iSNVs to P22 (D), P21(E), P10 (G), P13 (H), P20 (J) and P29 (L). Variants with frequency greater than 0.5 fixed as SNVs in the recipient.
Figure 4.

Reconstruction of CH1and CH3 transmission chains. (A) and (C) show epidemiological links between samples, while (B) and (D) show refined links after in corporating within-host diversity, bottleneck estimates, SNP distance and days between samples for CH1 and CH3, respectively. Bold green line connects pairs with greater than three shared iSNVs, SNP distance of ≤2, and days between samples less than ten. Dashed lines show pairs that shared greater than three iSNVs but days between samples was greater than ten or did not share any iSNVs but SNP distance was less than two and days between samples less than ten. Maroon lines show samples that did not share any iSNVs with the source in CH1 even though other recipients from the same source share multiple iSNVs with the source.
We also explored CH3 samples for co-occurring iSNVs as indicative of transmission events in the population. We leveraged pairwise sample comparisons (SNP distance and L1 Norm), bottleneck estimations and time when samples were acquired to explore putative transmission events within and between transmission clusters identified by phylogenetic analysis. While we found a statistically significant difference (R = 0.07, P < 0.001) between L1 Norm distance and time between samples as well as number of shared variants, we did not consider it as a good indicator of transmission as the distance between sample pairs separated by long periods of time (more than ten days) and therefore unlikely to be actual direct transmission pairs was relatively small compared with those separated by short periods of time (less than ten days) (Supplementary Fig. S4). Of the 2,824 potential source-recipient pairs given seventy four patients, most (2,474/2,824) did not share any iSNVs, 154 pairs shared a single iSNV while 63 had 2 and 133 pairs shared 3 or more iSNVs (Fig. 5A). In terms of nucleotide positions where iSNVs were shared, we observed a number of positions including 6,763, 16,376, 22,675, 24,034, 26,530, and 28,881/2/3 that exhibited strong signals for shared variants (Fig. 5B).
Figure 5.
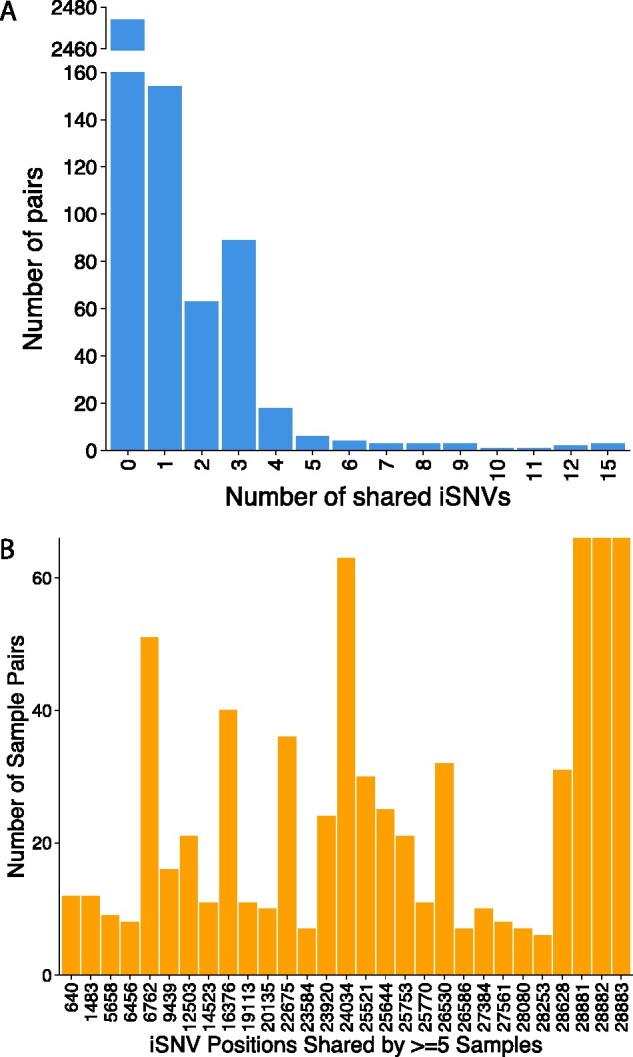
Putative iSNVs transmission events amongst CH3 samples. (A) Gapped barplot showing number of shared iSNVs amongst CH3 pairs and (B) at given nucleotide positions. Majority of pairs had no shared iSNVs while positions 28881/2/3 co-evolved as iSNVs in three samples and SNVs in twenty-three other samples. Positions 6,762, 16,376, 22,675, 24,034, and 26,530 showed strong signals for shared iSNVs and later fixed as SNVs.
In the CH3 sample outbreak, HW7 was identified as one of the likely sources of infection by the epidemiological investigation. We found shared iSNVs between HW7 and HWs eight and eleven suggestive of transmission events between these pairs with HW7 as the source. There was little support for HW7 as the source in other pairs. The potential direct link between HW7 and HW8 was not confirmed as the pair was separated by an SNP distance of 3, which was above the allowed threshold of 2 for pairs to be considered. HW8 and HW9 shared seven iSNVs, were separated by an SNP distance 0 and samples were collected the same day suggesting likely transmission events between the two. Considering the bi-directional bottleneck estimates between the pair, HW9 as identified was the potential source to HW8. We also found strong support for transmission events between HW19 and HW20 (Supplementary Table S4, Fig. 4D). The two were partners and shared upto fifteen iSNVs. Since the samples had been collected on the same day, we explored the bottleneck estimates in both directions. When HW19 was taken as the source, a bottleneck estimate of 85 was estimated and when HW20 was taken as the source, the estimate increased to 91, suggesting that HW19 was a more likely source of infection to HW20. HW25 also shared fifteen and twelve iSNVs with HW19 and HW20, respectively, suggesting possible transmission of the virus from either HW19 or HW20; however, upon comparing the SNP distance between the pairs, HW19 and HW25 were separated by an SNP distance of 3 while HW20 and HW25 were separated by an SNP distance of 2, as such, HW20 was considered the likely source of infection to HW25. Overall, these findings suggest that certain within-host variants in this study could have been transmitted to recipients; however, most within-host variants appear to have arisen independently after transmission.
3.4 Evaluation of the potential impact of false-positives
The presence of shared variants not related by transmission can bias conclusions from the results of transmission analyses and inflate the bottleneck estimates (Martin and Koelle 2021). We therefore explored the impact of masking suspicious variants by masking sixteen mutations that occurred in greater than ten samples (Supplementary Tables S8). First, we re-evaluated these variants against the criteria defined by Lythgoe et al. (2021) (i.e. consistent low-frequency, strand bias, or low reproducibility) and then following Sapoval et al. (2021), we confirmed that they did not occur near either end of the genome. For CH1, we observed an increase in some of the bottleneck estimates (paired t-test; t = 1.6952, P > 0.9), and sample pairs with iSNVs comprised mostly highly abundant variants were no longer supported. All shared variants between P7_P23 were lost. There was a drop in count of shared iSNVs between sample pairs HW4_P11 from 8 to 1, P3_10 dropped from 4 to 1 (Supplementary Tables S9). CH3, on the other hand, was barely affected as the number of shared variants and bottleneck estimates remained consistent (paired t-test; t = 6.8493, P < 0.001) (Supplementary Tables S10). The number of shared variants between HW19 and HW20 reduced from 15 to 13, and the bottleneck estimate from 85 to 77. Together these results highlight the potential of false-positive iSNVs to inflate bottleneck estimates.
3.5 Variant prioritisation and pervasion across outbreaks
Most of the variants expressed in viral sequences usually do not confer any advantage to the virus and therefore are lost; however, a small set of likely advantageous mutations can be positively selected for and fixed as the dominant variant in the population by selection pressure. Using the upset plot (Fig. 2D), we further captured the intersections between iSNVs and SNVs in the CH1 and CH3 datasets to identify frequently and universally occurring iSNVs that could be of potential significance. Three convergent SNVs occurred in CH1 and CH3 independently. We also found eighteen iSNVs identified in the CH1 outbreak that occurred as SNVs in at least one CH3 sample and forty-four iSNVs in the CH3 outbreak that occurred as SNVs in at least one CH1 sample. Furthermore, seven iSNVs that were present in both outbreaks were also present as SNVs in CH3 while two that were present in both outbreaks occurred as SNVs in CH1. Figure 2E shows the dominant variants across outbreaks. Shared variants prevalent in both outbreaks could also be explained by potential spatio-temporal overlap of the outbreaks i.e. the time when they occurred (Fig. 1) and distance between the two hospitals (approx. 4KM apart).
3.6 Selection pressure in the SARS-CoV-2 genome
To assess within-host (intrahost) viral genetic diversity for all samples, we estimated nucleotide diversity (π) as the mean number of single nucleotide differences per site for all protein-coding regions (Nei and Li 1979). Mean π was 2.88 × 10−4, ranging from 0 to 5.97 × 10−3 across samples and was significantly higher in CH1 (5.08 × 10−4) than in CH3 (1.84 × 10−4; P = 2.21 × 10−5; Mann–Whitney U test) (Supplementary Table S6). Diversity was also higher in CH1 than in CH3 samples at both non-synonymous (πN = 2.46 × 10−4 vs 0.84 × 10−4; P = 4.42 × 10−6) and synonymous (πS = 2.62 × 10−4 vs 0.99 × 10−4; P = 2.79 × 10−5) sites (Mann–Whitney U tests). This was true even when limiting to samples sequenced by the same laboratory (NHLS-IALCH, P < 0.00268; Mann–Whitney U test), suggesting this result is not a methodological artefact.
To infer selection pressures acting at the within-host level, we next compared πN to πS, with πN > πS (πN/πS > 1) being consistent with positive selection favoring amino acid changes, and πN < πS (πN/πS < 1) with purifying selection eliminating amino acid changes. Despite the differences in overall diversity between the two outbreaks, their πN/πS ratios were similar, with πN/πS = 0.94 for CH1 and 0.85 for CH3, both statistically indistinguishable from neutrality (P > 0.345; Wilcoxon signed rank tests) (Supplementary Table S6). This result is consistent with the documented preponderance among human viruses of purifying selection acting on viral genomes at the host population scale but the relaxation of selection within hosts (Holmes 2009).
Because disparate selection pressures are expected to act on different sites in a genome, we next computed πN and πS for individual genes and sliding windows within each gene to identify candidate targets of within-host positive selection. However, because our strict quality control criteria eliminated the majority of low-frequency iSNVs, we were underpowered to obtain reliable estimates of πN and πS for most individual samples. We therefore estimated the mean number of nonsynonymous and synonymous differences and sites for each codon across all samples (i.e. codon means rather than sample means), allowing us to identify selection pressures acting consistently across different hosts.
At the whole gene level, the strongest evidence for positive selection was observed for M (πN/πS = 12.46; P = 0.00238) and ORF7a (πN/πS undefined; P = 0.0146) in the CH1 outbreak, and for ORF3a for both the CH3 outbreak (πN/πS = 8.30; P = 0.0317) as well as all combined samples (πN/πS = 3.66; P = 0.0239) (Fig. 6; Supplementary Table S7). The strongest evidence for purifying selection was observed for S in the combined data (πN/πS = 0.48; P = 0.0390) and N in the CH1 outbreak (πN/πS = 0.29; P = 0.0965). Interestingly, despite evidence for purifying selection acting on N in the CH1 outbreak, the ratio for N was >1 (albeit insignificantly so) for the CH3 outbreak (2.16; P = 0.223), warranting investigation with a higher-powered dataset.
Figure 6.

Whole gene within-host nonsynonymous (πN) and synonymous (πS) nucleotide diversity in SARS-CoV-2 samples from the CH1 and CH3 outbreaks. Each gene/outbreak is shaded according to the normalized difference between mean nonsynonymous and synonymous differences per site (πN − πS) to indicate purifying selection (πN < πS; blue) or positive selection (πN > πS; red). Values of πN/πS range from a minimum of 0.007 (nsp9, CH1 outbreak; P = 0.257) to a maximum of 12.46 (M, CH1 outbreak; P = 0.00238), where significance was evaluated using Z-tests of the null hypothesis that πN − πS = 0 (10,000 bootstrap replicates, codon unit). Sites encoding two or more genes in different reading frames were excluded from analysis (e.g. ORF3a sites overlapping ORF3c, ORF3d, or ORF3b). Error bars represent the standard error, evaluated using 10,000 bootstrap replicates (codon unit).
To examine evidence for selection at the within-gene level, we analyzed sliding windows of thirty codons across each protein-coding gene to identify candidate targets of positive selection. Because of the limited number of variants, we took a conservative approach by combining all samples from both outbreaks, allowing us to identify sites undergoing consistent selection pressures in both outbreaks. This analysis yielded several windows for which πN exceeded both whole gene πS and the window’s πS (i.e. πN-SE(πN) > πS+SE(πS)), including regions in nsp2, nsp3, nsp4, nsp6, nsp8, nsp13, nsp14, nsp15, nsp16, E, M, ORF3a, and ORF7b (Supplementary Fig. S5). The longest region was codons 21–190 of nsp8 (length 170 codons), which also had the highest πN/πS = 36.1 (Table 4). Specific codons exhibiting non-synonymous diversity within these regions are listed in Table 4 and serve as a list of candidates for further study.
Table 4.
Candidate regions of positive selection within hosts.
| Gene producta | Codonsb | Length (codons) | π N (×104)c | π S (×104)c | π N/πS (P-value)c,d | Codons with nonsynonymous differencesb,e |
|---|---|---|---|---|---|---|
| nsp2 | 331–369 | 39 | 2.98 (±1.14) | 1.21 (±0.69) | 2.46 | 332, 336, 338, 340+, 345, 355, 359, 360, 362, 365 |
| (0.176) | ||||||
| nsp3 | 103–155 | 53 | 1.69 (±0.75) | 0.07 (±0.08) | 22.92 | 112+, 113, 126+, 132, 142, 143, 153 |
| (0.033)* | ||||||
| nsp3 | 220–255 | 36 | 1.09 (±0.37) | 0 (–) | – | 224, 230, 231+, 233, 236, 247, 249 |
| (0.003)** | ||||||
| nsp3 | 419–457 | 39 | 1.68 (±0.60) | 0 (–) | – | 422, 424+, 441, 442, 445, 448, 449, 457 |
| (0.005)** | ||||||
| nsp3 | 511–540 | 30 | 2.60 (±1.19) | 0.61 (±0.60) | 4.27 | 511, 517, 520, 523, 528 |
| (0.072) | ||||||
| nsp3 | 962–1,007 | 46 | 2.55 (±1.65) | 0.39 (±0.38) | 6.56 | 966, 980, 981, 985+, 991 |
| (0.206) | ||||||
| nsp3 | 1,156–1,274 | 119 | 3.31 (±1.24) | 0 (–) | – | 1175 , 1177, 1186, 1198, 1200, 1202, 1203, 1205, 1216, 1226, 1245, 1246, 1247 |
| (0.008)** | ||||||
| nsp3 | 1,433–1,493 | 61 | 1.87 (±0.71) | 0 (–) | – | 1437, 1449, 1451, 1462, 1464, 1475, 1481, 1482 |
| (0.008)** | ||||||
| nsp3 | 1,589–1,644 | 56 | 1.17 (±0.41) | 0.23 (±0.23) | 5.03 | 1595, 1597, 1599, 1615, 1617, 1618, 1620, 1641 |
| (0.049)* | ||||||
| nsp3 | 1,733–1,765 | 33 | 1.98 (±1.14) | 0.43 (±0.44) | 4.58 | 1738, 1748, 1760, 1761 |
| (0.223) | ||||||
| nsp3 | 1,774–1,824 | 51 | 2.05 (±1.06) | 0.47 (±0.46) | 4.40 | 1789, 1795+, 1796, 1803, 1804, 1807 |
| (0.186) | ||||||
| nsp4 | 140–173 | 34 | 3.76 (±1.48) | 0.61 (±0.61) | 6.16 | 140, 144, 148, 151, 152, 161, 162, 170 |
| (0.059) | ||||||
| nsp6 | 65–127 | 63 | 5.88 (±2.66) | 0.88 (±0.86) | 6.71 | 74, 76, 83, 84, 86, 90, 91, 94, 98, 104, 106+, 112, 119 |
| (0.077) | ||||||
| nsp6 | 169–206 | 38 | 7.83 (±6.23) | 0.86 (±0.84) | 9.08 | 189, 190, 195, 197 |
| (0.266) | ||||||
| nsp8 | 21–190 | 170 | 3.51 (±2.03) | 0.10 (±0.10) | 36.12 | 50 , 57, 59, 60, 85, 91, 92, 106, 107, 110, 112, 119, 129, 138, 141, 145, 159, 163, 174 |
| (0.097) | ||||||
| nsp13 | 565–594 | 30 | 17.55 (±11.66) | 0.83 (±0.87) | 21.09 | 565, 566, 586, 588 |
| (0.158) | ||||||
| nsp14 | 248–289 | 42 | 7.09 (±4.54) | 0.52 (±0.47) | 13.71 | 255+, 267, 269, 272, 274, 276, 278, 286, 289 |
| (0.151) | ||||||
| nsp15 | 83–120 | 38 | 0.82 (±0.41) | 0.08 (±0.08) | 10.46 | 92 , 97, 107, 112+ |
| (0.044)* | ||||||
| nsp15 | 236–337 | 102 | 5.10 (±3.35) | 0 (–) | – | 250, 256, 267, 270+, 282, 287, 321, 324, 327, 336, 337 |
| (0.130) | ||||||
| nsp16 | 85–115 | 31 | 1.76 (±0.84) | 0 (–) | – | 86, 91, 98, 104, 114 |
| (0.037)* | ||||||
| ORF3af | 97–136 | 40 | 5.99 (±3.11) | 1.32 (±1.30) | 4.55 | 100, 103, 117, 118, 121, 123, 125, 126, 128, 130 |
| (0.080) | ||||||
| E | 44–76 | 33 | 4.51 (±2.00) | 1.17 (±0.81) | 3.86 | 50, 52, 58, 60, 68, 71, 72 |
| (0.130) | ||||||
| M | 135–201 | 67 | 1.96 (±0.60) | 0 (–) | – | 154, 158, 160, 161, 163, 164, 167, 187, 189, 193, 196, 198 |
| (0.001)** | ||||||
| ORF7b | 1–38 | 38 | 0.28 (±0.28) | 0 (–) | – | 9 |
| (0.307) |
Genes are ordered 5′ to 3′ by start site in the genome.
Codons are numbered with respect to mature gene products, that is each nonstructural protein (nsp) is re-numbered starting at 1.
Undefined values are indicated with a horizontal line (–).
P-values refer to Z tests of the hypothesis that πN = πS, evaluated for the indicated region using 10,000 bootstrap replicates (codon unit) *P<0.05; **P<0.01.
The codon with the highest πN (best candidate) for each region is shown with underline and bold; codons with evidence for between-host pervasive and episodic positive selection and an increasing frequency trend (Pond 2020) are shown with a ‘+’.
Note that the hypothesized overlapping gene ORF3d occupies codons 44–102 of ORF3a.
4. Discussion
In the present study, we assessed the utility of studying within-host diversity to elucidate selection pressures within, and transmission events between, hosts. We confirmed that our method can improve the power of efforts to retrace transmission events during outbreaks. Specifically, the combination of within-host diversity and bottleneck estimation, SNP distance, and time series improved the resolution of transmission events between hosts in both outbreaks.
Of the fifteen putative source-recipient pairs from CH1, twelve shared more than three iSNVs, suggesting that transmission of iSNVs is indeed common with SARS-CoV-2. This was further supported by bottleneck analysis, which indicated transmission involving at least four virions. The three pairs CH1 putative source-recipient pairs sharing no iSNVs, may have either been incorrectly designated as such during that outbreak investigation (i.e. false categorization), or transmission events between these pairs may have invoved only a single genetic variant (i.e. transmission monophyly (Leitner 2019)). However, given the fact that certain variants are shared by other recipients from the same source, we considered these to be incorrectly designated pairs.
Furthermore, using shared iSNVs and bottleneck estimates between CH3 source–recipient pairs, we brought additional support to the epidemiologically inferred transmission patterns that were originally not clear from phylogenetic analysis, including transmission events between HW7 and HWs 10 and 11. Samples were taken from these three HWs within a ten-day period and all were infected with a predominating genetic variant that was genetically identical, but had minor frequency variants carrying three or more iSNVs. Although HW8 and HW9 shared iSNVs with HW7, they also had an SNP distance of 3 indicating either a higher evolutionary rate of the virus within the new host or the infection of the host with genetically distinct viruses from a different source. We also show that HW20 most likely infected both HW19 and HW25, evidenced by shared iSNVs and estimates of >12 transmitted variants. Interestingly, HW19 and HW24 who were sampled three days apart had an SNP distance of 3 and did not share any within-host variants even though both HWs worked in the same recovery room, suggestive of unrelated transmission events and emphasizing that even healthcare workers are most often infected in the community rather than by patients (Braun et al. 2021).
The narrow transmission bottlenecks observed in these outbreaks may be attributed to a small number of virions that crossed the host cell barrier and established infection, or to deleterious stochastic dynamics via elimination within the respiratory tract (Wang et al. 2020). Low numbers of variants transmitted could also be attributed to the adaptive dynamics theory of evolution, which assumes very limited genetic variation in pathogen populations and that a single pathogen strain will reach equilibrium before a new strain arises by mutation (Berngruber et al. 2013). Ultimately, our bottleneck estimates are consistent with results from other studies (Ghafari et al. 2020; Martin and Koelle 2021; Lythgoe et al. 2021) showing that shared variants related by transmission are characterized by low bottleneck estimates. We also observed some large bottleneck estimates in sample pairs that did not share any iSNVs and in sample pairs where the iSNV frequency was nearly equal between the source and recipient, especially noticeable in samples sharing less than two variants. Similar results have been reported by Popa et al. (2020), which upon further evaluation by Martin and Koelle (2021), appeared to be more likely the results of variants not shared by transmission.
In this study, we set a threshold of at least three shared minor alleles required to support a putative transmission event. This, however, will only hold when the transmission bottleneck is high and not in the event that the bottleneck is low or only the dominant strain is transmitted. This challenge is further exercabated by the stringent variant calling requirements to eliminate false-positive variants while retaining the true variants. As seen in this and other studies (Tonkin-Hill et al. 2020; Sapoval et al. 2021; Lythgoe et al. 2021; Martin and Koelle 2021), after application of quality control measures, most transmission pairs share only one to three minor alleles with many others sharing none. An alternative approach, steming from the understanding that variants not linked by transmission, will likely inflate bottleneck sizes would be that, a single variant passing all quality control criteria, shared at a low bottleneck estimate (e.g. 1–3) in the absence of fixed de novo variants in the recipient (Martin and Koelle 2021), together with strong epidemiological evidence for transmission could be considered linked by transmission. The main limitation to this approach is that the probability of a single variant being spurious is relatively high hence our choice of at least three. This, however, highlights the need for further research on robust techniques for inference of transmission events from shared minor variants under low transmission bottlenecks.
MAF thresholds are an important driver of bottleneck estimates. While MAFs less than two per cent cannot be separated from noise and should be eliminated altogether, higher thresholds result in the loss of true variants. For example, at an allele frequency of two per cent, Popa et al. (2020) reported bottleneck estimates of >50 virions for each transmission pair. Raising the cutoff to three per cent significantly reduced the bottleneck estimates. A further re-analysis of the same data by Martin and Koelle (2021) with the MAF threshold raised to six per cent resulted in a drastic drop in the bottleneck estimates to under 3. Indeed, raising the frequency cuttoff is a quick and efficient way to eliminate false-positive minor alleles; however, it also results in the loss of several potentially true minor alleles and also increases statistical uncertainity (Martin and Koelle, 2021). Instead the use of more effective filtration techniques such as position of the allele on the read, strand bias, number of reads supporting the allele may offer a more balanced criteria for identification and elimination of false positives while retaining true variants, and is encouraged.
Cautious application of masking can help eliminate false positive and strengthen the evidence for transmission events. Low frequency variants can arise de novo within the host rather than through transmission and when selected for can be prevalent across multiple hosts resulting in false signals for transmission. Low-frequency variants can also arise at sites vulnerable to in vitro generation of variants (Lythgoe et al. 2021). In this study, we evaluated the impact of eliminating suspicious variants. Indeed, some sample pairs that were related by only highly abundant low-frequency alleles showed no relation by minor alleles in the masked dataset (Supplementary Table S9). The pairs affected were from the first outbreak. Our rationale for retaining these minor alleles was based on the fact that this outbreak was homogeneous (i.e. occurred among inpatients in a span of two weeks before it was controlled), implying that the infections were highly related and therefore it is not surprising that these variants are common to these patients either through direct or indirect transmission. Furthermore, the close proximity and similar time frame of the second outbreak suggests that some cases between the two outbreaks could be related. Overall, we show that the careful application of masking can help reduce bias in transmission analyses by eliminating false positives.
In order to understand the impact of selection pressures on the patterns of variation represented in the transmission events, we assessed both mutational patterns and the frequency and diversity of within- (iSNVs) and between- (SNVs) host variants. In all samples and across outbreaks, we found an excess of A→G, C→U, U→C, U→A and G→A nucleotide changes in both iSNVs and SNVs (Fig. 2A), and, as expected based on its size, ORF1ab harboured most of the nonsynonymous and synonymous variants compared to other genes (Table 1). It also had the largest fraction of iSNV and SNV mutational patterns with higher numbers in the nsp3 and nsp12 encoding regions, followed by the S gene (Supplementary Figure S3F). However, adjusting for gene length, the highest concentration of variants occurred in the N, S and ORF3a (Table 1). In the S gene A→G, C→U and U→A mutations predominated, in nsp3 by C→U, A→G, and G→A mutations predominated while in nsp12 and nsp13 C→U mutations predominated, both for iSNVs and SNVs. These findings are consistent with previous studies that found an enrichment of C→U mutations in ORF1a (Di Giorgio et al. 2020; Sapoval et al. 2020). It has been suggested that the C→U mutation enrichment in the SARS-CoV-2 genome is likely driven by host response to counter the virus through the APOBEC and ADAR deaminase activity (Di Giorgio et al. 2020; Simmonds and Schwemmle 2020). These studies also note that mutation changes in A→G and U→C in the SARS-CoV-2 genome were mediated by the actions of ADARs, while G→A mutations were derived from APOBEC-mediated C-to-U deamination (Porath et al. 2014; Roth et al. 2019; Di Giorgio et al. 2020; Sapoval et al. 2020).
When assessing within-host nucleotide diversity (π) of SARS-CoV-2 in our samples, we found no significant deviation from neutrality at the whole-genome level for either the CH1 or the CH3 outbreaks. However, at the per-gene level, the πN/πS ratio differed by gene and sometimes by outbreak, with nsp13, nsp15, ORF3a, M, and ORF7a showing mild evidence for positive selection in at least one outbreak. To increase the resolution of this analysis, we also generated a list of candidate regions undergoing positive selection by examining sliding windows across each gene. Of particular interest are nsp3 codon 424 and nsp6 codon 106, which (1) occur within our candidate regions of within-host positive selection; (2) have the highest within-host πN value in their region; and (3) also show evidence of between-host pervasive and episodic positive selection and an increasing frequency trend in the selection analysis of Pond (2020). Although our dataset was underpowered to conduct a more fine-scale analyses, these results serve as an important starting point for further investigations into the possible targets of positive selection acting on the SARS-CoV-2 genome within and between hosts.
Our study is subject to several limitations. It is difficult to distinguish between the transmission of within-host variants and recurrent mutation of the same iSNV in independent hosts. Our quality control criteria yielded few iSNVs, severely limiting the power of our selection analysis; this could be amelioriated by increasing the number of samples in future studies, or by applying more powerful filtering criteria that allow more variants to be retained. Regions in which multiple genes overlap the same sites in different reading frames were excluded from analysis, because they can create artefactual signals of positive selection (Nelson, C. W. 2020b). Finally, our within-host sliding window analysis combined all variants from both the CH1 and CH3 outbreaks. While this allows the detection of selective pressures acting similarly in both outbreaks, it is possible that certain targets of selection experienced different pressures in each outbreak, for example, positive selection in CH1 but purifying selection in CH3.
In summary, we showed that integrating within-host diversity and bottleneck estimates in outbreak investigations can yield better resolution during transmission analyses by providing insights into both chains of infection and directions of transmission. We also showed a complex landscape of within-host diversity and evolution of SARS-CoV-2 during infection, with between-host purifying selection potentially explaining the small number of shared within-host variants (iSNVs) transmitted despite larger estimated viral founding populations. This study therefore enhanced our understanding of potential viral transmissions within and across SARS-CoV-2 cases and shed light on the use of within-host variants and bottleneck estimates to retrace the chains of viral transmission in a population. Results obtained from this study emphasize the need for additional research on the role of within-host variants in modulating antigenicity and pathogenicity to shed light on biological mechanisms driving the rapid spread and complex disease progression of SARS-CoV-2.
Supplementary Material
Acknowledgements
We wish to thank all laboratory personnel that have worked to genotype SARS-CoV-2 samples. We also thank Gerry Tonkin-Hill, Tyler Smith and Daniel B. Weissman for their assistance during this analysis. This study was funded by a research flagship grant from the South African Medical Research Council (MRC-RFA-UFSP-01- 2013/UKZN HIVEPI), the Technology Innovation Agency and the Department of Science and Innovation and National Human Genome Research Institute of the National Institutes of Health under Award Number U24HG006941. H3ABioNet is an initiative of the Human Health and Heredity in Africa Consortium (H3Africa). The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funders.
Data and code availability
Viral consensus genomes reported in this article have been deposited at Global Initiative on Sharing All Influenza Data (GISAID) (all accessions in Supplementary Table S1) (Shu and McCauley 2017) while the raw FastQ sequences have been deposited to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) (Leinonen et al. 2011) (Project Accession No. PRJNA636748). Analysis scripts are available at https://github.com/jsan4christ/within-host-diversity-manuscript-analysis-code
Authors contributions
SEJ, SN, RL, and TdO conceived and designed the analysis and SEJ, SN, AMK, EW, WS, CWN, RL, and TdO performed the analyses. SEJ, SN, AMK, HT, VF, JG, BC, SP, LS, MF, IG, EW, KK, CWN, AMK, WS, DPM, RL, and TdO have contributed to the interpretation and discussion of the results and writing of the article.
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest: None declared.
References
- Bajaj A., Purohit H. J. (2020) ‘Understanding SARS-CoV-2: Genetic Diversity, Transmission and Cure in Human’, Indian Journal of Microbiology, 60: 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berngruber T. W. et al. (2013) ‘Evolution of Virulence in Emerging Epidemics’, PLoS Pathogens, 9: e1003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun K. M. et al. (2021) ‘Viral Sequencing Reveals US Healthcare Personnel Rarely Become Infected with SARS-CoV-2 through Patient Contact’, MedRxiv.
- Butler D. J. et al. (2020) ‘Shotgun Transcriptome and Isothermal Profiling of SARS-CoV-2 Infection Reveals Unique Host Responses, Viral Diversification, and Drug Interactions’, bioRxiv. [DOI] [PMC free article] [PubMed]
- Cingolani P. et al. (2012a) ‘Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift’. Frontiers in Genetics, 3: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P. et al. (2012b) ‘A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila melanogaster Strain w1118; Iso-2; Iso-3’, Fly, 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleemput S. et al. (2020a) ‘Genome Detective Coronavirus Typing Tool for Rapid Identification and Characterization of Novel Coronavirus Genomes’, Bioinformatics, 36: 3552–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleemput S. et al. (2020b) SARS-CoV-2 Genome Assembly Pipeline with Genome Detective for Illumina and Oxford Nanopore Technologies. protocols.io: Kwazulu Natal Research and Innovation Sequencing Platform.
- Costello M. et al. (2018) ‘Characterization and Remediation of Sample Index Swaps by Non-Redundant Dual Indexing on Massively Parallel Sequencing Platforms’, BMC Genomics, 19: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio S. et al. (2020) ‘Evidence for Host-Dependent RNA Editing in the Transcriptome of SARS-CoV-2’, bioRxiv, 2020.03.02.973255. [DOI] [PMC free article] [PubMed]
- Du Plessis L., COVID-19 Genomics UK (COG-UK) Consortium, et al. (2021) ‘Establishment and Lineage Dynamics of the SARS-CoV-2 Epidemic in the UK’, Science, 371: 708–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N. R. et al. (2021) ‘Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in Manaus: Preliminary Findings’, Virological [Online]<https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586> accessed 12 January 2021. [Google Scholar]
- Fontanet A. et al. (2021) ‘SARS-CoV-2 Variants and Ending the COVID-19 Pandemic’, The Lancet, 397: 952–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafari M. et al. (2020) ‘Inferring Transmission Bottleneck Size from Viral Sequence Data Using a Novel Haplotype Reconstruction Method’, Journal of Virology, 94 (13) e00014-20; DOI: 10.1128/JVI.00014-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giandhari J. et al. (2020) ‘Early Transmission of SARS-CoV-2 in South Africa: An Epidemiological and Phylogenetic Report’, medRxiv: The Preprint Server for Health Sciences [Online] <http://europepmc.org/abstract/MED/32511505, 10.1101/2020.05.29.20116376>, <https://europepmc.org/articles/PMC7273273, https://europepmc.org/articles/PMC7273273?pdf=render>. [Accessed 2020/05//]. [DOI] [PMC free article] [PubMed]
- Gojobori T., Moriyama E. N., Kimura M. (1990) ‘Molecular Clock of Viral Evolution, and the Neutral Theory’, Proceedings of the National Academy of Sciences, 87: 10015–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G. et al. (2020) ‘New Insights of Emerging SARS-CoV-2: Epidemiology, Etiology, Clinical Features, Clinical Treatment, and Prevention’, Frontiers in Cell and Developmental Biology, 8: 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison P. W., Jordan G. E., Montgomery S. H. (2014) ‘SWAMP: Sliding Window Alignment Masker for PAML’, Evolutionary Bioinformatics, 10: EBO.S18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Deng Y., Li W. (2020) ‘Coronavirus Disease 2019: What We Know’? Journal of Medical Virology, 92: 719–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C. (2009) The Evolution and Emergence of RNA Viruses. New York: Oxford University Press. [Google Scholar]
- KRISP. (2020) Report into a Nosocomial Outbreak of Coronavirus Disease 2019 (COVID-19) at Netcare St. Augustine's Hospital [Online]. <https://www.krisp.org.za/news.php?id=421 accessed 20 June 2020.
- Lauring A. S. (2020) ‘Within-Host Viral Diversity: A Window into Viral Evolution’, Annual Review of Virology, 7: 63–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R., Sugawara H., Shumway M, & International Nucleotide Sequence Database Collaboration (2011) ‘The Sequence Read Archive’, Nucleic Acids Research, 39: D19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner T. (2019) ‘Phylogenetics in HIV Transmission: Taking Within-Host Diversity into Account’, Current Opinion in HIV and AIDS, 14: 181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas M. et al. (2008) ‘Viral Escape Mechanisms–Escapology Taught by Viruses’, International Journal of Experimental Pathology, 82: 269–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lythgoe K. A. et al. (2020) ‘Shared SARS-CoV-2 Diversity Suggests Localised Transmission of Minority Variants’, bioRxiv, doi: 10.1101/2020.05.28.118992. [DOI]
- Lythgoe K. A., on behalf of the Oxford Virus Sequencing Analysis Group (OVSG), et al. (2021) ‘SARS-CoV-2 within-Host Diversity and Transmission’, Science, 372: eabg0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. A., Koelle K. (2021) ‘Reanalysis of Deep-Sequencing Data from Austria Points towards a Small SARS-COV-2 Transmission Bottleneck on the Order of One to Three Virions’, BioRxiv. [Google Scholar]
- Mavian C. et al. (2020) ‘Regaining Perspective on SARS-CoV-2 Molecular Tracing and Its Implications’, medRxiv. [Google Scholar]
- Nakamura T. et al. (2018) ‘Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments’, Bioinformatics, 34: 2490–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M., Gojobori T. (1986) ‘Simple Methods for Estimating the Numbers of Synonymous and Nonsynonymous Nucleotide Substitutions’, Molecular Biology and Evolution, 3: 418–26. [DOI] [PubMed] [Google Scholar]
- Nei M., Li W. H. (1979) ‘Mathematical Model for Studying Genetic Variation in Terms of Restriction Endonucleases’, Proceedings of the National Academy of Sciences, 76: 5269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. W. et al. (2020a) ‘Dynamically Evolving Novel Overlapping Gene as a Factor in the SARS-CoV-2 Pandemic’, eLife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICD. 2020. Latest Confirmed Cases of COVID-19 in South Africa (31 Oct 2020) [Online]. <https://www.nicd.ac.za/latest-confirmed-cases-of-covid-19-in-south-africa-31-oct-2020/> accessed 31 Oct 2020.
- Pightling A. W. et al. (2019) ‘Within-Species Contamination of Bacterial Whole-Genome Sequence Data Has a Greater Influence on Clustering Analyses than between-Species Contamination’, Genome Biology, 20, 286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay S. et al. (2020) ‘Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic’, bioRxiv, 2020.06.10.144212. [DOI] [PMC free article] [PubMed]
- Pond S. (2020) Natural Selection Analysis of Global SARS-CoV-2/COVID-19 Enabled by Data from GISAID [Online]. <https://observablehq.com/@spond/revised-sars-cov-2-analytics-page> accessed 3 March 2021.
- Popa A. et al. (2020) ‘Genomic Epidemiology of Superspreading Events in Austria Reveals Mutational Dynamics and Transmission Properties of SARS-CoV-2’, Science Translational Medicine, 12: eabe2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath H. T., Carmi S., Levanon E. Y. (2014) ‘A Genome-Wide Map of Hyper-Edited RNA Reveals Numerous New Sites’, Nature Communications, 5: 4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’. Systematic Biology, 67: 901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, Virus Evolution, 2: vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) ‘Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations’, Virological, 9. [Google Scholar]
- Roth S. H., Levanon E. Y., Eisenberg E. (2019) ‘Genome-Wide Quantification of ADAR Adenosine-to-Inosine RNA Editing Activity’, Nature Methods, 16: 1131–8. [DOI] [PubMed] [Google Scholar]
- SAMRC. (2020) Report on Weekly Deaths in South Africa [Online]. <https://www.samrc.ac.za/reports/report-weekly-deaths-south-africa> accessed 31 Oct 2020.
- Sanjuan R., Moya A., Elena S. F. (2004) ‘The Distribution of Fitness Effects Caused by Single-Nucleotide Substitutions in an RNA Virus’, Proceedings of the National Academy of Sciences, 101: 8396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapoval N. et al. (2021) ‘SARS-CoV-2 Genomic Diversity and the Implications for qRT-PCR Diagnostics and Transmission’, Genome Research, 31: 635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapoval N. et al. (2020) ‘Hidden Genomic Diversity of SARS-CoV-2: Implications for qRT-PCR Diagnostics and Transmission’, bioRxiv, 2020.07.02.184481. [DOI] [PMC free article] [PubMed]
- Shen Z. et al. (2020) ‘Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients with Coronavirus Disease’, Clinical Infectious Diseases, 71: 713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., Mccauley J. (2017) ‘GISAID: Global Initiative on Sharing All Influenza Data - from Vision to Reality’, Euro Surveillance, 22(13):30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. PMID: 28382917; PMCID: PMC5388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P., Schwemmle M. (2020) ‘Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories’, mSphere, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siqueira J. D. et al. (2020) ‘SARS-CoV-2 Genomic and Quasispecies Analyses in Cancer Patients Reveal Relaxed Intrahost Virus Evolution’, bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel Leonard A. et al. (2017) ‘Transmission Bottleneck Size Estimation from Pathogen Deep-Sequencing Data, with an Application to Human Influenza a Virus’, Journal of Virology, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evol, 4: vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavare S. (1986) ‘Some Probabilistic and Statistical Problems in Analysis of DNA Sequences’, Lectures in Mathematics in the Life Sciences, 17: 57–86. [Google Scholar]
- Tegally H. et al. (2020) ‘Emergence and Rapid Spread of a New Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Lineage with Multiple Spike Mutations in South Africa’, MedRxiv. [Google Scholar]
- Tonkin-Hill G. et al. (2020) ‘Patterns of within-Host Genetic Diversity in SARS-CoV-2’, BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilsker M. et al. (2019) ‘Genome Detective: An Automated System for Virus Identification from High-Throughput Sequencing Data’, Bioinformatics, 35: 871–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. (2020) ‘Intra-Host Variation and Evolutionary Dynamics of SARS-CoV-2 Population in COVID-19 Patients’, bioRxiv, 2020.05.20.103549. [DOI] [PMC free article] [PubMed]
- Nelson C. W. et al. (2020b) ‘OLGenie: Estimating Natural Selection to Predict Functional Overlapping Genes’, Molecular Biology and Evolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2020) ‘COVID-19 Public Health Emergency of International Concern (PHEIC) Global Research and Innovation Forum’ [Online]<https://www.who.int/publications/m/item/covid-19-public-health-emergency-of-international-concern-(pheic)-global-research-and-innovation-forum>, accessed 03 August 2020.
- Wilm A. et al. (2012) ‘LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets’, Nucleic Acids Research, 40: 11189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfel R. et al. (2020) ‘Virological Assessment of Hospitalized Patients with COVID-2019’, Nature, 581: 465–9. [DOI] [PubMed] [Google Scholar]
- Yu G. (2020) ‘Using Ggtree to Visualize Data on Tree‐like Structures’, Current Protocols in Bioinformatics, 69. [DOI] [PubMed] [Google Scholar]
- Zhou Z.-Y., et al. (2020) ‘Worldwide Tracing of Mutations and the Evolutionary Dynamics of SARS-CoV-2’, biorxiv. [Google Scholar]
- Zhu N. et al. (2020) ‘A Novel Coronavirus from Patients with Pneumonia in China’, New England Journal of Medicine, 382: 727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Viral consensus genomes reported in this article have been deposited at Global Initiative on Sharing All Influenza Data (GISAID) (all accessions in Supplementary Table S1) (Shu and McCauley 2017) while the raw FastQ sequences have been deposited to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) (Leinonen et al. 2011) (Project Accession No. PRJNA636748). Analysis scripts are available at https://github.com/jsan4christ/within-host-diversity-manuscript-analysis-code