

Abstract
Recent advances in sample preparation and analysis have enabled direct profiling of protein expression in single mammalian cells and other trace samples. Several techniques to prepare and analyze low-input samples employ custom fluidics for nanoliter sample processing and manual sample injection onto a specialized separation column. While being effective, these highly specialized systems require significant expertise to fabricate and operate, which has greatly limited implementation in most proteomic laboratories. Here, we report a fully automated platform termed autoPOTS (automated preparation in one pot for trace samples) that uses only commercially available instrumentation for sample processing and analysis. An unmodified, low-cost commercial robotic pipetting platform was utilized for one-pot sample preparation. We used low-volume 384-well plates and periodically added water or buffer to the microwells to compensate for limited evaporation during sample incubation. Prepared samples were analyzed directly from the well plate with a commercial autosampler that was modified with a 10-port valve for compatibility with 30 μm i.d. nanoLC columns. We used autoPOTS to analyze 1–500 HeLa cells and observed only a moderate reduction in peptide coverage for 150 cells and a 24% reduction in coverage for single cells compared to our previously developed nanoPOTS platform. To evaluate clinical feasibility, we identified an average of 1095 protein groups from ~130 sorted B or T lymphocytes. We anticipate that the straightforward implementation of autoPOTS will make it an attractive option for low-input and single-cell proteomics in many laboratories.
Graphical Abstract
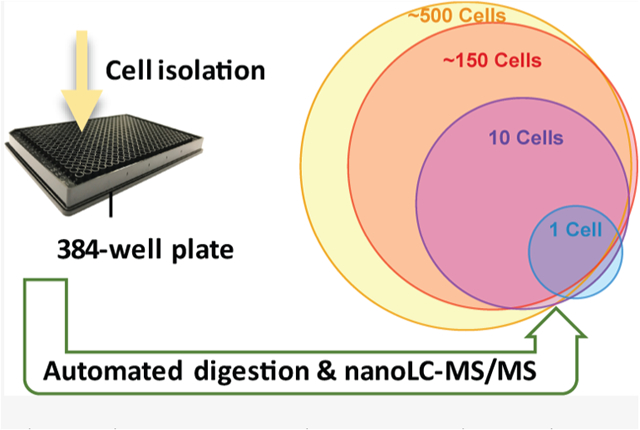
INTRODUCTION
Proteome profiling reveals insights into the physiology of biological systems with respect to expression, signaling, and interactions, driving advances in clinical research and molecular medicine. Mass spectrometry (MS)-based proteomic techniques can now provide unbiased identification and quantification of proteins in tissues with tremendous depth,1–3 yet the large sample input material typically required for analysis can exceed amounts obtained from clinical specimens. For example, fine needle aspiration biopsies or samples enriched by laser-capture microdissection or fluorescence-activated cell sorting (FACS) are generally challenging for conventional workflows. Sample input requirements have also limited our ability to study cellular processes at the level of individual cells and to profile protein expression across tissues with high spatial resolution.
A variety of approaches have been developed to reduce sample requirements for proteomic analyses,4–8 yet deep profiling of low-input samples has remained a challenge. Several recent advances have been combined to make such proteomic analysis of low-input samples more sensitive, robust, and with higher throughput. For example, analyzing TMT-labeled samples in the presence of a larger sample can increase the proteome coverage for single cells.9–11 Ultrasensitive separations are also tremendously beneficial, which have been achieved through nanoflow capillary electrophoresis,12,13 porous layer open tubular liquid chromatography (LC),14,15 or narrow-bore packed column LC.16 Gas-phase separation17 or fractionation using, for example, field-asymmetric high-field ion mobility spectrometry (FAIMS)18,19 can also dramatically increase the proteome coverage for (near) single-cell samples.
As another approach to further reduce sample input requirements, our group20 and others21,22 have miniaturized processing volumes to the nanoliter range. This provides the dual benefits of reduced surface exposure and increased sample concentrations to minimize adsorptive losses and increase the efficiency of reactions with trypsin and other reagents.23 The nanoPOTS (nanodroplet processing in one pot for trace samples) platform reported by our group, combined with ultrasensitive LC–MS, enabled thousands of proteins to be profiled from as few as 10 mammalian cells20 and hundreds of proteins from single cells.24 Recent improvements to the analytical workflow, including further miniaturization of the LC separations to operate at 20 nL/min16 and incorporating the FAIMS Pro interface for added selectivity, have increased the coverage to ~1100 protein groups per cell based solely on high-confidence MS/MS identifications.18 NanoPOTS is also compatible with isobaric labeling workflows for increased throughput10,25 and has been used for a number of single-cell10,16,18,24–27 and spatially resolved nanoproteomic studies.28–30 However, because nanoPOTS involves a custom-built robotic nanoliter pipetting system, custom-microfabricated “nanowell” chips, and numerous delicate manual steps during processing and analysis, dissemination of the platform to other laboratories has been limited.
We have previously addressed some of the shortcomings of the original nanoPOTS platform by upscaling the dispensed volumes to those of a conventional hand-held pipette,31 but this still required custom microfabricated glass nanowell chips and manual sample injection for LC/MS analysis. An autosampler has also been developed to automatically reconstitute and inject samples from a nanoPOTS chip for LC/MS analysis.27 However, this also utilized a customized robotic platform, again limiting dissemination. Here, we report on a fully automated platform termed autoPOTS (automated processing in one pot for trace samples), which utilizes an unmodified low-cost commercial liquid handling robot and a commercial autosampler to process and analyze low-input samples (Figure 1). The sample processing workflow closely follows that of our previously developed nanoPOTS protocol but with volumes upscaled to the low-microliter range for compatibility with the liquid handler and 384-well plates. A commercial autosampler modified with a 10-port valve retrieves >90% of the prepared sample, concentrates peptides on a solid-phase extraction (SPE) column while flushing salts and other contaminants, and injects the sample onto a 30 μm i.d. LC column operating at ~40 nL/min.
Figure 1.
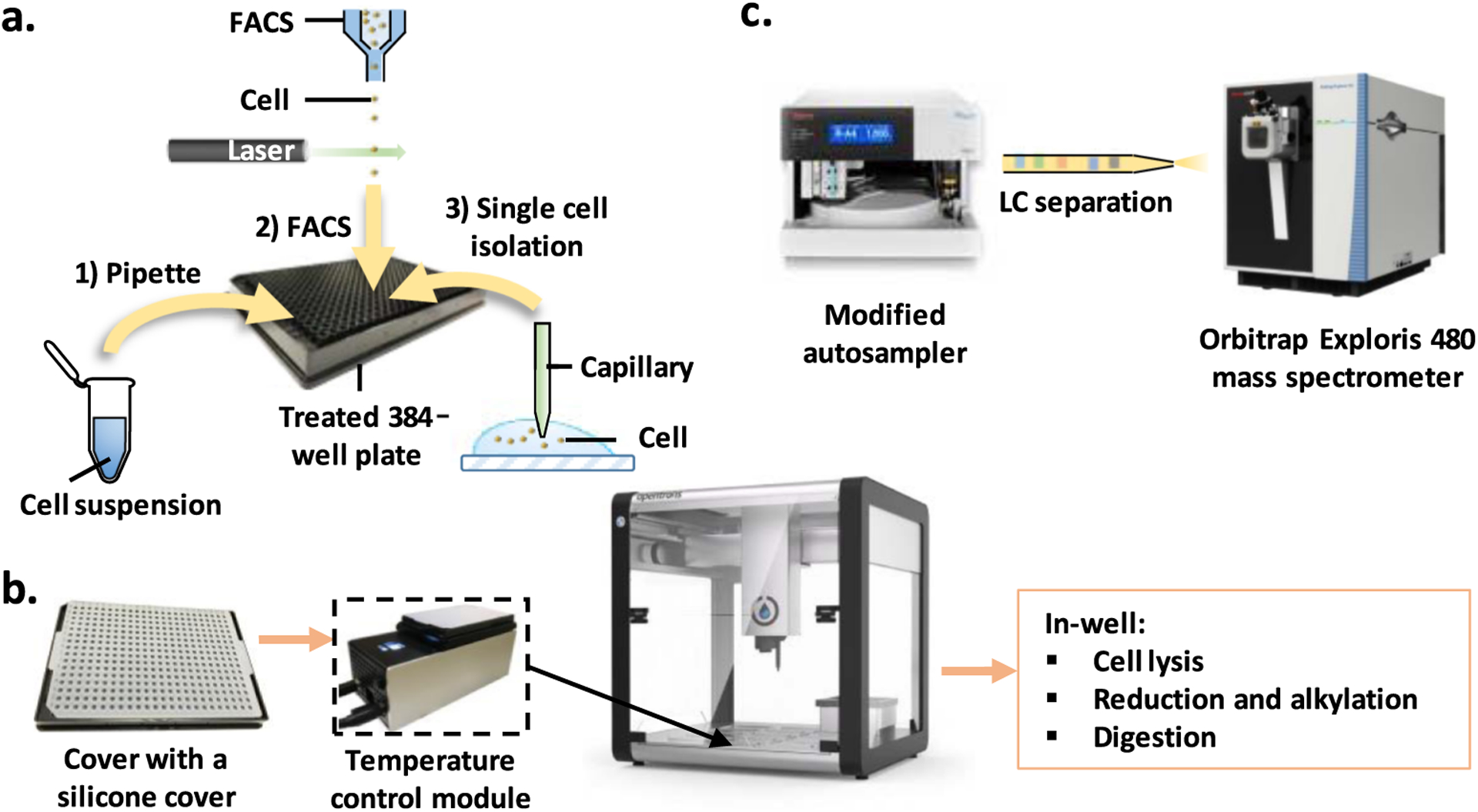
Workflow for automated sample processing and analysis of low-input samples. (a) Cells are dispensed into prefilled wells of a 384-well plate using any of the three sample isolation methods: (1) limiting dilution from cell suspension, (2) FACS, or (3) capillary-based micromanipulation. (b) After collecting cells, wells are sealed with a prescored silicone cover and placed on the OT-2 temperature control module. Cell lysis, protein reduction, alkylation, and digestion are performed in a one-pot workflow; buffer or water is added periodically to compensate for limited evaporation. (c) The well plate is loaded onto an autosampler for nanoLC-MS/MS analysis.
We detail the well plate surface treatment as well as compensation for limited evaporation through the sealed well plates during incubation steps at elevated temperatures. Proteome coverage is characterized for samples comprising 1–500 cultured HeLa cells, and we compare protein expression for populations of ~130 B or T lymphocytes from clinical blood samples. AutoPOTS was originally intended for processing and analyzing clinical samples containing hundreds of cells. We found that the platform performs quite comparably to nanoPOTS for such “mesoscale” samples while resulting in a ~25% reduction in peptide coverage at the single-cell level. Thus, autoPOTS should be suitable for a broad range of applications involving limited samples including single cells. While low-input proteomics remains technically challenging, the autoPOTS platform avoids microfabricated nanowells and custom robotic systems, which should facilitate implementation in many research and clinical settings.
EXPERIMENTAL SECTION
Reagents and Materials.
Ammonium bicarbonate (ABC) buffer (50 mM) was freshly prepared from 500 mM stock solution. Dithiothreitol (DTT), iodoacetamide (IAA), and formic acid (FA, LC–MS grade) were purchased from Thermo Fisher Scientific (Waltham, MA). LC–MS grade water and acetonitrile were purchased from Honeywell (Charlotte, NC). Trypsin and lys-C were products of Promega (Madison, WI). APC antihuman CD19, fluorescein isothiocyanate (FITC) antihuman CD3, and propidium iodide solution (PI) were purchased from BioLegend (San Diego, CA). All other unmentioned chemicals were from Sigma-Aldrich (St. Louis, MO). Low-volume 384-well microplates with a transparent, flat bottom were purchased from Corning (3544, Corning, NY). Polytetrafluoroethylene-coated silicone sealing mats were products of Arctic White (Bethlehem, PA), and adhesive storage foil was purchased from Eppendorf (Hauppauge, NY). All columns were packed with 3 μm Jupiter 300 Å C18-coated particles (Phenomenex, Torrance, CA) as described previously.16
Reproducibility and Accuracy of Automated Pipetting.
An OT-2 liquid handler (Opentrons, Brooklyn, NY) was calibrated as directed in software and the included 10 μL pipette was used for all pipetting operations. Fluorescein sodium salt (NaFl) was prepared in 50 mM sodium borate buffer and was used as an indicator to test pipetting reproducibility and accuracy. The fluorescent signal was read using a Synergy H4 Hybrid Reader (BioTek, Winooski, VT) with an excitation filter of 485/13.5 nm and an emission filter of 528/17.0 nm. The sensitivity of the plate reader was set to 120 following initial optimization. The linear response range of NaFl was determined using 10 μL aliquots of 0–500 μM NaFl, and a linear range up to 10 μM was found (Figure S1). After OT-2 pipetting, the 384-well microplate was centrifuged at 1500 rpm for 1 min to drive any condensed liquid from the mat and sidewalls into the well. The pipetting accuracy was assessed by directly comparing it with a freshly calibrated manual pipette (PIPETMAN L P20L, Gilson, Middleton, WI). For smaller volumes, 9, 8, and 7 μL of 1 μM NaFl were first added using a manual pipette into the wells and then 1, 2, and 3 μL of 10 μM NaFl were, respectively, added using the OT-2 to generate a total volume of 10 μL in each well. Standard solutions of equivalent final concentrations were prepared manually for comparison (Figure S2a and Table S1). Reproducibility of OT-2 pipetting for 4–10 μL volumes was tested using 10 μM NaFl with 12 replicates of each volume (Figure S2b). The impact of the pipetting speed on accuracy and reproducibility was also characterized (Table S2), and the default settings were used for subsequent experiments.
Evaporation Rate at Different Temperatures.
Standard addition was used to determine the evaporation rate of solutions in the well plates with the sealing mat affixed. Briefly, 5 μL of 1 μM NaFl was pipetted into each well through a prescored mat using OT-2. After centrifugation at 1500 rpm for 1 min, the microplate was placed on the OT-2 temperature module or in a refrigerator or freezer for defined intervals. After incubation, the microplate was brought to room temperature. 8 μL of preprepared standard solutions of NaFl (0, 0.625, 1.25, 2.5, and 3.75 μM) were pipetted into microplate wells. Then, 2 μL of the remaining solution from each well from the incubated microplate was sampled into the wells containing 8 μL of standard solutions to reach a final volume of 10 μL. A linear relationship was determined based on fluorescence signal intensity (I) for the added volume (Vs) of 10 μM NaFl (0, 0.5, 1, 2, and 3 μL, Figure S3). For a fitted linear relationship of I = mVs + b, the concentration following evaporation (c) was calculated as follows: , and the evaporation rate (v) was calculated as follows:
Cell Culture and Harvesting.
HeLa cells were obtained from ATCC (Manassas, VA) and cultured at 37 °C with 5% CO2 using Dulbecco’s Modified Eagle’s Medium (VWR, Radnor, PA) with 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were split every 2 days and harvested upon reaching ~70% confluency. Before use, cultured HeLa cells were collected and washed three times in phosphate-buffered saline (PBS) with centrifugation at 200 × g for 5 min each time. The cells were then resuspended in PBS to a final concentration of ~104 cells/mL and these suspensions were used to isolate single cells and supernatant by micromanipulation.16 Briefly, Fisherbrand microscope slides (part #: 15-188-48) were soaked in 1% bovine serum albumin (BSA) for 30 min, washed with water, and dried in air. 200 μL of cell suspension was dispensed onto a BSA-treated glass slide mounted on a SVM340 microscope (LabSmith, Livermore, CA). Single cells were aspirated from the slide along with 6 nL of supernatant using a capillary (200 μm i.d., 360 μm o.d., with a ~30 μm pulled tip) and dispensed into a 384-well microplate. Blank samples containing only the supernatant were similarly collected and dispensed into microwells. For 10-cell samples, cells were collected one at a time into the same capillary for a total volume of ~60 nL and then this cell suspension was transferred to the microplate. Successful transfer was validated using a microscope. For 150 and 500 HeLa cells, 150 and 500 nL of 106 cells/mL of cell suspension (as determined using a Bio-Rad TC20 automated cell counter, Hercules, CA) were added to the wells using a 200 μm i.d. capillary. The number of cells deposited into nanowells was verified by obtaining photomicro-graphs of each nanowell and counting the number of cells. The number of cells identified in each well is shown in Table S3.
Determination of FACS Accuracy.
We used a FACSAria Fusion sorter (BD Biosciences, San Jose, USA) to isolate lymphocyte cells. Before sorting, the accuracy of the instrument was tested using 10 μm diameter rainbow fluorescent beads (Bangs Laboratories, Fishers, Indiana). Purity mode was used to target 145 cells/well with side-scatter sorting. The sorted beads were observed and counted under a microscope using the DAPI channel.
B and T Lymphocyte Preparation and FACS.
Following standard clinical practice, blood was drawn from a healthy male donor into a 4 mL EDTA tube at the Brigham Young University Student Health Center (IRB Authorization X19045) and immediately placed on ice. The protocol for preparing the samples prior to FACS analysis can be found online at dx.doi.org/10.17504/protocols.io.bkvxkw7n and is briefly described below. Small samples of whole blood (200 μL for multiple-stained cells and 100 μL for unstained and single-stained cells used as sorting controls) were added to 4 mL of red blood cell lysis buffer (BioLegend, San Diego, CA) and incubated at room temperature for 3 min. The white blood cells were separated from the lysed red blood cells and extracellular debris by centrifugation at 250 × g for 5 min. The samples were decanted and white blood cells in the pellet were resuspended in 100 μL PBS. The following fluorescent stains were added to the sorting sample and the appropriate single-stain control: 5 μL of APC antihuman CD19 to stain B cells, 5 μL of FITC antihuman CD3 to stain T cells, and 10 μL of PI to stain ruptured cells and allow for the selection of intact cells. The FACS system requires unstained cells and single-stain control cells to accurately gate the cell populations, so in addition to the sorting sample cells with all three stains, cells were prepared with no stain, APC only, FITC only, and PI only. Cells were incubated at room temperature for 15 min to allow antibody binding. These were then washed by adding 2 mL of PBS to each tube, centrifuging at 250 × g for 5 min, and decanting. The cells were resuspended in 300 μL PBS and filtered using 40 μm filters prior to sorting. The fluorescently labeled cells were sorted into a 384-well microplate into 5 replicates of 145 cells each for both B and T cells.
OT-2 One-Pot Sample Preparation Workflow.
The sample preparation workflow was developed by upscaling reagent volumes used for the nanoPOTS workflow16,20 for compatibility with the OT-2-pipetting capabilities. A 384-well microplate was prewashed using three steps to reduce the polymer contaminants. First, the microplate was filled with water and sonicated for 15 min. The water was then replaced with mobile phase A (0.1% v/v FA in water) and the microplate was sonicated for another 15 min. After removing mobile phase A, 0.01% n-dodecyl β-d-maltoside (DDM) was pipetted into the wells and then removed. Before cell loading, 4 μL of 1% DDM was dispensed to the wells. Once cells were loaded into the wells by limiting dilution, micromanipulation, or FACS, the plate was covered with the sealing mat and sonicated for 5 min using a 200 W GT SONIC-R9 (Shenzhen, China) sonicator, followed by centrifugation at 1500 rpm for 1 min. The well plate was mounted on the OT-2 system and 1 μL of 25 mM DTT was dispensed in each well for reduction. During the 1 h incubation at 70 °C, the OT-2 dispensed 2 μL of ABC buffer to each well every 15 min to compensate for evaporative losses. The microplate was then cooled to 25 °C and 1 μL of 60 mM IAA was added and incubated for 30 min in the dark. After alkylation, proteins were predigested using 1 μL of lys-C for 3 h at 37 °C and then digested by adding 1 μL of trypsin and incubating for 12 h at 37 °C. The concentration of enzymes was 1 ng/μL for 0, 1, 10, and 150 cells, and 5 ng/μL for 500 cells. During the 37 °C incubation, 2 μL of H2O was added to each well every 3 h, and 1 μL was added at 12 h. After digestion with trypsin, 1 μL of 5% FA was dispensed and the reaction was incubated at room temperature for 1 h. All pipetting steps were fully automated using the OT-2 platform and only two Python programs were used (Figure 2). After sample preparation, the well plate was centrifuged at 1500 rpm for 2 min, the silicone mat was removed, and a foil adhesive mat was used for long-term storage at −20 °C prior to LC–MS analysis.
Figure 2.
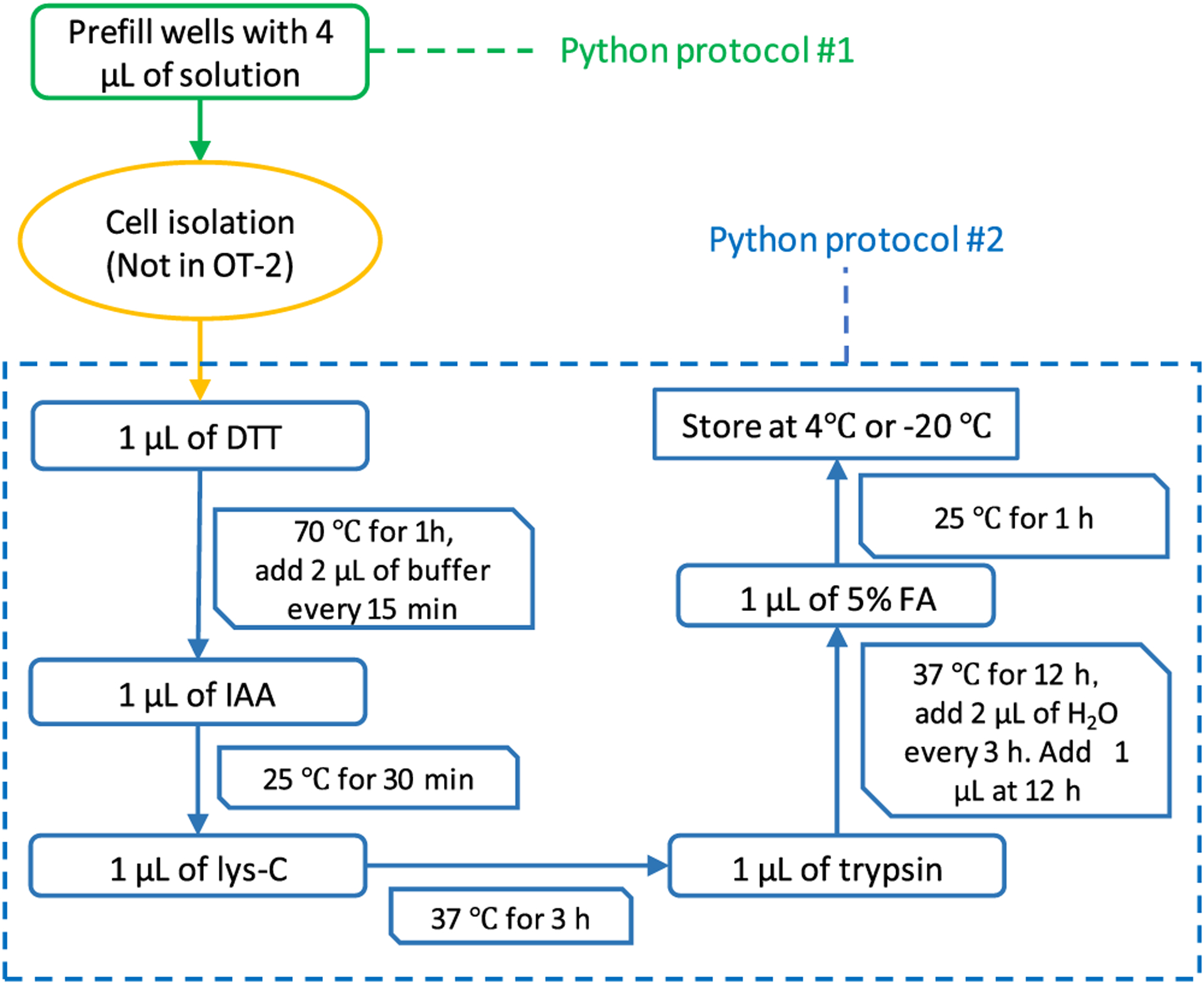
AutoPOTS sample preparation workflow. Python protocols 1 and 2 are performed in the OT-2, while cell isolation is performed separately.
Autosampler Modification and Operation.
An UltiMate WPS-3000 TPL autosampler (Thermo Fisher, Waltham, MA) with 2.4 μL injection needle was modified with a 10-port Nanovolume valve (part #C72MX-4570, VICI, Houston, TX) replacing the standard 6-port valve to allow sample clean-up and enrichment with a SPE column (Figure 3). The SPE column was a 5-cm-long, 100 μm i.d. fused silica capillary packed with 3 μm Jupiter particles (Phenomenex, Torrance, CA) and fritted on both ends using a Kasil formamide frit kit (Next Advance, Troy, NY) according to manufacturer’s instructions. The sample loop was made from a 22-cm-long, 200 μm i.d. fused silica capillary. The autosampler was coupled with an UltiMate 3000 RSLCnano pump (Thermo Fisher Scientific, Waltham, MA). The sample-containing 384-well microplate was thawed if stored, frozen, and centrifuged for 1 min at 1500 rpm before being loaded into the autosampler. The temperature inside the autosampler chamber was set to 4 °C. The “SampleHeight” and “Offset 384-Wellplate” settings in software were selected to allow the sample needle to gently touch the bottom of wells. A sample vial containing 15 mL of mobile phase A was also loaded into the autosampler. A “user-defined program” (UDP) method was created for injection as follows: (1) set the UdpInjectValve to load; (2) use UdpDraw to aspirate 10 μL of mobile phase A from the vial; (3) use UdpDraw to aspirate 5.5 μL of sample (plus a limited amount of air) from a well (specific wells are set in the Xcalibur software sample running queue); (4) use UdpDraw to aspirate an additional 2.9 μL of mobile phase A from the vial; (5) set the UdpInjectValve to inject; (6) add a UdpInjectMarker step; (7) add a UdpMixNeedleWash step to move the needle back to the waste container; and (8) wash the needle with 80 μL of mobile phase A using UdpBufferLoopWash.
Figure 3.
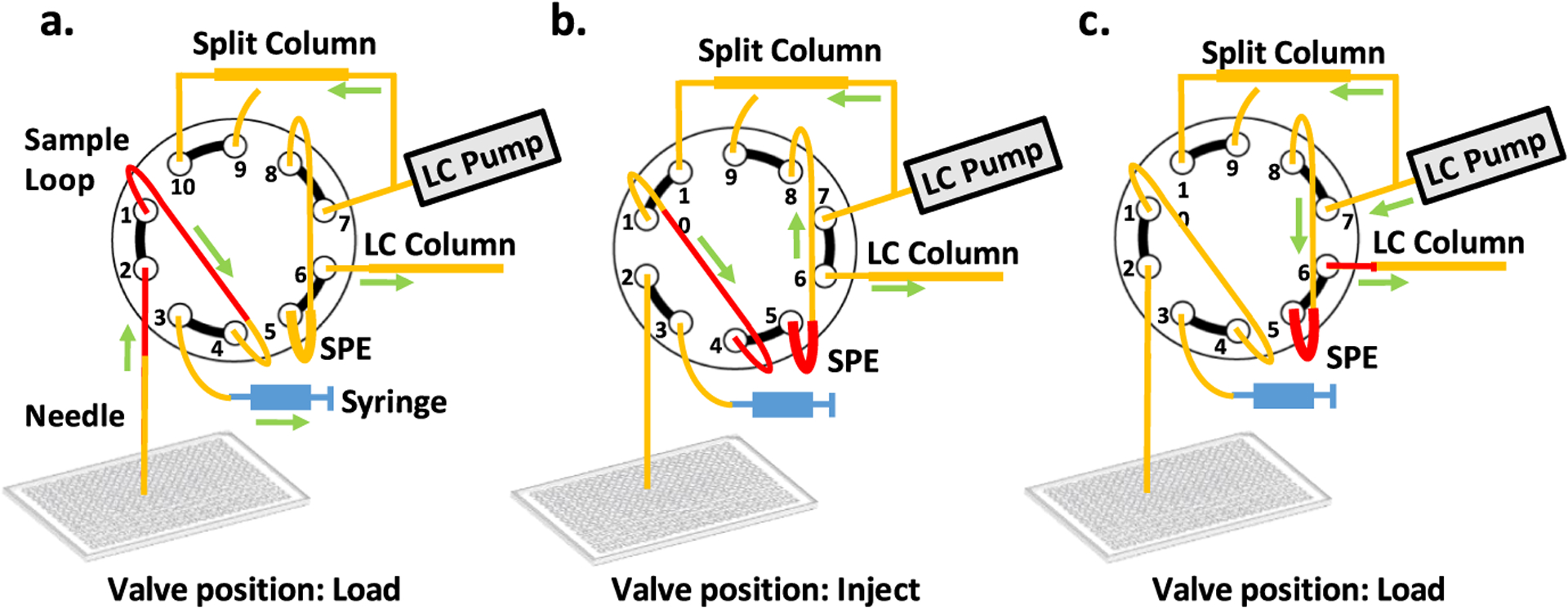
Modified autosampler for low-input sample injection and analysis. A 10-port nanovolume valve replaces the standard 6-port valve and is connected a 30 μm i.d. analytical column, an SPE column, a split-flow column, a 7 μL sample loop, an injection needle, and the autosampler syringe. (a) Sample is loaded into the sample loop. (b) The valve is switched to the injection position and flow from the split column drives the sample from the sample loop to the SPE column. (c) The valve is switched back to the load position and the sample is separated by gradient-elution reversed-phase nanoLC at a flow rate of ~40 nL/min.
NanoLC-MS/MS.
A home-packed nanoLC column of 30 μm i.d. (45 cm long) and a split flow column of 75 μm i.d. (30 cm long) were packed with the same particles as the SPE column and connected to the valve as shown in Figure 3. Other connecting tubing is 20 μm i.d. fused silica capillary and the outer diameter of all fused silica tubing is 360 μm. A 10 μm i.d. chemically etched emitter32 was butt-coupled to the separation column as described previously.16 The flow rate through the separation column was ~40 nL/min with a total LC pump flow rate of 0.3 μL/min and a pressure of ~340 bar. To load samples from the sample loop to the SPE column, an increased flow rate of 0.32 μL/min was used for the first 35 min (valve position “inject” in Figure 3). From 39 to 42 min, the flow rate was changed to 0 nL/min. At 42 min, the valve was changed back to “load” (Figure 3) in the script editor and the flow rate of 0.3 μL/min was restored for separation. The percentage of mobile phase B was increased from 2 to 8% over 5 min. A 100 min linear gradient of 8 to 22% mobile phase B (0.1% FA in acetonitrile), followed by a 20 min linear gradient of 22–45% mobile phase B. Then, 90% mobile phase B was used to clean the column for 5 min before the column was re-equilibrated for 20 min with 2% mobile phase B.
An Orbitrap Exploris 480 (Thermo Fisher) mass spectrometer with the Nanospray Flex ion source was used. The electrospray voltage was set to 2.0 kV and the temperature of the ion transfer tube was set to 200 °C. For MS1, the Orbitrap resolution was set to 120,000 (at m/z 200) with the normalized AGC target of 100%. The maximum injection time was set to 250 ms for 1 and 10 HeLa cells and to 100 ms for 150 and 500 HeLa cells. Precursor ions of +2 to +6 charge were selected for MS2 analysis. The exclusion duration was set to 90 s for 1 and 10 HeLa cells, and to 60 s for 150 or more HeLa cells. The intensity threshold was set to 5 × 103 for single-cell samples and 1 × 104 for other samples. For MS2, the HCD collision energy was 30% and the isolation window was 1.6. Orbitrap resolution and maximum injection times were varied based on the sample size as follows. The maximum injection times were 500, 250, 100, and 50 ms for 1, 10, 150, and 500 HeLa cells, respectively. Resolution was 60,000 for 1 and 10 cells, 30,000 for 150 cells, and 15,000 for 500 cells. For primary immune cells, the settings corresponding to 10 HeLa cells were applied.
Data Processing.
To compare the protein coverage with our previous nanoPOTS work, the raw files of HeLa cells were first processed with MaxQuant 1.6.7.0 for feature detection, searching, and quantification. MS/MS spectra were searched against a FASTA file obtained through UniProt (Swiss-Prot, reviewed, and downloaded July 2020). The minimum peptide length was six. Equivalent cell loadings were grouped for label-free quantitation. All other settings were default, including the PSM and protein FDR set to 0.01. Protein identifications are reported as protein groups, meaning that proteins lacking a unique peptide are collapsed. Potential contaminants (contaminant protein identifications defined and downloaded in http://www.coxdocs.org/doku.php?id=maxquant:start_downloads.htm) and reverse peptides, as well as those only identified as modified proteins, were filtered out from further analysis. The results were further visualized with an online Venn diagram tool (https://www.meta-chart.com/venn).
MSFragger33 with FragPipe was used to search against the same human proteome database for both HeLa cells and primary immune cells using default settings other than enabling quantification. The false discovery rate was set at 0.01 for ionFDR, peptideFDR, and proteinFDR. Protein identification for MSFragger uses the ProteinProphet algorithm. The identified proteins in MSFragger did not contain trypsin or keratin contaminants. Match between runs was not used for MaxQuant or MSFragger analysis. Further analysis was conducted in iPython Jupyter notebooks.34 Data were normalized by log 2 transformation of the entire dataset. To account for batch effects between runs, values from each run were adjusted by the median value, resulting in a log 2-scale dataset centered around zero. The data were adjusted back to an unlogged scale for coefficient of variation (CV) calculations. Gene set analysis was performed using Enrichr.35,36 The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021882.
RESULTS AND DISCUSSION
An overview of the fully automated sample processing and analysis workflow is shown in Figure 1. After prefilling the wells of a 384-well microplate with cell lysis solution, cells are loaded into the wells using one of three methods: dilution from suspension, micromanipulation, or FACS. The well plate is sonicated for 5 min with the sealing mat installed to aid in cell disruption. The sample-containing well plate is then loaded onto the temperature control module of the OT-2 platform where the one-pot sample processing protocol takes place without user interaction. The protocol includes reagent dispensing, incubation, and periodic addition of water or buffer to compensate for limited evaporation through the sealing mat. Following sample preparation, the 384-well microplate is transferred to an autosampler for ultrasensitive nanoLC-MS/MS analysis.
We selected a low-volume (50 μL total capacity) low-binding 384-well microplate with a transparent, flat bottom to minimize nonspecific adsorption and for compatibility with plate readers and microscopes. As provided by the manufacturer, intense polymer contamination peaks were observed in the resulting chromatograms (Figure S4). These were substantially reduced by precleaning the well plates as described in the Experimental Section, yet some contamination persisted. The residual contamination has not significantly impacted the overall performance, but a different well plate having desirable surface and optical properties, combined with an appropriate sealing mat, may ultimately prove more suitable for autoPOTS.
Low-Volume Pipetting Performance and Evaporation through Sealing Mats.
Having selected an appropriate well plate and sealing mat combination, we sought to determine whether the Opentrons OT-2 near its low-volume limit of 1 μL37 could deliver reagents with sufficient accuracy and precision to enable reproducible preparation of low-input proteome samples. Of the many automated liquid handling systems available on the market today, the OT-2 is particularly attractive because of its low price (currently, $5000 plus the cost of accessories), which should be accessible to many laboratories. We characterized the performance of the OT-2 from 1–10 μL using a first-generation P10 single-channel pipette (Table S1) and found very close agreement with the manufacturer specifications.37 The CV was 5.6% at the lower limit of 1 μL, which we considered sufficiently reproducible for our purposes. While this work was performed using the first-generation P10 pipette and not the newer P20 pipette, the manufacturer reports an identical pipetting performance at 1 μL,37 so the two pipettes are expected to be interchangeable.
Sample preparation for bottom-up proteomics typically includes hours-long incubations at elevated temperatures. In our one-pot workflow for low-input samples,20,31 cells are lysed and proteins are reduced in one step using a mixture of DDM and DTT at 70 °C for 1 h. Alkylation takes place at 25 °C for 30 min, followed by a 3 h predigestion with lys-C and 12 h trypsin digestion at 37 °C. During the high-temperature or long-term incubation steps, evaporation is not negligible, even with a sealing mat in place. To this end, we determined the evaporation rate at all temperatures used for sample preparation and storage (Table 1). For this evaluation, a new silicone sealing mat was applied to an empty well plate and 5 μL of NaFl solution was added through the prescored sealing mat using the OT-2. Adhesive sealing foils were also tested for long-term storage at 4 °C and −20 °C (Table 1). Greater evaporation rates were observed for the outermost wells of the array, so samples were excluded from these wells. Moreover, when set to cool, the thermoelectric temperature control module exhausts a significant amount of heat, which causes the temperature to rise in the chamber. As such, the observed evaporation rate was actually twice as high at 4 °C than at 25 °C, so special attention is required if the cooling function is used with the temperature module. For longer term low-temperature storage, the silicone mat should be replaced with an adhesive sealing foil, which had reduced evaporation rates of ~0.41 μL/day at 4 °C and 0.14 μL/day at −20 °C.
Table 1.
Evaporation Rates at Various Temperatures Used for autoPOTS Preparation and Storage. Temperature was Controlled using the OT-2 Temperature Module if not Otherwise Specified
| temperature (°C) | approximate evaporation rate | sealing mat |
|---|---|---|
| 70 | 0.13 μL/min | Silicone |
| 37 | 0.62 μL/h | Silicone |
| 25 | 0.12 μL/h | Silicone |
| 4 | 0.23 μL/h | Silicone |
| 4 (refrigerator) | 0.06 μL/h | Silicone |
| 4 (refrigerator) | 0.41 μL/day | Foil |
| −20 (freezer) | 1 μL/week | Foil |
Automated Sample Preparation.
Having determined the evaporation rates at various temperatures used for sample processing, we developed a sample preparation protocol that closely follows the nanoPOTS workflow but with larger dispensed volumes and periodic addition of buffer or water to compensate for the evaporation. ABC buffer decomposes into volatile components at elevated temperatures, so we replenished with ABC for the 70 °C cell lysis/protein reduction step and with water for all other incubations. The OT-2 is controlled using scripts developed using the Opentrons Python Protocol API Version 2 (https://docs.opentrons.com/v2/). For autoPOTS, two protocols were employed. The first protocol adds 4 μL of 1% DDM solution to each well before cell loading and sonication. Once samples are loaded into wells and the sealing mat is in place, the second protocol controls all remaining sample preparation steps. Using the workflow shown in Figure 2, the final sample volume is ~6 μL. The speed of the OT-2 system is limited to 10 pipetting operations/min such that fewer than 384 samples can be prepared at one time, as limited by the duty cycle for the 70 °C incubation step of the protocol. To minimize pipette tip consumption, one tip was used for sample replicates (e.g., all samples comprising 10 HeLa cells), with a buffer wash between each sample. After preparation, an additional volume of mobile phase A may be added to compensate for anticipated evaporative losses during sample storage.
Automated Sample Injection.
Our previous nanoPOTS20 and μPOTS31 protocols utilized manual sample injection for LC–MS/MS, which was labor intensive and technically challenging as it required the breaking and reforming of high-pressure connections for each sample. To reduce the potential for human error and increase the reproducibility, we modified a commercial autosampler for sample introduction. The original design of the autosampler includes a six-port valve to aspirate the sample into a sample loop using a syringe, and direct loading of the sample onto an LC column using a high-pressure pump. However, as our one-pot prepared samples contain salts, lipids, and cell debris, there was concern that direct injection of such samples could limit the lifetime of both the column and the electrospray emitter. Moreover, the 30 μm i.d. column used here operated at ~40 nL/min such that direct loading of the 7 μL sample loop would require ~3 h. We thus modified the autosampler with a 10-port valve (Figure 3) to incorporate an SPE for sample precleaning and concentration. Sample loading onto an SPE column typically requires an auxiliary loading pump, but we connected a 75 μm i.d. packed split flow column to the 10-port valve such that its outflow drove the sample onto the SPE. This small modification both simplified the operation and avoided the need for a dedicated loading pump. It did, however, limit the sample loading flow rate to that of the split column, which in this case was ~280 nL/min for a loading time of ~35 min. Loading time could be reduced without impacting the separation performance by employing a wider bore or a shorter split flow column, but with some additional mobile phase consumption. Other details of operation are shown in Figure 3 and described in the Experimental Section. From our ~6 μL autoPOTS-prepared samples, we injected 5.5 μL into the sample loop while the remaining ~0.5 μL of sample could not be loaded because of the geometry of the well plate, resulting in a sample utilization efficiency of ~90%. Maximizing the injected sample amount increased the likelihood of aspirating air bubbles along with the sample. By setting the syringe to aspirate the sample at a low flowrate of 0.2 μL/s, the sample could be aspirated into the sample loop despite the introduction of some air bubbles into the line. With this approach, even in cases of unexpected evaporation, sample volumes as low as 3 μL could still be collected and loaded.
AutoPOTS Performance for Low-Input Proteome Profiling.
We used the autoPOTS workflow for label-free proteome profiling of 1–500 HeLa cells. For comparison with our previous nanoPOTS work,20 peptide and protein coverage were analyzed with MaxQuant and resulted in an average of 104, 605, 6047, 15728, and 26565 peptides identified from 0, 1, 10, 150, and 500 cells, respectively (Figure S5). Average MS/MS protein identifications increased from 163 for 1 cell to 3293 for 500 cells, as shown in Supporting Information Table E1. On comparing the coverage with past studies that used the same cell type and data analysis settings, and similar conditions for LC and MS, we observe a 24% reduction in peptide coverage relative to nanoPOTS at the single-cell level24 and just 12% for ~150 cells.20 In contrast, sample preparation in a total volume of 25 μL using untreated plastic vials was reduced relative to nanoPOTS by 90% for 10 cells and 32% for 150 cells,20 thus underscoring the benefits of surface treatment and volume reduction to the extent possible with autoPOTS.
By using more sensitive algorithms of MSFragger,33,38 we identified an average of 301, 1461, 2679, and 3347 protein groups from 1, 10, ~150, and ~500 cells, respectively (Figure 4 and Supporting Information Table E1). Most of the proteins found in 1, 10, and 150 cells are included in the 500-cell group (Figure 4c). Also, FACS-sorted samples containing 10–500 HeLa cells showed a similar coverage (Figure S6). The results of HeLa cells indicate a narrowing of the performance gap between the specialized nanoPOTS platform and the fully automated, low-cost, and broadly accessible autoPOTS. While it is not likely that this gap will ever close completely given the increased surface exposure of the present platform relative to nanoliter preparation, the moderate reduction in coverage, particularly for low-nanogram samples, make autoPOTS ideally suited for many low-input applications.
Figure 4.
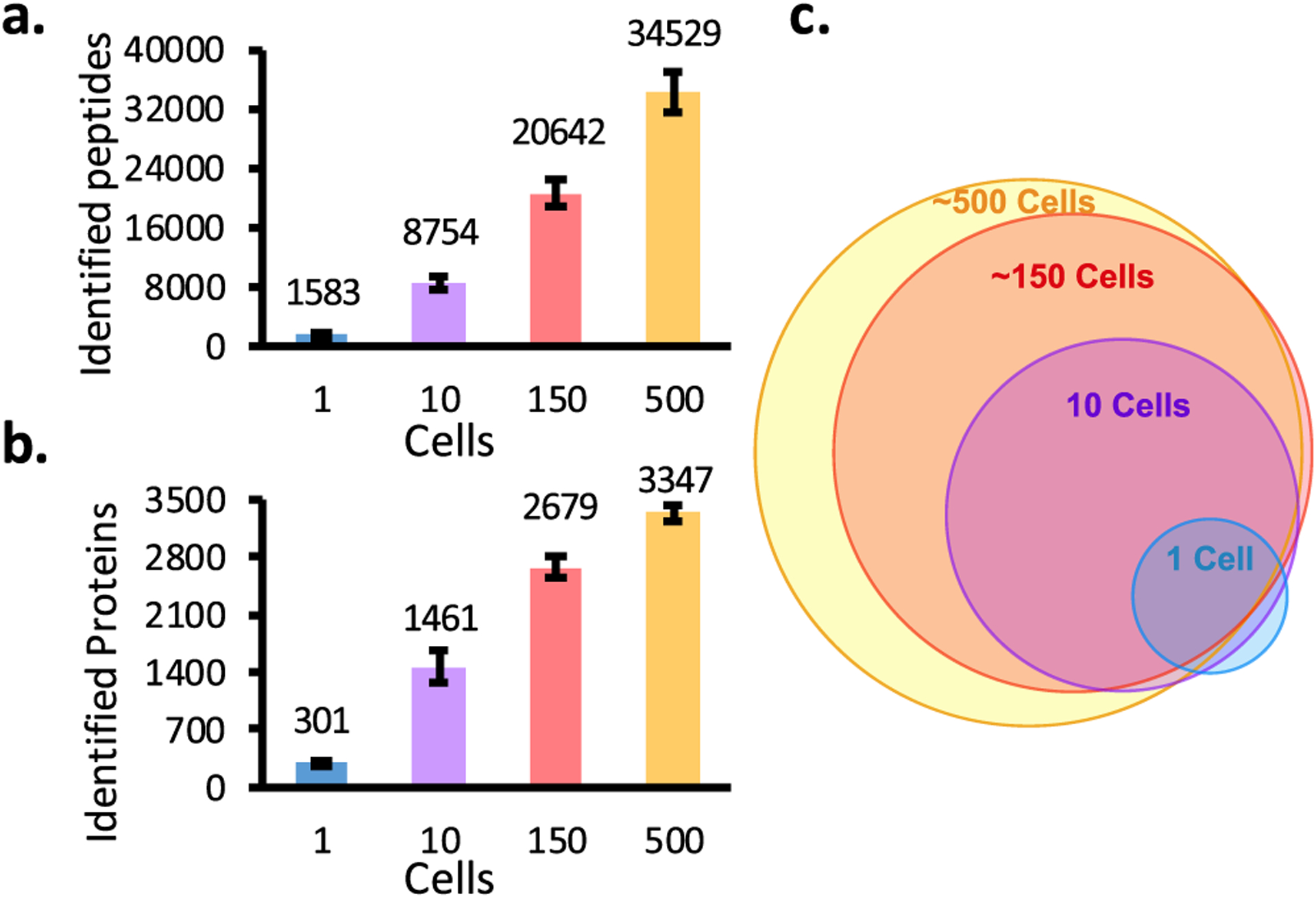
Number of peptides (a) and proteins (b) identified by MS/MS from 1, 10, ~150, and ~500 HeLa cells. (c) Overlap of protein groups identified in two of three replicates from various sample loadings.
AutoPOTS for Clinical Samples.
We applied the autoPOTS workflow to the analysis of B and T lymphocytes collected from a healthy donor and analyzed the data using MSFragger. Prior to cell sorting, we tested the efficiency of our FACS instrument using fluorescent beads. With the same settings used for cell sorting, an average of 129 beads was collected per well with the target set at 145 (Table S4), indicating a sorting efficiency of ~89%. We sorted 5 replicates of 145 B and T cells for sample processing and analysis by autoPOTS. Because of the extremely small size of lymphocytes (125 ± 15 μm3, BNID 11143931) compared to that of HeLa cells (1198–4290 μm3, BNID 103725),39 we expected these samples to provide a similar protein yield to that of 10 HeLa cells, and MS acquisition parameters were selected accordingly. Average proteome coverage from all the sorted ~130 lymphocyte samples was 1095 protein groups. Technical reproducibility among the replicates was high (average Spearman correlation coefficient between replicates was 0.92), and B cells and T cells had distinct protein profiles (Figure 5a). Gene set enrichment analysis using a cell-type classifier35,40 clearly identified these datasets as hematopoietic and lymphoblastoid cells (Table S5). Gene set enrichment using Gene Ontology41 and Wikipathways42 (Tables S6 and S7) identified pathways specific to immune function (e.g., proteasome degradation, MHC protein complex binding, B receptor signaling, interferon signaling, and so on) as well as metabolic and cell maintenance functions (mRNA binding, ribosomes, TCA cycle, translation factor activity, and so on).
Figure 5.
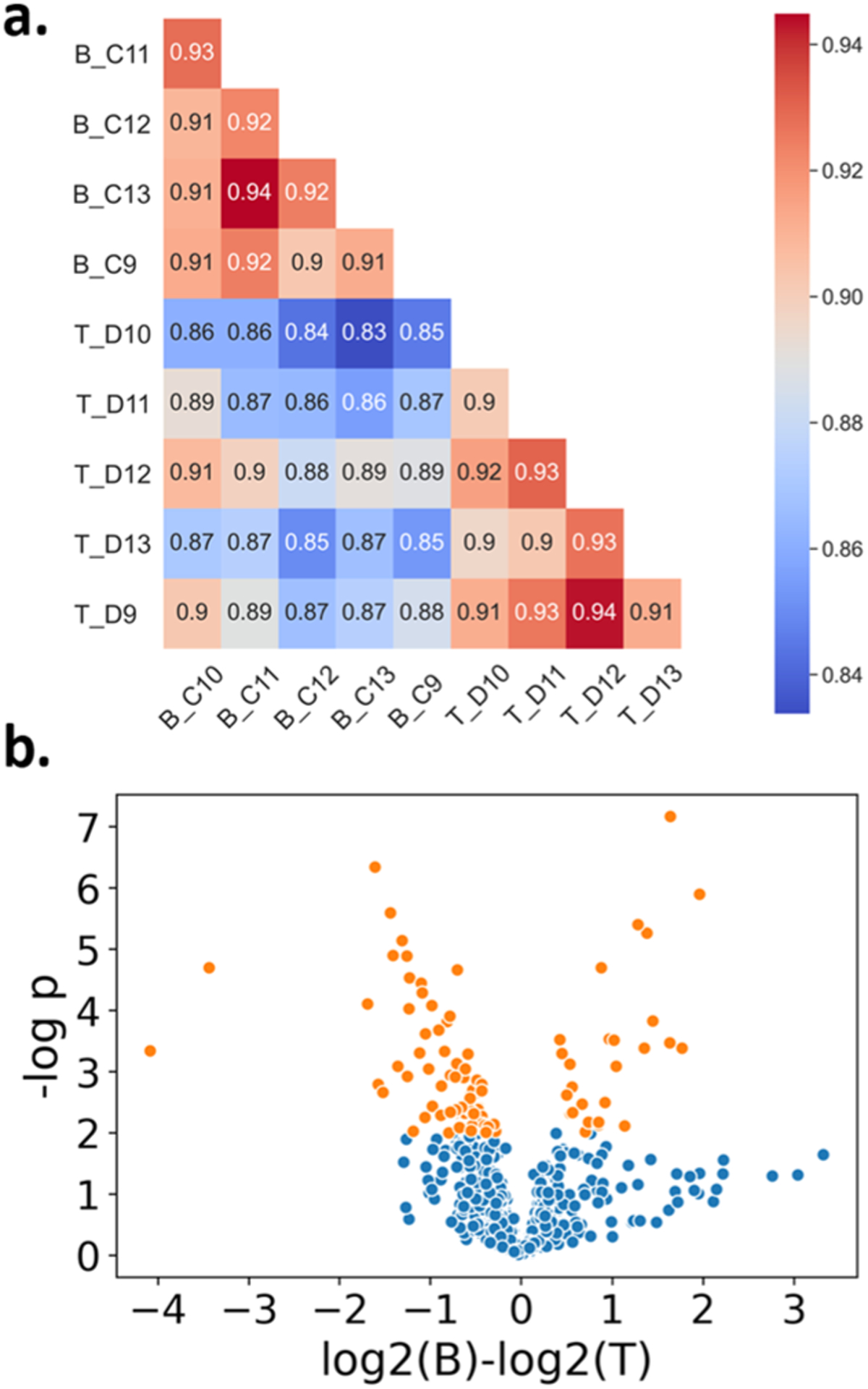
Proteome profiling of ~130 human B and T lymphocytes. (a) Spearman correlations of each sample. (b) Significant proteins (orange) identified using the t-test (p-value > 0.01) from the 961 protein groups identified in at least three of five replicates for each cell type.
To illustrate the ability to distinguish between these similar lymphocyte subpopulations, we identified proteins that were selectively expressed or enriched in each cell type. 71 proteins were identified in at least three B cell replicates and were absent in the T cell data. There were 55 proteins identified in at least three T cell replicates, which were absent in the B cell data. We next identified proteins that were differentially expressed between the two cell types. 961 proteins were identified in at least three replicates of both B and T cells and were compared for differential expression (Figure 5b). 69 protein groups were significantly more abundant in T cells and 26 were more abundant in B cells (t-test, p < 0.01). These are available in Supporting Information Table E2. From these highly expressed and cell-type-specific proteins, we find numerous examples confirming the known biological function of each type of immune cell. For example, B cells are responsible for antibody production,43 so it is unsurprising that the immunoglobulin heavy constant chain, IGHM, was strongly and exclusively identified in B cells. Classic markers such as CD20, a B-lymphocyte antigen involved in development and activation,44–46 were also found in their expected subtype. T cell markers CD2 (T-cell surface antigen), CD5 (T-cell surface glycoprotein), and CD6 (T-cell differentiation antigen) expressed distinctly in T cells47–49 were strongly identified in all T cell replicates and completely absent in B cell replicates.
CONCLUSIONS
This work describes a one-pot sample processing workflow for low-input proteomic samples that is fully automated for both sample preparation and LC–MS analysis. A low-cost OT-2 pipetting robot was used to process samples in a total volume of ~6 μL in a 384-well microplate. The reproducibility and accuracy of OT-2 pipetting and evaporation rates at different temperature were characterized to develop the workflow. The platform was evaluated in terms of proteome coverage using samples containing 1–500 HeLa cells. Compared with nanoPOTS preparation and similar LC–MS analysis, we observed a moderate reduction of proteome coverage of 24 and 12%, respectively, for 1 and ~150 cells. Moreover, a commercial autosampler was used and modified with a 10-port valve for compatibility with narrow-bore columns operating at ~40 nL/min. The automated workflow can substantially reduce contamination and human error during sample preparation and sample analysis compared with μPOTS. We also demonstrated the application of the developed workflow by comparing the proteomes of B and T lymphocytes with only ~130 cell input. Each lymphocyte is approximately 20 times smaller than typical HeLa cells, yet the workflow was able to identify an average of 1095 proteins. Importantly, the identified proteins are characteristic of general lymphocytes as well as specific B or T cell function, illustrating the precision and applicability of the system for clinically relevant samples. While autoPOTS worked well for the samples described here, the limited sample cleanup may be insufficient for larger or more lipid-rich samples, which could potentially lead to column or SPE blockage. Such increased sample loadings should be carefully evaluated for column compatibility and larger-bore columns may be required. AutoPOTS may be further improved for greater coverage by optimizing the well plate surface treatment and also digestion conditions, including reaction times, enzyme concentrations, and temperature. In its current form, this robust and automated method using only commercially available instrumentation should facilitate the implementation of low-input label-free proteomics in many laboratories.
Supplementary Material
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award numbers R33CA225248 (R.T.K.), R01GM138931 (R.T.K.), R01CA235575 (R.T.K.), and U24CA210972 (S.H.P.). Y.L. gratefully acknowledges support from the BYU Department of Chemistry and Biochemistry through the Roland K. Robins Fellowship and from the BYU Simmons Center for Cancer Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c04240.
Calibration curve for fluorescein samples; accuracy and reproducibility of OT-2 pipetting vs manual pipetting; effect of pipetting speed and changing tips; determination of evaporation rates from well plates; microscopy image of the cell suspension for accurate cell counting; peptide and protein IDs of sampled cells; FACS-sorting accuracy; chromatograms of HeLa protein digest standard; number of peptides and proteins identified by MS/MS analysis; and gene set enrichment analysis using various methods (PDF)
Identification of HeLa cells (XLSX)
Identification of lymphocytes (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c04240
The authors declare no competing financial interest.
Contributor Information
Yiran Liang, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
Hayden Acor, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
Michaela A. McCown, Department of Biology, Brigham Young University, Provo, Utah 84602, United States
Andikan J. Nwosu, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Hannah Boekweg, Department of Biology, Brigham Young University, Provo, Utah 84602, United States.
Nathaniel B. Axtell, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Thy Truong, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
Yongzheng Cong, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
Samuel H. Payne, Department of Biology, Brigham Young University, Provo, Utah 84602, United States;.
Ryan T. Kelly, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States;.
REFERENCES
- (1).Nagaraj N; Wisniewski JR; Geiger T; Cox J; Kircher M; Kelso J; Pääbo S; Mann M Mol. Syst. Biol 2011, 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Beck M; Schmidt A; Malmstroem J; Claassen M; Ori A; Szymborska A; Herzog F; Rinner O; Ellenberg J; Aebersold R Mol. Syst. Biol 2011, 7, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang D; Eraslan B; Wieland T; Hallström B; Hopf T; Zolg DP; Zecha J; Asplund A; Li L. h.; Meng C; et al. Mol. Syst. Biol 2019, 15, No. e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wiśniewski JR; Zougman A; Nagaraj N; Mann M Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- (5).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Mol. Syst. Biol 2014, 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sielaff M; Kuharev J; Bohn T; Hahlbrock J; Bopp T; Tenzer S; Distler UJ Proteome Res 2017, 16, 4060–4072. [DOI] [PubMed] [Google Scholar]
- (7).Hughes CS; Moggridge S; Müller T; Sorensen PH; Morin GB; Krijgsveld J Nat. Protoc 2019, 14, 68–85. [DOI] [PubMed] [Google Scholar]
- (8).Kulak NA; Pichler G; Paron I; Nagaraj N; Mann M Nat. Methods 2014, 11, 319–324. [DOI] [PubMed] [Google Scholar]
- (9).Budnik B; Levy E; Harmange G; Slavov N Genome Biol 2018, 19, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dou M; Clair G; Tsai C-F; Xu K; Chrisler WB; Sontag RL; Zhao R; Moore RJ; Liu T; Pasa-Tolic L; et al. Anal. Chem 2019, 91, 13119–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Specht H; Emmott E; Petelski AA; Huffman RG; Perlman DH; Serra M; Kharchenko P; Koller A; Slavov N bioRxiv 2019, 665307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lombard-Banek C; Moody SA; Nemes P Angew. Chem., Int. Ed 2016, 55, 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang Z; Qu Y; Dovichi NJ TrAC, Trends Anal. Chem 2018, 108, 23–37. [Google Scholar]
- (14).Yue G; Luo Q; Zhang J; Wu S-L; Karger BL Anal. Chem 2007, 79, 938–946. [DOI] [PubMed] [Google Scholar]
- (15).Li S; Plouffe BD; Belov AM; Ray S; Wang X; Murthy SK; Karger BL; Ivanov AR Mol. Cell. Proteomics 2015, 14, 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cong Y; Liang Y; Motamedchaboki K; Huguet R; Truong T; Zhao R; Shen Y; Lopez-Ferrer D; Zhu Y; Kelly RT Anal. Chem 2020, 92, 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dou M; Chouinard CD; Zhu Y; Nagy G; Liyu AV; Ibrahim YM; Smith RD; Kelly RT Anal. Bioanal. Chem 2019, 411, 5363–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cong Y; Motamedchaboki K; Misal SA; Liang Y; Guise AJ; Truong T; Huguet R; Plowey ED; Zhu Y; Lopez-Ferrer D; et al. Chem. Sci 2021, DOI: 10.1039/D0SC03636F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Greguš M; Kostas JC; Ray S; Abbatiello SE; Ivanov AR Anal. Chem 2020, 92, 14702–14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; et al. Nat. Commun 2018, 9, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shao X; Wang X; Guan S; Lin H; Yan G; Gao M; Deng C; Zhang X Anal. Chem 2018, 90, 14003–14010. [DOI] [PubMed] [Google Scholar]
- (22).Li Z-Y; Huang M; Wang X-K; Zhu Y; Li J-S; Wong CCL; Fang Q Anal. Chem 2018, 90, 5430–5438. [DOI] [PubMed] [Google Scholar]
- (23).Kelly RT Mol. Cell. Proteomics 2020, 19, 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhu Y; Clair G; Chrisler WB; Shen Y; Zhao R; Shukla AK; Moore RJ; Misra RS; Pryhuber GS; Smith RD; et al. Angew. Chem., Int. Ed 2018, 57, 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tsai C-F; Zhao R; Williams SM; Moore RJ; Schultz K; Chrisler WB; Pasa-Tolic L; Rodland KD; Smith RD; Shi T; et al. Mol. Cell. Proteomics 2020, 19, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhu Y; Podolak J; Zhao R; Shukla AK; Moore RJ; Thomas GV; Kelly RT Anal. Chem 2018, 90, 11756–11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Williams SM; Liyu AV; Tsai C-F; Moore RJ; Orton DJ; Chrisler WB; Gaffrey MJ; Liu T; Smith RD; Kelly RT; et al. Anal. Chem 2020, 92, 10588–10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhu Y; Dou M; Piehowski PD; Liang Y; Wang F; Chu RK; Chrisler WB; Smith JN; Schwarz KC; Shen Y; et al. Mol. Cell. Proteomics 2018, 17, 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liang Y; Zhu Y; Dou M; Xu K; Chu RK; Chrisler WB; Zhao R; Hixson KK; Kelly RT Anal. Chem 2018, 90, 11106–11114. [DOI] [PubMed] [Google Scholar]
- (30).Piehowski PD; Zhu Y; Bramer LM; Stratton KG; Zhao R; Orton DJ; Moore RJ; Yuan J; Mitchell HD; Gao Y; et al. Nat. Commun 2020, 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Xu K; Liang Y; Piehowski PD; Dou M; Schwarz KC; Zhao R; Sontag RL; Moore RJ; Zhu Y; Kelly RT Anal. Bioanal. Chem 2019, 411, 4587–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kelly RT; Page JS; Luo Q; Moore RJ; Orton DJ; Tang K; Smith RD Anal. Chem 2006, 78, 7796–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI Nat. Methods 2017, 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Payne S Lymphocyte Data https://github.com/PayneLab/Lymphocytes1 (accessed Aug 7, 2020).
- (35).Kuleshov MV; Jones MR; Rouillard AD; Fernandez NF; Duan Q; Wang Z; Koplev S; Jenkins SL; Jagodnik KM; Lachmann A; et al. Nucleic Acids Res 2016, 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen EY; Tan CM; Kou Y; Duan Q; Wang Z; Meirelles G; Clark NR; Ma’ayan A BMC Bioinf 2013, 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Opentrons. Opentrons Electronic Pipettes https://opentrons.com/publications/Opentrons-Pipette-White-Paper.pdf (accessed Aug 7, 2020).
- (38).Yu F; Haynes SE; Teo GC; Avtonomov DM; Polasky DA; Nesvizhskii AI Mol. Cell. Proteomics 2020, 19, 1575–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Milo R; Jorgensen P; Moran U; Weber G; Springer M Nucleic Acids Res 2010, 38, D750–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Schmidt T; Samaras P; Frejno M; Gessulat S; Barnert M; Kienegger H; Krcmar H; Schlegl J; Ehrlich H-C; Aiche S; et al. Nucleic Acids Res 2018, 46, D1271–D1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ashburner M; Ball CA; Blake JA; Botstein D; Butler H; Cherry JM; Davis AP; Dolinski K; Dwight SS; Eppig JT; et al. Nat. Genet 2000, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Slenter DN; Kutmon M; Hanspers K; Riutta A; Windsor J; Nunes N; Mélius J; Cirillo E; Coort SL; DIgles D; et al. Nucleic Acids Res 2018, 46, D661–D667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lebien TW; Tedder TF Blood 2008, 112, 1570–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Pavlasova G; Mraz M Haematologica 2020, 105, 1494–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bubien JK; Zhou LJ; Bell PD; Frizzell RA; Tedder TF J. Cell Biol 1993, 121, 1121–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Polyak MJ; Li H; Shariat N; Deans JP J. Biol. Chem 2008, 283, 18545–18552. [DOI] [PubMed] [Google Scholar]
- (47).Gimferrer I; Farnós M; Calvo M; Mittelbrunn M; Enrich C; Sánchez-Madrid F; Vives J; Lozano FJ Biol. Chem 2003, 278, 8564–8571. [DOI] [PubMed] [Google Scholar]
- (48).Ginaldi L; Farahat N; Matutes E; De Martinis M; Morilla R; Catovsky DJ Clin. Pathol 1996, 49, 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Li S; Juco J; Mann KP; Holden JT Am. J. Clin. Pathol 2004, 121, 268–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.