

Abstract
The optimal extraction of information from untargeted metabolomics analyses is a continuing challenge. Here, we describe an approach that combines stable isotope labeling, liquid chromatography– mass spectrometry (LC–MS), and a computational pipeline to automatically identify metabolites produced from a selected metabolic precursor. We identified the subset of the soluble metabolome generated from phenylalanine (Phe) in Arabidopsis thaliana, which we refer to as the Phe-derived metabolome (FDM) In addition to identifying Phe-derived metabolites present in a single wild-type reference accession, the FDM was established in nine enzymatic and regulatory mutants in the phenylpropanoid pathway. To identify genes associated with variation in Phe-derived metabolites in Arabidopsis, MS features collected by untargeted metabolite profiling of an Arabidopsis diversity panel were retrospectively annotated to the FDM and natural genetic variants responsible for differences in accumulation of FDM features were identified by genome-wide association. Large differences in Phe-derived metabolite accumulation and presence/absence variation of abundant metabolites were observed in the nine mutants as well as between accessions from the diversity panel. Many Phe-derived metabolites that accumulated in mutants also accumulated in non-Col-0 accessions and was associated to genes with known or suspected functions in the phenylpropanoid pathway as well as genes with no known functions. Overall, we show that cataloguing a biochemical pathway’s products through isotopic labeling across genetic variants can substantially contribute to the identification of metabolites and genes associated with their biosynthesis.
An isotopic labeling and LC–MS pipeline to identify metabolites produced from Phe and its integration with genome-wide association identifies genes associated with the phenylpropanoid pathway.
Introduction
Hundreds of thousands of different chemical compounds are estimated to exist within the ∼350,000 species of flowering plants. This metabolite diversity contributes greatly to plant adaptation and fitness (Pichersky and Lewinsohn, 2011). For example, various specialized metabolites influence pollination by providing flowers with color and scent, defend against pathogens and herbivores through their toxicity, contribute to abiotic stress tolerance and modify the physical characteristics of the plant body through their hydrophobicity and structural rigidity.
Advances in the resolution and sensitivity of analytical techniques permit the detection and measurements of a greater amount of plant chemical diversity. In particular, untargeted mass spectrometry (MS) coupled with liquid chromatography (LC–MS) can detect and quantify, in relative terms, thousands of metabolites and “metabolite features” (MS peaks generated by fragmentation and/or adduct formation in the MS source) within a single analytical run. This information allows for the detection of differences in all mass features across different genetic or environmental contrasts. However, in untargeted metabolomics, the only information collected on a metabolite is its mass-to-charge ratio (m/z), retention time, relative abundance, and any in-source-generated fragmentation products. While untargeted MS techniques are powerful in resolving a metabolome and identifying differences between genotypes or treatments, this information alone is rarely sufficient to assign chemical identities to metabolites or their features. Moreover, any subsequent chemical formula determination and structural identification for metabolites of interest proceeds via low-throughput approaches such as analysis of MS/MS fragmentation patterns and nuclear magnetic resonance spectroscopy. Knowledge of the precursor of a compound of interest would significantly reduce the structure space that would have to be considered when identifying metabolites.
Precursor–product relationships and metabolic pathways have been studied using both radioactive isotopes (Brown and Neish, 1955, 1956; Benson et al., 1950; Roughan et al., 1980) and stable isotopes, with the advent of highly accurate MS (Weng et al., 2012; Allen et al., 2015; Wang et al., 2018). In most labeling studies, a few metabolites of known mass and identity are tracked, despite the fact that dozens to hundreds of other metabolites will also incorporate the label. Several computational programs have been developed to complement isotopic labeling studies and identify labeled metabolites and metabolite features in LC and GC MS datasets (e.g. DLEMMA and MISO [Feldberg et al., 2009; Feldberg et al., 2018; Dong et al., 2019] X13CMS [Huang et al., 2014], MIA [Weindl et al., 2016], geoRge [Capellades et al., 2016], and MetExtract [Bueschl et al., 2012; Bueschl et al., 2017; Doppler et al., 2019]). Here, we describe the development and implementation of a new XCMS-based (Smith et al., 2006) analytical pipeline to detect isotopically labeled metabolite features in untargeted MS datasets. We applied our method (named Pathway of Origin Determination in Untargeted Metabolomics or PODIUM) to identify metabolites incorporating ring-labeled [13C]-phenylalanine (Phe) in stems of WT Col-0 and nine mutants in core enzymes of Arabidopsis thaliana phenylpropanoid metabolism. In addition, we show that the library of Phe-derived MS features can be applied in genome-wide association (GWA) studies to identify genes involved in the biosynthesis of known and yet-uncharacterized Phe-derived metabolites.
Results
A [13C6]-Phe isotopic labeling strategy identifies soluble metabolites derived from phenylalanine in Arabidopsis stems
We developed an isotopic labeling strategy and computational tool to identify MS features that have incorporated an isotopically labeled precursor. This approach adds important information to LC–MS analyses that can be used to filter metabolomics data sets to focus on a metabolic pathway and metabolites derived from a metabolic precursor of interest. The Arabidopsis phenylpropanoid pathway was chosen to develop and evaluate this method because [13C6]-Phe is rapidly incorporated into endogenous substrate pools (Wang et al., 2018), most of the reactions in the canonical pathway have been resolved, and many Arabidopsis soluble phenylpropanoid metabolites have already been identified (Fraser and Chapple, 2011; Vanholme et al., 2012). Thus, the results of our study could be benchmarked by comparison to existing data on genes, enzymes, and metabolites. If successful, this method should identify known players involved in this metabolic pathway and by extension, could be expected to identify novel genes and metabolites when applied to the investigation of more poorly explored pathways.
Phe-derived metabolite features were identified by comparing mass-to-charge ratios (m/z) and retention times for mass features collected and quantified by LC–MS from tissues fed with either a [13C6]-Phe or [12C]-Phe precursor (Figure 1). To specifically identify the precursor-derived mass features, we identified peak-pairs. Peak-pairs are defined as co-eluting MS features that have a difference in m/z corresponding to the number of isotopically labeled carbons in the labeled precursor relative to natural 12C-form. To help eliminate false positives caused by co-eluting metabolites or experimental artifacts, only those peak-pairs whose labeled peak occurred at significantly higher levels in the 13C-fed versus 12C-fed samples were retained. In the end, the user is given .csv files containing the m/z, retention time windows, and ion abundance for all identified MS features across all samples that were put through the pipeline (i.e. the standard XCMS output), and also a file containing the same set of information but only for only identified 12C-13C peak-pair clusters. Detailed information about the program we developed can be found in Supplemental File S1.
Figure 1.

Summary of the pipeline to feed, detect, and positively identify metabolites derived from an isotopically labeled precursor. A, [13C6]-Phe and [12C]-Phe are fed to biologically equivalent stem tissue. B, Metabolites are extracted and separated on LC–MS and peaks are identified with XCMS. All peaks are scanned for MS features consistent with an incorporated [13C6]-Phe. C, Peak-pairs are identified. M301T200 (named as such because it has a [M-H]− m/z of 301 and retention time of 200 s) is a Phe-derived feature because: (1) At 200 s, a peak 6 Da larger than M301 (red color and named M307T200) is detected in [13C6]-Phe-fed tissue. (2) M307T200 in [13C6]-Phe-fed tissue is significantly more abundant than M307T200 in [12C]-Phe-fed tissue. Sketch of Arabidopsis stem was downloaded from FigShare (Bouché, Frédéric [2018]: https://doi.org/10.6084/m9.figshare.7159949.v1).
We evaluated the effectiveness of our labeling and analytical strategy for phenylpropanoids in Arabidopsis stems by examining the degree of labeling in four representative metabolites derived from different branches of the pathway. All mass features throughout this paper are referred to by their negative ion mode (which includes [M-H]− and any adduct ions) m/z ratio and retention time using a C18 reverse-phase column. For example, in wild-type Col-0, the pathway intermediate p-coumaric acid has an [M-H]− m/z value of 163 and elutes at 714 s. The mass feature is therefore referred to as Phe_M163T714 (the “Phe_” prefix denotes that this feature is found in the FDM, as opposed to the GWA dataset to be described later). The pool of p-coumaric acid was labeled in the presence of [13C6]-Phe and the six heavy carbon atoms caused the labeled form to have a m/z ratio of 169 (Phe_M169T714). We found that peaks in a peak-pair vary in their relative abundance depending upon pre-existing metabolite abundances and turnover rates (Figure 2). The ion counts for Phe_M169T714 were 100-fold higher than the background in the [12C]-Phe fed sample, indicating that Phe_M169T714 was derived from [13C6]-Phe (Figure 2, A). The Phe_M169T714 isotope-labeled form of the p-coumaric acid almost completely replaced the 12C form of the metabolite in the [13C6]-Phe fed sample during the 24 h feeding experiment. This extensive labeling is consistent with rapid turnover of p-coumaric acid as an intermediate of phenylpropanoid metabolism. Our pipeline also effectively detected less efficient labeling of compounds. For example, the abundant flavonol-glycoside kaempferol-3-rhamnoside-7-rhamnoside (Phe_M577T729) and the hydroxycinnamate ester sinapoylmalate (Phe_M339T736) co-eluted with +6 Da features (Phe_M583T729 and Phe_M345T736, respectively) in labeled tissues, indicating that they were derived from [13C6]-Phe, as expected. The lower relative accumulation of the 13C-form for those metabolites, in contrast to p-coumaric acid, was consistent with large pre-existing pools of flavonols and sinapoylmalate in stems, to which a proportionately modest number of 13C-derived molecules were added during the feeding period. Knowing that there are compounds built from multiple Phe-derived subunits, we also searched for +12 m/z peak-pairs. Guaiacyl (8-O-4) feruloylmalate is a neolignan thought to be produced from the conjugation of two different Phe-derived intermediates, coniferyl alcohol and feruloylmalate. Accordingly, the unlabeled guaiacyl (8-O-4) feruloylmalate (Phe_M505T750) co-eluted with + 6 Da feature (Phe_M511T750) and a +12 Da feature (Phe_M517T748) in the [12C]-Phe-fed sample. These isotopomers correspond to guaiacyl (8-O-4) feruloylmalate where one or both of the Phe-derived components was derived from [13C6]-Phe. These results demonstrate that our computational approach detects and quantifies the labeling pathway intermediates and end-products in a way that reflects underlying metabolic processes.
Figure 2.
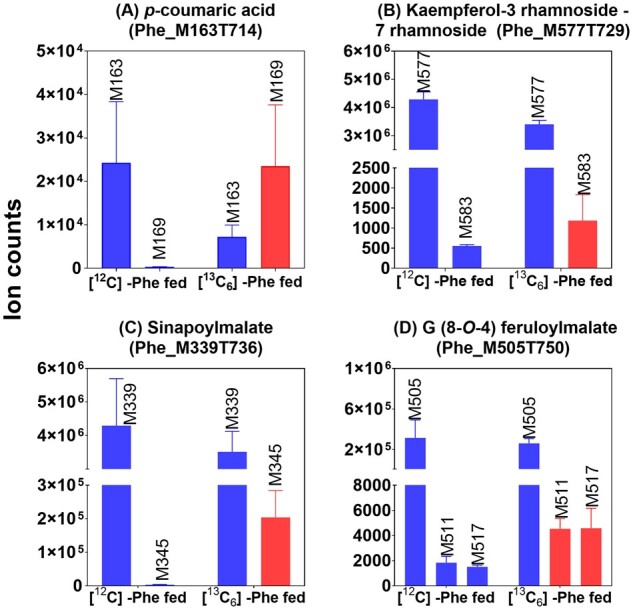
Labeling of four known Phe-derived compounds. Each panel shows the abundance of the indicated Phe-derived metabolite in the [12C]-Phe and [13C6]-Phe-fed wild-type Col-0 stems. Blue bars represent [12C]-derived metabolites and red bars represent the corresponding metabolite(s) identified as incorporating one or two [13C6]-Phe molecules. Error bars indicate ±sd for three biological replicates.
Based on our current understanding of Phe metabolism, most of the peak-pairs detected by application of the pipeline should exhibit an m/z difference of 6 (or higher multiples of 6 such as 12, or 18) due to incorporation of the entire phenyl ring into products. To evaluate this specificity, we set the program to also detect peak-pairs that exhibited an m/z difference of 1–12. As expected, most peak-pairs detected were indeed +6 (Supplemental Figure S1). Because the data were not deisotoped, many of the +4 and +5 peak-pairs detected could be attributed to the pairing of a natural +1 and +2 isotopologue of a [12C6]-Phe-derived compound to a feature that incorporated a [13C6]-Phe-derived ring. To estimate this type of isotopologue pairing, +1 and +2 isotopologues were predicted for the [12C6]-Phe-derived features using CAMERA (Kuhl et al., 2012), and then compared against the lists of +5 and +4 peak-pairs. There were 390 M + 1 and 108 M + 2 isotopologues predicted by CAMERA among the [12C6]-Phe-derived features. Of those isotopologues, 372 and 66 were captured as the [12C] compound in a +5 peak-pair and +4 peak-pair, respectively. Similarly, 695 of the 1094 + 7 peak-pairs were the result of pairing a natural Phe-derived compound to a newly synthesized [13C6]-Phe-derived compound that contained an additional 13C in its Phe sidechain (Supplemental Data Set S1). While isotopologues can account for some of the non +6 pairings, the remaining peak-pairs exhibiting mass differences other than 6 may correspond to unknown products produced from the catabolism of Phe, or false-positive detection of co-eluting compounds of differing masses.
The Phe-derived metabolomes of -type Col-0 differ from those of enzymatic and regulatory mutants of the pathway
The FDM was established in 10 different lines of the Col-0 accession (Supplemental Table S1). In addition to Col-0 -type plants, nine mutants with known alterations in phenylpropanoid accumulation were labeled and profiled. These included plants harboring hypomorphic alleles that affect enzymatic steps in the pathway including reduced epidermal fluorescence (ref) 3-3, which contains a mutation in CINNAMATE 4-HYDROXYLASE (C4H; Schilmiller et al., 2009), omt-1, which is a T-DNA knockout mutant of CAFFEIC ACID/5-HYDROXYFERULIC ACID O-METHYLTRANSFERASE 1 (OMT; Goujon et al., 2003), ccr1, a T-DNA null mutant of CINNAMOYL-COA REDUCTASE 1 (CCR; Mir Derikvand et al., 2008), and fah1-2, a loss-of-function mutant of FERULATE 5-HYDROXYLASE (F5H; Chapple et al., 1992). The tt4-2 mutant, which produces no flavonoids because it is null for CHALCONE SYNTHASE (CHS), was used to identify these metabolites within the profiled sets (Burbulis et al., 1996). In addition to these single mutants, multiple mutants lacking activities encoded by multiple paralogs were also profiled. These included a triple mutant with T-DNA insertions in three of the four p-COUMAROYL COA LIGASE (4CL) genes, 4cl1 4cl2 4cl3 (Li et al., 2015), and the cadC cadD double mutant which contains T-DNA insertions in two CINNAMYL ALCOHOL DEHYDROGENASE (CAD) genes required for the synthesis of cinnamyl alcohols (Anderson et al., 2015b). The med5a med5b double mutant, which is null for both MED5 subunit paralogs of the MEDIATOR transcriptional complex (Bonawitz et al., 2012, 2014) exhibits enhanced flux into the phenylpropanoid pathway and also restores growth to the severely dwarfed ref8–1 hypomorphic mutant of P-COUMARATE 3'-HYDROXYLASE (C3'H) without reversing its chemical phenotype (Franke et al., 2002; Bonawitz et al., 2012). This feature permits the analysis of the ref8 mutant’s chemistry without the complications of radically different growth. The med5a med5b mutant was also used to evaluate the consequences of enhanced pathway flux and as a control for the ref8–1 med5a med5b triple mutant to study the effect of reduced C3′H activity.
In total, 28,136 MS features were identified across the 10 genotypes by our isotope-detection peak picking protocol. Of these, 2,829 were predicted by our peak-pairing method to contain all six carbons from the aromatic ring of Phe, and 448 features were predicted to be derived from multiple Phe molecules (Table 1 and Supplemental Data Set S2). Because samples were run in negative ion mode, metabolites that had a positive charge (e.g. anthocyanins) were not detected. In addition to intact metabolites derived from Phe, the library also contains fragments and adducts of intact Phe-derived metabolites that were formed in the MS source that met the peak-pairing criteria described above.
Table 1.
Phe-derived metabolite features collected in wild-type Col-0 Arabidopsis and nine phenylpropanoid pathway mutants
| Total features collected | Total features after removal of +1 and +2 natural isotopologues | Features incorporating one [13C6]-Phe | Features incorporating two [13C6]-Phe’s | Features incorporating three [13C6]-Phe’s | Features incorporating four [13C6]-Phe’s |
|---|---|---|---|---|---|
| 2,829 | 2,294 | 2,294 | 406 | 39 | 3 |
As stated above, the enzymatic and regulatory mutants used in this study produce many Phe-derived soluble metabolites that differ quantitatively or in terms of presence/absence from wild-type Col-0 (Fraser and Chapple, 2011; Vanholme et al., 2012). To test whether our pipeline detected these differences, we applied orthogonal projections to latent structures discriminant analysis (OPLS-DA; Bylesjö et al., 2006) to the 30 12C-Phe-fed samples (10 genotypes with 3 replicates each) based on the ion abundance of every predicted Phe-derived metabolite feature. In the OPLS-DA score plot (Figure 3), most mutant genotypes occupied distinct spaces across the two components with clear clustering of the three replicates. This pattern suggests that the method is reproducible in detecting Phe-derived MS-features and that the Phe-derived features vary in their accumulation between the different genotypes.
Figure 3.
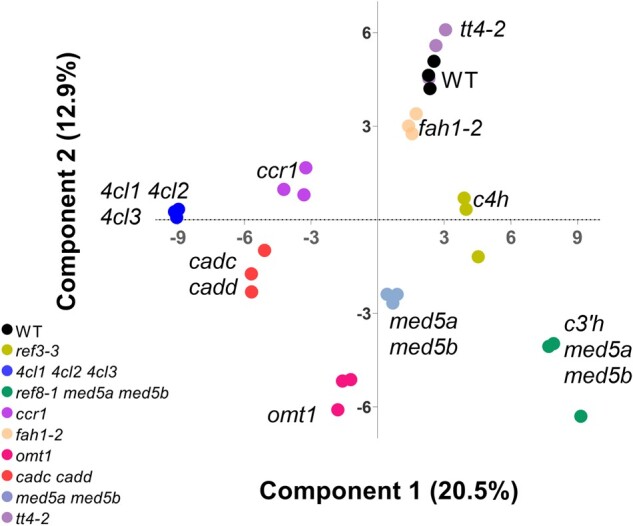
Orthogonal partial least squares discriminant analysis (OPLS-DA) scores plot showing the effect of genotype on the accumulation of Phe-derived metabolite features. The different genotypes are labeled and distinguished by color, and each dot within each genotype represents a biological replicate (n = 3). The values below each axis report the percentage of the variance explained by the first two components. The plot was computed using the annotated Phe-derived features from samples that were fed with [12C]-Phe.
One benefit to measuring a suite of metabolites derived from a specific biochemical pathway is that changes in carbon allocation to a pathway in response to enzymatic or regulatory perturbations can be assessed. To this end, we tabulated relative changes in the total ion counts and individual feature counts in each phenylpropanoid pathway mutant and compared them with wild type. We note that the abundance of Phe-derived MS-features may be influenced by the excess Phe provided during labeling, and different Phe-derived compounds may ionize differently. Nevertheless, the aggregated ion counts for Phe-derived metabolite features from samples that were fed with 12C-Phe was significantly higher in most of the mutants relative to their wild-type controls (Figure 4). Thus, perturbations in many phenylpropanoid-related genes cause Phe-derived pathway intermediates and end products to be redirected to metabolites that are absent or of low abundance in the wild type. However, this is not true for omt1, or tt4-2 and fah1-2, even though they lack flavonoid glycosides and sinapoylmalate, respectively, two classes of abundant Phe-derived metabolites. We also tested whether PODIUM optimally extracted likely Phe-derived MS features, relative to all the MS features captured. Indeed, mutants with a large number of Phe-derived features that differed in abundance relative to wild type (Figures 4, 5) also contained the fewest non-Phe-derived MS features that were different in abundance from wild type (Supplemental Figure S2). Next, we examined differences in ion counts for individual Phe-derived metabolite features in each mutant compared with wild type (Figure 5). Mutants that accumulated more total Phe-derived metabolite features (ref3–3, 4cl1 4cl2 4cl3, ref8–1 med5, ccr1, cadc cadd, med5) also contained multiple features that accumulated to higher levels than in the wild type. This finding is in general agreement with previous observations that some phenylpropanoid-pathway mutants produce novel compounds that are not detected in wild type (Fraser and Chapple, 2011; Vanholme et al., 2012; Bonawitz et al., 2014). Consistent with the total-ion counts, tt4-2, fah1-2, and omt1 did not accumulate as many novel features as the other mutants.
Figure 4.

Aggregate abundance of Phe-derived metabolite features in each genotype. Genotypes significantly different from wild type are denoted by the stars above each bar as determined by one-way ANOVA (****P-value < 0.0001; ***P-value of 0.002; **P-value of 0.0043; ns = not significantly different from wild type) corrected for multiple comparisons using Dunnett’s test. Error bars indicate ±sd of three biological replicates. The plot was computed using the annotated Phe-derived features from samples that were fed with [12C]-Phe.
Figure 5.

Abundance of individual Phe-derived metabolite features in wild-type and mutant genotypes. Colored dots in each panel depicts the average accumulation (n = 3) of a single metabolite feature in a mutant in comparison to its accumulation in wild type (black dots). Features are ordered (left to right) based on their abundance in wild type. Error bars are not plotted, to simplify visualization. The plot was computed using the annotated Phe-derived features from samples that were fed with [12C]-Phe. The full FDM can be found in Supplemental Data Set S2.
Labeling of mutants identifies ion abundance differences in Phe-derived metabolites
We next evaluated whether individual Phe-derived metabolites known to be produced in wild-type Col-0 or are characteristic of mutant genotypes were captured by our labeling. We were able to provide tentative identities to 498 MS features as Phe-derived metabolites using multiple criteria. Specifically, Phe-derived metabolites were annotated if their m/z (±15 ppm) and relative retention time values were consistent with known Phe-derived compounds in Arabidopsis and the characterized mutants if they co-eluted with characteristic daughter ions produced through in-source MS1 fragmentation, and following post hoc MS/MS analysis of selected metabolites from unlabeled wild-type Col-0 plants (Supplemental File S2 and Supplemental Data Set S2; Afendi et al., 2012; Vanholme et al., 2012; Morreel et al., 2014; Sundin et al., 2014; Dima et al., 2015). Metabolite diversity across the mutants was then evaluated by assigning 94 of the best characterized metabolites to eight different structurally diverse groups (oligolignols/lignans/neolignans; flavonol glycosides; and conjugates of benzenoids, cinnamates, coumarates, ferulates, 5-hydroxyferulates, or sinapates).
Metabolite abundances for each of the eight groups were compared between the mutant genotypes by summing the ion counts for all features belonging to a particular class (Figure 6). In general, the abundances of metabolites from each class agreed with previous characterizations of the mutants (Fraser and Chapple, 2011; Vanholme et al., 2012; Bonawitz et al., 2014). Specifically, loss of C4H, 4CL, C3'H, CCR1, and OMT1 resulted in the production of hydroxycinnamic acid (HCA) conjugates (i.e. HCA conjugated to glucose or malate) that were not abundant in wild type. This included accumulation of cinnamoyl conjugates in ref3–3, coumaroyl derivatives in 4cl and c3'h (i.e. ref8–1 med5a med5b) mutants, feruloyl conjugates in ccr1, and 5-hydroxyferuloyl hexose in omt1 (Figure 6). Reduction of C4H activity in ref3–3 also resulted in the accumulation of suspected benzenoids (e.g. hydroxybenzoic acid glucoside) presumably from a competing reaction that chain-shortens cinnamic acid that is no longer being used by C4H (Widhalm and Dudareva, 2015). Sinapoyl conjugates, primarily sinapoylmalate, were reduced in most mutants that contain perturbations in enzymes required for the synthesis of sinapic acid. Exceptions to this included the weak C4H allele, ref3–3 (Schilmiller et al., 2009), and ccr1, which may be functionally redundant with CCR2 (Mir Derikvand et al., 2008). Sinapoyl conjugates increased in the cadC cadD double mutant, presumably due to decreased conversion of hydroxycinnamaldehydes into monolignols and redirection to sinapic acid synthesis. MED5 is a negative transcriptional regulator of phenylpropanoid pathway genes and regulatory factors, and its loss of function caused the accumulation of sinapoylmalate and other Phe-derived products not abundant in wild type, such as 5-hydroxyferuloyl hexose, and neolignans (Bonawitz et al., 2014; Kim et al., 2020). In the ref8–1 med5a med5b triple mutant, the med5a med5b phenylpropanoid hyperaccumulation phenotype persisted but was accompanied by loss of C3′H-dependent metabolites and accumulation of coumaroyl derivatives.
Figure 6.

Total ion counts for 94 selected Phe-derived metabolites across different genotypes. Each panel shows the summed ion counts (±sd; n = 3) for one or more tentatively identified Phe-derived metabolites belonging to a specific metabolic class. Metabolites were identified based matching their m/z values to Arabidopsis Phe-derived metabolite libraries. The identity of some metabolites was also separately confirmed by their MS/MS fragmentation pattern performed post hoc. The plots were computed using the annotated Phe-derived features from samples that were fed with [12C]-Phe. A list of metabolites used in this figure can be found in Supplemental Data Set S2.
Hierarchal clustering of Phe-derived metabolite features in mutant genotypes identifies metabolites of similar biosynthetic origins
The variation in all Phe-derived metabolite features in the different mutant genotypes, relative to wild type, was visualized following hierarchical clustering (Figure 7). In principle, MS features that are derived from metabolites produced by the same branch of the phenylpropanoid pathway will co-vary in two or more genotypes and will co-cluster. For example, omt1 and fah1-2 each lack an enzyme critical to the production of sinapic acid. Those mutants cluster together (y-axis), and there is a strong reduction in a group of co-clustering (x-axis) MS features that contain known sinapate esters. Nevertheless, these two genotypes are distinguished by the clustering algorithm because omt1 accumulates a group of metabolites that includes 5-hydroxyferuloyl hexose, which is a metabolite that is not produced in fah1 (Chapple et al., 1992).
Figure 7.

Dendrogram illustrating log2 fold changes in Phe-derived metabolite features in pathway mutants compared to wild type. A, Dendrogram for the subset of metabolites assigned a tentative identity based on m/z ratio and Phe in structure. B, Dendrogram including all Phe-derived MS-features. For both plots, Phe-derived metabolite features were grouped by the complete linkage method for hierarchical clustering in R (hclust) based on their average log2-fold difference in ion counts compared with wild type. For each metabolite feature, the difference from wild type is described by a color scale relative to wild type (blue = down, white = no change, red = up). Metabolites with a putative identity are denoted by colors and numbered (x-axis) and in (B) representative metabolites for each class are labeled on top of the x-axis. The plots were computed using the annotated Phe-derived features from samples that were fed with [12C]-Phe.
In addition to applying hierarchical clustering to the identified metabolites, we also clustered the hundreds of Phe-derived metabolite features that did not match a soluble phenylpropanoid identified in a metabolite library (Figure 7, B). Many of those unknown features may be uncharacterized metabolites produced from Phe, and thus their co-clustering with known MS features in mutants can provide information about their biosynthesis and structure. This resource can be found in Supplemental Data Set S3.
In addition to the potential identification of novel intact metabolites, hierarchical clustering can also help identify MS-induced artifacts, such as isotopologues, adducts, and in-source fragmentation of intact MS features. CAMERA, a metabolomics tool widely used to identify and eliminate artifacts, applies multiple criteria, including identical LC retention times and ion abundance, to group MS features (Kuhl et al., 2012). CAMERA was not very effective when applied to the FDM because it was unable to distinguish many distinct but co-chromatographing Phe-derived metabolites. For example, sinapoylmalate and feruloylmalate both elute between 737 and 739 s but were incorrectly identified as a single feature by CAMERA. Because CAMERA uses chromatographic and spectral information, and hierarchical clustering uses genetic variance, we applied them sequentially to see if this complementary information about MS features improved the accuracy of the identification of parent ions and their Phe-derived daughter ions. The metabolite dendrogram was split by k-means clustering into 40 groups and MS features in each cluster were then processed using the shared retention time information provided by CAMERA. This grouping approach was evaluated by determining the variance in retention time for MS features within each k-means cluster following CAMERA annotation.
For groups of chemically distinct metabolites that share genetic control, the retention times of features within each of the 40 k-means clusters were highly variable, indicating that each k-means cluster also contains MS features derived from distinct metabolites. Grouping of MS features that share retention times within each k-means cluster using CAMERA annotations further partitioned MS features in each k-means cluster into 2–15 subgroups. The expectation is that most of these subgroups within a k-means cluster will contain a single parental ion and multiple fragments or adducts consistent with fragmentation of the parental ion. For example, sinapoylmalate and feruloylmalate were in separate k-means clusters and known Phe-derived fragments of those two metabolites were clustered with the correct parent metabolite. Applying this process to the entire dataset and retaining one feature per subgroup (putatively identified as the parent ion), collapsed the total number of Phe-derived MS features in the library from 2,294 to 1,337 (Supplemental Table S2). Thus, biochemical pathway mutants combined with pathway-of-origin labeling and shared retention time data improved our data processing pipeline by allowing us to reduce the complexity of the MS data while avoiding erroneously collapsing metabolite features that are derived from distinct compounds.
Functional gene–metabolite relationships can be identified by combining pathway-of-origin annotations with metabolic GWA studies
We next assessed the value of applying the FDM to retrospectively classify metabolites and MS features within independently processed untargeted MS datasets. We established a metabolome containing 3,906 MS features derived from the analysis of the stems of 422 Arabidopsis natural accessions. The MS features were used as traits in GWA analyses in combination with approximately 1.6 million single-nucleotide polymorphisms (SNPs) that had a minor-allele frequency greater than 5% in the chosen accession population. All of the mass features collected from natural accessions with m/z ratios between 120 and 950 and retention times between 250 and 900 s were paired with their corresponding mass feature in the FDM. Although similar chromatographic approaches were used for both metabolomes, because they were established approximately 2 years apart, the m/z ratio and retention times of mass features were not identical and had to be paired within m/z ratio (±15 ppm) retention time windows. The precision of the dataset pairing was verified by manually checking that the most abundant features in Col-0 in the GWA dataset and wild-type Col-0 in the FDM were paired. Differences in retention time and m/z between those abundant features were then used to validate the pairings of the remaining features (Supplemental Data Set S4). In the end, we retrospectively annotated 176 metabolite features in the natural accessions as derived from Phe and identified tens of thousands of SNPs associated with Phe-derived MS features.
This retrospective annotation identified both intact parental ions and MS-induced artifacts and fragmentation ions (e.g. sinapoylmalate and known Phe-derived daughter ions of sinapoylmalate). In the previous Phe-labeling experiments, we used hierarchical clustering based on genotype and shared retention times to collapse many of Phe-derived MS features into a putative parent ion. Here, in a conceptually similar approach, we tested whether the association tests in the GWA could be used to identify likely parental metabolites by identifying groups of metabolite features that co-chromatograph and associate to the same SNPs. To permit comparison of SNP-to-metabolite associations without interference from too many false positive tests, we used tables of associations with P-values less than 10−4 for the comparisons. The differential accumulation of sinapoylmalate and feruloylmalate and their respective daughter ions was again used to illustrate the effectiveness of this approach to collapse the MS features into likely metabolites. Specifically, in numerous natural accessions, a group of Phe-derived metabolite features eluted between 716 and 718 s (Figure 8, A) that included sinapoylmalate (M339T717) and feruloylmalate (M309T718), Phe-containing daughter ions of sinapoylmalate (i.e. m/z 149, 164, 223), feruloylmalate (i.e. m/z 193, 134), and their respective +1 and +2 isotopologues (m/z 340, 341, or m/z 310; Figure 8, B). In total, greater than 20% of the SNPs that associated with sinapoylmalate and its known fragments (with a P-value of less than 9.99 × 10−5), or feruloylmalate and its fragments, were shared. There were no shared associations between sinapoylmalate and feruloylmalate and their fragments (Figure 8, C). Based on the success of collapsing features associated with sinapoylmalate and feruloylmalate, we applied this approach to all 176 predicted Phe-derived features in the GWA dataset. Groups were predicted as instances where two or more Phe-derived features share 5% of the same SNP associations and elute within 5 s of one another. This approach identified 33 feature groups containing 2–16 MS features, and 36 MS features with no apparent fragments (Supplemental Data Set S5). Within each of the 33 metabolite feature groups, a putative parent metabolite was selected as the ion that matched a known Arabidopsis Phe-derived metabolite, or the feature with the largest m/z ratio and/or largest ion abundance. We do note that this process may be limited by ion suppression from co-eluting compounds that may inaccurately associate dissimilar MS features to a common SNP, and by the fact that a parent ion is difficult to predict from MS1 information alone. Nevertheless, by analyzing shared SNP associations, the list of Phe-derived compounds was reduced from 176 to 69 features, 42 of which had m/z values that matched a known Phe-derived metabolite (Supplemental Data Set S4).
Figure 8.

GWA and isotopic labeling can resolve distinct metabolites and fragments and isotopologues that co-chromatograph. A, Extracted ion chromatogram from stems showing all MS features that co-elute with sinapoylmalate (SM) and feruloylmalate (FM). Identified SM and FM fragments are highlighted in red and blue, respectively. B, MS/MS fragmentation of SM (M-H of 339) and FM (M-H of 309). C, The percentage of SNPs that the parental ions, fragments, and isotopologues of SM and FM share.
Phe-derived metabolites vary across Arabidopsis natural accessions
Variation in the 69 predicted parental Phe-derived metabolite features was assessed in the stems of 422 Arabidopsis natural accessions (Figures 9, 10 and Supplemental Data Set S6). The population-average ion count for each predicted Phe-derived MS feature was compared with the reference accession Col-0 and the accession that accumulated the most and least of each respective MS feature (Figure 9). This comparison shows greater than five-fold variation in ion abundance between the highest and lowest accumulating accessions. Figure 10 further illustrates this variation for specific metabolites and metabolite classes (e.g. benzenoids; neolignans; flavonoids; 5-hydroxyferulate; and coumaroyl-, feruloyl-, or sinapoyl-containing metabolites). For many metabolites, notably coumaroyl hexose, feruloyl containing metabolites, and 5-hydroxyferuloyl hexose, a small number of accessions accumulated relatively high levels in comparison to most other accessions. By contrast, sinapoylmalate is present at high levels in almost all accessions but is almost completely absent in two accessions (ICE120 and ICE107). The absence of sinapoylmalate has been observed in other Arabidopsis accessions and results from deletion mutations in the gene responsible for the transesterification of sinapate from sinapoylglucose to malate (Li et al., 2010a). Consistent with these previous studies, ICE20 and ICE107 also exhibit elevated sinapoylglucose (Supplemental Data Set S6).
Figure 9.
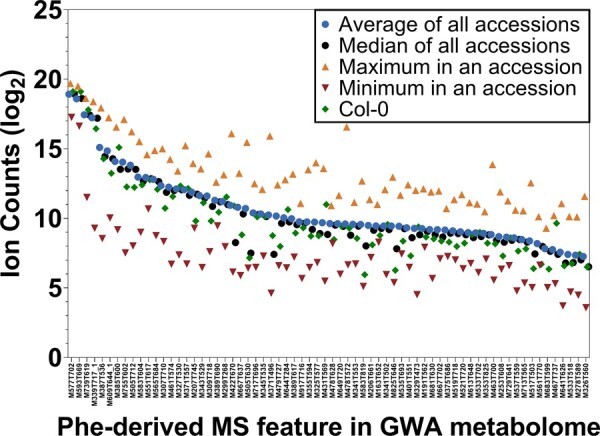
Abundance ranges of Phe-derived metabolite features in 422 Arabidopsis natural accessions. Sixty-nine metabolite features in the GWA dataset predicted to be Phe-derived are designated on the x-axis. Features are ordered based on the average abundance and error bars were excluded for improved visualization. Full data can be found in Supplemental Data Set S5.
Figure 10.

Abundances of selected Phe-derived metabolites across 422 Arabidopsis natural accessions. Plots of average (n = 3) accumulation of tentatively identified Phe-derived metabolites from a specific class for each natural accession (represented by the black dots). Error bars were excluded for improved visualization. Tentative identities of the neolignans (A–M) are given in Supplemental Data Set S6. Flavonol glycosides are identified by their m/z ratio and retention time because multiple similar structures could result in identical m/z ratios.
GWA identifies genetic variation contributing to Phe-metabolite variation
To identify loci encoding variation in phenylpropanoid metabolism, we queried the GWA dataset for SNPs associated to Phe-derived metabolite features at a P-value less than 10−4. Using that criterion, the 69 predicted parental Phe-metabolites formed approximately 59,000 individual SNP–metabolite associations with 50,675 SNPs (Figure 11, A and Supplemental Data Set S7). The number of parental MS features associating to each SNP ranged from 1 to 10 (Figure 11, B). MS features that associate to a particular SNP may be related either by shared genetic control or because they represent unidentified fragments or adducts. As these are all predicted to be Phe-derived, we sought to determine if variation in these metabolites was associated with genes with known or suspected functions in the phenylpropanoid pathway. Associations were placed into three groups: (1) associations to the core phenylpropanoid pathway genes used to construct the FDM, (2) associations between metabolites and SNPs linked to genes that have an experimentally verified or suspected function in the phenylpropanoid pathway (Vanholme et al., 2012), or (3) strong associations to genes with no previously established function in the phenylpropanoid pathway.
Figure 11.

Associations between Phe-derived metabolite features and SNPs linked to core Phe-pathway genes. A, Manhattan plot showing associations between all predicted parental Phe-derived metabolites and all SNPs to which they associate. A single Phe-derived MS feature may associate to multiple SNPs, and a single SNP in the plot may associate to multiple Phe-derived MS features. B, The number of parental Phe-derived MS features that associate to each SNP. C, Manhattan plot showing associations between all predicted parental Phe-derived metabolites and a selection of core phenylpropanoid-pathway genes. D, Ion abundances for metabolite features that associate to SNPs linked to a phenylpropanoid pathway gene (panel C) that when mutated causes the metabolite feature to change in abundance relative to wild type. Each panel is labeled with the name of the gene to which the metabolite feature associates. The red color in the bars represents the accumulation of the metabolite feature in the mutant, and blue bars reports the accumulation in wild type (n = 3 ±SD). Metabolite features with a putative identification based on accessed metabolite libraries are M327T530, caffeyl alcohol hexoside; M371T557, 5-hydroxyferuloyl hexose. E, Ion abundance of unknown MS feature M253T608 across all natural accessions (black dots).
SNPs linked to 11 of the 13 genes used in the mutant labeling experiments formed 243 associations to 34 Phe-derived metabolite features (Figure 11, C and Supplemental Data Set S8, S9). Seven of these metabolite features that were affected by SNPs linked to eight core phenylpropanoid pathway genes were also significantly altered in their accumulation in one or more of the phenylpropanoid mutants (Figure 11, D). The associations to 4CL were of particular interest because of the greater than 10-fold increase in the abundance of associative Phe-derived MS features in the 4cl1 4cl2 4cl3 triple mutant. One of the mutant-induced metabolite features, M253T608, was associated to SNPs linked to all four 4CL genes. Although its structure is unknown, the ion abundance of M253T608 was enriched in only 10 accessions (not including Col-0; Figure 11, E), suggesting that natural genetic variation in 4CLs generates hypomorphic alleles that lead to M253T608 accumulation. There were also several SNPs linked to CADC that associated with variation in M327T530, which is a MS feature that has a mass consistent with a caffeyl alcohol hexoside. Finally, there was a very strong association between SNPs linked to OMT1 and 5-hydroxyferuloyl hexose (M371T557). This association was previously identified in an Arabidopsis GWA with leaf-derived metabolites (Wu et al., 2018), indicating that the same factors influence its variation in a variety of tissues.
The dataset was also queried for associations to SNPs linked to genes involved in the phenylpropanoid pathway other than those represented by mutants in the feeding experiments. We compiled a list of 210 genes with a putative or functionally verified role in the phenylpropanoid pathway (Vanholme et al., 2012). Among the associations at a P-value of 10−4 or lower, 3,918 unique SNP-metabolite feature associations were linked (five genes up and five genes down from SNP) to 205 of the 210 predicted phenylpropanoid pathway genes (Supplemental Data Set S7). Because of the large number of associations, and to reduce false positive discoveries, we focused on 27 independent regions that contained a phenylpropanoid-pathway-related gene which associated to a Phe-derived metabolite feature where at least one SNP association had a P-value of less than 10−8 (Supplemental Table S3). This list included the functionally verified examples of associations to 4CL and OMT1, and a publication-verified association between flavonoid glycosyltransferase UGT78D1 (AT1G30530; Jones et al., 2003) and a flavonoid identified as kaempferol 3,7-di-O-α-l-rhamnoside (M577T702). The list was largely populated by genes with experimentally unverified functions, or associations to unknown Phe-derived metabolites. For example, the strongest association was between UGT72E3, an enzyme associated with monolignol glycosylation (Lim et al., 2005) and unknown metabolite M431T569. The second strongest association was between SNPs located across a GDSL lipase gene cluster (AT1G28580 to AT1G28670) and the accumulation of feruloylmalate (M309T718). This enzyme cluster contains sinapoylcholine esterase (Clauss et al., 2008), suggesting that it, or another enzyme in the cluster, may be involved in the metabolism of other hydroxycinnamate esters, such as feruloylmalate, in stems. Another strong association was between a cluster of four genes encoding putative 2-oxoglutarate-dependent dioxygenases and annotated as flavonol synthases (FLS). Within this cluster, only FLS2 has been shown to affect flavonol production, whereas FLS3 through FLS5 in Col-0 are missing critical functional residues (Preuss et al., 2009). Interestingly, the SNPs linked to the tandemly duplicated FLS2 FLS3 FLS4 and FLS5 genes were associated not with flavonols but with five Phe-derived metabolites, three of which tentatively identified neolignans: guaiacyl (8-O-4) caffeyl alcohol hexose (M505T630), guaiacyl (8-5) feruloyl hexose (M533T702), and guaiacyl (8-5) guaiacyl hexose (M565T684; Supplemental Document S2).
Finally, we examined associations to SNPs linked to genes that were not included in our list of putative phenylpropanoid pathway genes (Supplemental Data Set S7). While there are many thousands of potential causative associations, we specifically searched for low P-value associations to genes that could have a role in phenylpropanoid metabolism based on sequence similarity or co-expression. Of particular interest was an association to SNPs in the tandemly duplicated uncharacterized alcohol dehydrogenases (ALDHs) AT1G22430 and AT1G22440 were strongly linked to Phe-derived MS features preliminarily identified as neolignans: guaiacyl (8-O-4) feruloyl hexose (M551T617), guaiacyl (8-O-4) ferulic acid (M389T690), and guaiacyl (8-O-4) guaiacyl hexose (M583T604; Supplemental Document S2). Moreover, the uncharacterized ALDH enzymes are similar to CADC and CADD and according to ATTEDII (Obayashi et al., 2018) are co-expressed with the phenylpropanoid genes UGT72E2, and pinoresinol reductase 2, both of which are involved in the synthesis of neolignans (Lim et al., 2005; Nakatsubo et al., 2008). Thus, this association appears to identify a previously unknown gene associated with the metabolism of neolignans. While the associations described here are statistically strong, Col-0-derived T-DNA knockouts across the GDSL-lipase cluster (SALK_082907, SALK_094358, SALK_013628), FLS cluster (SALK_023235 and SALK_050041), and AT1G22440 (SALK_00800 and SALK 030343) did not alter the metabolic phenotype, suggesting that the enzymes may function differently in the Col-0 relative to other accessions or have a redundant partner in Arabidopsis and require higher order knockouts to observe a phenotype.
Discussion
Untargeted LC–MS provides the technical capacity to detect hundreds of thousands of metabolites. The only parameters collected in these experiments are m/z ratio, retention time, and ion abundance. Without additional information, these data cannot provide chemical identity. The large structural space occupied by plant metabolites means that this limited information is often not sufficient to fully inform downstream analyses enabled by metabolite identification including the association of metabolites with the enzyme activities, the construction of biosynthetic pathways, and the identification of genes that encode and control these pathways. In fact, it could be argued that we know more about pathways that produce metabolites that can be identified by means other than MS, such as UV absorption or fluorescence, because of the ease with which these metabolites can be detected in a complex extract. The motivation behind our study was to add an analogous dimension of information to MS data that enable the organization of a subset of collected MS features based upon their precursor of origin.
In this manuscript, we describe the successful implementation of a pipeline that accomplishes such a task. Overall, our study identified (1) the biochemical origins for hundreds of Phe-derived mass features, many of which have been previously unannotated and uncharacterized, (2) the Phe-derived metabolomes of nine mutants in the phenylpropanoid pathway, (3) global changes in the soluble metabolic output of the phenylpropanoid pathway when it is perturbed, (4) variation in the FDM for natural accessions of Arabidopsis and identification of putative causal genes through GWA, and (5) mass and retention time for these metabolites that can be used by other researchers to retrospectively annotate Phe-derived metabolites in other untargeted MS datasets. To accomplish this, we developed a new program (PODIUM) that can identify MS features that incorporated fed-isotopic labels within untargeted MS datasets. Simply feeding and identifying MS features in a single reference wild type by this method generates a pathway-specific metabolite library. The addition of a genetic component, via a collection of natural accessions or loss-of-function mutants increased the size of this library and its utility to detect structural and biosynthetic relationships between co-varying MS features. Thus, using genotype as a complementary informational dimension improved the identification of metabolites and candidate genes associated with their synthesis when this approach is combined with GWA.
We chose the well-studied phenylpropanoid pathway and Arabidopsis to test this approach because of the widely available genetic tools and biochemical information. We found that labeling metabolic pathway mutants that have strong or null mutations in single-copy genes and genes that influence a large number of products helped in describing the metabolic space occupied Phe-derived metabolites. In addition, a priori information about the pathway enabled us to evaluate whether metabolites in mutants exhibited the expected changes relative to wild type and allowed us to predict MS feature identity using untargeted MS1 data. Nevertheless, the pipeline does not depend upon extensive prior information or the use of mutants, and we show that identifying pathway specific metabolites across a panel of genetically diverse members of the same species, such as Arabidopsis accessions, aided in the identification of metabolites associated with naturally occurring polymorphisms in core pathway genes in the interrogated pathway. Thus, while the same genetic resources may not be available for other metabolic pathways and plant species, we anticipate that this approach can still be extended to other metabolic pathways, plant species, and even to users conducting research on prokaryotes, fungi, and animals.
Isotopic labeling as a tool to identify biochemical pathway-specific metabolites
In plant biochemistry, both radioactive and stable isotope labeling have been used to determine the metabolic precursors and help elucidate the structure of plant metabolites (Benson et al., 1950; Brown and Neish, 1955, 1956; Roughan et al., 1980; Giavalisco et al., 2009, 2011; Weng et al., 2012; Glaser et al., 2014; Wang et al., 2018; Tsugawa et al., 2019). Arabidopsis has been grown under constant 13CO2, 15N, or 34S to determine its entire element-specific metabolome (Giavalisco et al., 2009, 2011; Glaser et al., 2014; Tsugawa et al., 2019). In addition, biosynthetic pathways for specialized metabolites derived from fatty acids, isoprenoids, and amino acids have been identified by feeding plants substrates such as labeled glucose, acetate, methionine, lysine, Phe, tyrosine, and tryptophan (Halkier and Du, 1997; Lichtenthaler et al., 1997; Weng et al., 2012; Allen et al., 2015; Doppler et al., 2019). Identification of mass features that incorporate a heavy isotope can be done by manually scanning ion chromatograms or by applying computational pipelines, such as through the method we describe here. For applications such as metabolic flux analysis, manual curation of MS data is usually sufficient because the labeling pattern of only a few known intermediates and end products are desired for the flux models (e.g. Wang et al., 2018). By contrast, manual inspection of isotope mass shifts in mass spectral data is impractical for the hundreds to thousands of metabolite features that are expected to be labeled from a metabolic precursor, such as an amino acid. Several MS-data processing programs have been developed to automatically identify mass features that incorporated a fed heavy isotope within untargeted MS1 datasets (Feldberg et al., 2009; Bueschl et al., 2012, 2017; Huang et al., 2014; Capellades et al., 2016; Dong et al., 2019). Our isotope detection program is conceptually similar to those previously described methods, but with some modifications. For example, our method requires separate feeding of tissues with a light and heavy isotope, employs a custom peak-pairing algorithm built on top of XCMS (Smith et al., 2006) to automatically identify labeled MS features, and tests for enrichment of the heavy isotope signal in the samples that were fed with the heavy precursor. Where our method differs from other methods is that it was designed to detect labeled MS features across multiple genotypes, and that it searches for a single type of mass shift at a time which allows for the detection of labeled MS features that meet specific criteria, such as preservation of a labeled structural motif in the precursor (e.g. the +6 labeled phenyl ring of Phe). This contrasts with tracking all metabolites that exhibit any form of a mass shift in the labeled sample, such as occurs in geoRGE and X13CMS (Huang et al., 2014; Capellades et al., 2016). Other approaches have used multiple distinctly labeled precursors to enable automated chemical formula prediction (e.g. DLEMMA and MISO; Feldberg et al., 2009; Dong et al., 2019). While interpretation of multiple labels can provide additional structural information, using a single labeled precursor simplifies the labeling step and the interpretation of labeling patterns, as well as reducing the number of statistical tests needed to detect a labeled metabolite because only one peak-pair is identified. In addition, our method is highly stringent in its detection of multiple labeled metabolites because it requires there to be a labeled peak present for every incorporated precursor molecule. For example, a M + 12 compound derived from Phe must also have a coeluting +6 compound in order to be considered valid.
In total, our pipeline predicted almost 3,000 metabolite features derived from Phe in stems of wild type and nine phenylpropanoid pathway mutants. However, since it has been estimated that upward of 90% of the mass features collected through MS may be artifacts (i.e. fragments and adducts of true metabolites as well as LC/MS signal noise; Mahieu and Patti, 2017), the number of intact Phe-derived metabolites is likely much less than 3,000. Here, we show that it is possible to identify artifacts and collapse multiple MS features into a single peak by adding genetic dimensions to this pipeline. Specifically, we demonstrated that co-chromatographing MS features that are derived from Phe and also co-vary in a specific mutant genotype are likely derived from an identical parental metabolite and that fragments as well as isotopologues can be located by SNP associations identified through GWA. Applying these genetic dimensions predicted that almost 50% of the MS features were in fact artifacts from a measured Phe-derived metabolite. Despite the power to collapse multiple MS features with this process, post hoc MS/MS analysis is still required to accurately determine specific parent and daughter ions among co-chromatographing features.
Even after the MS feature reduction approaches (Supplemental Table S2), the accumulation of over a thousand unknown phenylpropanoids was affected by both natural and induced variants at genes known to encode phenylpropanoid biosynthetic enzymes. We anticipate that the genetic dimensions can also aid in the structural identification of uncharacterized and novel metabolites. We show that clustering similarly accumulating Phe-derived MS features that do not share the same retention time can provide basic branch-of-origin information and preliminary structural information that would be missed if only wild type was examined. For example, MS features produced exclusively in the ref3–3 mutant are likely cinnamate derived, MS features that are lost in fah1 are likely sinapate derived, and MS features lost in tt4 are derived through the flavonoid branch. Along the same lines, if multiple Phe-derived MS features associate to the same set of SNPs it suggests they depend upon a common enzyme for their synthesis and may share some structural similarity. Indeed, a recent GWA analysis on soluble maize metabolites identified structurally related hydroxycinnamate-esters with strong associations to identical SNPs in a gene encoding a citrate synthase enzyme (Zhou et al., 2019). Similarly, we identified multiple putative neolignans that strongly associate to SNPs in a CAD-like alcohol dehydrogenase and a flavanol synthase gene cluster, suggesting that their synthesis commonly depends upon these uncharacterized loci.
Evaluating differences in phenylpropanoid accumulation in wild type and pathway mutants
Although the majority of Phe goes toward production of the insoluble extracellular polymer lignin, a proportion of the flux is responsible for synthesizing a wide array of soluble products, many of which help protect against biotic and abiotic stresses (Vanholme et al., 2012; Wang et al., 2018). Previously, Vanholme et al. (2012) identified and quantified differences in approximately 200 Phe-derived metabolites in wild type and 10 mutants in the pathway, 4 of which were shared with our analysis (f5h, ccr1, c4h, and omt1). However, the identification of Phe-derived compounds in that work was focused on compounds that had a characteristic UV absorbance or could be identified by MS/MS analysis. Our labeling-derived library expands the set of known phenylpropanoids in Arabidopsis approximately six-fold and allows for a global examination of Phe metabolism. This included the exploration of changes in Phe allocation and identification of previously unknown compounds that are accumulated when other steps in the pathway are altered by mutation. Our global assessment of the enzyme mutants found that six of them (ref3–3, 4cl1 4cl2 4cl3, ref8–1, ccr1, cadC cadD) accumulated significantly more soluble metabolites than wild type, whereas omt1, tt4-2, and fah1-2 did not. There is no difference in lignin deposition between wild type and tt4-2 and fah1-2 (Meyer et al., 1996; Li et al., 2010b), whereas the mutants that exhibited an increase in total soluble Phe-derived metabolites generally produce less lignin than wild type (Fraser and Chapple, 2011; Vanholme et al., 2012; Bonawitz et al., 2014). Thus, it seems likely that a small spillover of carbon from lignin allocation into soluble metabolites in mutants with impeded lignin biosynthesis would lead to higher levels of typical metabolites and the accumulation of novel ones. Vanholme et al. (2012) similarly showed that mutants that produce less lignin also upregulate metabolic pathways that supply monolignols and accumulate additional soluble glycosylated phenylpropanoids. Transcriptional feedback mechanisms that down-regulate phenylpropanoid metabolism in fah1 may also have a role in preventing the altered accumulation of soluble phenylpropanoids in that genotype (Anderson et al., 2015a).
The FDM of the med5 mutant illustrates the value of regulatory mutants in identifying pathway-specific metabolites. The med5 mutant over-produces Phe-derived MS features that wild type produces but does not produce the novel metabolites present in ref3–3, 4cl1 4cl2 4cl3, ccr1, or omt1. The use of the med5 ref8–1 triple mutants allows plants harboring ref8–1 to produce a stem that could be fed with Phe (Bonawitz et al., 2014) thereby revealing the effects of blocking this step. The loss of the C′3H enzyme in ref8–1 resulted in more total Phe-derived ions; however, ref8–1 had a metabolite profile similar to 4cl1 4cl2 4cl3 because they block flux through a similar branch of the pathway. This result further supports the hypothesis that med5 regulates Phe flux at PAL (Kim et al., 2020) and that mutants in which lignin monomer biosynthesis is blocked accumulate novel metabolites not present in wild-type controls.
Retrospective identification of phenylpropanoids by GWA identifies pathway specific gene–metabolite relationships
A long-term objective of this work is to identify genes that influence phenylpropanoid biosynthesis through GWA. Specialized metabolic traits are often controlled by few large effect loci; thus, a GWA approach is particularly suited to identify new genes directly influencing these pathways (Wu et al., 2016, 2018). GWA studies with Arabidopsis metabolites identified statistically strong SNP associations (i.e. P-value of lead SNP to metabolite is < 1.0E−08) linked to enzymes belonging to specialized metabolism that were later verified by experimental analysis. These include the identification of metabolites induced by abiotic stress (Wu et al., 2018), discovery of new enzymes for the glycosylation and acylation of flavonoids absent in Col-0 (Ishihara et al., 2016; Tohge et al., 2016), identification of differences in the glycosylation of dihydroxybenzoic acids (Li et al., 2014; Chen and Li, 2017), genes involved in glucosinolate biosynthesis (Chan et al., 2011), and identification of previously unknown amino acid metabolism (Strauch et al., 2015).
Despite the potential to discover novel biochemistry through GWA, it can be challenging to identify genes that control metabolic traits. For example, a single SNP can associate to a group of unrelated MS features, and multiple SNPs can associate with the same MS feature (Atwell et al., 2010; Korte and Farlow, 2013). Thus, without additional data on the potential biological relevance of a GWA result, data analysis can be slowed due to the testing of many candidate genes, and follow-up studies can be biased toward a few known metabolites. This issue can be mitigated by annotation of the biochemical pathway to which the MS features belong. As we demonstrate here with natural variation in phenylpropanoids, identification of MS features as being Phe-derived can improve confidence in selecting candidate genes for further study. We identified associations between known phenylpropanoid pathway genes and known and unknown Phe-derived metabolites, some of which were verified in Col-0 knockouts lines. The most statistically significant were an association between 5-hydroxyferuloyl hexose production and OMT1 that was previously identified in Arabidopsis leaves (Wu et al., 2018) and unknown Phe-derived metabolites that associate to 4CL genes. In addition, this approach located associations between phenylpropanoids and genes with no previously known relationships or experimentally verified functions in the phenylpropanoid pathway. In fact, all of the SNP–FDM associations with a P-value < 1.0e−15 were linked with predicted Phe-derived metabolite genes. Without the basic knowledge of the pathway to which the metabolite belongs, we would not be able to assign these strong associations to linked phenylpropanoid enzymes. For pathways that are less well described, the list of candidate genes could be filtered based on computationally derived annotations or co-expressed genes sets. For example, we identified an association of feruloylmalate to SNPs in a gene cluster that contains an enzyme that metabolizes a related metabolite, sinapoylcholine, in developing seeds, two separate groups of neolignans that strongly associate to SNPs linked to a flavonol synthase-like gene cluster, and an uncharacterized CAD-like alcohol dehydrogenase that is co-expressed with phenylpropanoid-related genes. Together, these results demonstrate that selection of candidate genes affecting metabolites identified by a GWA approach can be greatly aided by knowing at least the metabolic origin of the associative metabolites that is provided by our isotopic labeling approach.
Materials and methods
Plant material and growth conditions
The A. thaliana plants used in the Phe feeding were grown in Redi-Earth Plug and Seedling Mixture (Sun Gro Horticulture) augmented with Scotts Osmocote Plus controlled-release fertilizer (Hummert International). Potted seeds were cold treated at 4°C for 5 days and then moved into a growth chamber (Percival) and grown under a 16-h light/8-h dark photoperiod with a light intensity of 100 μE m−2 s−1 supplied by a combination of halogen and fluorescent bulbs and at a constant temperature of 22°C. The FDM was established in wild-type Col-0 and nine lines with that contain mutations in enzymes of the pathway (Supplemental Table S1).
The Arabidopsis accessions used to generate the GWA dataset were grown as described (Strauch et al., 2015). These accessions were planted in triplicate using a restricted randomization design to distribute genotypes across trays and minimize environment and genotype confounding effects. Three Col-0 plants were planted in each flat at three fixed positions and used to assess variation between flats. All accessions were grown on a single bench in a growth room at 22°C and 50% humidity under long-day conditions (16-h light, 8-h dark) for 7 days. All plants were then moved to 4°C for 8 weeks under 16-h light and 8-h dark cycles to vernalize the plants and induce flowering. Following this treatment, plants were returned to a growth room at 22°C and 50% humidity under long-day conditions (16-h light, 8-h dark) for 28 days. Of the 440 accessions planted, 422 had stems long enough to collect metabolites at this time. The top 10 cm of each bolted inflorescence was cut from the plant, flash frozen by placement in an ethanol-dry ice slurry and then stored at −70°C until metabolite extraction.
Phenylalanine feeding
Phe feeding was performed similarly to Wang et al. (2018). Briefly 4–5-week-old plants were removed from the soil, washed with water, and the top 15 cm of the stem was cut off with double-edged razor blade under water. For each of the three biological replicates, three cut stems from separate plants were placed in 1.5 mL Eppendorf tubes containing 1 mL of ammonia-free Murashige and Skoog medium and either 1 mM [12C] l-Phe (Sigma) or 1 mM ring-[13C6] labeled l-Phe (Cambridge Isotope Laboratories, Cat No. CLM-1055). Stem feeding was done for 24 h under constant light, with new tubes and fresh media being substituted after the first 12 h. At the end of the feeding, each replicate was rinsed with water and patted dry. The basal 5 mm of the stem was removed to mitigate the effects of any localized wounding at the cut site and the next basal 3 cm section of stem was weighed, and flash frozen in liquid nitrogen and stored at −70°C until metabolite extraction.
Metabolite extraction and LC–MS analysis of soluble metabolites
For both datasets, soluble metabolites were extracted from frozen stems in 50% methanol (v/v) at a concentration of 100 mg fresh mass mL−1 at 65°C for 2 h, vortexing every 30 min. Samples were then centrifuged for 5 min at 13,000 × g, and the soluble fraction was transferred to a new tube. For the FDM, samples were concentrated in a speed vacuum at 30°C and the dried extract was then re-dissolved in 50% methanol (v/v) at 10% of the original volume. All extracts were stored at −20°C until LC–MS analysis.
Chromatographic separations were performed using an Agilent 1100 HPLC system (Agilent Technologies, Palo Alto, CA, USA) with a Shimadzu Shim-pack XR-ODS (3.0 × 75 mm × 2.2 µm) separation column and a 5-µL injection volume. A binary solvent system was used where solvent A was 0.1% aqueous formic acid (v/v) and solvent B was 0.1% formic acid (v/v) in acetonitrile. Initial conditions of 98:2 A:B were held for 1 min, followed by linear gradients to 94:6 at 5 min, 54:46 at 15 min, 5:95 at 21.5 min, and a 5:95 hold for 2 min. The column was then re-equilibrated by returning to 98:2 over 1 min and holding for 4 min, for a total analytical run time of 28.5 min. The mobile phase flow rate was 0.6 mL min−1 and the column was maintained at 30°C. Following separation, the column effluent was introduced via negative electrospray ionization (ESI) into an Agilent 6210 time-of-flight mass spectrometer. The following settings were used for the ESI and MS: capillary voltage of 3.2 kV; N2 gas temperature of 350°C; drying gas flow rate of 11 L/min; nebulizer gas pressure of 55 psi; fragmentor voltage of 125 V; skimmer voltage of 60 V; octopole RF of 250 V; mass range 80–1,000 m/z. Mass accuracy was enhanced by infusing Agilent Reference Mass Correction Solution (G1969-85001) throughout each run. Data from each run were centroided and converted to .m/zm/zData format using Agilent MassHunter Qualitative Analysis (v B.06) before analysis by our pipeline.
Isotopic labeling method and computational analysis
The isotopic labeling method involved feeding two biologically equivalent samples with either a labeled or unlabeled precursor, extracting the desired metabolites and then analyzing each sample via LC–MS. This treatment creates a unique MS signature for each metabolite derived from the labeled precursor in the label-fed samples in the form of paired peaks. Paired peaks are defined as metabolite features that coelute and differ in m/z by the mass difference between the labeled and unlabeled precursor. To automate identification of labeled MS features, we developed an R package called PODIUM. The program leverages the unique mass for mass features derived from the heavy isotope to separate them from the rest of the MS signals. The program then compares the labeling pattern for each metabolite against the corresponding data from the non-label-fed samples via an unpaired, one-tailed t test that tests whether the labeled peak in a given peak-pair is significantly greater in the label-fed samples. In doing this, the program can rigorously determine whether the observed labeling pattern is derived from the labeled precursor or the result of random biological variation. Supplemental File S1 contains additional information regarding the peak-pairing algorithm, the settings we applied to generate the FDM, and installing and running the PODIUM package. The full program and sample MS data can be accessed through our GitHub page (https://github.com/chapple-lab/podium).
LC–MS data processing and GWA analysis
Stem metabolite features used for GWA were processed according to the same procedure described in Strauch et al. (2015). Briefly, metabolite features in the accessions were identified using XCMS (Smith et al., 2006) without deisotoping or adduct detection (Supplemental Data Set S10). The SNPs used for mapping were derived from a combination of SNP array and resequencing data (Atwell et al., 2010; Platt et al., 2010; Cao et al., 2011; Horton et al., 2012) followed by imputation using BEAGLE (v3; Browning and Browning, 2011). The resequencing of 80 accessions (Cao et al., 2011) and other accessions obtained from the 1,001 genomes project page resulted in full coverage data for 244 of the accessions used in this study (Atwell et al., 2010). The remaining 196 accessions had genotypes from a SNP array consisting of 211,781 SNPs that corresponded to sequenced SNPs (Horton et al., 2012). Genotypes for all missing positions were imputed using BEAGLE. These genotypes were filtered to remove SNP positions with a minor allele frequency less than 5%, resulting a data set with 1.6 million (1.6M) SNPs that were used in the GWA. Of the 466 genotypes we generated SNP data for, MS features from 422 accessions were used for GWA. Associations were calculated using the Efficient Mixed-Model Association eXpedited procedure. EMMAx corrects for population structure by calculating a kinship matrix and including this matrix in a linear model as a covariate (Kang et al., 2010). To create a database of possible associations, all SNP-to-metabolite associations returning P-values less than 10−4 were recorded. This permitted querying the set of associations for candidate gene associations, and pathway level candidate testing, without a high false-negative rate. False negatives, i.e. failure to score association due to an inappropriately strict statistical cutoff, would present a major impediment to linking metabolite features and a lack of overlap between SNPs would be assessed, incorrectly, as a lack of shared control between metabolic features. In total, from all the mass features, 3,595 detected features had at least one SNP which returned a P-value of less than 10−4.
Accession numbers
Sequence data can be found under the following Arabidopsis Genome Initiative accession numbers: C4H/REF3 (AT2G30490), 4CL1 (AT1G51680), 4CL2 (AT3G21240), 4CL3 (AT1G65060), 4CL4 (AT3G21230), C3′H/REF8 (AT2G40890), CCR1 (AT1G15950), F5H/FAH1 (AT4G36220), CADC (AT3G19450), CADD (AT4G34230), OMT (AT5G54160), CHS/TT4 (AT5G13930), MED5a/RFR1 (AT3G23590), and MED5b/REF4 (AT2G48110).
Supplemental data
Supplemental Figure S1. The number of peak-pairs detected for m/z differences of M + 1 to M + 12.
Supplemental Figure S2. PODIUM optimally extracts likely Phe-derived MS features.
Supplemental Table S1. Mutants used in this study
Supplemental Table S2. Number of Phe-derived MS features detected after each filtering procedure
Supplemental Table S3. Top associations between a Phe-derived MS feature and genes with known or suspected functions in the phenylpropanoid pathway
Supplemental Data Set S1 . Likely sources of non +6 peak-pairs.
Supplemental Data Set S2. Information about metabolite features identified as Phe derived.
Supplemental Data Set S3. Grouping of Phe-derived metabolite features to detect fragments, artifacts, and identify putative parental metabolites.
Supplemental Data Set S4. Pairing of metabolite features between the FDM and GWA datasets.
Supplemental Data Set S5. Summary of rationale for collapsing Phe-derived MS features by shared SNP associations.
Supplemental Data Set S6. Phe-derived MS feature abundance across Arabidopsis natural accessions.
Supplemental Data Set S7. Arabidopsis SNP markers associations to stem Phe-derived MS features.
Supplemental Data Set S8. Arabidopsis SNP markers in or linked to core phenylpropanoid pathway genes and their associations to Phe-derived MS features.
Supplemental Data Set S9. All Phe and predicted non-Phe SNP–MS-feature associations to core phenylpropanoid pathway genes.
Supplemental Data Set S10. Ion intensity values for MS features detected across Arabidopsis natural accessions.
Supplemental Data Set S11. Supporting ANOVA and T test results for Figure 4 and Supplemental Figure S2.
Supplemental File S1. Description of the PODIUM pipeline.
Supplemental File S2. MS/MS spectra for selected phenylalanine-derived metabolites.
Supplementary Material
Acknowledgments
The authors thank Dr. Bruce Cooper (Bindley Bioscience Center, Purdue University) for assistance in acquisition of the LC–MS metabolite profiling data. They also acknowledge Joanne Cusumano and Dr. Yi Li (both of Purdue University) for their contributions in preparing metabolite samples used for GWA.
Funding
This work was supported by the U.S. Department of Energy, Office of Science (BER), Grant DE-SC0020368 (C.C. and B.D.) and by the U.S. Department of Energy, Office of Science (BES), through Grant DE-FG02-07ER15905 (C.C.). J.P.S. was supported in part by a United States Department of Agriculture National Institute of Food and Agriculture postdoctoral grant 2018-08121/1019231.
Conflict of interest statement. None declared.
Contributor Information
Jeffrey P Simpson, Department of Biochemistry, Purdue University, West Lafayette, IN 47907, USA; Purdue University Center for Plant Biology, West Lafayette, IN 47907, USA.
Cole Wunderlich, Department of Biochemistry, Purdue University, West Lafayette, IN 47907, USA.
Xu Li, Plants for Human Health Institute, North Carolina State University, Kannapolis, NC 28081, USA; Department of Plant and Microbial Biology, North Carolina State University, Raleigh, NC 27695, USA.
Brian Dilkes, Department of Biochemistry, Purdue University, West Lafayette, IN 47907, USA; Purdue University Center for Plant Biology, West Lafayette, IN 47907, USA.
Clint Chapple, Department of Biochemistry, Purdue University, West Lafayette, IN 47907, USA; Purdue University Center for Plant Biology, West Lafayette, IN 47907, USA.
J.P.S., C.W., X.L., B.D., and C.C. conceived of the project. All authors contributed to conducting the experiments and analysis and discussion of the results. J.P.S., C.W., B.D., and C.C. wrote the manuscript.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell/pages/General-Instructions) are: Clint Chapple (chapple@purdue.edu) and Brian Dilkes (bdilkes@purdue.edu).
References
- Afendi FM, Okada T, Yamazaki M, Hirai-Morita A, Nakamura Y, Nakamura K, Ikeda S, Takahashi H, Altaf-Ul-Amin M, Darusman LK, et al. (2012) KNApSAcK family databases: integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol 53: e1. [DOI] [PubMed] [Google Scholar]
- Allen DK, Bates PD, Tjellstrom H (. 2015) Tracking the metabolic pulse of plant lipid production with isotopic labeling and flux analyses: past, present and future. Prog Lipid Res 58: 97–120 [DOI] [PubMed] [Google Scholar]
- Anderson NA, Bonawitz ND, Nyffeler K, Chapple C (. 2015a) Loss of FERULATE 5-HYDROXYLASE leads to mediator-dependent inhibition of soluble phenylpropanoid biosynthesis in Arabidopsis. Plant Physiol 169: 1557–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson NA, Tobimatsu Y, Ciesielski PN, Ximenes E, Ralph J, Donohoe BS, Ladisch M, Chapple C (. 2015b) Manipulation of guaiacyl and syringyl monomer biosynthesis in an Arabidopsis cinnamyl alcohol dehydrogenase mutant results in atypical lignin biosynthesis and modified cell wall structure. Plant Cell 27: 2195–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwell S, Huang YS, Vilhjalmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, et al. (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson AA, Bassham JA, Calvin M, Goodale TC, Haas VA, Stepha W (. 1950) The path of carbon in photosynthesis. V. Paper chromatography and radioautography of the products. J Am Chem Soc 72: 1710–1718 [Google Scholar]
- Bonawitz ND, Kim JI, Tobimatsu Y, Ciesielski PN, Anderson NA, Ximenes E, Maeda J, Ralph J, Donohoe BS, Ladisch M, et al. (. 2014) Disruption of mediator rescues the stunted growth of a lignin-deficient Arabidopsis mutant. Nature 509: 376–380 [DOI] [PubMed] [Google Scholar]
- Bonawitz ND, Soltau WL, Blatchley MR, Powers BL, Hurlock AK, Seals LA, Weng JK, Stout J, Chapple C (. 2012) REF4 and RFR1, subunits of the transcriptional coregulatory complex mediator, are required for phenylpropanoid homeostasis in Arabidopsis. J Biol Chem 287: 5434–5445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Neish AC (. 1955) Shikimic acid as a precursor in lignin biosynthesis. Nature 175: 688–689 [DOI] [PubMed] [Google Scholar]
- Brown SA, Neish AC (. 1956) Studies of lignin biosynthesis using isotopic carbon. V. Comparative studies on different plant species. Can J Biochem Phys 34: 769–778 [PubMed] [Google Scholar]
- Browning BL, Browning SR (. 2011) A fast, powerful method for detecting identity by descent. Am J Hum Genet 88: 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueschl C, Kluger B, Berthiller F, Lirk G, Winkler S, Krska R, Schuhmacher R (. 2012) MetExtract: a new software tool for the automated comprehensive extraction of metabolite-derived LC/MS signals in metabolomics research. Bioinformatics 28: 736–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueschl C, Kluger B, Neumann NKN, Doppler M, Maschietto V, Thallinger GG, Meng-Reiterer J, Krska R, Schuhmacher R (. 2017) MetExtract II: a software suite for stable isotope-assisted untargeted metabolomics. Anal Chem 89: 9518–9526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulis IE, Iacobucci M, Shirley BW (. 1996) A null mutation in the first enzyme of flavonoid biosynthesis does not affect male fertility in Arabidopsis. Plant Cell 8: 1013–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylesjö M, Rantalainen M, Cloarec O, Nicholson JK, Holmes E, Trygg J (. 2006) OPLS discriminant analysis: combining the strengths of PLS-DA and SIMCA classification. J Chemometr 20: 341–351 [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Gunther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C et al. (2011) Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet 43: 956–963 [DOI] [PubMed] [Google Scholar]
- Capellades J, Navarro M, Samino S, Garcia-Ramirez M, Hernandez C, Simo R, Vinaixa M, Yanes O (. 2016) geoRge: a computational tool to detect the presence of stable isotope labeling in LC/MS-based untargeted metabolomics. Anal Chem 88: 621–628 [DOI] [PubMed] [Google Scholar]
- Chan EK, Rowe HC, Corwin JA, Joseph B, Kliebenstein DJ (. 2011) Combining genome-wide association mapping and transcriptional networks to identify novel genes controlling glucosinolates in Arabidopsis thaliana. PLoS Biol 9: e1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple CC, Vogt T, Ellis BE, Somerville C (. 1992) An Arabidopsis mutant defective in the general phenylpropanoid pathway. Plant Cell 4: 1413–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HY, Li X (2017) Identification of a residue responsible for UDP-sugar donor selectivity of a dihydroxybenzoic acid glycosyltransferase from Arabidopsis natural accessions. Plant J 89: 195–203 [DOI] [PubMed] [Google Scholar]
- Clauss K, Baumert A, Nimtz M, Milkowski C, Strack D (. 2008) Role of a GDSL lipase-like protein as sinapine esterase in Brassicaceae. Plant J 53: 802–813 [DOI] [PubMed] [Google Scholar]
- Dima O, Morreel K, Vanholme B, Kim H, Ralph J, Boerjan W (. 2015) Small glycosylated lignin oligomers are stored in Arabidopsis leaf vacuoles. Plant Cell 27: 695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Feldberg L, Aharoni A (. 2019) Miso: an R package for multiple isotope labeling assisted metabolomics data analysis. Bioinformatics 35: 3524–3526 [DOI] [PubMed] [Google Scholar]
- Doppler M, Bueschl C, Kluger B, Koutnik A, Lemmens M, Buerstmayr H, Rechthaler J, Krska R, Adam G, Schuhmacher R (. 2019) Stable isotope-assisted plant metabolomics: combination of global and tracer-based labeling for enhanced untargeted profiling and compound annotation. Front Plant Sci 10: 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldberg L, Dong Y, Heinig U, Rogachev I, Aharoni A (. 2018) DLEMMA-MS-imaging for identification of spatially localized metabolites and metabolic network map reconstruction. Anal Chem 90: 10231–10238 [DOI] [PubMed] [Google Scholar]
- Feldberg L, Venger I, Malitsky S, Rogachev I, Aharoni A (. 2009) Dual labeling of metabolites for metabolome analysis (DLEMMA): a new approach for the identification and relative quantification of metabolites by means of dual isotope labeling and liquid chromatography–mass spectrometry. Anal Chem 81: 9257–9266 [DOI] [PubMed] [Google Scholar]
- Franke R, Humphreys J, Hemm MR, Daenault JW, Ruegger MO, Cusumano JC, Chapple CC (. 2002) The Arabidopsis REF8 gene encodes the 3-hydroxylase of phenylpropanoid metabolism. Plant J 2002: 33–45 [DOI] [PubMed] [Google Scholar]
- Fraser CM, Chapple C (2011) The phenylpropanoid pathway in Arabidopsis. Arabidopsis Book; 9: e0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giavalisco P, Kohl K, Hummel J, Seiwert B, Willmitzer L (. 2009) 13C isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography–mass spectrometry-based metabolomic research. Anal Chem 81: 6546–6551 [DOI] [PubMed] [Google Scholar]
- Giavalisco P, Li Y, Matthes A, Eckhardt A, Hubberten HM, Hesse H, Segu S, Hummel J, Kohl K, Willmitzer L (2011) Elemental formula annotation of polar and lipophilic metabolites using (13) C, (15) N and (34) S isotope labelling, in combination with high-resolution mass spectrometry. Plant J 68: 364–376 [DOI] [PubMed] [Google Scholar]
- Glaser K, Kanawati B, Kubo T, Schmitt-Kopplin P, Grill E (. 2014) Exploring the Arabidopsis sulfur metabolome. Plant J 77: 31–45 [DOI] [PubMed] [Google Scholar]
- Goujon T, Sibout R, Pollet B, Maba B, Nussaume L, Bechtold N, Lu F, Ralph J, Mila I, Barriere Y et al. (2003) A new Arabidopsis thaliana mutant deficient in the expression of O-methyltransferase impacts lignins and sinapoyl esters. Plant Mol Biol 51: 973–989 [DOI] [PubMed] [Google Scholar]
- Halkier BA,, Du L (. 1997) The biosynthesis of glucosinolates. Trends Plant Sci 2: 425–431 [DOI] [PubMed] [Google Scholar]
- Horton MW, Hancock AM, Huang YS, Toomajian C, Atwell S, Auton A, Muliyati NW, Platt A, Sperone FG, Vilhjalmsson BJ et al. (2012) Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat Genet 44: 212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Chen YJ, Cho K, Nikolskiy I, Crawford PA, Patti GJ (. 2014) X13CMS: global tracking of isotopic labels in untargeted metabolomics. Anal Chem 86: 1632–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Tohge T, Viehover P, Fernie AR, Weisshaar B, Stracke R (. 2016) Natural variation in flavonol accumulation in Arabidopsis is determined by the flavonol glucosyltransferase BGLU6. J Exp Bot 67: 1505–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Messner B, Nakajima J, Schaffner AR, Saito K (. 2003) UGT73C6 and UGT78D1, glycosyltransferases involved in flavonol glycoside biosynthesis in Arabidopsis thaliana. J Biol Chem 278: 43910–43918 [DOI] [PubMed] [Google Scholar]
- Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, Eskin E (. 2010) Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42: 348–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Zhang X, Pascuzzi PE, Liu CJ, Chapple C (. 2020) Glucosinolate and phenylpropanoid biosynthesis are linked by proteasome-dependent degradation of PAL. New Phytol 225: 154–168 [DOI] [PubMed] [Google Scholar]
- Korte A, Farlow A (. 2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl C, Tautenhahn R, Bottcher C, Larson TR, Neumann S (. 2012) CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal Chem 84: 283–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Bergelson J, Chapple C (. 2010a) The Arabidopsis accession Pna-10 is a naturally occurring sng1 deletion mutant. Mol Plant 3: 91–100 [DOI] [PubMed] [Google Scholar]
- Li X, Bonawitz ND, Weng JK, Chapple C (. 2010b) The growth reduction associated with repressed lignin biosynthesis in Arabidopsis thaliana is independent of flavonoids. Plant Cell 22: 1620–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Svedin E, Mo H, Atwell S, Dilkes BP, Chapple C (. 2014) Exploiting natural variation of secondary metabolism identifies a gene controlling the glycosylation diversity of dihydroxybenzoic acids in Arabidopsis thaliana. Genetics 198: 1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kim JI, Pysh L, Chapple C (2015) Four isoforms of Arabidopsis 4-coumarate:CoA ligase have overlapping yet distinct roles in phenylpropanoid metabolism. Plant Physiol 169: 2409–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler HK, Schwender J, Disch A, Romhmer M (. 1997) Biosynthesis of isoprenoids in higher plant chloroplasts proceeds via a mevalonate-independent pathway. FEBS Lett 400: 271–274 [DOI] [PubMed] [Google Scholar]
- Lim EK, Jackson RG, Bowles DJ (. 2005) Identification and characterisation of Arabidopsis glycosyltransferases capable of glucosylating coniferyl aldehyde and sinapyl aldehyde. FEBS Lett 579: 2802–2806 [DOI] [PubMed] [Google Scholar]
- Mahieu NG, Patti GJ (. 2017) Systems-level annotation of a metabolomics data set reduces 25000 features to fewer than 1000 unique metabolites. Anal Chem 89: 10397–10406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Cusumano JC, Somerville C, Chapple C (. 1996) Ferulate-5-hydroxylase from Arabidopsis thaliana defines a new family of cytochrome P450-dependent monooxygenases. Proc Natl Acad Sci U S A 96: 6869–6874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir Derikvand M, Sierra JB, Ruel K, Pollet B, Do CT, Thevenin J, Buffard D, Jouanin L, Lapierre C (2008) Redirection of the phenylpropanoid pathway to feruloyl malate in Arabidopsis mutants deficient for cinnamoyl-CoA reductase 1. Planta 227: 943–956 [DOI] [PubMed] [Google Scholar]
- Morreel K, Saeys Y, Dima O, Lu F, Van De Peer Y, Vanholme R, Ralph J, Vanholme B,, Boerjan W (. 2014) Systematic structural characterization of metabolites in Arabidopsis via candidate substrate-product pair networks. Plant Cell 26: 929–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsubo T, Mizutani M, Suzuki S, Hattori T, Umezawa T (. 2008) Characterization of Arabidopsis thaliana pinoresinol reductase, a new type of enzyme involved in lignan biosynthesis. J Biol Chem 283: 15550–15557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Aoki Y, Tadaka S, Kagaya Y, Kinoshita K (. 2018) ATTED-II in 2018: a plant coexpression database based on investigation of the statistical property of the mutual rank index. Plant Cell Physiol 59: e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichersky E, Lewinsohn E (2011) Convergent evolution in plant specialized metabolism. Annu Rev Plant Biol 62: 549–566 [DOI] [PubMed] [Google Scholar]
- Platt A, Vilhjalmsson BJ, Nordborg M (. 2010) Conditions under which genome-wide association studies will be positively misleading. Genetics 186: 1045–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss A, Stracke R, Weisshaar B, Hillebrecht A, Matern U, Martens S (. 2009) Arabidopsis thaliana expresses a second functional flavonol synthase. FEBS Lett 583: 1981–1986 [DOI] [PubMed] [Google Scholar]
- Roughan GP, Holland R, Slack RC (. 1980) The role of chloroplasts and microsomal fractions in polar-lipid synthesis from [1-14C] acetate by cell-free preparations from spinach (Spinacia oleracea) leaves. Biochem J 188: 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilmiller AL, Stout J, Weng JK, Humphreys J, Ruegger MO, Chapple C (. 2009) Mutations in the cinnamate 4-hydroxylase gene impact metabolism, growth and development in Arabidopsis. Plant J 60: 771–782 [DOI] [PubMed] [Google Scholar]
- Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G (2006) XCMS: processing mass spectrometry data for metaboite profiling using nonlinear peak alignment, matching, and identification. Anal Chem 78: 779–787 [DOI] [PubMed] [Google Scholar]
- Strauch R, Svedin E, Dilkes B, Chapple CC, Li X (. 2015) Discovery of a novel amino acid racemase through exploration of natural variation in Arabidopsis thaliana. Proc Natl Acad Sci U S A 112: 11726–11731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundin L, Vanholme R, Geerinck J, Goeminne G, Hofer R, Kim H, Ralph J, Boerjan W (. 2014) Mutation of the inducible ARABIDOPSIS THALIANA CYTOCHROME P450 REDUCTASE2 alters lignin composition and improves saccharification. Plant Physiol 166: 1956–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohge T, Wendenburg R, Ishihara H, Nakabayashi R, Watanabe M, Sulpice R, Hoefgen R, Takayama H, Saito K, Stitt M, et al. (. 2016) Characterization of a recently evolved flavonol-phenylacyltransferase gene provides signatures of natural light selection in Brassicaceae. Nat Commun 7: 12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugawa H, Nakabayashi R, Mori T, Yamada M, Takahashi M, Rai A, Sugiyama R, Yamamoto H, Nakaya T, Yamazaki M et al. (2019) A cheminformatics approach to characterize metabolomes in stable-isotope-labeled organisms. Nat Methods 16: 295–298 [DOI] [PubMed] [Google Scholar]
- Vanholme R, Storme V, Vanholme B, Sundin L, Christensen JH, Goeminne G, Halpin C, Rohde A, Morreel K, Boerjan W (. 2012) A systems biology view of responses to lignin biosynthesis perturbations in Arabidopsis. Plant Cell 24: 3506–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Guo L, Jaini R, Klempien A, Mccoy RM, Morgan JA, Dudareva N, Chapple C (. 2018) A (13)C isotope labeling method for the measurement of lignin metabolic flux in Arabidopsis stems. Plant Methods 14: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindl D, Wegner A, Hiller K (. 2016) MIA: non-targeted mass isotopolome analysis. Bioinformatics 32: 2875–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J-K, Li Y, Mo H, Chapple CC (. 2012) Assembly of an evoultionarily new pathway for alpha-pyrone biosynthesis in Arabidopsis. Science 337: 960–964 [DOI] [PubMed] [Google Scholar]
- Widhalm JR, Dudareva N (. 2015) A familiar ring to it: biosynthesis of plant benzoic acids. Mol Plant 8: 83–97 [DOI] [PubMed] [Google Scholar]
- Wu S, Alseekh S, Cuadros-Inostroza A, Fusari CM, Mutwil M, Kooke R, Keurentjes JB, Fernie AR, Willmitzer L, Brotman Y (. 2016) Combined use of genome-wide association data and correlation networks unravels key regulators of primary metabolism in Arabidopsis thaliana. PLoS Genet 12: e1006363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Tohge T, Cuadros-Inostroza A, Tong H, Tenenboim H, Kooke R, Meret M, Keurentjes JB, Nikoloski Z, Fernie AR et al. (2018) Mapping the Arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Mol Plant 11: 118–134 [DOI] [PubMed] [Google Scholar]
- Zhou S, Kremling KA, Bandillo N, Richter A, Zhang YK, Ahern KR, Artyukhin AB, Hui JX, Younkin GC, Schroeder FC et al. (2019) Metabolome-scale genome-wide association studies reveal chemical diversity and genetic control of maize specialized metabolites. Plant Cell 31: 937–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.