

Abstract
Tauopathies are a class of neurodegenerative disorders characterized by neuronal and glial inclusions composed of the hyperphosphorylated tau protein. Four-repeat (4R)-tauopathies are characterized by the presence of inclusions containing only tau isoforms with 4 microtubule-binding domain repeats. The most prevalent pathological entities in this group are corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP). Though distinct pathological entities, CBD and PSP share multiple biochemical and genetic features suggesting overlapping pathophysiology. We report the case of a patient with an 18-year clinical course consistent with behavioral variant frontotemporal dementia. The neuropathological assessment revealed unclassifiable frontotemporal lobar degeneration with tau-immunoreactive inclusions sharing features of both CBD and PSP. Whole-genome sequencing revealed a unique combination of pleiotropic genetic risk variants associated with both PSP and CBD that may have collectively predisposed this individual to higher vulnerability to 4R-tauopathies. These findings support the observation that CBD and PSP share genetic co-expression networks that influence neurodegenerative pathogenesis common to 4R tauopathies.
Keywords: corticobasal degeneration, progressive supranuclear palsy, genetics, pleiotropy, neuropathology, frontotemporal dementia, atypical
Introduction
Tauopathies are a heterogeneous group of neurodegenerative diseases with prominent tau pathology in the neuronal and glial cells of the central nervous system. Tau is an abundant microtubule-associated protein in the cytosol of neurons and, to a much lesser extent, glial cells that plays a central role in microtubule dynamics by regulating assembly, dynamic behavior, and the spatial organization of the cell cytoskeleton1. In tauopathies, tau protein becomes hyperphosphorylated and forms abnormal fibrillary aggregates. Tau exists in 6 isoforms based on the presence of 0, 1, or 2 sequence inserts in the amino-terminus of the protein and inclusion or exclusion of the second of four microtubule-binding potential repeat domains (MTBD). Tauopathies are classified by the predominance of the various tau isoforms found in cytoplasmic inclusions; inclusions predominantly composed of tau with 3 MTBDs (i.e., 3R-tauopathies), 4 MTBDs (i.e., 4R-tauopathies), or a combination of 3R and 4R tau2. The two most prevalent 4R-tauopathies are progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), which are distinct pathological entities with pathognomonic hallmarks. Both PSP and CBD may represent the neuropathological substrate of various clinical syndromes such as PSP-Steele-Richardson-Olszewski’s syndrome3, corticobasal syndrome4, behavioral variant frontotemporal dementia5, and nonfluent variant primary progressive aphasia6. The terms PSP and CBD should not be used to refer to the name of the clinical syndromes that these pathological entities can underlie, though unfortunately, the mutual use of similar terminology by pathologists and clinicians has historically nurtured this misconception.
As a clinicopathologic entity, PSP was first described in 1964 as a progressive neurodegenerative disorder in a cohort of nine patients with progressive vertical supranuclear gaze palsy, pseudobulbar palsy, retrocollis, and axial rigidity, and neuropathological evidence of neurofibrillary tangles (NFTs) and gliosis in the basal ganglia, brainstem structures, and cerebellar dentate nucleus7. Clinical and pathological criteria of PSP, however, were not developed until 1994 and subsequently revised in 1996 by the National Institute of Neurological Disorders and Stroke3,8. The International Parkinson and Movement Disorder Society (MDS) has recently standardized the definition of several PSP phenotypes as a combination of four core PSP-related clinical features (ocular motor dysfunction, postural instability, akinesia, and cognitive dysfunction) with three levels of diagnostic certainty9.
The current neuropathologic criteria for the diagnosis of PSP have remained largely unchanged since 19963. They define definite histopathological PSP as a high density of NFTs and neuropil threads in at least three of the following areas: pallidum, subthalamic nucleus, substantia nigra, or pons. These changes are accompanied by low to high density of pathology in at least three of the following areas: striatum, medulla, oculomotor complex, or dentate nucleus. Fulfillment of these criteria in a patient with PSP-compatible history, after exclusion of ischemic and degenerative lesions diagnostic of other disorders, defines definite PSP. The characteristic glial inclusions, “tufted astrocytes” (TA), were described two years following this consensus criterion10. Since then, the TAs of PSP alongside the pathological characteristics of tau aggregation and the pathological diversity of PSP have been used to help differentiate PSP from other 4R tauopathies11.
CBD was first described as “corticodentatonigral degeneration with neuronal achromasia” in three cases in 196812, while the classic clinical presentation, known as a corticobasal syndrome (CBS), would not be described until 198913. CBS is described as varying combinations of levodopa-unresponsive parkinsonism, asymmetric akinesia/rigidity, limb/oculomotor apraxia, dystonia, cortical sensory deficits, myoclonus, and alien limb phenomenon13–15. The erroneous construct of assigning a single clinical presentation to a distinct pathological entity is perhaps best exemplified by the interchangeable use of CBD and CBS terminology in scientific literature through time. It comes as no surprise that the perpetuation of this misconception has led to the frequent antemortem clinical misdiagnosis of CBD in up to 25–56% of cases4,16–21. Current consensus criteria describe four CBD phenotypes: corticobasal syndrome (CBS), frontal behavioral-spatial syndrome (FBS), nonfluent/agrammatic variant of primary progressive aphasia (nfvPPA), and progressive supranuclear palsy syndrome (PSPS). These clinical CBD phenotypes are then combined to create two sets of criteria: clinical research criteria that are more specific, but less sensitive, to underlying CBD (“Probable CBD”), and broader criteria that are more sensitive, but less specific (i.e., higher chance of false positives), to underlying CBD (“Possible CBD”).4.
The current neuropathologic criteria for the diagnosis of CBD is based on the 2002 Office of Rare Diseases of the National Institutes of Health neuropathologic criteria. The minimal criteria necessary for neuropathological diagnosis of CBD include cortical and striatal tau-positive neuronal and glial lesions, especially astrocytic plaques (AP) and thread-like lesions, in both white matter and gray matter, along with focal cortical and substantia nigra neuronal loss, together with the presence of ballooned neurons22.
Despite being distinct pathological entities, CBD and PSP share considerable clinical and pathological overlap. Recent observations of the varied clinical and pathological presentations of these disorders have shed light on shared tau-related biochemical and genetic co-expression networks responsible for their neurodegenerative pathogenesis23–26. Therefore, it is important to reevaluate and differentiate the features and pathogenesis of these two conditions for both diagnostic and therapeutic efforts. We report the case of a patient who developed clinical features consistent with behavioral variant frontotemporal dementia (bvFTD), whose autopsy demonstrated an unclassifiable tau pathology with intermediate features of CBD and PSP. We discuss in detail the aspects of his clinical presentation and neuropathology, as well as pleiotropic genetic risk factors, to highlight the features that might have played a role in the determinism of these unique pathological findings.
Case presentation
A 47-year-old right-handed male presented to the UCSF Memory and Aging Center clinic with an 11-year history of gradual and progressive decline in behavior and cognition. The participant was enrolled in an ongoing research project on 4-repeat tauopathies at UCSF, for which he provided written informed consent in accordance with the Declaration of Helsinki.
Though never formally diagnosed, his family reported that since childhood, he had exhibited symptoms that they felt mimicked autistic spectrum disorder (ASD) and attention-deficit/hyperactivity disorder (ADHD), as well as cyclothymic behavior that did not significantly interfere with his functioning. At the age of 36, he began exhibiting questionable judgment with diminished social interrelatedness. At the age of 40, he began reneging on family and work responsibilities. By age 41, he began exhibiting dysfunctional attention/working memory and cognitive inflexibility that interfered with his work responsibilities resulting in his job termination. He later struggled to maintain new employment, became socially inappropriate, impulsive, and developed more severe attention/executive impairment. By age 46, he became obsessive, apathetic, sexually inappropriate, and developed abnormal hyperoral behavior that interfered with his ability to complete instrumental activities of daily living.
Neurological examination at age 47 showed a blunted affect, hypomimia, and behaviors mildly inappropriate to the context. Throughout the interview, he was cooperative yet very impulsive, frequently interrupting the interviewer and occasionally laughing. His speech was fluent at a regular rate and volume. Muscle tone was within normal limits without pyramidal and extrapyramidal features. Segmental strength was full, and muscle stretch reflexes (MSRs) were normal. No fasciculations or Babinski signs were noted. Laboratory workup, including serological evaluation (vitamin B12, Folate, TSH, T4, Methylmalonic acid, homocysteine), was unrevealing. He was diagnosed with bvFTD based on his early behavioral disinhibition, impulsivity, apathy, and loss of sympathy/social interrelatedness5.
Fourteen months later, his behavioral outbursts, social/sexual inappropriateness, and repetitive stereotypic behavior had necessitated his relocation to an assisted living facility. His speech was then characterized by pseudobulbar dysarthria, speech apraxia, and echolalic/perseverative. Extraocular movements (initiation, amplitude, and speed) were intact; however, he had difficulty maintaining focus and exhibited diminished impulse inhibition on anti-saccade testing. Muscle tone was increased in his upper extremities in an extrapyramidal fashion without lateralization. Generalized muscle bulk and strength was full and symmetric. His fine motor movements were mildly uncoordinated, and his writing was micrographic. There was bilateral dysdiadochokinesia, limb-kinetic apraxia, and ideomotor apraxia without lateralization. MSRs were normal. No fasciculations or Babinski signs were noted. Small steps and generalized bradykinesia characterized the gait.
Following this last examination, his speech became progressively more dysarthric and spastic, and he began developing swallowing difficulty by the age of 51. By the age of 52, his speech had become incomprehensible, and he lost the ability to walk without assistance. At the age of 53, he became bedridden, and he eventually died of respiratory failure at age 54, following an estimated disease duration of 18 years.
Other medical history included depression, hypertension, insomnia, and obstructive sleep apnea managed by CPAP. His medications included lisinopril, bupropion, trazodone, and sertraline. He never smoked or used illicit drugs and occasionally drank alcohol. His family had a mixed Russian/Polish/Ashkenazi heritage with a history remarkable for unspecified late-onset cognitive decline in both grandfathers, obsessive-compulsive disorder (OCD) in his father and child, and another child with ASD.
Neuropsychological Testing
The patient completed two formal neuropsychological evaluations27 through our research program at ages 47 and 49 (14 months apart). Table 1 shows raw scores from comprehensive neuropsychological testing at both time points.
Table 1.
Neuropsychological performance at ages 47 and 49 (14 months apart)
| Cognitive Test Scores | Age 47 | Age 49 | Normative Reference* | ||
|---|---|---|---|---|---|
| M (SD) | Mdn (IQR) | ||||
| Global | CDR | ||||
| Global | 0.5 | 1.0 | - | - | |
| Sum of Boxes | 4.0 | 7.0 | - | - | |
| Behavior | 2.0 | 3.0 | - | - | |
| Language | 0.5 | 1.0 | - | - | |
| MMSE | 26 | 22 | 29.2 (1.0) | 29 (29, 30) | |
| Memory | CVLT-SF (9-word list) | ||||
| Immediate Recall (T1-T4) | 18 | 15 | 28.9 (3.1) | 29 (27, 32) | |
| 30” Delay | 6 | 5 | 7.7 (1.3) | 8 (7, 9) | |
| 10’ Delay | 7 | 5 | 7.2 (1.5) | 7.5 (6, 8) | |
| Recognition (d’) | 3.5 | 1.5 | 3.1 (0.5) | - | |
| Benson Figure | |||||
| 10’ Delay | 13 | 15 | 12.7 (2.4) | 13 (11, 14) | |
| Recognition | Correct | Correct | - | ||
| Executive Function | Digit Span | ||||
| Forward | 5 | 4 | 7.3 (1.2) | 7 (6, 8) | |
| Backward | 3 | 3 | 5.7 (1.3) | 6 (5, 7) | |
| Modified Trails | |||||
| Correct Lines (max=14) | 14 | 5 | - | - | |
| Time” (max=120”) | 25” | 120” | 24.2 (10.3) | 22 (17, 29) | |
| Errors | 1 | 12 | - | - | |
| Design Fluency (designs/min) | |||||
| Total Correct | 4 | 2 | 12.6 (3.4) | 12 (10, 15) | |
| Repetition Errors | 7 | 17 | - | ||
| D-Words (words/min) | |||||
| Total Correct | 4 | 3 | 15.9 (4.6) | 16 (13, 19) | |
| Errors | 1 | 7 | - | - | |
| Animal Fluency (words/min) | |||||
| Total Correct | 9 | 6 | 24.0 (5.7) | 24 (20, 28) | |
| Errors | 0 | 3 | - | - | |
| Spatial/Construction | MMSE Pentagons | Correct | Incorrect | - | |
| Benson Figure Copy | 16 | 14 | 15.6 (0.9) | 16 (15, 16) | |
| VOSP Number Location | 10 | 8 | 9.4 (0.9) | 10 (9, 10) | |
| Language | WRAT-4 Word Reading | 63 | 61 | 64.1 (3.3) | 65 (61, 67) |
| BNT-15 | 15 | 15 | 14.1 (1.1) | 15 (14, 15) | |
| PPVT (max=16) | 16 | 15 | - | - | |
| Comprehension (max=5) | 5 | 2 | - | - | |
| Repetition (max=5) | 5 | 4 | - | - | |
| Verbal Agility (max=6) | 6 | 3 | - | - | |
Normative reference values derived from a cohort of demographically-matched, cognitively healthy adults: (N=74–100 across tests, age=48.2±9.2 years old, education=17.0±2.3 years, 100% male, 100% white/Caucasian). Both mean±SD [M (SD)] and median with interquartile range [Mdn (IQR)] are provided where possible.
| Prompt | Patient Response |
|---|---|
| The cat ate the caterpillar | “Cat ate the cata-pilla” |
| Justin is taller than Henry | “Justin is taller than He-ry” |
| A teacher bought three pairs of gloves | “Peacher bought three pairs of gloves” |
| We walked to the lake and then to the store | “We walked to the lake and then to the store” |
| The rabbit was given to the child by a fireman | “Rabbit was given to the child by a fireman” |
His first visit occurred approximately ten years after the onset of behavioral symptoms. Behaviorally, he was perseverative with a lack of social/emotional engagement, as well as mildly impulsive and socially inappropriate. On cognitive testing, he had profound difficulties with attention, working memory, and all fluency tasks (lexical, semantic, and design). He notably produced seven repetition errors on design fluency. He had a poor immediate recall of a 9-word list on memory testing but intact delayed recall and recognition for both the word list and a complex figure. Language and visuospatial/constructional skills were also intact, and he denied mood symptoms. The patient’s wife’s responses on informant-report measures (Supplemental Table 1) suggested that she had noticed significantly increased social withdrawal and decreased interpersonal warmth compared to his baseline. She also reported he had mildly reduced emotional empathy but a marked loss of cognitive empathy (i.e., an inability to take others’ perspectives in real-life situations). On performance-based testing, his ability to perform basic theory of mind reasoning remained intact in both cognitive and emotional domains. While his ability to name emotional states was intact, he struggled to differentiate sincere versus sarcastic conversational exchanges.
Around 14 months later, he behaviorally showed worsening perseverations, motor stereotypies, impulsivity, and social inappropriateness. New behaviors observed during testing included stimulus-boundedness and distractibility. Executive function scores worsened further, and perseverative repetition errors were prominent. Delayed recall on memory testing remained relatively intact, particularly for a complex figure, but he produced many false positive errors on word list recognition. Sentence comprehension, repetition abilities, and verbal agility worsened while naming and single-word comprehension remained intact. Phrase repetition errors included mild speech apraxia and agrammatism. Visuospatial/constructional skills remained relatively intact, and he denied mood symptoms. Additionally, the patient had not only shown a precipitous drop in interpersonal warmth from his baseline behavior but also showed new impairments in interpersonal dominance and self-assurance. On performance-based theory of mind testing, he now evidenced new impairments inferring others’ non-emotional (i.e., “cognitive theory of mind”) and emotional (i.e., “emotional theory of mind”) beliefs. He also developed new difficulties naming emotions that were not evident at the first visit.
Overall, he presented first with significant executive dysfunction and relative sparing of other cognitive domains. At follow-up, executive functioning worsened, and he also developed mild difficulties with phrase repetition and sentence comprehension. Delayed recall, visuospatial/constructional skills, and semantic knowledge remained relatively intact. These findings, coupled with his behavioral history, supported his behavioral variant frontotemporal dementia diagnosis.
Neurodiagnostic and Genetic Testing
The patient underwent an MRI of the brain without contrast at age 44 that showed very mild dorsolateral-predominant frontoparietal volume loss (Figure 1A). A repeat MRI of the brain without contrast performed at age 47 showed significant change with marked right greater than left dorsolateral-predominant frontoparietal volume loss with focal atrophy of the orbitofrontal, caudate, and medial temporal cortex (Figure 1B). A repeat MRI of the brain without contrast at age 49 showed significant progression of volume loss with severe right greater than left frontotemporal volume loss and pronounced atrophy of the caudate nuclei and anterior insulae (Figure 1C). Brain FDG-PET at age 48 showed striking dorsal greater than ventral prefrontal hypometabolism with less prominent dorsal parietal hypometabolism (Figure 1D).
Figure 1.
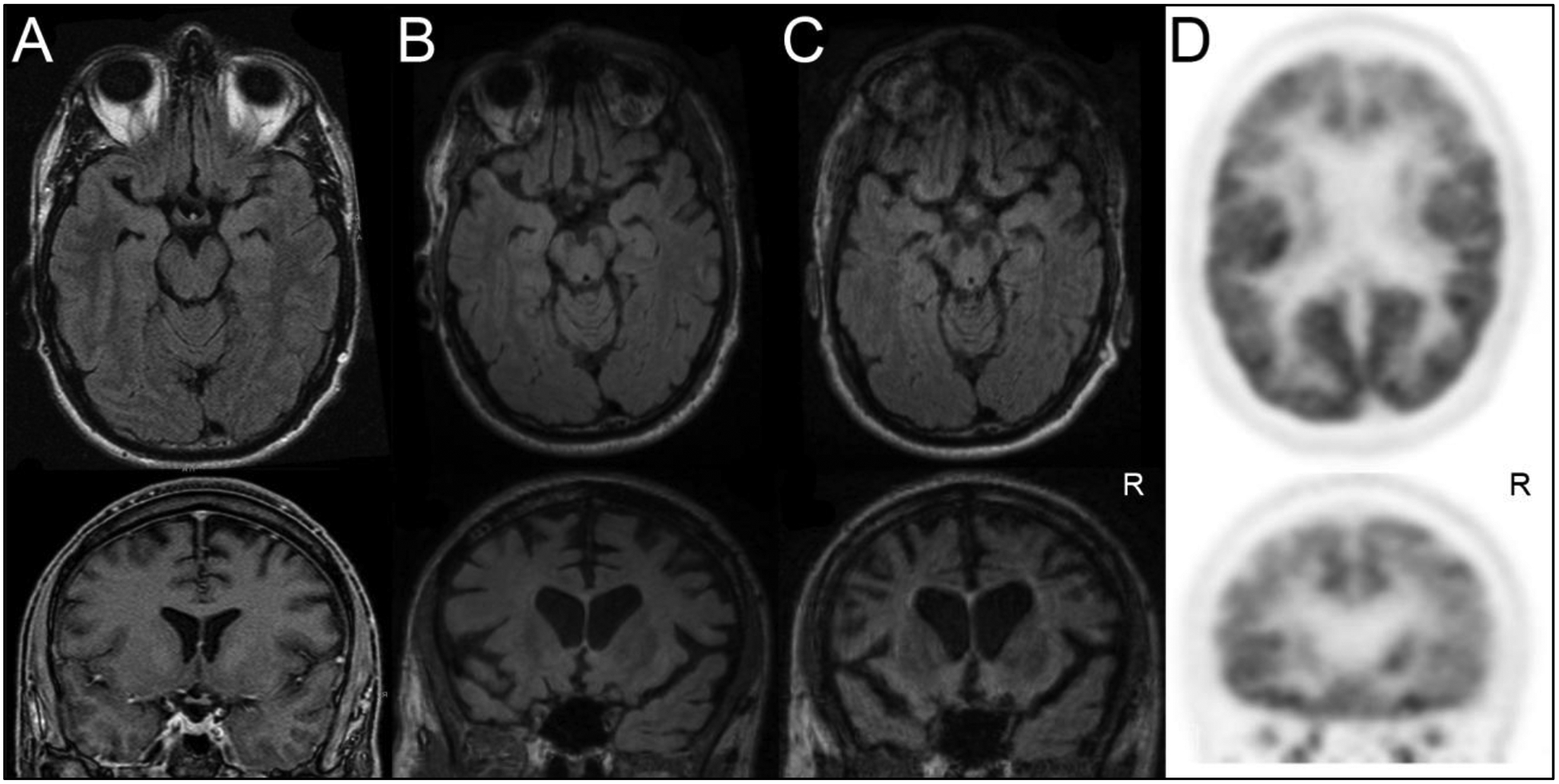
Serial T2 FLAIR Axial/Coronal Brain MRIs are shown at the ages (a) 44, (b) 47, and (c) 49. Serial imaging demonstrates interval worsening right greater than left dorsolateral-predominant frontoparietal volume loss with focal atrophy of the orbitofrontal, caudate, and the medial temporal region. (d) FDG-PET brain imaging performed at age 48 shows predominant dorsal greater than ventral prefrontal hypometabolism
Analysis of dementia-related risk genotypes revealed an APOE genotype of E3/E328. No pathogenic or predicted deleterious mutations in MAPT, GRN, C9ORF72, TARDBP, FUS, APP, PSEN1, or PSEN2 were identified, and the patient did not carry the rare TREM2 R47H risk variant. However, whole-genome sequencing revealed that the patient did carry multiple genetic risk factors. Established common genetic risk variants were identified for PSP (STX6 rs57113693)29,30, CBD (lnc-KIF13B-1 rs643472)18, and both PSP-CBD (MAPT H1c haplotype rs242557)18,29,30. Additional suggestive risk variants were identified for PSP (ASAP1 rs2045091 and DUSP10 rs12125383)30 and CBD (TSPEAR rs875125)18. Finally, a recently documented pleiotropic risk variant for both PSP and CBD was identified in EGFR (rs759162)31 (Table 2). We did not identify any structural variants of MAPT in the WGS data. We did not investigate the possibility of MAPT duplications.
Table 2.
Genetic risk variants for PSP and CBD identified in the patient.
| Variant | Chr | Position (Base Pair) | Genotype | MAF | Gene | Associated Phenotype | Odds Ratio | |
|---|---|---|---|---|---|---|---|---|
| Established risk variants | rs57113693 | 1 | 180952294 | C/T | 0.40 | STX6 | PSP29,30 | 1.35 |
| *rs242557 | 17 | 44019712 | A/G | 0.39 | MAPT | PSP + CBD18,29,30 | 1.57 | |
| rs643472 | 8 | 29153777 | C/T | 0.24 | lnc-KIF13B-1 | CBD18 | 1.82 | |
| Suggestive risk variants | rs2045091 | 8 | 1310s75859 | T/C | 0.16 | ASAP1 | PSP30 | 1.25 |
| rs12125383 | 1 | 222168434 | A/G | 0.21 | DUSP10 | PSP30 | 1.28 | |
| rs875125 | 21 | 46129686 | A/C | 0.08 | TSPEAR | CBD18 | 1.22 | |
| Pleotropic risk variants | rs759162 | 7 | 55228173 | C/T | 0.28 | EGFR | PSP + CBD31 | NA |
SNP tags MAPT H1c haplotype. The patient confirmed negative for H2 haplotype via tag SNP rs8070723. Chr, chromosome; MAF, minor allele frequency as reported in GnomAD European population. The allele in red is the risk-associated allele
Neuropathological evaluation
The brain and the spinal cord were collected at autopsy and processed as previously described32. The brain weighed 1088 grams. The right cerebral/cerebellar hemispheres, whole brainstem, and spinal cord were fixed in 10% neutral buffered formalin. Macroscopically, there was severe atrophy of the dorsolateral prefrontal and orbitofrontal cortex, and anterior temporal pole with severe lateral ventricular enlargement. There was a narrowing of the cerebral cortex and atrophy of the deep nuclei, especially in the caudate nucleus and the amygdala. The hippocampus was normal in bulk, and there was no evidence of microinfarcts. The brain stem tegmentum showed severe atrophy, especially in the dorsal pons. The substantia nigra demonstrated moderately decreased pigmentation. There was significant atrophy of the dentate nucleus.
Microscopically, there was superficial cortical microvacuolation most prominently in the middle/superior frontal gyrus and subgenual cingulate cortex, and to a lesser extent, in the mild cingulate, temporal, postcentral, angular, anterior orbital, and middle insular cortices. Astrogliosis was prominent in the middle frontal gyrus and superior sulcus, and moderate in the frontal and cingulate cortices. Severe gliosis was seen in the substantia nigra, dentate nucleus, and brainstem. Neuronal loss (higher than 50% but lower than 75%) was observed in the substantia nigra (Supplemental Table 1). There were numerous ballooned neurons in the anterior cingulate gyrus, subgenual cingulate gyrus, and amygdala with severe white matter rarefaction of degenerative etiology in the middle frontal gyrus, inferior frontal gyrus, and superior frontal sulcus. (Figure 2A).
Figure 2.
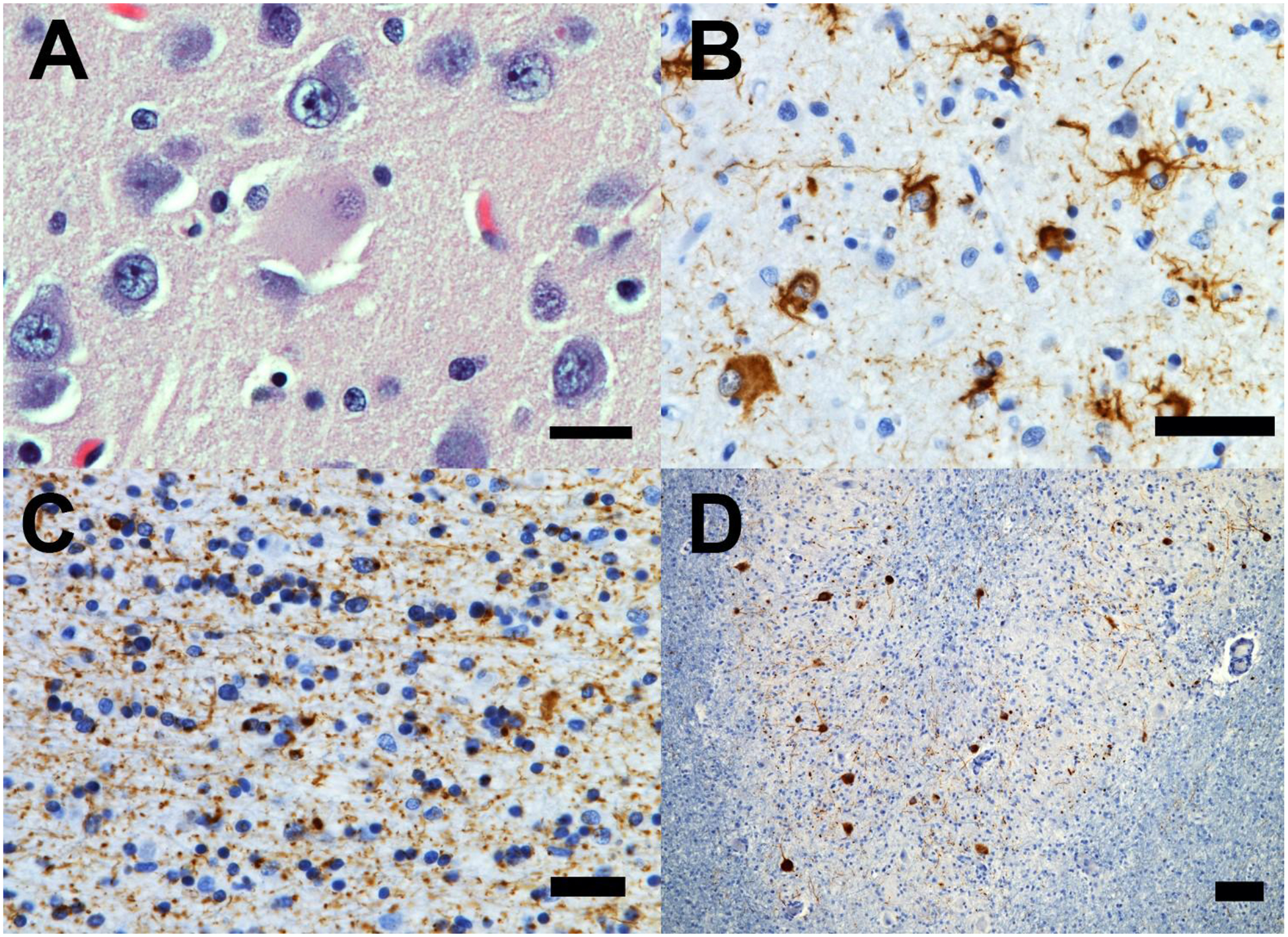
(a) Ballooned neurons (here an example in the cingulate gyrus) with glossy cytoplasm, and eccentric nucleus were found in large numbers in the most affected cortical areas. (b) Atypical tufted-like astrocytes with enlarged cytoplasm and thick, proximal to the nuclei tau-immunoreactive inclusions were seen in large number in the most affected cortical regions (here a few representatives examples in the superior frontal gyrus) (c) Severe white matter tau pathology with oligodendroglial coiled bodies and extensive tau-immunoreactive neuritic pathology in the superior frontal gyrus. (d) Low-magnification image of the dentate gyrus showing diffuse/granular tau-immunoreactive neuronal cytoplasmic and dendritic inclusion. Stains: A = hematoxylin/eosin; B,C, D: CP13 (pS202Tau). Bars: A, B, C = 25 microns; D = 250 microns.
Immunohistochemistry (IHC) for phosphorylated tau (CP13) showed globose tangles in large numbers in the amygdala, moderate numbers in substantia nigra, tectum, periaqueductal gray, and subgenual cingulate cortex, and small numbers in the ventral striatum, subthalamic nucleus, and angular gyrus. Neuronal cytoplasmic inclusions (NCI) were predominately diffuse/granular inclusions found in a large proportion of neurons in the anterior cingulate cortex, middle frontal gyrus, ventral striatum, subgenual cingulate cortex, inferior temporal gyrus, amygdala, dentate gyrus, CA1/subiculum, and dentate nucleus. Moderate numbers of NCI were found in the subthalamic nucleus, substantia nigra, periaqueductal gray, median raphe, pontine nuclei, anterior orbital gyrus, and angular gyrus, and small numbers were found in the tectum, dorsal raphe, oculomotor nucleus, trochlear nucleus, frontal pole, CA3–4, CA2, entorhinal cortex, precentral gyrus, and postcentral gyrus.
Abundant tau-immunoreactive (IR) tufted astrocytes were observed (Figure 2B). They were found in large numbers in the frontal pole, anterior orbital gyrus, anterior cingulate cortex, middle frontal gyrus, ventral striatum, subgenual cingulate cortex, inferior temporal gyrus, amygdala, precentral gyrus, postcentral gyrus, and angular gyrus, and in the periaqueductal gray. Moderate numbers of tufted astrocytes were seen in the tectum, CA1/subiculum, and entorhinal cortex, and small numbers were observed in the subthalamic nucleus, dorsal raphe, median raphe, and CA2. The majority of the tufted astrocytes had larger cell bodies and thicker processes than the ones commonly observed in PSP and were intensively tau-IR. There were no neurofibrillary tangles, Pick bodies, astrocytic plaques, thorny astrocytes in any region examined.
There was severe white matter tau pathology in neurites in the ventral striatum, and to a lesser extent in the anterior orbital gyrus, anterior and subgenual cingulate gyri, middle frontal gyrus (Figure 2C), inferior temporal gyrus, precentral and postcentral gyri, and angular gyrus, and mild in frontal pole, dentate gyrus, entorhinal gyrus, and dentate nucleus (Figure 2D).
There was severe neuropil threads pathology throughout the most affected cortical regions, and a moderate number of tau-IR oligodendroglial coiled bodies were predominately seen in the anterior orbital gyrus, middle frontal gyrus, ventral striatum, subgenual cingulate cortex, and inferior temporal gyrus and in smaller numbers in the subthalamic nucleus, substantia nigra, tectum, periaqueductal gray, frontal pole, anterior cingulate cortex, amygdala, precentral gyrus, postcentral gyrus, and angular gyrus.
IHC for phosphorylated 3-repeat tau (RD3) did not show neurofibrillary tangle pathology of Alzheimer’s disease in any region (Braak stage 0). Gallyas silver stain, useful for differentiating 4R tauopathies, was positive for argyrophilic astrocytes, effectively ruling out globular glial tauopathy (GGT). Beta-amyloid IHC showed rare amyloid plaque pathology restricted to the striate cortex and the hippocampus (Thal phase 2). These findings were consistent with an incidental diagnosis of low Alzheimer’s Disease Neuropathological Changes (ADNC) A1, B0, C0. IHC for TDP-43, FUS, and alpha-synuclein were negative33. There was no significant arteriolosclerosis or microinfarctions found in any region examined.
Discussion
We report the case of a patient who developed progressive disinhibition, impulsivity, apathy, and loss of social interrelatedness/empathy beginning at age 36, followed by stereotyped compulsive behavior and hyperorality by age 48. Later he developed progressive pseudobulbar dysarthria, speech apraxia, parkinsonism, and limb-kinetic/ideomotor apraxia leading to choking, mutism, and death by age 54. The clinical presentation fulfills the research diagnostic criteria for bvFTD5. Neuroimaging showed asymmetric right-sided predominant dorsolateral-parietal volumetric loss with pronounced focal cortical atrophy of the orbitofrontal, anterior insula, and mild temporal cortices with severe caudate/amygdala atrophy. The predicted underlying neuropathology at the time of clinical examination was either TDP-43 pathology, given the pronounced asymmetric anterior temporal atrophy, or FTLD-FUS given the early age at onset and the severe caudate atrophy. Neuropathologically, the present case shared typical features of CBD and PSP but did not fulfill the necessary research criteria for either.
Neuropathological examination displayed the unusual coexistence of both features of PSP, most notably the presence of abundant though atypical TAs, and features of CBD such as frequent cortical ballooned neurons, and extensive neuritic white matter pathology, which is uncharacteristic of PSP and more typical of CBD22,26. The 2002 Office of Rare Diseases of the National Institutes of Health neuropathologic criteria for CBD includes at a minimum “cortical and striatal tau-positive neuronal and glial lesions, especially astrocytic plaques and thread-like lesions in both white matter and gray matter, along with neuronal loss in focal cortical regions and in the substantia nigra”22. Our case had no identifiable astrocytic plaques. While APs and TAs may coexist34, they are regarded as highly characteristic and distinctive between CBD and PSP10,11,26. Though the definite histopathological criteria for PSP has remained largely unchanged since the NINDS-SPSP of 19963, TAs alongside other pathological characteristics of tau aggregation are used to help differentiate PSP from other 4R tauopathies11. Additionally, despite the identical composition of tau isoforms, different proteolytic processing of abnormal tau occurs in these two diseases. Arai et al. demonstrated through immunoblots of sarkosyl‐insoluble brain extracts, 33kDa bands predominated in the low molecular weight tau fragments in PSP. In contrast, two closely related bands of approximately 37kDa predominated in CBD. Interestingly, atypical cases had immunoblots that demonstrated both a 33kDa band and a 37kDa doublet35. In the present case, TAs were present, though atypical given the extent of tau protein immunoreactivity and the thickness of the astrocytic processes consistent with activated astrogliosis. The extensive presence of ballooned neurons observed in this case is more characteristic of CBD. Ballooned neurons are consistently found in the affected cortices of CBD, whereas they are much less commonly observed in PSP22,36. These neuropathological features do not allow for a definitive pathological diagnosis of this case within the established criteria for known tauopathies resulting in our primary diagnosis of frontotemporal lobar degeneration with tau-immunoreactive inclusions (FTLD-tau) unspecified.
Though clinically and pathologically distinct neurodegenerative disorders, the growing wealth of genetic data and analysis has offered insight into shared pathobiology underlying disease risk for both CBD and PSP. In 1998, the discovery of multiple microtubule-associated protein tau (MAPT) mutations in FTDP-17 provided the first evidence that changes in tau alone could cause neurodegenerative disease37–39. Two years later, a case series of 18 subjects recognized that the H1 MAPT haplotype was significantly overrepresented in patients with CBD, a haplotype already reported in association with PSP24. This genetic overlap was confirmed in a later case series of 57 cases25. Subsequently, multiple genome-wide association studies (GWAS) have confirmed that single nucleotide polymorphisms (SNPs) within the H1 haplotype of the MAPT locus are associated with increased risk for CBD, PSP, and FTLD18,29,31,40. After a decade of GWAS, many questions regarding CBD and PSP etiology and pathophysiology remain unanswered; however, a series of recent pleiotropy analysis studies have shed light on the genetic link underlying these disorders.
The term pleiotropy is derived from the Greek words pleio, which means “many,” and tropic, which means “affecting.” Genes that affect multiple, apparently unrelated, phenotypes are thus called pleiotropic genes. GWAS of numerous disorders and phenotypes have both expanded the pool of variants associated with degenerative conditions and provided insight into genetic pleiotropy. A GWAS comparing 1,114 autopsy-confirmed cases of PSP to 3,287 control subjects found significant genetic variations in three regions: EIF2AK3, STX6, and MOBP29. These findings were later supported by GWAS of pathologically confirmed CBD and PSP cases that identified SNPs within MOBP that were associated with greater disease risk18. In a recent study, Yokoyama et al. systematically assessed the genetic overlap and shared gene loci between tauopathies to identify SNPs jointly associated with CBD, PSP, and FTD31. Pleiotropy analysis identified multiple overlapping common genetic risk factors and shared molecular pathways between the three tauopathies with up to 800-fold genetic enrichment in CBD with PSP SNPs. Although MAPT and MOBP represented the most consistent and reliable findings, Yokoyama et al. detected novel shared risk signals within CXCR4, EGFR, and GLDC. Taken together, these various GWAS have identified multiple common variants associated with CBD and PSP, namely 17 variants in 14 genes (STX6, EIF2AK3, MOBP, MAPT, RUNX2, SLCO1A2, DUSP10, SP1, ASAP1, WDR63, SOS1, lnc-KIF13B-1, PRAKG2, TSPEAR)18,29–31. Each of these tauopathy-associated pleiotropic variants may be associated with a unique neuroanatomic gene expression signature that may influence regional and neuronal selective vulnerability and thus disease phenotype30. The biological roles of these different candidates, coupled with their differential expression patterns in the brain, support the idea that these genetic risk factors may promote specific neuroanatomical patterns of tauopathy when observed in different combinations. Although no single common variant may be informative clinically, the combination of these risk variants may, in part, drive tauopathy-susceptible degeneration in the form of CBD, PSP, or FTD.
In this case report, the patient’s GWAS identified multiple PSP specific variants (rs57113693 STX6, rs2045091 ASAP1, rs12125383 DUSP10), CBD specific (rs643472 lnc-KIF13B-1, rs875125 TSPEAR), and overlapping PSP-CBD (rs242557 MAPT, rs759162 EGFR) genetic variants contributing to disease risk. Functionally, these genes represent multiple shared risk loci for several neuroanatomically relevant gene co-expression networks that are enriched for several neurodevelopmental pathways. Together, these results suggest that these multiple genetic variants contributed to pleiotropic risk for tauopathy with mixed CBD-PSP features. Genetic pleiotropy may be associated with the subject’s premorbid behavior and family’s neuropsychiatric history, although this is speculative41. Many of these aforementioned genetic risk variants have been associated with various neuropsychiatric presentations. Mutations of growth factors such as EGFR have been linked to neurodevelopmental abnormalities resulting in the clinical manifestations of ADHD and ASD42 with high-functioning ASD showing a significant reduction in serum levels of EGF as compared with controls43. Rare variants in AGAP1 (GTPase-activating protein involved in membrane trafficking and cytoskeleton dynamics) have been associated with autism44. Mutations in STX6 (SNARE protein involved in synaptic transmission) have been associated with schizophrenia45 and ASD46. Collectively, these findings suggest that the distinctive neurodevelopmental phenotype and neurodegenerative regional vulnerability of this patient may relate to the various neuropsychiatric phenotypes expressed by the patient and members of his family.
Future research should further investigate these observations by comparing the varied clinical presentations of mixed or overlapping PSP and CBD pathological features to known PSP/CBD genetic risk variants. A comprehensive approach may include calculating a genetic risk score (GRS) of known variants in a weighted manner that may be prospectively used to screen clinically ambiguous cases as it has been done in early-onset PSP47.
Supplementary Material
Acknowledgments
We thank the research participant and his family for their generous contribution to science.
Funding
This work was funded by the National Institute on Aging P01AG019724, P30AG062422, K08AG052648, R01AG048234, R01AG038791, R01AG032289, R01AG048234, the Larry L. Hillblom Foundation, and the Rainwater Charitable Foundation.
Footnotes
Conflict of Interest/ Disclosure statement
The authors have no conflicts of interest related to this publication.
The authors whose names are listed immediately below certify that they have NO affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in this manuscript.
James Rini, MD. MPH.
Contributor Information
James Rini, Behavioral Neurology, Memory and Aging Center, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
Breton Asken, Neuropsychology, Memory and Aging Center, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
Ethan Geier, Memory and Aging Center, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
Katherine Rankin, Memory and Aging Center, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
Joel Kramer, Memory and Aging Center, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
Adam Boxer, Behavioral Neurologist, Memory and Aging Center, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
Bruce Miller, Director, Memory and Aging Center, Co-Director, Global Brain Health Institute, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
Jennifer Yokoyama, Memory and Aging Center, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
Salvatore Spina, Behavioral Neurologist, Memory and Aging Center, University of California, San Francisco, 675 Nelson Rising Lane, Suite 190, San Francisco, CA 94158.
REFERENCES
- 1.Barbier P, Zejneli O, Martinho M, et al. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front Aging Neurosci. 2019;11. doi: 10.3389/fnagi.2019.00204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irwin DJ. Tauopathies as Clinicopathological Entities. Parkinsonism Relat Disord. 2016;22(0 1):S29–S33. doi: 10.1016/j.parkreldis.2015.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9. doi: 10.1212/wnl.47.1.1 [DOI] [PubMed] [Google Scholar]
- 4.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503. doi: 10.1212/WNL.0b013e31827f0fd1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456–2477. doi: 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. doi: 10.1212/WNL.0b013e31821103e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steele JC, Richardson JC, Olszewski J. PROGRESSIVE SUPRANUCLEAR PALSY. A HETEROGENEOUS DEGENERATION INVOLVING THE BRAIN STEM, BASAL GANGLIA AND CEREBELLUM WITH VERTICAL GAZE AND PSEUDOBULBAR PALSY, NUCHAL DYSTONIA AND DEMENTIA. Arch Neurol. 1964;10:333–359. doi: 10.1001/archneur.1964.00460160003001 [DOI] [PubMed] [Google Scholar]
- 8.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994;44(11):2015–2019. doi: 10.1212/wnl.44.11.2015 [DOI] [PubMed] [Google Scholar]
- 9.Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord Off J Mov Disord Soc. 2017;32(6):853–864. doi: 10.1002/mds.26987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komori T, Arai N, Oda M, et al. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol (Berl). 1998;96(4):401–408. doi: 10.1007/s004010050911 [DOI] [PubMed] [Google Scholar]
- 11.Yoshida M Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathol Off J Jpn Soc Neuropathol. 2014;34(6):555–570. doi: 10.1111/neup.12143 [DOI] [PubMed] [Google Scholar]
- 12.Rebeiz JJ, Kolodny EH, Richardson EP. Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol. 1968;18(1):20–33. doi: 10.1001/archneur.1968.00470310034003 [DOI] [PubMed] [Google Scholar]
- 13.Gibb WR, Luthert PJ, Marsden CD. Corticobasal degeneration. Brain J Neurol. 1989;112 ( Pt 5):1171–1192. doi: 10.1093/brain/112.5.1171 [DOI] [PubMed] [Google Scholar]
- 14.Bak TH, Hodges JR. Corticobasal degeneration: clinical aspects. Handb Clin Neurol. 2008;89:509–521. doi: 10.1016/S0072-9752(07)01247-X [DOI] [PubMed] [Google Scholar]
- 15.Riley DE, Lang AE, Lewis A, et al. Cortical-basal ganglionic degeneration. Neurology. 1990;40(8):1203–1212. doi: 10.1212/wnl.40.8.1203 [DOI] [PubMed] [Google Scholar]
- 16.Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999;53(4):795–800. doi: 10.1212/wnl.53.4.795 [DOI] [PubMed] [Google Scholar]
- 17.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain J Neurol. 2002;125(Pt 4):861–870. doi: 10.1093/brain/awf080 [DOI] [PubMed] [Google Scholar]
- 18.Kouri N, Ross OA, Dombroski B, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 2015;6. doi: 10.1038/ncomms8247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ling H, O’Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain J Neurol. 2010;133(Pt 7):2045–2057. doi: 10.1093/brain/awq123 [DOI] [PubMed] [Google Scholar]
- 20.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology. 1997;48(1):119–125. doi: 10.1212/wnl.48.1.119 [DOI] [PubMed] [Google Scholar]
- 21.Murray R, Neumann M, Forman MS, et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology. 2007;68(16):1274–1283. doi: 10.1212/01.wnl.0000259519.78480.c3 [DOI] [PubMed] [Google Scholar]
- 22.Dickson DW, Bergeron C, Chin SS, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935–946. doi: 10.1093/jnen/61.11.935 [DOI] [PubMed] [Google Scholar]
- 23.Buée L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol Zurich Switz. 1999;9(4):681–693. doi: 10.1111/j.1750-3639.1999.tb00550.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Maria E, Tabaton M, Vigo T, et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol. 2000;47(3):374–377. doi: [DOI] [PubMed] [Google Scholar]
- 25.Houlden H, Baker M, Morris HR, et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56(12):1702–1706. doi: 10.1212/wnl.56.12.1702 [DOI] [PubMed] [Google Scholar]
- 26.Litvan I, Grimes DA, Lang AE, et al. Clinical features differentiating patients with postmortem confirmed progressive supranuclear palsy and corticobasal degeneration. J Neurol. 1999;246 Suppl 2:II1–5. doi: 10.1007/bf03161075 [DOI] [PubMed] [Google Scholar]
- 27.Kramer JH, Jurik J, Sha SJ, et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol Off J Soc Behav Cogn Neurol. 2003;16(4):211–218. doi: 10.1097/00146965-200312000-00002 [DOI] [PubMed] [Google Scholar]
- 28.Geier EG, Bourdenx M, Storm NJ, et al. Rare variants in the neuronal ceroid lipofuscinosis gene MFSD8 are candidate risk factors for frontotemporal dementia. Acta Neuropathol (Berl). 2019;137(1):71–88. doi: 10.1007/s00401-018-1925-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Höglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43(7):699–705. doi: 10.1038/ng.859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen JA, Chen Z, Won H, et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol Neurodegener. 2018;13(1):41. doi: 10.1186/s13024-018-0270-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yokoyama JS, Karch CM, Fan CC, et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol (Berl). 2017;133(5):825–837. doi: 10.1007/s00401-017-1693-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spina S, Brown JA, Deng J, et al. Neuropathological correlates of structural and functional imaging biomarkers in 4-repeat tauopathies. Brain J Neurol. 2019;142(7):2068–2081. doi: 10.1093/brain/awz122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol (Berl). 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katsuse O, Iseki E, Arai T, et al. 4-repeat tauopathy sharing pathological and biochemical features of corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol (Berl). 2003;106(3):251–260. doi: 10.1007/s00401-003-0728-8 [DOI] [PubMed] [Google Scholar]
- 35.Arai T, Ikeda K, Akiyama H, et al. Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol. 2004;55(1):72–79. doi: 10.1002/ana.10793 [DOI] [PubMed] [Google Scholar]
- 36.Togo T, Dickson DW. Ballooned neurons in progressive supranuclear palsy are usually due to concurrent argyrophilic grain disease. Acta Neuropathol (Berl). 2002;104(1):53–56. doi: 10.1007/s00401-002-0520-1 [DOI] [PubMed] [Google Scholar]
- 37.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- 38.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95(11):6469–6473. doi: 10.1073/pnas.95.11.6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’souza J, Megginson WL. The Financial and Operating Performance of Privatized Firms during the 1990s. J Finance. 1999;54(4):1397–1438. doi: 10.1111/0022-1082.00150 [DOI] [Google Scholar]
- 40.Vandrovcova J, Anaya F, Kay V, Lees A, Hardy J, de Silva R. Disentangling the role of the tau gene locus in sporadic tauopathies. Curr Alzheimer Res. 2010;7(8):726–734. doi: 10.2174/156720510793611619 [DOI] [PubMed] [Google Scholar]
- 41.Hodgkin J Seven types of pleiotropy. Int J Dev Biol. 1998;42(3):501–505. [PubMed] [Google Scholar]
- 42.Galvez-Contreras AY, Campos-Ordonez T, Gonzalez-Castaneda RE, Gonzalez-Perez O. Alterations of Growth Factors in Autism and Attention-Deficit/Hyperactivity Disorder. Front Psychiatry. 2017;8. doi: 10.3389/fpsyt.2017.00126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki K, Hashimoto K, Iwata Y, et al. Decreased serum levels of epidermal growth factor in adult subjects with high-functioning autism. Biol Psychiatry. 2007;62(3):267–269. doi: 10.1016/j.biopsych.2006.08.001 [DOI] [PubMed] [Google Scholar]
- 44.Wassink TH, Piven J, Vieland VJ, et al. Evaluation of the chromosome 2q37.3 gene CENTG2 as an autism susceptibility gene. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet. 2005;136B(1):36–44. doi: 10.1002/ajmg.b.30180 [DOI] [PubMed] [Google Scholar]
- 45.Sanders AR, Göring HHH, Duan J, et al. Transcriptome study of differential expression in schizophrenia. Hum Mol Genet. 2013;22(24):5001–5014. doi: 10.1093/hmg/ddt350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaste P, Klei L, Sanders SJ, et al. A genomewide association study of autism using the Simons Simplex Collection: Does reducing phenotypic heterogeneity in autism increase genetic homogeneity? Biol Psychiatry. 2015;77(9):775–784. doi: 10.1016/j.biopsych.2014.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jabbari E, Woodside J, Tan MMX, et al. The genetic and clinico-pathological profile of early-onset progressive supranuclear palsy. Mov Disord. 2019;34(9):1307–1314. doi: 10.1002/mds.27786 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.