

Abstract
Summary: Inflammatory myofibroblastic tumors include a diverse group of lesions characterized by inflammatory cell infiltration and variable fibrotic responses. Occurrence in the temporal bone is unusual. We present CT and MR imaging findings of an inflammatory myofibroblastic tumor of the temporal bone in a 26-year-old patient with repeated paroxystic episodes of rotatory vertigo that occurred over a few days. CT and MR imaging revealed a homogeneously enhancing soft-tissue mass of the right mastoid with bone erosion of the tegmen and extensive dural thickening. The mass resembled a malignant tumor, and the patient underwent an extended mastoidectomy through a retroauricular approach. Microscopic examination showed myofibroblastic spindle cells with mixed inflammatory infiltrate, and the pathologic diagnosis was inflammatory myofibroblastic tumor.
Earlier reports of inflammatory myofibroblastic tumor, also known as inflammatory pseudotumor, were confined to cases involving the lung or pleura (or both), until examples were described in the abdomen, skin and soft tissues, spleen, heart, bladder, upper respiratory tract, mediastinum, and kidney (1). Inflammatory myofibroblastic tumors of the head and neck occur most commonly in the orbits and rarely in the skull base. Other areas of involvement include the larynx, the paranasal sinuses, the cervical esophagus, and the soft tissues of the neck. They include a heterogeneous group of lesions characterized by inflammatory cell infiltration and variable fibrotic reaction. Inflammatory myofibroblastic tumors are now a widely recognized entity with a generally favorable prognosis, although rare examples of malignant transformation and even distant metastases have occurred (2). Inflammatory pseudotumors of the middle and inner ear have been described, to our knowledge, in only four cases in the English literature (3, 4). We herein describe the clinical features, radiologic and histopathologic findings, and treatment outcomes.
Case Report
A 26-year-old black man was admitted to the Department of Otolaryngology of the University of Brescia in May 2002 for repeated paroxystic episodes of rotatory vertigo that occurred over a few days. The attacks lasted a few minutes each and were unleashed by sudden head movements; they were accompanied by intense neurovegetative signs, without neurologic signs or symptoms. Laboratory findings were within normal ranges, except for erythrocyte sedimentation rate (73 mm/h) and seroproteic electrophoresis (decreased albumin, 47.3%; increased gamma globulins, 29.0%).
The patient underwent a complete neuro-otologic evaluation. Micro-otoscopy revealed the eardrums to be normal, and clinical vestibular examination revealed a left horizontal spontaneous nystagmus (grade I) with rotatory component. Pure tone audiometry showed a sensorineural hearing loss at high frequencies in the right ear and a modest conductive component at lower frequencies; middle ear impedance testing ascertained a flat tympanogram and an elevated threshold of contralaterally evoked acoustic stapedial reflexes in the same ear. Vestibular examination with electronystagmography, performed on the second day after admission, failed to detect any spontaneous nystagmus. Recording of the auditory brain stem response showed increased wave III and V latencies on the affected side, indicating a possible retrocochlear disease.
High-resolution CT scans of the temporal bone in the axial and coronal planes, performed to rule out a suspected expansive lesion, showed an erosive mass, involving the right mastoid and extending anteriorly into the attic, without erosion of the ossicles (Fig 1). The tegmen was dehiscent, and the residual pneumatized mastoid was not aerated. CT also showed erosive changes of the cortex of the lateral semicircular canal and of the sigmoid sinus plate. No signs of labyrinthine fistula were observed.
Fig 1.

High resolution bone algorithm CT scans. Erosive expansile lesion of the right temporal bone, involving the mastoid antrum, with complete erosion of the tegmen (short arrows) and partial erosion of the cortex of the lateral semicircular canal (arrowheads) can be seen. The lesion extends throughout the attic, sparing the ossicular chain (long arrow).
A, Axial view.
B, Coronal view.
MR imaging at 1.5 T was performed the same day. T2-weighted fast spin-echo axial and coronal sections (4000/100 [TR/TE]), T1-weighted spin-echo images (500/15), T1-weighted axial and coronal sections, and 2D time-of-flight MR venograms (25/9; flip angle, 30 degrees) were obtained after IV injection of gadopentetate dimeglumine (0.1 mmol/kg; Magnevist, Shering, Germany).
The lesion was characterized by low signal intensity in T2-weighted and by intermediate signal intensity in T1-weighted images, with marked enhancement after the administration of contrast material (Fig 2). MR imaging showed dural enhancement and thickening starting at the petrous bone, involving the right edge of the tentorium, and extending in the temporoparietal region (Fig 2D). No edema was observed in the temporal white matter. The mass reached the otic capsule but without any signal intensity abnormality of the membranous labyrinth. MR venography showed a mild indentation on the right sigmoid sinus (Fig 2E).
Fig 2.
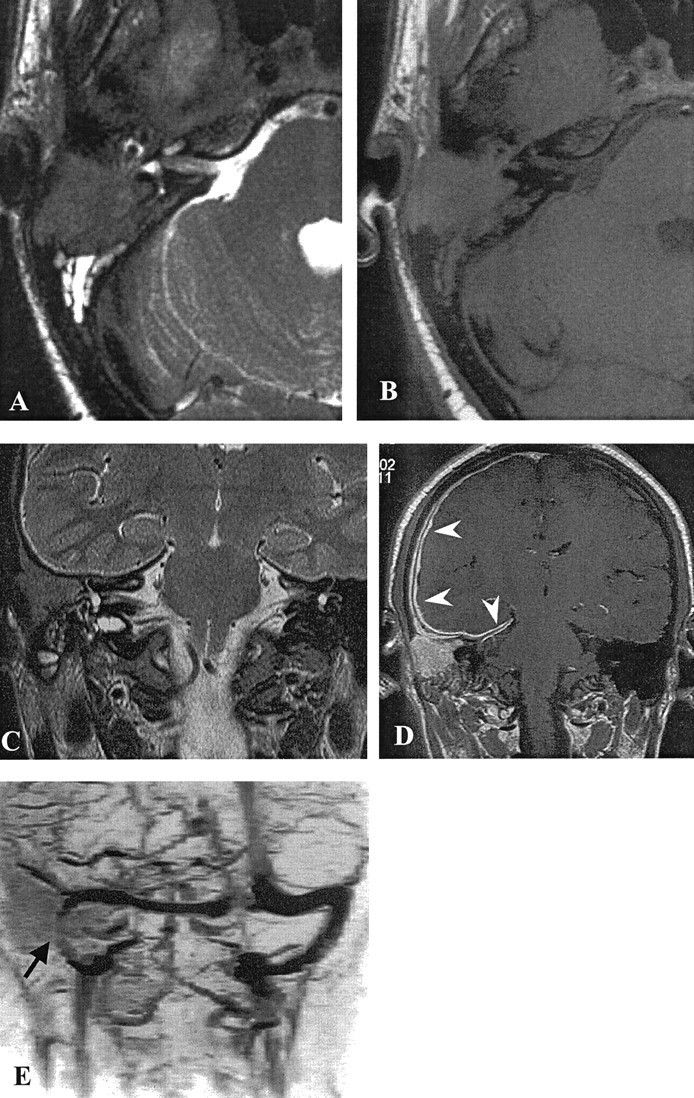
MR images.
A, Axial view T2-weighted MR image reveals a low intensity lesion occupying the right mastoid with associated hyperintense debris.
B, Axial view T1-weighted image. The lesion is homogeneously isointense to brain.
C, Coronal view T2-weighted MR image shows infiltration of the otic capsule, but the signal intensity of the lateral and superior semicircular canals is normal.
D, Axial view contrast-enhanced T1-weighted MR image shows homogeneous enhancement of the lesion, with an extensive regular thickening and enhancement of the dura of the convexity and of the right edge of the tentorium (arrowheads).
E, MR venogram shows patency of the right sigmoid sinus, which is slightly reduced in size (arrow).
An antibiotic multidrug protocol was promptly IV administered: Ceftazidime (1 g × 3), Vancomycin (500 mg × 2), and Netilmycin (300 mg). The results of a CSF examination were negative for bacterial colonization.
Based on the erosion of the tegmen and imaging findings suggesting a vascularized tumor, the decision was made to perform an extended mastoidectomy through a retroauricular approach to remove the lesion and protect the patient from the risk of meningitis. The cortical bone appeared crisp, tender, and completely involved by an osteolytic lesion. Intraoperative frozen sections were obtained and documented an inflammatory pseudotumor. After identification of the sigmoid sinus and of the middle and posterior cranial fossa dura, the lesion was dissected from the facial nerve. The ossicular chain appeared intact and mobile. The wall of the lateral semicircular canal was thin and irregular without fistulization. The posterior wall of the outer external canal was drilled to the level of the fallopian canal. An inflammatory reaction of the dural meningeal layer of the middle cranial fossa that appeared strictly adherent to the lesion was observed. Careful cleavage of the mass from the dura was achieved and its exenteration completed. The tegmen defect was repaired by means of composite flexible hydroxyapatite sheath reinforced with a 3 × 2 cm patch of dehydrated homologous fascia temporalis. No neurologic deficits were observed at the end of the surgical procedure.
CT performed on the first postoperative day showed residual dural meningeal enhancement. Microscopically, the lesion showed proliferating stromal cells oriented in well-formed intersecting fascicles or haphazardly distributed, with mixed inflammatory infiltrate (Fig 3A). Plasma cells predominated, but aggregates of lymphocytes were also present (Fig 3B).
Fig 3.
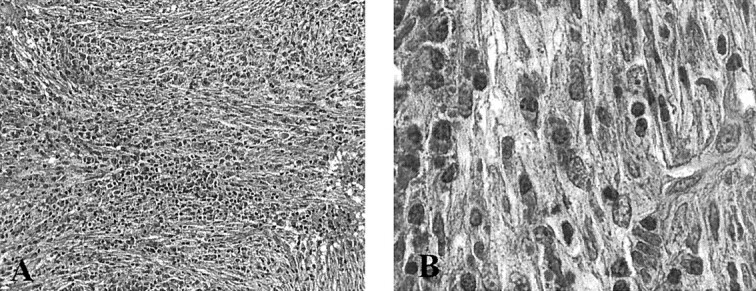
Microscopic images.
A, Proliferation of spindle cells oriented in intersecting fascicles or haphazardly distributed, accompanied by numerous plasma cells and small lymphocytes (original magnification, ×200).
B, Myofibroblasts show minimal nuclear pleomorphism (original magnification, ×1000)
The spindle cells showed minimal nuclear pleomorphism and very low mitotic activity, with diffuse immunoreactivity for vimentin and muscle-specific actin. These cells were negative for Epstein-Barr virus, tested with both in situ hybridization for Epstein-Barr virus-encoded RNA and immunostaining for Epstein-Barr virus latent membrane protein.
The final histopathologic diagnosis was inflammatory myofibroblastic tumor. The postoperative course was uneventful, and the patient was discharged 14 days after surgery. He was to continue a regimen of antibiotic therapy.
Discussion
Inflammatory myofibroblastic tumor has been described as occurring in most organs and anatomic sites with few exceptions. Terms generally considered synonymous with inflammatory myofibroblastic tumor include inflammatory pseudotumor, plasma cell granuloma, and pseudosarcomatous myofibroblastic lesion. The term inflammatory myofibroblastic tumor was initially proposed and the lesion originally described by Pettinato et al (5) in 1990, in their study of 20 lesions of the lung. It was later designated as a postinflammatory tumor. The most proper definition, formalized in the 1994 World Health Organization classification of soft-tissue tumors, is inflammatory myofibroblastic tumor, which refers to “a tumor composed of differentiated myofibroblastic spindle cells usually accompanied by numerous plasma cells and/or lymphocytes” (6).
The cause and pathogenesis of the inflammatory myofibroblastic tumor are largely unknown, although reports of postsurgical, posttraumatic, and postinfectious cases have prompted some speculation that an initially reactive process can change into an overt neoplastic disease (7). A subset of inflammatory myofibroblastic tumors, particularly those in the spleen and liver, harbor the Epstein-Barr virus in spindle cells (8).
Inflammatory myofibroblastic tumor of the head and neck is most often encountered as a benign chronic inflammatory lesion of the orbit without invasion of the bony walls (9), but it has also been reported to occur in extraorbital locations such as the maxillary sinus, infratemporal fossa, nasopharynx, pterygopalatine fossa, and skull base (10–13).
In their series, Mulder et al (3) describe three cases of fibroinflammatory pseudotumors of the ear in adult patients, which seem to have several features in common with our case. Their symptoms were otalgia (n = 2), otorrhea (n = 2), vertigo (n = 2), and hearing loss (n = 3). All cases were characterized by destruction of the inner ear, with variable degrees of erosion of semicircular canals and cochlea and disruption of the ossicular chain occurring in only one-third of the cases. All lesions appeared hypointense on T2-weighted images, with marked enhancement after the administration of contrast material. Signal intensity loss was detected in the cochlea in one case. Similar imaging findings were reported in a case of a large enhancing mass destroying the petrous bone and extending in the posterior and middle cranial fossae in a female neonate (4).
In all cases, the histopathologic findings indicated a moderately cellular process with a variable admixture of spindle cells and inflammatory infiltrate with minimal nuclear pleomorphism and mitotic activity. At histopathologic examination, the inflammatory myofibroblastic tumor presents macroscopically as a solid, elastic pink-whitish mass with a characteristic fibroinflammatory appearance. The microscopic features of the inflammatory myofibroblastic tumor are dominated by differentiated vimentin-positive myofibroblastic spindle cells, a variably prominent collagenous stroma, and an inflammatory component consisting mainly of lymphocytes and plasma cells. Any one of these cellular or stromal elements and growth pattern may vary in relative proportions from one inflammatory myofibroblastic tumor to another or from one microscopic field to the next in the same tumor. Three basic histologic patterns are recognized: myxoid/vascular, compact spindle cell, and hypocellular fibrous patterns.
Other tumors composed of myofibroblasts and fibroblasts pose significant challenges in differential diagnosis because of their morphologic overlap with inflammatory myofibroblastic tumor. These include both benign and malignant tumors and tumor-like conditions. The less cellular, more sclerotic form may be mistaken for aggressive fibromatosis. The prominent inflammatory component is the critical distinguishing feature. The cellular form of inflammatory myofibroblastic tumor may be mistaken for fibrosarcoma or other spindle cell malignancies. Recognition of the prominent inflammatory component and the lack of significant cytologic atypia are helpful diagnostic features. However, there is undoubtedly morphologic and clinical overlap between inflammatory myofibroblastic tumor and inflammatory fibrosarcoma. The question of distinction between the two rests on whether they are truly distinct or part of a neoplastic continuum of myofibroblastic proliferations with increasing cellular atypia and aggressiveness. Our case showed histologic benign features: low cellularity, no nuclear atypia, and only occasional typical mitoses. In our patient, the lesion caused an extensive lytic defect of the mastoid and tegmen without erosion of the ossicular chain or of the inner ear.
CT is usually insufficient for defining the inherent structure of the lytic lesion, although the relationships with the inner ear and the otic capsule define its destructive or expansive nature, discriminating, for instance, chronic inflammatory disease from bone metastases. Against the diagnosis of cholesteatoma of the middle ear was the location of the osteolytic defect in the mastoid antrum without widening of the aditus and the homogeneous enhancement on contrast-enhanced T1-weighted images, together with low signal intensity on T2-weighted images. Cholesterol granuloma, which is usually bright on all spin-echo sequences, was also excluded on the basis of the MR imaging signal intensity pattern (14).
In our patient, sparing of the ossicular chain was considered in contrast with the typical behavior of a destructive lesion, such as a primary bone tumor or metastasis. The marked contrast enhancement could also have suggested a highly vascular tumor, such as a paraganglioma, although the lesion was not originating in the tympanic cavity and did not show the typical permeative bone changes.
According to previous reports (9–13), it is very difficult, both clinically and radiologically, to determine whether an infiltrating soft-tissue mass located in the temporal bone, with bone destruction, is an inflammatory myofibroblastic tumor or a malignant neoplasm.
Although bone remodeling rarely occurs in the orbits, in extraorbital head and neck locations, such as the maxillary sinus or the skull base, inflammatory myofibroblastic tumor exhibits more aggressive behavior, with bone involvement clearly shown by CT as localized bone changes, such as erosion, remodeling, and sclerosis (10, 13).
Common MR imaging findings in all reported cases of inflammatory myofibroblastic tumor of the head and neck are low signal intensity in T2-weighted images and homogeneous contrast enhancement. This hypointensity can be explained by the relative lack of mobile protons within fibrotic lesions. However, low signal intensity on T2-weighted images, indicating either elevated cellularity or a high nuclear-to-cytoplasmic ratio, is also typical of malignant neoplasms.
Plasma cell granulomas have been reported to occur in the temporal bone and have the same radiologic appearance as the inflammatory myofibroblastic tumor. They are generally included in the heterogeneous family of inflammatory pseudotumors, because they are primarily composed of mature plasma cells in a fibrous stroma, although some authors think they represent a different histologic entity (15).
The very rare malignant neoplasms involving the temporal bone are most commonly represented by squamous cell carcinoma, basal cell carcinoma, and parotid tumors. In a series of 15 patients with temporal bone malignancies, including basal cell carcinoma, squamous cell carcinoma, adenocarcinoma, osteosarcoma, and temporal bone secondary involvement by primary parotid malignancies, the most common sites of bone destruction shown by CT were the external auditory canal, middle ear, and mastoid (16). Because of the attenuated bone of the otic capsule, invasion of the inner ear is very uncommon. These tumors usually spread by direct extension and tend to be isointense to brain in T1- and T2-weighted images, enhance homogeneously with dural enhancement in most cases, and are practically indistinguishable from a chronic inflammatory lesion with a fibrotic component.
In our case, MR imaging also showed extensive dural thickening and enhancement, which involved the middle cranial fossa, the right tentorial margin, and the parietal convexity and persisted after surgery. Dural thickening and enhancement represent an unspecific reactive change to any adjacent dural lesion and can also manifest with malignant neoplasms. They seem to be a common finding in inflammatory myofibroblastic tumor of the skull base, as reported by Han et al (13) who observed intracranial dural involvement in all patients with fibrosing inflammatory pseudotumors of the skull base.
MR imaging and CT play key roles for complete clinical evaluation and subsequent treatment options. The administration of high dose corticosteroid is the primary treatment for inflammatory myofibroblastic tumor, and response to medical therapy is thought to approximately parallel the acuity of the inflammatory process. Acute lesions typically respond to high doses of corticosteroid, but chronic lesions, which tend to have more fibrosis, generally do not respond to medical therapy. In their series, however, Han et al (13) found no relationship between the duration of signs and symptoms and the degree of fibrosis.
In our opinion, surgery is indicated, especially in a case of extensive lytic defect of the tegmina, because the patient is at risk of developing meningitis or intracranial suppurative complications. Total removal of the disease can be achieved, and sealing of the defect is required. Although no long-term follow-up reports on the outcome of surgery for inflammatory myofibroblastic tumor of the temporal bone are available, according to the surgical results in all the other head and neck locations, the prognosis is favorable and recurrence is considered exceptional.
Conclusion
Inflammatory myofibroblastic tumors of the temporal bone are very uncommon and may mimic malignant tumors. CT and MR imaging are both required to thoroughly assess the extent of bone destruction and the inherent structure of the lesion. The pattern of a soft-tissue mass characterized by low signal intensity on T2-weighted images and marked contrast enhancement with a very peculiar destruction of the mastoid and sparing of the inner ear and of the ossicular chain is contraindicative of an aggressive tumor and likely suggests a chronic inflammatory lesion. Despite their benignity they are locally destructive lesions that require prompt treatment. Early diagnosis may facilitate complete resection without significant morbidity.
References
- 1.Coffin CM, Watterson J, Priest JR, et al. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor): a clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 1995;19:859–872 [DOI] [PubMed] [Google Scholar]
- 2.Maier HC, Sommers SC. Recurrent and metastatic pulmonary fibrous/histiocytoma/plasma cell granuloma in a child. Cancer 1987;60:1073–1076 [DOI] [PubMed] [Google Scholar]
- 3.Mulder JJ, Cremers WR, Joosten F, Wiersma A, van den Broek P. Fibroinflammatory pseudotumor of the ear: a locally destructive benign lesion. Arch Otolaryngol Head Neck Surg 1995;121:930–933 [DOI] [PubMed] [Google Scholar]
- 4.Pollack IF, Hamilton RL, Fitz C, Kassam A, Snyderman CH. Congenital reactive myofibroblastic tumor of the petrous bone: case report. Neurosurgery 2001;48:430–435 [DOI] [PubMed] [Google Scholar]
- 5.Pettinato G, Manivel JC, De Rosa N, Dehner LP. Inflammatory myofibroblastic tumor (plasma cell granuloma): clinicopathologic study of 20 cases with immunohistochemical and ultrastructural observations. Am J Clin Pathol 1990;94:538–546 [DOI] [PubMed] [Google Scholar]
- 6.Weiss SW. Histological Typing of Soft Tissue Tumors. 2nd ed. Berlin: Springer-Verlag;1994. :48
- 7.Chan JK. Inflammatory pseudotumor: a family of lesions of diverse nature and etiologies. Adv Anat Pathol 1996;3:156–169 [Google Scholar]
- 8.Arber DA, Kamel OW, van de Rijn M, et al. Frequent presence of the Epstein-Barr virus in inflammatory pseudotumor. Hum Pathol 1995;26:1093–1098 [DOI] [PubMed] [Google Scholar]
- 9.Weber AL, Romo LV, Sabates NR. Pseudotumor of the orbit: clinical, pathologic, and radiologic evaluation. Radiol Clin North Am 1999;37:151–168 [DOI] [PubMed] [Google Scholar]
- 10.Som PM, Brandwein MS, Maldjian C, Reino AJ, Lawson W. Inflammatory pseudotumor of the maxillary sinus: CT and MR findings in six cases. AJR Am J Roentgenol 1994;163:689–692 [DOI] [PubMed] [Google Scholar]
- 11.De Vuysere S, Hermans R, Sciot R, Crevits I, Marchal G. Extraorbital inflammatory pseudotumor of the head and neck: CT and MR findings in three patients. AJNR Am J Neuroradiol 1999;20:1133–1139 [PMC free article] [PubMed] [Google Scholar]
- 12.Ribeiro AC, Joshi VM, Funkhouser WK, Mukherji SK. Inflammatory myofibroblastic tumor involving the pterygopalatine fossa. AJNR Am J Neuroradiol 2001;22:518–520 [PMC free article] [PubMed] [Google Scholar]
- 13.Han MH, Chi JG, Kim MS, et al. Fibrosing inflammatory pseudotumors involving the skull base: MR and CT manifestations with histopathologic comparison. AJNR Am J Neuroradiol 1996;17:515–521 [PMC free article] [PubMed] [Google Scholar]
- 14.Mafee MF, Kumar A, Heffner DK. Epidermoid cyst (cholesteatoma) and cholesterol granuloma of the temporal bone and epidermoid cysts affecting the brain. Neuroimaging Clin N Am 1994;4:561–578 [PubMed] [Google Scholar]
- 15.Janicki PT, Adams JS, McElveen JT Jr. Plasma cell granuloma of the temporal bone. Am J Otol 1996;17:123–126 [PubMed] [Google Scholar]
- 16.Horowitz SW, Leonetti JP, Azar-Kia B, Fine M, Izquierdo R. CT and MR of temporal bone malignancies primary and secondary to parotid carcinoma. AJNR Am J Neuroradiol 1994;15:755–762 [PMC free article] [PubMed] [Google Scholar]