

Abstract
In the quest for new antibiotics, two novel engineered Cationic Antimicrobial Peptides (eCAPs) have been rationally designed. WLBU2 and D8 (all 8 valines are the D-enantiomer) efficiently kill both Gram-negative and –positive bacteria, but WLBU2 is toxic and D8 non-toxic to eukaryotic cells. We explore protein secondary structure, location of peptides in six lipid model membranes, changes in membrane structure and pore evidence. We suggest that protein secondary structure is not a critical determinant of bactericidal activity, but that membrane thinning and dual location of WLBU2 and D8 in the membrane headgroup and hydrocarbon region may be important. While neither peptide thins the Gram-negative lipopolysaccharide outer membrane model, both locate deep into its hydrocarbon region where they are primed for self-promoted uptake into the periplasm. The partially α-helical secondary structure of WLBU2 in a red blood cell (RBC) membrane model containing 50% cholesterol, could play a role in destabilizing this RBC membrane model causing pore formation that is not observed with the D8 random coil, which correlates with RBC hemolysis caused by WLBU2 but not by D8.
Keywords: engineered Cationic Antimicrobial Peptides (eCAPs), Gram-negative and Gram-positive bacteria, protein/lipid interaction, x-ray diffuse scattering (XDS), neutron reflectivity (NR)
Graphical Abstract
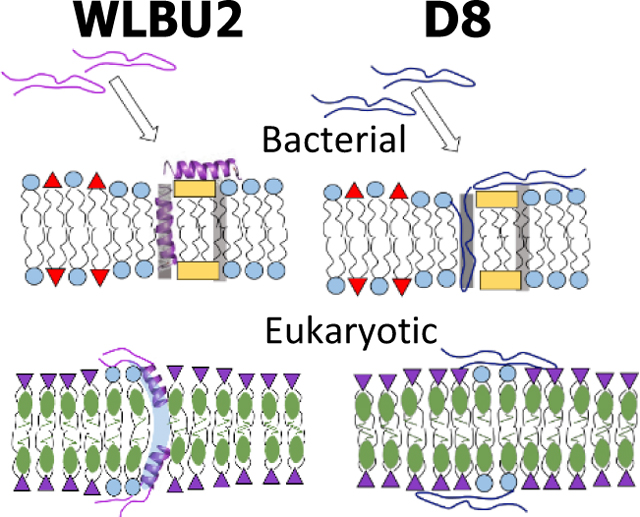
We use four bacterial and two eukaryotic lipid model membranes to explore secondary structure of two engineered Cationic Antimicrobial Peptides (eCAPs) and their locations within these models. Six synergistic biophysical methods determined protein secondary structure, changes in thickness of model membranes, locations of eCAPS in the models and evidence for a pore structure.
Introduction
Antibiotics have long made the world safer; the use of well-known drugs such as penicillin derivatives and fluoroquinolones allows surgical interventions that save lives without risk of the infection that was so prevalent before the early 20th century. However, due to growing multi-drug resistance in many bacterial strains, the need for better antibiotics is urgent [1]. By taking inspiration from nature [2],3, engineered cationic antimicrobial peptides (eCAPs) attack the negatively charged bacterial membrane 4 rather than a metabolic pathway as with traditional antibiotics. Thus development of resistance can take as long as four weeks 5 instead of a few days 6 to occur.
Natural antimicrobial peptides (AMPs) like colistin and LL-37 have been well studied 5, 7–11. Some natural AMPs kill eukaryotic as well as bacterial cells, or they kill only one type of bacteria or only in specified environments. Colistin, for example, only kills Gram-negative (G(−)) but not Gram-positive (G(+)) bacteria and displays a decrease in antimicrobial activity when in the presence of divalent cations 12. While many methods have been used to design eCAPs in an effort to improve selectivity, rational Design takes into consideration hydrophobicity, hydrophobic moment, length of peptide, secondary structure, number and types of amino acids, and net charge 2, 13. Although AMPs can cause cellular toxicity, several AMPs are now in use clinically: colistin, gramicidin and daptomycin with many more in clinical trials14.
Despite the success of eCAPs, the mechanisms by which they kill bacteria are still controversial. While some AMPs have intracellular targets in addition to a membrane perturbation 15–18, in this work we focus on the non-specific membrane target. Previously there had been speculation that AMPs form pores through membranes19–23. Barrel-stave pore formation in lipid bilayers of the natural AMP alamethicin has been observed using neutron and X-ray scattering 24,25 and toroidal pores have also been observed with other natural peptides like melittin 26 and magainin. It has been assumed that eCAPs also form pores, but the evidence to validate this assumption is meager 27,28. Indeed, the evidence against pore formation is mounting: Wimley et al. showed that the ion release rate from a lipid vesicle with a singular pore was about 1000 X slower4 than the release rates that are known for ion channels 29.
Besides the visible pore models, the “carpet model” from Shai et al., proposes that AMPs coat the surface of the membrane like a carpet, and then dismantle regions of the lipid membrane 30,31. Another model from Bechinger is the “detergent model” 32,33. In this model the AMPs act like detergents and thereby degrade the membrane at high concentrations by forming transient small pores in the membrane. Despite the popularity of these theories a domain theory has also been proposed by the Epand laboratory that can predict the cytotoxic range based on lipid clustering caused by antimicrobial agents34. We have recently suggested that colistin kills bacteria by the formation of domains with different material moduli 35, which could lead to an increase in membrane permeability along the domain walls. The domain theory requires a smaller perturbation than the carpet, detergent or pore models in that observable holes or pores are not required.
In this work we used six biophysical methods to investigate the structural interactions of two eCAPs, WLBU2 and D8, when interacting with four bacterial and two eukaryotic model membranes, in an effort to yield structural insights into the bactericidal and toxicity mechanisms. This study is the structural counterpart to our study of material moduli which suggested that WLBU2 and D8 kill both G(−) and G(+) via domain formation that creates instabilities along the domain walls 36. WLBU2 and D8 were engineered using rational design where hydrophobicity, hydrophobic moment, net charge, length of peptide, secondary structure and types of amino acids were considered 2, 5, 12, 37,38. While the highly charged (+13), linear 24-mer WLBU2 contains 3 types of amino acids (arginine, valine and tryptophan), in D8 all 8 valines are represented by the D-enantiomer. The peptide sequence is: RRWVR RVRRW VRRVV RVVRR WVRR (see also Fig. S1). Both peptides retain their efficacy in physiological saline 2, 36 and WLBU2 kills bacteria in high salt conditions such as in the cystic fibrosis epithelial airway39. X-ray diffuse scattering (XDS), neutron reflectometry (NR), circular dichroism (CD), nuclear magnetic resonance (NMR), differential scanning calorimetry (DSC) and molecular dynamics (MD) simulation were employed. All methods employ fully hydrated conditions, such as occur in the human body.
Results
We describe the structural effects of the addition of WLBU2 and D8 to four bacterial and two eukaryotic lipid model membranes, as investigated with six different biophysical techniques. The six model membranes used were G(−) inner membrane (IM) (POPE:POPG:TOCL 7:2:1 molar ratio), G(+) (POPG:DOTAP:POPE:TOCL 6:1.5:1.5:1), G(−) outer membrane (OM) (lipopolysaccharide model (LPS:DLPG 1:9) (see also Fig. S2) and di-deoxy-manno-octulosonic acid (KDO2)), typical eukaryotic cell Euk23 (POPC:POPE:chol 5:1:1.8), and red blood eukaryotic cell Euk50 (POPC:POPE:chol 5:1:6). While KDO2 is a rare, rough mutant of LPS, it was employed since it is a simple system that is amenable to MD simulation containing six lipid chains as in LPS, but only four sugar moieties in its headgroup. Each technique illuminates a different aspect of the structural change of the bilayer caused by the peptides and the location of the peptides in the model membranes. The sum of these changes yields a detailed picture of the interactions of the peptides with the model membranes as a basis for a mechanistic understanding of their mode of action.
XDS.
Fig. 1A–C show examples of x-ray diffuse scattering (XDS). The sample is a stack of lipid membranes mimicking the eukaryotic cell containing 23 mole % cholesterol (Euk23). The lobes of diffuse data are produced by fully hydrated, fluctuating lipid membranes. As the peptide WLBU2 is added at increasing concentration (A->B->C), the sample fluctuations increase 36, thus diminishing the uppermost lobes. Lamellar scattering produces the discrete green spots and reflects the D-spacing (due to the distance between membranes in a stack). XDS data of this type are used to calculate form factors, which in turn are used to calculate electron density profiles (EDPs) 40–42, (see also Fig. S3 for details of data analysis). The form factors for the XDS data shown in Fig. 1A–C are displayed in Fig. 1D, where there is a shift to higher qz values as the concentration of WLBU2 increases (black→red→green traces). This indicates a thinning of the membrane due to WLBU2 binding. Form factors for all of the model membranes with both eCAPS are shown in Fig. S4. The EDP for the control data (Fig. 1A) is shown in Fig. 1E. We do not show EDPs with the peptides incorporated since peptide location in the membrane is better determined using neutron reflectivity (NR, see Fig. 3). Bilayer component groups are indicated in the caption to Fig. 1E.
Figure 1.

Examples of background-subtracted XDS data. Red indicates negative, while white indicates positive intensity values, and green indicates the greatest intensity. (A) Euk 23 control, (B) 500:1 Euk23:WLBU2 molar ratio, (C) 100:1 Euk23:WLBU2. (D) Form factor data for the 3 samples shown in A,B,C. (E) Electron density profile from the control Euk23 sample, shown in A. Component groups are: Phosphate (blue), GlycerolCarbonyl (red), CHCH2 hydrocarbon (green), CH3 methyl trough (brown), Water (cyan) and Total (black). D-spacings: A. 104 Å, B. 100 Å, C. 85 Å. X-ray scans were performed at 37 °C.
Figure 3.

Component volume occupancy vs. distance from substrate for tethered bilayers and bilayer-associated peptide obtained by NR. Component groups are: gold-covered silicon wafer (orange), lipid tether (green), headgroups (light blue), hydrocarbons (dark blue), water (gray), peptide (red). 68% confidence limits of the peptide envelope are also shown in red. (A) G(−) IM/WLBU2 (500:1), (B) G(−) IM/D8 (500:1), (C) G(+)/WLBU2 (500:1), (D) G(+)/D8 (500:1), (E) Euk23/WLBU2 (250:1), (F) Euk23/D8 (250:1), (G) Euk50/WLBU2 (500:1), (H) Euk50/D8 (600:1), (I) G(−) OM (LPS:DLPG (1:3))/WLBU2 molar ratio (500:1), (J) G(−) OM (LPS:DLPG (1:3))/D8 (500:1), (K) G(−) OM (KDO2:DLPG (1:11))/WLBU2 (500:1),(L) G(−) OM (KDO2:DLPG (1:11))/D8 (500:1). Lipid:peptide molar ratios are shown in parentheses. Experiments were carried out at 37°C.
The total headgroup (combined phosphate and glycerol/carbonyl) peak-to-peak distance (DHH, black) and hydrocarbon full-width at half-maximal (2DC, green) are two measures of the membrane thickness. The EDP also determines the area per lipid (AL) when the lipid and peptide volumes are measured in a separate experiment. A summary of the XDS structural results for all of the membranes used in this study interacting with both WLBU2 and D8 is shown in Fig 2.
Figure 2.

Area per lipid (AL) (black) and half hydrocarbon thickness DC (red), of bacterial and eukaryotic membrane models with the addition of either peptide. WLBU2 (solid symbols), D8 (open symbols). (A) G(−) inner membrane (IM) model, (B) G(+) membrane model, (C) KDO2, (D) LPS model, (E) Euk23 model, (F) Euk50 model, (G) Minimum inhibitory concentrations (MIC) in aPseudomonas aeruginosa (G(−)), bMethicillin-resistant Staphylococcus aureus (G(+)), cPeripheral blood mononuclear cell toxicity (%) measured by propidium iodide in flow cytometry at 50 μM peptide, % RBC lysis (adapted from (36)). Standard deviation of AL is ± 1.0 Å2 and DC is ± 0.5 Å. X-ray and cell studies were performed at 37 °C.
As shown in Fig. 2, the effects of both peptides on AL and DC are dependent on the respective lipid membrane type. While many lipid:peptide molar ratios were characterized, results for only a low and high concentration of peptide are shown, since our material parameter results indicated that at low concentration in G(−) and G(+) membrane models, the peptides stiffen the membrane, while at high concentration they soften the membrane 36, suggesting that domains with different material moduli are juxtaposed, leading to unstable domain walls between them. It was of interest to find a a structural correlate for these material parameter results. For G(−) IM (Fig. 2A) and G(+) cell membrane model (Fig. 2B), there is a steady increase in AL and steady decrease in DC when interacting with both WLBU2 and D8. Thus, rather than a non-monotonic structural correlation to the material properties 36, the correlation of membrane softening with membrane thinning at high concentration (75:1 lipid:peptide molar ratio) may be more significant. The membrane thinning also correlates with efficient bactericidal activity (low MIC ~3 μM values) for both peptides as measured with the G(−) bacteria P. aeruginosa and with the G(+) bacteria methicillin-resistant S. aureus and shown in Fig. 2G, Suggesting that membrane thinning may be involved in bactericidal activity. For the outer membrane (OM) models of G(−) bacteria, there is a small decrease in AL and increase in membrane thickness (KDO2, Fig. 2C), while for LPS model there is no change in either structural parameter (Fig. 2D). While the OM is the first point of contact of eCAPs 43, it is the inner membrane (IM) that determines its killing efficiency, since the IM is the ultimate protective barrier for the bacteria 44. There was slight toxicity to peripheral blood mononuclear cells (PBMC) by D8, and more so for WLBU2 (Fig. 2G). These toxicity results may not be directly comparable to the structural effects of Euk23 (Fig. 2E), since PBMC’s contain ~33% cholesterol 45 while Euk23 contains 23% cholesterol, and differences between the peptides’ activity may arise with increasing membrane cholesterol content. For the sixth RBC membrane model, Euk50, Fig. 2F shows a small decrease in AL and increase in membrane thickness for both peptides, while the lysis of RBCs is strongly peptide dependent (Fig. 2G). This could indicate that beyond a threshold cholesterol concentration that membrane thickness and peptide toxicity are no longer correlated. A related result was obtained by Ramamoorthy et al. using dye leakage experiments where raft-containing model membranes were susceptible to AMPs, while membranes containing mostly LO (liquid-ordered) lipids with a higher concentration of cholesterol were protected46. Their result is similar to the case of D8 in our study, but not to that of WLBU2 which is toxic to RBC model membranes containing 50 mole % cholesterol.
NR.
While XDS can accurately measure area/lipid and hydrocarbon thickness, it is difficult to pinpoint the location of a 24-mer peptide position in the membrane due to the low electron contrast between lipid and protein. In order to determine the peptide’s position more accurately, neutron reflectivity is used, where there is a greater variability in scattering length density for the light elements compared to x-rays. A gold-covered silicon wafer containing a lipid tether is used to support a single bilayer with embedded proteins. Previous measurements (data not shown) indicated that the same result is obtained if the peptide is embedded in the lipid membrane or added through the aqueous phase; all of the present results are for embedded peptides. Fig. 3 summarizes example NR data; in most cases, duplicates or triplicates were carried out. For the outer membrane (OM) models of G(−) bacteria, LPS alone or KDO2 alone, full coverage on the tethered wafer was not achieved, so DLPG with the same lipid chain length was added to stabilize these membranes (see Fig. S5,6 and Tables S1–6).
Fig. 3 is a graphical summary of the membrane location of both WLBU2 and D8 in all six model membranes from NR measurements; these NR results are quantitated in Tables S1–S6. The red envelope is the peptide location, shown with 68% confidence limits and the other membrane components are described in the caption to Fig. 3. In both G(−) IM (A,B) and G(+) models (C,D), both peptides are located near the outer headgroup and also deep into the hydrocarbon region. In Euk23 (E, F), the primary location for both peptides is in the carbonyl/glycerol region, just below the headgroups, referred to as the lipid-water interface of the membrane. For Euk50 (G, H), both peptides are located in the periphery of the headgroup region near the bulk solvent, with a slightly greater penetration for WBLU2 compared to D8. For the G(−) OM LPS model (I,J), there is a quite deep penetration for both WLBU2 and D8, with little of either peptide in the headgroup region, which is different from the G(−) IM model (A,B). For the G(−) OM KDO2 model, the peptides locate primarily in the headgroup region, with only a small penetration to the headgroup in the inner leaflet.
CD.
In order to assess the secondary structure of both peptides in aqueous and membrane environments, circular dichroism spectroscopy was carried out. The secondary structure was then analyzed using a shape analysis of the four motifs: α-helix, β-sheet, β-turn and random coil (see SI for details.) While this analysis was successful for WLBU2, it was not for D8, since D8 contains 1/3 of its amino acids as the D-enantiomer, where the four motifs for D-amino acids are inverted 47,48. The analyzed CD results shown here apply only to WLBU2. Fig. 4A shows the ellipticity of WLBU2 in water (red trace) and in G(−) IM (black trace). In water or 15 mM PBS (where M = moles/L), WLBU2’s secondary structure was largely random coil with some β-sheet. The helix content increased as the molar ratio of WLBU2:lipid decreased, as shown in Fig. 4B. For all lipid model membranes, a molar ratio of 1:10 WLBU2:lipid was used since the signal-to-noise was excellent and peptide aggregation was minimized. The α-helical content differed for the different model membranes as shown in Fig. 4C. G(−), G(+) and KDO2 induced the highest α-helix formation, while both eukaryotic models had the lowest amount of α-helix (see also Tables S8–S14).
Figure 4.

CD results of WLBU2 in aqueous solution, pH 7 and with lipid membrane models. (A) G(−) IM/WLBU2 in G(−) IM (black trace) and WLBU2 in water (red trace), (B) Secondary structure results of WLBU2 in G(−) IM model with decreasing WLBU2:lipid molar ratio in 15 mM PBS, (C) Secondary structure results of 10 μM WLBU2 in 15 mM PBS in six model membranes at 10:1 lipid:peptide molar ratio. R2 indicates the goodness of the fit to the Brahms and Brahms data set49. All CD experiments were carried out at 37 °C. The errors on the percentages are 3–5%.
NMR.
Fig. 5A–F shows static 31P NMR spectra as both peptides are added to two lipid model membranes: G(−) IM and Euk50. In G(−) IM either POPE or POPG contained perdeuterated sn-1 chains, but only POPE results are shown since POPG yielded similar results, indicating a homogeneous sample. Fig. 5A–F indicate that these samples are in the fluid, liquid crystalline phase. For additions of both WLBU2 and D8 to G(−) IM (Fig. 5A–C), the chemical shift anisotropy Δσ, which corresponds to the width of 31P NMR spectrum, increased, indicating a reduced amplitude of headgroup motion or altered head-group orientation. In Fig. 5A the 31P NMR spectrum consists of a major component with Δσ ~34 ppm (the simulated contribution shown in dark blue, the experimental NMR spectrum in red).
Figure 5.

(A-F) Static 31P NMR spectra of two model membranes. (A-C) G(−) IM using sn-1 chain deuterated POPE-d31. (A) G(−)IM control, (B) G(−)IM/WLBU2, (C) G(−)IM/D8, (D-F) Euk50 using sn-1 chain deuterated POPC-d31. (D) Euk50 control, (E) Euk50/WLBU2, (F) Euk50/D8, (G) Summary of Δσ for the major components in A-F, errors are ~ ±1 ppm. (H-M) Static 2H NMR powder spectra. (H-J) G(−) using sn-1 chain deuterated POPE-d31. (H) G(−)IM control, (I) G(−)IM/WLBU2, (J) G(−)IM/D8, (K-M) Euk50 using sn-1 chain deuterated POPC-d31. (K) Euk50 control, (L) Euk50/WLBU2, (M) Euk50/D8. (N-O) 13C chemical shift differences of Cα-Cβ in ppm for the labelled amino acids R5, W10 and V15. (N) WLBU2 and D8 in G(−)IM model. (O) WLBU2 and D8 in 15 mM PBS (open symbols) or with 50% (v:v) TFE (solid symbols). α-helix (black bar), random coil (white bar) and β-sheet (light grey bar). All NMR experiments were carried out at 37°C; lipid:peptide molar ratios were 30:1.
Furthermore, a second minor component (light blue) and a small isotropic peak (orange) were observed. Fig. 5D–F show 31P NMR spectra when both peptides are added to the Euk50 mixture. There is almost no change in Δσ, indicating that unlike with G(−) IM model the headgroup motion/orientation in Euk50 does not change. However, in Fig. 5E, a sharp isotropic peak (near 0 ppm) indicates that a fraction of the lipids reorients isotropically with respect to the magnetic field on a fast time scale (τ << 100 ms). As this isotropic peak behavior is not seen in the control Euk50 or when D8 is added, it indicates a different interaction mechanism for WLBU2 with this RBC model. Fig. 5H–M show the measured static 2H NMR, confirming the fluid phase for both models. In the G(−) IM mixtures, samples were either per-deuterated POPE-d31 or POPG-d31 (see also Fig. S7). When either peptide was added to G(−) IM or Euk50 the quadrupolar splitting was reduced. Such a reduction in lipid chain order leads to a decrease in acyl chain length of ~0.8 Å (reduction in membrane hydrophobic thickness by 1.6 Å) in the G(−) IM membrane and even less in the Euk50. Interestingly, a sharp isotropic peak appeared at 0 kHz only in the WLBU2 containing sample, similar to the 31P NMR result, indicating a fast reorientation of the lipids over a highly curved membrane structure. As in the 31P NMR spectra, this did not occur in control Euk50 or when D8 was added to Euk50.
For secondary structure, 13C labeled amino acids were used in the peptides. For WLBU2, 3 amino acids were labeled: R5, W10 and V15, and for D8, 2 residues were labeled: R5 and W10. A homonuclear single quantum coherence spectroscopy - total correlation spectroscopy (HSQC-TOCSY) NMR spectrum acquired in 15 mM PBS was used to confirm that both WLBU2 and D8 adopt a random coil structure in 15 mM PBS. For peptides embedded in lipids, 13C solid state NMR DARR spectra under magic angle spinning conditions were used. The obtained secondary structures are shown in Fig. 5N,O. The Cα-Cβ chemical shift indicates that while WLBU2 adopts a largely α-helical content in G(−) IM model, D8 remains in a random coil conformation (Fig. 5N). The same results were obtained in the RBC Euk50 model membrane (data not shown). TFE was then used in an attempt to induce helix formation in both peptides in 15 mM PBS. As shown in Fig. 5O, while WLBU2 transitioned from random to α-helix upon addition of TFE, D8 remained in a random coil configuration. See also Fig. S8–10 and Tables S15–17 for additional NMR results.
DSC.
Fig. S11 shows the DSC heating scans with either WLBU2 (A,C,E,G) or D8 (B,D,F,H) embedded in DPPC. While DPPC is not present in typical eukaryotic or bacterial cells, it was used to investigate changes to its well-studied main phase transition. As shown in Fig. S11AB, both peptides caused a dramatic shift downwards in temperature of the main melting TM at even a small lipid:peptide molar ratio (1000:1), and in addition a double peak formed. This suggests that the gel phase is destabilized, and that the ripple phase is removed, since there is a merger of the pre-transition (at ~35oC in control DPPC) with the main transition (TM = 41.4oC in control DPPC). At the highest molar ratio (50:1) both peptides lowered TM by ~3oC. Both peptides caused the total enthalpy of melting of the combined peak 1 and peak 2 to decrease, and the full width at half maximum (FWHM) to increase. These changes indicate a loss of cooperativity of the lipid chains during melting due to both peptides. Details of the peak fitting that produced these results are shown in Fig. S12, S13.
MD Simulation.
Fig. 6A displays a snapshot near the end of the 400 nanosecond CHARMM36 simulation of WLBU2 interacting with a KDO2 membrane. WLBU2 was first constrained as a helix 10 Å from the membrane and then all constraints were removed. As shown, WLBU2 remains helical, bound to the surface of the KDO2 bilayer, with the arginine residues facing towards the octulosonic acid residues. Additional MD details are shown in Fig. S14.
Figure 6.

(A) MD simulation of WLBU2 (shown as an α-helical ribbon) on the surface of a KDO2 membrane. Helix colors: R, red, V, yellow, W, blue. Octulosonic acid residues (red sticks), lipid chains (purple sticks), sodium ions (green spheres), water (grey dots). (B-E) Schemes of peptide/lipid interactions. (B) WLBU2 (magenta) interacting with G(−) IM model with POPE (blue circles), POPG (red triangles), TOCL (yellow rectangles). The WLBU2 random coil transitions to α-helical, with some inserted and some in the headgroup region. (C) D8 (dark blue) interacting with G(−) IM model as a random coil, partially inserted and partially in the headgroup region. The G(−) IM models are thinned by both peptides. Shaded stripes in (B) and (C) represent walls between domains with different bending moduli 36. A similar scenario is predicted for both peptides interacting with the G(+) model (not shown). (D) WLBU2 interacting with Euk50 model with POPC (purple inverted triangles), POPE (blue circles) and cholesterol (olive ellipses). WLBU2 with a partial α-helical content perturbs a PE headgroup, which could cause a transient pore to open (light blue streak). (E) D8 (dark blue) as a random coil interacting with the Euk50 model locates on the surface. The Euk50 models are thickened by both peptides.
Discussion
The question we are asking is, are there structural requirements for an efficient eCAP? The Antibacterial Peptide Database (APD, http://aps.unmc.edu/AP/main.php) lists 3138 antibacterial peptides as of November, 2019. Of these, 34 entries are diastereomers, containing D-amino acids (AAs). Inclusion of D-AAs prolongs the lifetime of AMPs in vivo, since they are less susceptible to proteases 50. In the current study two eCAPs with an identical amino acid sequence except for their D-valine content were compared using six biophysical techniques to focus on the structural consequences of adding eight D-enantiomeric amino acids out of 24 total. Our study demonstrates several intriguing findings, including secondary structure determination, effect of membrane thickness and lipid area, location of peptides in the membrane, and membrane perturbations. We will consider these findings in terms of bactericidal activity and eukaryotic cell toxicity separately.
Bactericidal activity.
While WLBU2 is primarily random coil in aqueous environments, it transforms to primarily α-helical when in contact with G(−) IM and G(+) model membranes (Fig. 4, Fig. 5N), which suggests that the α-helical conformation is required for its bactericidal activity as it was measured for magainin and analogues 51. CD ellipticity for D8 could not be analyzed, but NMR found it to be in a random coil configuration in both aqueous phase and in membranes (Fig. 5N). Even TFE, which is known to induce α-helical conformation, could not induce α-helix in D8 (Fig. 5O). Since both peptides are equally effective at killing both G(−) and G(+) bacteria 52, it suggests that, at least for this linear, cationic 24-mer, that secondary structure is not a critical determinant of bactericidal efficacy. A similar result was previously obtained for the cytolysin pardaxin where only one-tenth of AAs as the D-enantiomer caused a reduction in helicity but retention of antibacterial activity 53. However, Seelig et al. used isothermal titration calorimetry to show that AMP binding to membranes decreases as 1/10 D-amino acids are incorporated which also causes helicity to decrease 54. The α-helical content is smallest (41%) with WLBU2 in LPS with its six lipid chains and significant carbohydrate extensions. The OM must be traversed before an eCAP can encounter the last protective barrier, the IM, where it could gain α-helical content.
If not secondary structure, what is the important structural criterion for bactericidal activity? Our XDS data have shown that both WLBU2 and D8 thin both G(−) and G(+) model membranes by ~1.5 Å for WLBU2 and ~2.7 Å for D8 (2DC in Fig. 2A,B). A similar thinning for WLBU2 in the G(−) membrane model (~1.6 Å) was also observed by 2H NMR (Table S16). Our previous publication showed that both WLBU2 and D8 stiffen G(−) IM and G(+) model membranes at low concentration and then soften model membranes at high concentration which suggested that domain formation with different material moduli could cause leakage along domain walls, thus killing the bacteria 36. Other investigators have observed lipid domain formation due to positively charged peptides interacting with negatively charged phospholipids 34,55. While we do not observe non-monotonic changes in membrane thickness or area/lipid at low and high peptide concentrations as for the elasticity results, the ultimate thinning of the membranes at high peptide concentration could be important as a membrane perturbation. This thinning is accompanied by an increase in area/lipid (Fig. 2A,B) and also a change in headgroup orientation and/or dynamics (Fig. 5A–C). We have observed a similar thinning for the cationic (+7) cell-penetrating HIV-1 Tat peptide where Tat translocated across PE-containing model membranes 56. For the OM models, there is a small increase in membrane thickness. While the outer membrane is the first to an encounter an eCAP 43, it is the inner membrane that is the ultimate protective barrier for the bacteria 44, so this lack of thinning in the OM suggests that another mechanism is important for its function.
Besides membrane thinning, peptide location could also play a crucial role. NR indicates that both WLBU2 and D8 are located not only in the headgroup but also deep into the hydrocarbon interior in G(−) and G(+) membrane models (Fig. 3A,B,E,F). This suggests that at least some penetration of the eCAPs is involved in bactericidal activity. However, the eCAPs also locate in the headgroup region, where they are most likely involved in binding to the negatively charged lipid headgroups (POPG, TOCL). If domain formation with different material moduli 36 is important, then some of the eCAPS may penetrate into the domain walls between lipid types, thus helping to destabilize the domain walls (shown in the scheme in Fig. 6B,C). In the LPS OM membrane model, both peptides locate deep into the hydrocarbon region. This location would allow self-promoted uptake 43, 57–60 where the eCAPs would then permeate through the peptidoglycan layer to the IM. For the OM model KDO2, NR indicates that both peptides remain primarily in the headgroup region which was also obtained by MD simulation (shown in Fig. 6A). Since KDO2 is a rare, rough mutant of LPS, this eCAP headgroup location may not be typical for the OM of most G(−) bacteria. We have shown previously that the abundance of carbohydrate residues in the LPS headgroup causes increased membrane fluctuations, which could facilitate peptide entry into the hydrocarbon interior 35. While there is not a great benefit in using deuterated peptides since the peptide has sufficient contrast with the hydrogenated lipid chains and with the lipid headgroups due to solvent exchange, in future experiments, the volume occupancy resolution of the peptide in the bilayer can be improved by performing a second set of experiments with either deuterated peptide or deuterated lipid chains, which would be simultaneously analyzed with the data set using hydrogenated material. In addition to membrane thinning in G(−) and G(+) models, and a dual peptide location, both peptides perturb lipid packing demonstrated by DSC (Fig. S12) even at the low concentration of 1000:1 DPPC:peptide. The perturbation was more gradual with WLBU2, but at the highest concentration of 50:1 there was a similar decrease in TM and enthalpy of melting caused by both peptides (Fig. S12C, D). This suggests that initially WLBU2 interacts with a smaller number of lipids than does D8, but that ultimately both peptides perturb the zwitterionic lipid DPPC similarly.
Eukaryotic cell toxicity.
There are two main differences between eukaryotic cells and bacterial cells: both G(−) and G(+) contain negative charges on their membrane exteriors, while negative charges are generally not exposed on the surface of eukaryotic cells. The second difference is that only ~10% bacteria contain sterols in their cell membranes 61. While secondary structure did not play a role in bactericidal killing of WLBU2 and D8, it may be important for toxicity in eukaryotic cells. We observed a dramatic difference in RBC lysis, with 0% lysis for D8 and 14% lysis for WLBU2 (Fig. 2G). A partial α-helical conformation of WLBU2 may be necessary for this perturbation. A similar result was observed for the diastereomers of pardaxin 53 and melittin 62.
There was no thinning of the Euk50 membrane with either peptide. Instead, we observed a small thickening which cannot explain the dramatic difference in cell toxicity between WLBU2 and D8. For the Euk23 model, a thinning of ~2.6 Å was observed where both WLBU2 and D8 show some toxicity to the corresponding PBMCs (Fig. 2G). It is likely that the correlation of membrane thinning with bactericidal activity or cell toxicity only holds below a threshold level of cholesterol (~20%). Membrane thinning might be a result of the amount of peptide that is membrane-associated times an interaction parameter.
In Euk23, both peptides are located in the interfacial region, directly beneath the headgroups (Fig. 3C, D). This indicates that an interior peptide location is also correlated with cell toxicity in this model as it was for bactericidal activity. For the RBC membrane model, Euk50, there was a slightly deeper penetration of WLBU2 compared to D8 into the hydrocarbon region. This location correlates with WLBU2’s high toxicity and no toxicity for D8 in RBCs (Fig. 2G).
An important clue in the mechanism of toxicity was from the NMR observation of a sharp component in the 31P NMR and 2H NMR spectra when WLBU2 interacted with the Euk50 model (Fig. 5E, L). Such an isotropic peak indicates the rapid orientation of the entire lipid, such as occurs on a highly curved surface or pore. This sharp component was not observed with D8 in Euk50 in either result (Fig. 5F, M), nor was it observed when WLBU2 or D8 interacted with the G(−) membrane model (Fig. 5B,C,I,J). The latter result supports our hypothesis that bactericidal activity of WLBU2 and D8 does not involve pore formation 36. It also suggests that pore formation may be the cause of RBC toxicity for WLBU2 but not D8 (shown in the scheme in Fig. 6D,E). Alternatively, the isotropic signal could also be caused by a partial micellization of the membrane 63.
Conclusion
This work provides evidence that the secondary structure, location of peptide in the membrane, membrane thickness and headgroup perturbation are eCAP and lipid dependent. WLBU2 is primarily random coil in aqueous phase and primarily α-helical in G(−) IM, G(+) and KDO2 membranes, but only partially α-helical in both eukaryotic membranes and LPS model. D8, with all 8 valines as the D-enantiomer, retains a random coil configuration in G(−) IM and Euk50, as well as in TFE. Since both peptides are equally bactericidal to both G(−) and G(+) bacteria, this demonstrates that secondary structure is not critical for this eCAP’s bactericidal activity. More important is the membrane thinning that occurs due to both peptides and dual location of both peptides in the hydrocarbon interior as well as in the headgroup region for G(−) IM and G(+) models, which could indicate peptide insertion into domain walls between domains of different stiffness 36. As for toxicity to eukaryotic cells, the partially α-helical structure of WLBU2 may play a role in destabilizing the RBC model, causing dynamic pore formation as part of the mechanism of the difference in eCAP toxicities seen in RBCs. Previously, we showed that while WLBU2 softens the RBC model, D8 stiffens it 36. This is correlated with a partial hydrocarbon penetration with WLBU2 and lack of hydrocarbon penetration with D8 in the RBC Euk50 membrane model.
Supplementary Material
Acknowledgements
The authors thank Belita Opene (UMaryland-Baltimore) for purifying the PA01 LPS, Prof. Emeritus John F. Nagle, Megan Roche, Yasmene Elhady and Diamond Moody for help with the data collection at CHESS, and Dr. Arthur Woll for his help at the G1 CHESS beamline. This work is based upon research conducted at Carnegie Mellon University and at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation (NSF) under award No. DMR-1332208. Parts of this research were performed at the NIST Center for Nanoscale Science. Certain commercial materials, equipment, and instruments are identified in this work to describe the experimental procedure as completely as possible. In no case does such an identification imply a recommendation or endorsement by NIST, nor does it imply that the materials, equipment, or instrument identified are necessarily the best available for the purpose. The neutron work was supported by the Department of Commerce through its Measurement Science and Engineering Program (70NANB13H009), the National Institute of Standards and Technology (NIST) Center for Neutron Research Comprehensive Grant Program (70NANB17H299), and the NIST IMS Program “Precision Measurement for Integral Membrane Proteins”. Additional support for this work was from National Institutes of Health (NIH) R01AI133351 (STN, YPD), NIH R01GM125917 (STN, BD), Carnegie Mellon SURF (AK, AS), CONICET, Programma de Becas Externas (FGD), NIH R01GM123169 (AP) and National Science Foundation (NSF) ACI-1548562 (AP, JCG), NIH R01GM101647 (FH), German Research Foundation (DFG) HU 720/15-2 (DH).
References
- [1].WHO, “Antimicrobial resistance WHO Global Report on surveillance”, can be found under http://www.who.int/drugresistance/documents/surveillancereport/en/, 2014.
- [2].Deslouches B, Phadke SM, Lazarevic V, Cascio M, Islam K, Montelaro RC, Mietzner TA, Antimicrob Agents Chemother 2005, 49, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].de Breij A, Riool M, Cordfunke RA, Malanovic N, de Boer L, Koning RI, Ravensbergen E, Franken M, van der Heijde T, Boekema BK, Kwakman PHS, Kamp N, El Ghalbzouri A, Lohner K, Zaat SAJ, Drijfhout JW, Nibbering PH, Sci Transl Med 2018, 10, 1–14. [DOI] [PubMed] [Google Scholar]
- [4].Wimley WC, ACS Chem Biol 2010, 5, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Burns JL, Montelaro RC, Antimicrob Agents Chemother 2015, 59, 1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pray L, Nature Education 2008, 1, 30. [Google Scholar]
- [7].Yang L, Ludtke SJ, He K, Heller WT, Harroun TA, Huang HW, Biophys J 1997, 72, Tu331-Tu331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ludtke SJ, He K, Heller WT, Harroun TA, Yang L, Huang HW, Biochemistry 1996, 35, 13723–13728. [DOI] [PubMed] [Google Scholar]
- [9].Liu YF, Nagle JF, Phys Rev E 2004, 69, 040901(1–4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen FY, Lee MT, Huang HW, Biophys J 2003, 84, 3751–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mathew B, Nagaraj R, PloS one 2017, 12, e0175858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Mietzner TA, Montelaro RC, Antimicrob Agents Chemother 2013, 57, 2511–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen YX, Mant CT, Farmer SW, Hancock REW, Vasil ML, Hodges RS, J Biol Chem 2005, 280, 12316–12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ghosh C, Sarkar P, Issa R, Haldar J, Trends in Microbiology 2019, 27, 323–338. [DOI] [PubMed] [Google Scholar]
- [15].Ho YH, Shah P, Chen YW, Chen CS, Mol Cell Proteomics 2016, 15, 1837–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brogden KA, Nat Rev Microbiol 2005, 3, 238–250. [DOI] [PubMed] [Google Scholar]
- [17].Sani M-A, Separovic F, Accounts of Chemical Research 2016, 49,1130–1138. [DOI] [PubMed] [Google Scholar]
- [18].Lee T-H, Hofferek V Separovic F, Sani M-A, Separovic F, Reid GE, Aguilar M-I, Current Opinion in Chemical Biology 2019, 52, 85–92. [DOI] [PubMed] [Google Scholar]
- [19].Bechinger B, Journal of Peptide Science 2015, 21, 346–355. [DOI] [PubMed] [Google Scholar]
- [20].Ramamoorthy A, S Thennarasu Lee DK, Tan AM, Maloy L, Biophysical Journal 2006, 91, 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Henzler Wildman KA, Lee DK, Ramamoorthy A, Biochemistry 2003, 42, 6545–6558. [DOI] [PubMed] [Google Scholar]
- [22].Hallock KJ, Lee DK, Ramamoorthy, Biophysical Journal 2003, 64, 3052–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bhunia A, Domadia PN, Torres J, Hallock KJ, Ramamoorthy A, The Journal of Biological Chemistry 2010, 285, 3883–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pan JJ, Tristram-Nagle S, Nagle JF, J Membrane Biol 2009, 231, 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].He K, Ludtke SJ, Worcester DL, Huang HW, Biophys J 1996, 70, 2659–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pantarotto D, Bianco A, Pellarini F, Tossi A, Giangaspero A, Zelezetsky I, Briand JP, Prato M, J Am Chem Soc 2002, 124, 12543–12549. [DOI] [PubMed] [Google Scholar]
- [27].Dura JA, Pierce DJ, Majkrzak CF, Maliszewskyj NC, McGillivray DJ, Losche M, O’Donovan KV, Mihailescu M, Perez-Salas U, Worcester DL, H White S, Rev Sci Instrum 2006, 77, 74301– 743011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kirby BJ, Kienzle PA, Maranville BB, Berk NF, Krycka J, Heinrich F, Majkrzak CF, Curr Opin in Colloid & Interface Sci 2012, 17, 44–53. [Google Scholar]
- [29].Hille B, Prog. Biophys. A Mol Biol 1970, 21, 3–32. [DOI] [PubMed] [Google Scholar]
- [30].Gazit E, Miller IR, Biggin PC, Sansom MS, Shai Y, J Mol Biol 1996, 258, 860–870. [DOI] [PubMed] [Google Scholar]
- [31].Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y, Biochemistry 1992, 31, 12416–12423. [DOI] [PubMed] [Google Scholar]
- [32].Bechinger B, Biochim Biophys Acta - Biomembranes 1999, 1462, 157–183. [DOI] [PubMed] [Google Scholar]
- [33].Bechinger B, Lohner K, Biochim Biophys Acta - Biomembranes 2006, 1758, 1529–1539. [DOI] [PubMed] [Google Scholar]
- [34].Matsuzaki K, Sugishita K, Ishibe N, Ueha M, Nakata S, Miyajima K, Epand RM, Biochemistry 1998, 37, 11856–63. [DOI] [PubMed] [Google Scholar]
- [35].Dupuy FG, Pagano I, Andenoro K, Peralta MF, Elhady Y, Heinrich F, Tristram-Nagle S, Biophys J 2018, 114, 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kumagai A, Dupuy FG, Arsov Z, Elhady Y, Moody D, Ernst RK, Deslouches RC Montelaro Y. Di P, Tristram-Nagle S, Soft Matter 2019, 15, 1860–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Deslouches B, Islam K, Craigo JK, Paranjape SM, Montelaro RC, Mietzner TA, Antimicrob Agents Chemother 2005, 49, 3208–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Deslouches B, Gonzalez IA, DeAlmeida D, Islam K, Steele C, Montelaro RC, Mietzner TA, J Antimicrob Chemother 2007, 60, 669–72. [32] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lashua LP, Melvin JA, Deslouches B, Pilewski JM, Montelaro RC, Bomberger JM, J Antimicrob Chemoth 2016, 71, 2200–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kučerka N, Liu YF, Chu NJ, Petrache HI, Tristram-Nagle S, Nagle JF, Biophys J 2005, 88, 2626–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lyatskaya Y, Liu Y, Tristram-Nagle S, Katsaras J, Nagle JF, Phys Rev E Stat Nonlin Soft Matter Phys 2001, 63, 011907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu Y, Nagle JF, Phys Rev E Stat Nonlin Soft Matter Phys 2004, 69, 040901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hancock RE, Trends Microbiol 1997, 5, 37–42. [DOI] [PubMed] [Google Scholar]
- [44].Lehrer RI, Barton A, Daher KA, Harwig SS, Ganz T, Selsted ME, J Clin Invest 1989, 84, 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gottfried EL, J Lipid Res 1967, 8, 321–327. [PubMed] [Google Scholar]
- [46].McHenry AJ, Sciacca MFM Brender JR, Ramamoorthy A, Biochimica et Biophysica Acta, 2012, 1818, 3019–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wade D. Boman A, Wahlin B, Drain CM, Andreu D, Boman HG, Merrifield RB, Proc. Natl. Acad. Sci USA, 1990, 87, 4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bessalle R, Kapitkovsky A, Gorea A, Shalit I, Fridkin M, FEBS Lett, 1990, 274, 151–155. [DOI] [PubMed] [Google Scholar]
- [49].Brahms S, Brahms J, J Mol Biol 1980, 138, 149–178. [DOI] [PubMed] [Google Scholar]
- [50].Papo N, Oren Z, Pag U, Sahl HG, Shai Y, J Biol Chem 2002, 277, 33913–33921. [DOI] [PubMed] [Google Scholar]
- [51].Chen HC, Brown JH, Morell JL, Huang CM, FEBS Letters 1988, 236, 462–466. [DOI] [PubMed] [Google Scholar]
- [52].Di YP, Chen QL,C, Montelaro RC, Doi Y, Deslouches B, Enhanced therapeutic index of an antimicrobial peptide in mice by increasing safety and activity against multidrug-resistant bacteria. Science Advances 2019, (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shai Y, Oren Z, J Biol Chem 1996, 271, 7305–7308. [DOI] [PubMed] [Google Scholar]
- [54].Wieprecht T, Apostolov O, Beyermann M, Seelig J, Journal of Molecular Biology 1999, 294, 785–794. [DOI] [PubMed] [Google Scholar]
- [55].Mason AJ, Martinez A, Glaubitz C, Danos O, Kichler A, Bechinger B. The FASEB Journal 2005,express article 10.1096/fj.05-4293fje. [DOI] [PubMed] [Google Scholar]
- [56].Neale C, Huang K, Garcia AE, Tristram-Nagle S, Membranes 2015, 5, 473–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hancock RE, Annu Rev Microbiol 1984, 38, 237–264. [DOI] [PubMed] [Google Scholar]
- [58].Hancock RE, Lancet 1997, 349, 418–422. [DOI] [PubMed] [Google Scholar]
- [59].Hancock RE, Clin Infec Dis 1998, 27 Suppl 1, S93–99. [DOI] [PubMed] [Google Scholar]
- [60].Hancock RE, Chapple DS, Antimicrob Agents Chemother 1999, 43, 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wilkinson SG, Microbial Lipids, Vol. I, Academic Press, San Diego, CA, 1988, p. 964. [Google Scholar]
- [62].Oren Z, Shai Y, Biochemistry 1997, 36, 1826–1835. [DOI] [PubMed] [Google Scholar]
- [63].Lohner K, Staudegger E, Prenner EJ, Lewis RNAH, Kriechbaum M, Degovics G, McElhaney RN, Biochemistry 1999, 38, 16514–16528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.