

Abstract
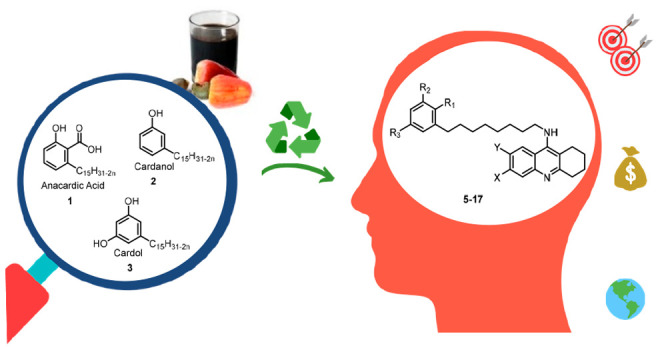
The multifactorial nature of Alzheimer’s disease (AD) is a reason for the lack of effective drugs as well as a basis for the development of “multi-target-directed ligands” (MTDLs). As cases increase in developing countries, there is a need of new drugs that are not only effective but also accessible. With this motivation, we report the first sustainable MTDLs, derived from cashew nutshell liquid (CNSL), an inexpensive food waste with anti-inflammatory properties. We applied a framework combination of functionalized CNSL components and well-established acetylcholinesterase (AChE)/butyrylcholinesterase (BChE) tacrine templates. MTDLs were selected based on hepatic, neuronal, and microglial cell toxicity. Enzymatic studies disclosed potent and selective AChE/BChE inhibitors (5, 6, and 12), with subnanomolar activities. The X-ray crystal structure of 5 complexed with BChE allowed rationalizing the observed activity (0.0352 nM). Investigation in BV-2 microglial cells revealed antineuroinflammatory and neuroprotective activities for 5 and 6 (already at 0.01 μM), confirming the design rationale.
Introduction
Dementia has grown as a major health and societal challenge nowadays, and its impact will be even more profound as the global population continues to age. The number of patients affected by dementia—with Alzheimer’s disease (AD) being the most frequent type—will increase from 35 million to an astonishing 135 million by 2050.1 Already 60% of them live in low- and middle-income countries, but by 2050, this will rise to 71%.1
The current lack of a cure is magnifying the problems of AD. While drugs (three acetylcholinesterase inhibitors (AChEI) and memantine) exist, they can only alleviate symptoms of dementia but are not able to halt the progression of the degenerative process. Often referred to as the “valley of death”, there is a large gap between basic research and translation to novel therapeutics. More than 200 drug candidates have failed in late-stage clinical trials, with a success rate of 0.4%, as compared with about 20% for cancer drugs.2 Over more than 400 clinical trials, there has been only one novel agent approved for AD since 2003 (GV-971 was approved in China in 2019 and is available only in China).3
As cases increase in populous countries like India, Brazil, and Indonesia, dementia will be an even more complex problem, especially in terms of an equitable access to treatments. Thus, there is a need to develop new medicines that are not only effective but also accessible, with no financial constraint.
The multifactorial nature of AD has been called into question as one of the factors contributing to the current lack of an effective drug therapy. The complex biology of AD is difficult to reduce to a single target whose modulation will impact the broad spectrum of pathologies and symptoms.4 More likely, AD is thought to be caused by a systemic breakdown of brain physiological networks.5,6 These have evolved to be very robust and redundant so that they are relatively insensitive to perturbations, with modulation by currently available single-target drugs having only a small, temporary effect. Conversely, treatments directed to multiple targets of the network would appear to have more chance of success.5,7,8
In 2008, we were among the first to propose single-molecule polypharmacology we named “multi-target-directed ligands” (MTDLs), as opposed to the available single-target drugs.9 Still today, these multifunctional molecules are considered a valuable option to effectively treat AD10−12 and similarly complex neurodegenerative diseases.10
In the years, we have realized that we are called not only to create drugs more adequate to face AD complexity but also to do so in a sustainable fashion so that the tools we develop are not only benign for the environment but also affordable and accessible to all the people and health systems that need them.
With this motivation, we have recently explored the possibility of developing new pharmacological tools for AD starting from cashew nutshell liquid (CNSL), an inexpensive and inedible food waste.13,14
Herein, we report the first sustainable MTDLs derived from CNSL, obtained by applying a rational framework combination approach.
Results and Discussion
Design
The concept of sustainability in its different nuances has been percolating the pharma’s activities for the past two decades.15 It has been embodied especially in the use of nontoxic solvents, biocatalytic processes, and waste minimization. Another still underexplored, yet increasingly important opportunity, is the use of biomass and waste feedstocks as a starting material for the development of new biologically active compounds and drugs.16 According to the #7 principle of Green Chemistry, using renewable resources, like microbial or plant biomass, offers a real alternative to traditional petrochemical intermediates.16
In addition to the clear environmental advantages, we believe that once properly optimized, the production of drugs from an inexpensive waste material may generate more affordable medicines and contribute to the goal of achieving Universal Health Coverage.17
With these concepts in mind, we undertook the task of developing an MTDL for AD starting from CNSL. Although we have already developed acetylcholinesterase (AChE)13 and histone deacetylase (HDAC)14 inhibitors with therapeutic potential for AD, this is the first time that a CNSL-derived MTDL has been rationally designed. To do so, we exploited a framework combination approach,8 which is the gold standard for creating new MTDLs, starting from compounds (or their pharmacophores) with the desired activity toward two targets of interest. A plethora of such hybrid molecules has been developed (for a recent review of the field, see the book by Decker18). The conjugation of tacrine with a second pharmacophoric moiety, pioneered by Pang et al.,19 is still an area of active research and development.12,20 As a key element of originality, this is the first report where one of the parent frameworks is derived from biomass.
Our starting point was the remarkable anti-inflammatory activity showed by anacardic acid21 (1, Figure 1), a major constituent of CNSL. CNSL is an abundant byproduct derived from cashew (Anacardium occidentale L.) processing as well as a renewable source of long-chain phenols, i.e., anacardic acids (1), cardanols (2), and cardols (3), each present as a mixture of (un)saturated enomers. In traditional folk medicine, CNSL has also been reported to have several medicinal properties, including anti-inflammatory, analgesic, and antitumoral effects, in addition to indications as a larvicide.22 We were particularly intrigued by the anti-inflammatory properties of 1a.23 These properties were recently found even superior to those of the anti-inflammatory drugs acetylsalicylic acid and dexamethasone in an in vitro cellular model.23 Particularly, 1a has been shown to exert an immunoprotective effect by decreasing the expression of inflammatory genes.23
Figure 1.

Design strategy toward hybrids 5–17 (see Table 1 for individual structures), starting from CNSL constituents 1–3 and tacrine templates 4a–4c.
Mounting evidence indicates that inflammation has a causal role in AD pathogenesis, which is not limited to the neuronal compartment but involves strong interactions with immunological mechanisms in the brain.24 Accordingly, neuroinflammation is an appealing target for therapeutic intervention in AD.25
Thus, we reasoned that a hybrid of the well-established tacrine and tacrine analogues 4a–4c (Figure 1) with that of 1–3 may furnish new MTDLs combining cholinesterase inhibition and antineuroinflammatory activity in a single molecule. The notion that the cholinergic system acts as an anti-inflammatory brake and that anticholinesterase drugs may positively modulate this process26 underlines a cross-talk between the two pathways and further supports our rationale. In fact, for maximizing efficacy, an MTDL should be directed to networked targets with established connectivity.27
As anticipated, 4a–4c represent particularly effective frameworks for the design of MTDLs for AD.28−30 This is for several reasons: (i) tacrine is a drug with micromolar activity toward both AChE and butyrylcholinesterase (BChE); (ii) this dual inhibition is positive considering the importance of both cholinesterases in AD pathology;31 (iii) its high ligand efficacy allows combination with a second framework without exceedingly increasing molecular weight;32 and (iv) the potential hepatotoxicity linked to the tacrine scaffold could be overcome by conjugation with a second framework.29
As a peculiar consideration, it should be noted that tacrine and derivatives are easily synthesizable through environment-friendly and economical approaches (e.g., catalysis with cheap and nontoxic catalysts and solvent-free reactions).33
Considering that the long alkyl chain (C15) of 1–3 might impair drug-like properties of the final compounds because of an excessive lipophilicity and an increase in molecular weight, our initial efforts focused on shorter (C8)-chain derivatives. In addition, we have assessed preliminary physicochemical properties and predicted blood–brain barrier (BBB) permeation of 1a in a parallel artificial membrane permeability assay (PAMPA). The experimentally found pKa value of 4.2 is the borderline with respect to the estimated pKa limits for CNS penetration (between 4 and 10).34 Furthermore, we could not determine log P and effective permeability coefficient (Pe) because of solubility problems (Table S1). On this basis, we decided to mainly focus on methylated derivatives (esters and ethers) and prepared the series of hybrids 5–17 (Figure 1).
Chemistry
Compounds 5–17 were synthesized following the convergent approach depicted in Scheme 2, using functionalized CNSL C8 components (21a–21d) and tacrine analogues (4a–4c) as starting reagents. The synthetic route was developed with an eye on green chemistry principles (microwave (MW)-assisted, less hazardous or solvent-free reactions, use of continuous-flow reactors) to improve the overall process sustainability.
Scheme 2. Synthesis of Final Compounds 5–17.
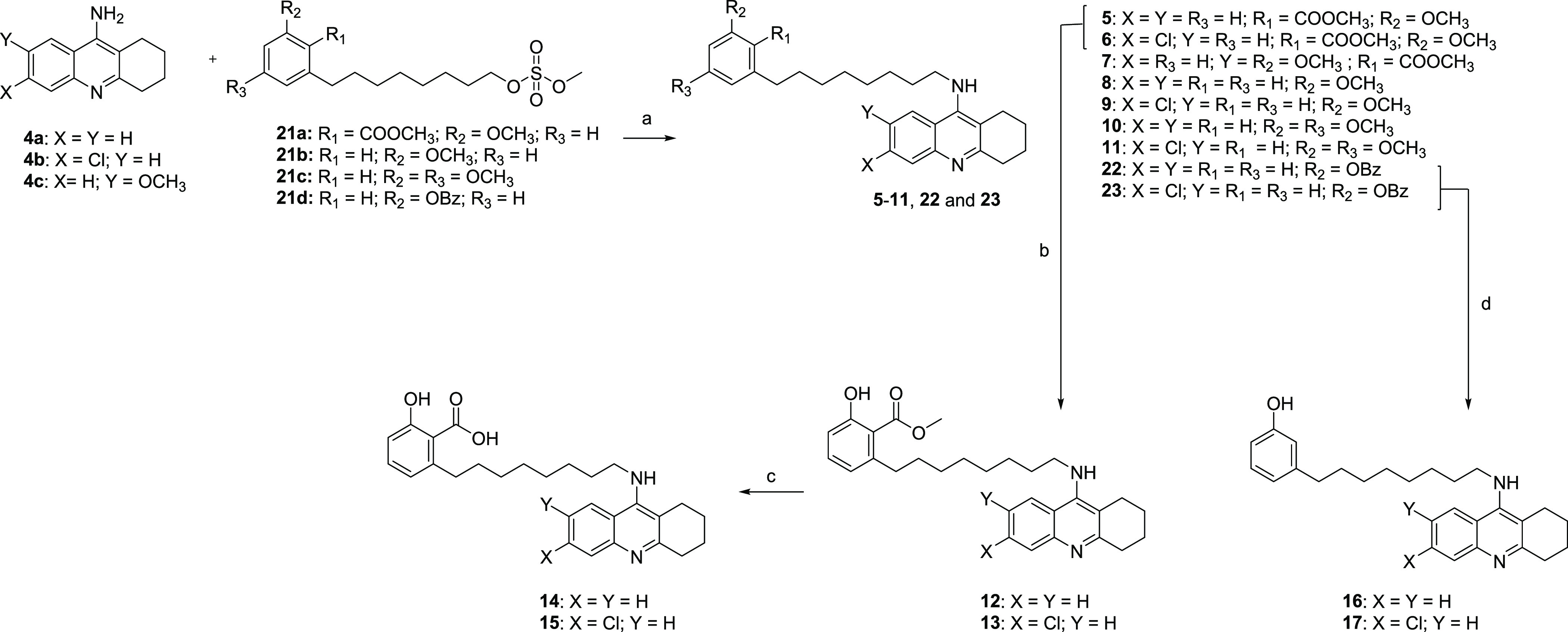
Reagents and conditions: (a) KOH, DMSO, MW: 120 °C, and 12 min (20–34%); (b) BBr3, 0 °C to rt, DCM, and 40 min (25%); (c) KOH, MeOH/H2O, MW: 100 °C, and 10 min (54–84%); and (d) H-Cube H2, 5 bar, and Pd/C 10% (41–45%).
A mixture of the unsaturated natural components 1–3 was extracted from technical CNSL and isolated in good yield. After the extraction, 1–3 were methylated on acid and phenolic groups to obtain compounds 18a–18c, respectively. Next, intermediates 18a–18c underwent oxidative cleavage by ozonolysis and reduction with sodium borohydride of the resulting secondary ozonides to the corresponding alcohols, to give the C8 derivatives 19a–19c, respectively (Scheme 1). To investigate the role of free phenolic and benzoic acid functions, cardanol 2 was transformed into its corresponding C8 primary alcohol, without undergoing the methylation step. Thus, before the ozonolysis reaction, 2 was acetylated with acetic anhydride to give a mixture of intermediates, which, after deprotection with HCl, provided 19d in good yield. To differentiate the reactivity between the phenolic and aliphatic alcohol groups in 19d, the phenol was protected as its benzyl ether 20. The functionalized mesylate intermediates 21a–21d were obtained by the reaction with methanesulfonyl chloride starting from 19a–19c and 20. In parallel, tacrine derivatives 4a–4c were synthesized following the reported one-pot and solvent-free reaction of cyclohexanone with 2-aminobenzonitrile under zinc chloride catalysis.35 With the functionalized CNSL compounds in hand, the convergent synthesis based on an aliphatic substitution between 21a–21d and tacrine derivatives 4a–4c (Scheme 2) was applied. To synthesize derivatives 5–11, 22, and 23, we developed an MW-assisted nucleophilic substitution procedure, which led to increases in yields and shortening of reaction time (from 12 h to 12 min), compared to a conventional heating protocol. Despite its utility in a nucleophilic substitution reaction, DMF is clearly not compatible with the drive toward more sustainable and environment-friendly medicinal chemistry development. Therefore, we opted for the safer DMSO.36 To investigate the role of free phenolic and benzoic acid functions, ether derivatives 5 and 6 were demethylated by using BBr3. Then, the methyl esters of 12 and 13 were hydrolyzed with KOH in MeOH/H2O under MW irradiation, to give 14 and 15, respectively. The benzylic intermediates 22 and 23 were deprotected using Pd/C 10% as the catalyst in a continuous-flow hydrogenation reactor, to obtain the free phenolic compounds 16 and 17, respectively. To note, continuous-flow processing has proved numerous advantages in terms of sustainability (cost, equipment size, energy consumption, waste generation, safety, and efficiency) over a traditional batch strategy.37
Scheme 1. Synthesis of CNSL Mesylate Intermediates 21a–21d.
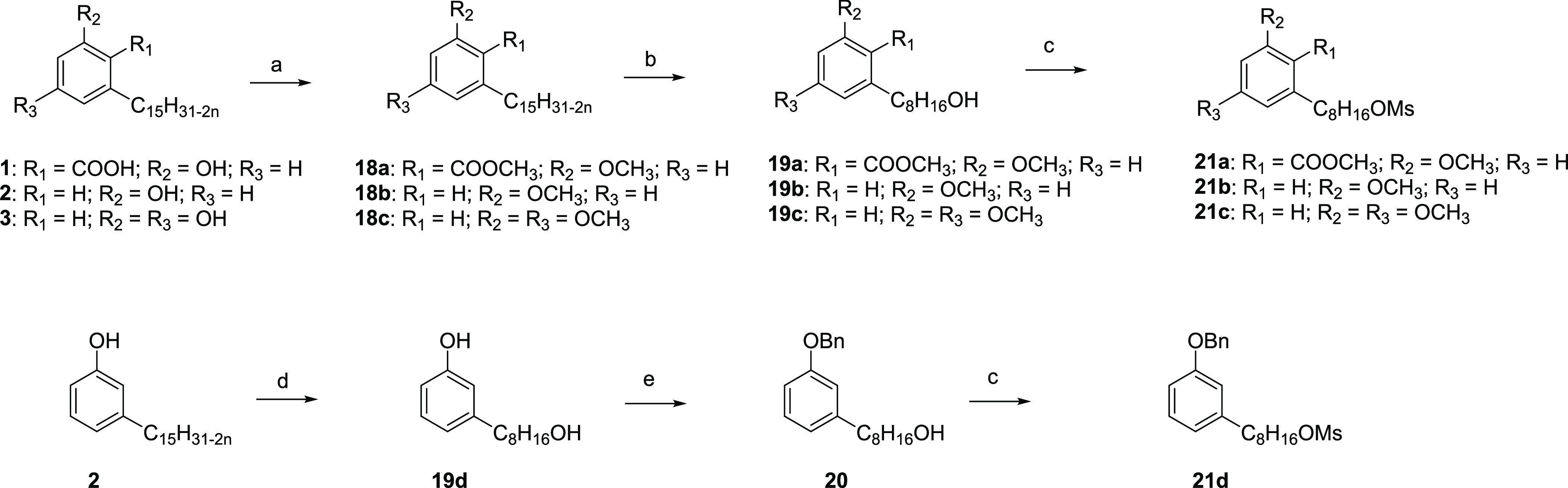
Reagents and conditions: (a) K2CO3 and acetone; MeI, 24 h, and 110 °C (66–80%); (b) O3, DCM/MeOH, and 0 °C; NaBH4, 24 h, and rt (60–70%); (c) methanesulfonyl chloride, TEA, DCM, 12 h, and rt (60–85%); (d) acetic anhydride, MW: 450 W, and 3 min; O3, DCM/MeOH, and 0 °C; NaBH4, 16 h, and rt; HCl conc. (67%); and (e) benzyl bromide, K2CO3, acetone, 12 h, and reflux (92%).
Biology
To investigate the multitarget profile of the newly synthesized hybrids, they were screened for their cholinesterase and neuroinflammatory activities at enzymatic and cellular levels, respectively.
hAChE and hBChE Inhibition Assays
To verify whether hybrids 5–17 could retain the anticholinesterase activity of the parent compounds 4a–4c, we evaluated their inhibitory potency against human recombinant AChE (hAChE) and BChE from human serum (hBChE) by using the method of Ellman et al.38 IC50 values for all compounds were calculated and are reported in Table 1 in comparison with reference compounds 4a–4c. In agreement with the literature data, we found that unsubstituted tacrine 4a shows a slight selectivity toward hBChE, 4b is a more potent hAChE inhibitor thanks to the positive effect of the chlorine atom,39 while the presence of a methoxy substituent (4c) is detrimental for both AChE and BChE inhibitions, albeit potentially positive in terms of metabolism/toxicity.40 Remarkably, all hybrids 5–17 showed an improved activity with respect to the parent compounds 4a–4c (hAChE: 1.2- to 13.5-fold for tacrine derivatives and from 1.1- to 5.7-fold for Cl-tacrine derivatives; hBChE: 1.5- to 1300-fold for tacrine derivatives and from 4.2- to 1900-fold for Cl-tacrine derivatives), supporting the effectiveness of the framework combination design strategy. However, the AChE/BChE inhibitory profiles exhibit varying trends; thus, the SARs will be discussed separately for the two enzymes.
Table 1. Cytotoxicity in HepG2 Cells and AChE and BChE Activities by 5–17 and Reference Compounds 4a–4c and 1ab.

Results are expressed as the mean of at least three experiments.
*Significance was determined by ANOVA; Dunnett’s multiple comparison test p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
All the hybrids were effective AChE inhibitors with potencies spanning two orders of magnitude (from nanomolar to single-digit micromolar) and with activity trends highly depending on the appended tacrine moiety. The most potent inhibitor was the dimethylated anacardic acid/Cl-tacrine hybrid 6 (IC50 = 2.54 nM), whereas the less active one was the dimethylated anacardic acid derivative carrying an OMe-tacrine unit 7 (IC50 equals to 1260 nM). This reinforces the finding that a Cl-tacrine moiety is more effective than an OMe-tacrine in recognizing the catalytic active site of the enzyme. As a confirmation of the tacrine scaffold as a driving force for AChE binding, within homogeneous subsets of hybrids, potency ranks in the following order: Cl-tacrine > tacrine > OMe-tacrine derivatives. As for the CNSL framework, methoxy-cardanols 8 and 9 and methoxy-cardols 10 and 11 exhibited similar profiles, suggesting that a second methoxy function on the aromatic ring does not provide additional interactions. Methylation of the anacardic acid salicylic moiety (both carboxylic and phenolic functions) was beneficial for affinity as dimethylated derivatives 5 and 6 and methyl esters 12 and 13 were more effective inhibitors than free anacardic acids 14 and 15.
However, the most striking results were observed for hBChE inhibition, with three hybrids (5, 6, and 12) exhibiting subnanomolar potencies. Particularly, compared to AChE, an inverted rank order of potency was noted, with tacrine derivatives being more potent than Cl-tacrine counterparts, paralleling the selectivity trend of the parent compounds tacrine (4a) versus Cl-tacrine (4b). Indeed, this result is consistent with previous finding, showing that the insertion of a chlorine atom in the position 6 of the tacrine nucleus is detrimental for BChE inhibition because of steric hindrance.39 The presence of the anacardic acid framework seems important for enhancing the interaction with the enzyme. In fact, the anacardic acid subset of hybrids (5, 6, and 12–15) was more potent than the corresponding cardanol (8, 9, 16, and 17) and cardol ones (10 and 11). Again, methylation of the anacardic acid at both the carboxylic and phenolic functions does make a favorable contribution to the activity as dimethylated 5 and 6 and methyl ester 12 were more potent than the free acid derivatives 14 and 15. Particularly, 5, 6, and 12 were the top-ranked inhibitors of the series. Their exceptionally high activity toward hBChE (5, IC50 = 0.0352 nM; 6, IC50 = 0.265 nM; and 12, IC50 = 0.177 nM) is not completely unexpected as similar tacrine heterodimers carrying trimethoxy-substituted benzene units were found to be very potent inhibitors of BChE.41
BChE has traditionally been considered a surrogate for AChE in cholinergic neurotransmission; however, an increasing number of studies point to a key, unique role for BChE in AD.42 Actually, AChE levels are decreased by ∼50% in AD brains, whereas BChE levels increase by as much as 900% during disease progression.43 BChE is also associated with peculiar AD biomarkers, including Aβ oligomers and plaques as well as neurofibrillary tangles.44 However, the mechanisms underlying BChE involvement in AD progression are not completely understood. Thus, the development of potent and selective BChE inhibitors would improve understanding of the role of BChE in the aetiology of AD and lead to a wider variety of treatment options. Finally, inhibition of BChE seems to be associated to less severe side effects.44
In this respect, our most potent inhibitor (5) compares favorably with BChE inhibitors showing high potency and selectivity over AChE,45−48 including some multifunctional inhibitors recently reported.49−51 The high potency and selectivity found for 5 made this hybrid of interest for further studies.
Crystal Structure of hBChE in Complex with 5
In order to rationalize at a molecular level the excellent inhibitory potency of 5, we solved its crystal structure in complex with hBChE. Crystallization, data collection and processing, and general structural analysis are reported in the Supporting Information.
Our structure revealed that 5 accommodates in the active-site gorge of hBChE without noticeable conformational adaptation of residues with respect to the unliganded parent structure (pdb entry: 1p0i). The tacrine moiety of 5 is in the same position as tacrine in the hBChE–tacrine complex (pdb entry: 4bds) with an rmsd of 0.26 Å.52 The key interactions are identical: the saturated cycle is embedded in the water molecule network, aromatic stacking with Trp82 (3.7 Å interplanar distance), hydrogen bonding between the pyridine nitrogen N26 and the main chain carbonyl of His438 (3.1 Å), and hydrogen bonding between the nitrogen atom N18 and a water molecule of the conserved water molecule network (2.9 Å) (Figure 2A,B). It is worth noting that the position of the tacrine moiety and its main stabilizing interactions are also identical in AChE in complex with alkylated tacrines or derivatives like huprines.52,53 Electron density for the C8 linker was not well defined in the first refinement cycles but is clearly visible in the polder map of the final structure when omitting the ligand (Figure 2A,B). This allows to constrain the orientation of the dimethyl anacardate moiety with the aromatic ring fitting into the groove of the acyl-binding pocket defined by residues Trp231, Leu286, Val288, Phe398, and Phe329, as commonly observed for other aromatic ligands,54 and the methoxy group pointing toward Trp231 (3.2 Å plane to methyl distance). The orientation of the methyl carboxylate substituent was more difficult to identify. The large peak of positive electron density in the initial |Fo| – |Fc| map corresponding to the dimethyl anacardate moiety is extending into the oxyanion hole defined by main chain NH of residues Gly116, Gly117, and Ala199. This density was initially modeled as a water molecule because it is usual to find one at this spot, bridging the catalytic serine to oxyanion hole residues, as seen for example in the hAChE–galantamine complex (pdb: 4ey6).55 Thus, we modeled the methyl carboxylate with its carbonyl oxygen (atom O8) pointing toward the water molecule in the oxyanion hole (Figure 2D). However, the interatomic distances between the water molecule and Ser198Oγ or O8 appeared to be unrealistically low (2.0 and 2.1 Å, respectively). So, we discarded this model. Alternatively, we made the hypothesis that the methyl carboxylate had been hydrolyzed knowing that benzoate esters are potential substrates of hBChE.56 However, modeling the substituent in its hydrolyzed carboxylate form and the water molecule in the oxyanion hole led to similar unrealistic interatomic distances (Figure 2E). Modeling of the molecule covalently bond to Ser198Oγ, i.e., benzoylated catalytic serine, or without a water molecule in the oxyanion hole also gave very poor unrealistic models.
Figure 2.

X-ray structure of 5 in complex with hBChE. Side view (A) and top view (B) of 5 in the active-site gorge of hBChE. The gorge is represented in white semitransparent cartoon with key residues as sticks and carbon atoms in green. The ligand is represented in ball and stick with carbon atoms in cyan. Oxygen atoms are represented in red, nitrogen atoms in blue, and sulfur atoms in yellow. Crystallographic water molecules are represented in red spheres and the dense hydrogen bond network in dashed lines. A 2.5 σ polder map57 calculated by omitting the ligand is represented as a blue mesh. Close-up view of the final model (C), inverted methyl carboxylate model (D), and hydrolyzed model (E) of the dimethyl anacardate moiety. Key interatomic distances are represented in dashed lines with values in Å.
Finally, modeling the methyl carboxylate with the methyl group in the oxyanion hole led to the most realistic structure (Figure 2C). In this model, O6 of the methyl ester is at close distance from C24 of the saturated ring (3.4 Å), and C7 is at the C–H hydrogen bond distance from Ser198Oγ (2.8 Å). Ser198 is an outlier in the Ramachandran plot (phi = 30° and psi = −100°), thus in an unfavorable conformation. Such an energy strained conformation has already been reported for this catalytic residue.58 Despite our efforts, a 6 σ positive peak remains unexplained in the |Fo| – |Fc| map, located in between Ser198Oγ, C7 of the methyl ester group, Gly116N, and Gly117N, suggesting that the final model is not perfect. The structure of the hBChE–5 complex confirmed that tacrine is perfectly adapted to bind to the choline-binding pocket of hBChE. Disorder of the eight-carbon linker seen in the electron density suggests that it could be optimized by reducing its length. A five-carbon linker seems a good compromise between length and flexibility and could inspire further medicinal chemistry. The dimethyl anacardate subpart has a complementary shape to the acyl-binding pocket and oxyanion hole, which allow for optimized van der Waals interactions.
Altogether, the multiple interactions of the two aromatic moieties with the two main pockets of the active-site gorge completely enlighten the high affinity of compound 5 for hBChE.
Cell-Based Screening
In parallel, a screening cascade with a set of assays was developed to identify those hybrids effectively modulating neuroinflammation at a cellular level. As the first step, the cytotoxicity profiles on the human liver cancer HepG2 (Table 1), human neuron-like SH-SY5Y (Figure S1), and murine microglial BV-2 (Figure S2) cell lines were determined. This was aimed to select those compounds endowed with an adequate safety–efficacy profile, a critical requirement for early-stage AD drug discovery.59 In fact, the failure of some AD candidates can be attributed to toxicity following drugs’ chronic administration.59 Particularly, in geriatric AD patients, due to aging, comorbidity, and subsequent polytherapy, there is an increased risk of drug–drug interactions and hepatotoxicity. Furthermore, tacrine (4a) has been withdrawn from the market for its severe side effects, the most notorious being hepatotoxic. Notwithstanding, this major clinical issue has been overcome in several cases and successful examples of tacrine hybrids with no hepatotoxicity have been reported.60,61 Thus, hepatotoxicity of 5–17 was preliminary assessed using a HepG2-based in vitro system, in comparison with parent compounds 1a and 4a. The HepG2 cell line has been extensively employed as a suitable in vitro system thanks to its homogeneous and consistent cellular features. We applied an experimental protocol we utilize in our laboratory, which shows 75–80% of residual cell viability for tacrine treatment (10 μM), following an incubation of 24 h.62 To further progress those molecules potentially devoid of hepatotoxicity, we set up an initial cutoff of >80% cell viability at 10 μM. Based on the collected data as well as on the cholinesterase inhibitory profiles (Table 1), compounds 5, 6, 9, 12–15, and 17 were selected as the most promising to continue the study. Neurotoxicity was evaluated by the MTT viability assay by treating human differentiated SH-SY5Y cells with increasing concentrations (0.1–1 μM) of the selected compounds for 24 h (Figure S1). Encouragingly, all the compounds were not significantly neurotoxic at both tested concentrations, being cell viability comparable or even slightly and significantly increased with respect to control cells.
The potential cytotoxicity of compounds 5, 6, 9, 12–15, and 17 was also evaluated on BV-2 microglial cells. BV-2 cells were treated with increasing concentrations (0.1–1 μM) of the selected compounds for 24 h, and cell viability was measured by the MTT assay (Figure S2). At the lowest concentration (0.1 μM), the compounds were not cytotoxic as cell viability of treated cells was comparable to the controls. At 1 μM, only compounds 5, 6, and 9 showed a slight, although significant, cytotoxicity.
Antineuroinflammatory Activity
To assess the antineuroinflammatory potential, we subjected the less toxic compounds of our library (5, 6, 9, 12–15, and 17) to phenotype-based screening in murine microglial BV-2 cells. The discovery that patients with AD show increased levels of inflammatory mediators and the association among AD risk genes and the innate immune functions indicates a major role for neuroinflammation in AD pathogenesis.63 A key player in the neuroinflammatory response is microglia. Microglia are considered resident macrophages in the brain that provide a first line of defense in the central nervous system (CNS).64 Under physiological conditions, microglia play an important role in the development, structural formation, and functional regulation of the nervous system.65−67 After exogenous stimulation or microenvironment changes in the brain, resting microglia transform to an activated phenotype that leads to neuronal tissue damage and to the expression of neuroinflammation-related genes.68,69 Activated microglia release a large number of inflammatory factors, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), by activating some signaling pathways, such as the Toll-like receptor 4 (TLR4), nuclear factor kappa B (NF-κB), and mitogen-activated protein kinase (MAPK) pathways.70,71 Moreover, activated microglia increase the expression of inducible nitric oxide synthase (iNOS)68 and COX-2.72 Therefore, inhibiting excessive microglial activation and reducing the production of proinflammatory mediators are potential avenues for controlling neuroinflammation in AD.
To study the anti-inflammatory activity of the selected compounds, BV-2 cells were exposed to bacterial endotoxin lipopolysaccharide (LPS) for 24 h. LPS is widely used as a proinflammatory agent as it induces inflammatory reactions both in primary microglia and BV-2 cells.73,74 The fact that 90% of the genes modulated by LPS in BV-2 cells are also induced in primary microglia and both show similar reaction patterns makes BV-2 cells a valid substitute for primary microglia in many experimental settings.75 Based on the cytotoxicity results (Figures S1 and S2) as well as the nanomolar cholinesterase potencies (Table 1), the experiments were carried out using concentrations in the 0.01–0.1 μM range.
Thus, BV-2 cells were treated with 5, 6, 9, 12–15, and 17 for 24 h and subsequently exposed to 100 ng/mL LPS for further 24 h (Figure 3). As assessed by the MTT assay, LPS treatment induced a significant reduction of cell viability. Anacardic acid (13–15) and cardanol (17) hybrids showed no ability to counteract LPS-induced damage (cell viability of treated cells was comparable to cells exposed to LPS).
Figure 3.

Anti-inflammatory effects of 5, 6, 9, 12–15, and 17 against LPS in BV-2 cells. BV-2 cells were treated with increasing concentration of the selected compounds (0.01–0.1 μM) for 24 h and exposed to 100 ng/mL LPS for further 24 h, and cell viability was evaluated by the MTT assay. Each bar represents means ± SEM of at least four independent experiments. Data were analyzed by one-way ANOVA followed by Tukey’s test. ○p < 0.05 compared to CTRL; *p < 0.05 compared to LPS.
On the other hand, dimethylated anacardic acids 5 and 6, methyl-cardanol 9, and anacardic acid methyl ester 12 significantly counteracted LPS-induced cell death. In particular, while 12 showed a moderate, although significant, effect already at 0.01 μM, 9 afforded total protection against LPS only at the highest tested concentration (0.1 μM). Remarkably, both dimethylated anacardic acid derivatives 5 and 6 (irrespective of the tacrine-appended moiety) restored cell viability to normal levels already at the very low concentration of 0.01 μM.
Of note, 5 and 6 were the most effective compounds not only in counteracting LPS-induced damage but also in inhibiting hBChE and hAChE, respectively. For this reason, their anti-inflammatory profile was investigated more in depth by evaluating their ability to modulate the expression of major neuroinflammatory cytokines, i.e., IL-1β, TNF-α, and mediators, i.e., iNOS and COX-2.
IL-1β and TNF-α are proinflammatory neurotoxic cytokines that contribute to neuronal dysfunction and neuronal loss in AD.76 iNOS is responsible of the formation of NO, one of the main cytotoxic mediators participating in the innate immune response in mammals. iNOS is not usually expressed in the brain. However, activated microglia are a major cellular source of iNOS. The excessive release of NO by activated microglia correlates with the progression of neurodegenerative disorders. Similarly, COX-2 has been associated with neurotoxicity, and inhibition of COX-2 induction reduces brain injury and delays the progress of neurodegenerative diseases.77
BV-2 cells were treated with 5 and 6 for 24 h and exposed to LPS 100 ng/mL for further 24 h, and the expression of the inflammatory mediators was evaluated by real-time polymerase chain reaction (RT-PCR) (Figure 4). Parent compounds 1a and 4a were added in the experimental setting as positive and negative controls, respectively. Intriguingly, parent compound 1a chemically resembles salicylic acid. Interestingly, de Souza et al. demonstrated that it has higher anti-inflammatory activity than acetylsalicylic acid in vitro.23 Based on these considerations, we chose 1a instead of salicylic/acetylsalicilic acids as a positive control. LPS significantly increased the expression of TNF-α, IL-1β, COX-2, and iNOS compared to control cells. As expected, 4a was not effective, while 1a, 5, and 6 efficiently suppressed the transcription of COX-2 and iNOS genes and proinflammatory cytokines. Of note, both 5 and 6 showed a higher anti-inflammatory activity than 1a. In fact, 6 was able to significantly reduce the expression of all the tested inflammatory mediators, 5 significantly downregulated IL-1β, COX-2, and iNOS (and not TNF-α), while 1a reduced only the expression of IL-1β and COX-2. The higher anti-inflammatory activity of 5 and 6 compared to 1a seems to support the effectiveness of the applied framework combination approach.
Figure 4.

Expression of proinflammatory cytokines and enzymes in LPS-induced BV-2 cells treated with 1a, 4a, 5, and 6. BV-2 cells were treated with 1a, 4a, 5, and 6 (0.1 μM) for 24 h and exposed to 100 ng/mL LPS for further 24 h, and real-time PCR was performed to detect TNF-α, IL-1β, iNOS, and COX-2 mRNA levels. Data are expressed as relative abundance vs CTRL. Each bar represents means ± SEM of three independent experiments. Data were analyzed with a one-way ANOVA followed by Tukey’s test. *p < 0.05 compared to LPS.
Notably, evidence indicates the cholinergic system as a mediator of neuroimmune interactions26 and BChE as an important player in regulating intrinsic inflammation and activity of cholinoceptive glial cells.43 Thus, it could be speculated that the higher activity of 5 and 6 compared to 1a could be due to their concomitant modulation of both pathways. However, the possibility that it could be due to the different cell bioavailability of the two compounds cannot be ruled out.
Our results on 1a’s anti-inflammatory activity are partially in agreement with the data of de Souza et al.,23 showing that 1a (LDT11) is able to significantly reduce the expression of TNF-α, iNOS, COX-2, NF-κB, IL-1β, and IL-6. This discrepancy could be related to the different concentrations used (50 μM vs 0.1 μM). In addition, de Souza et al. investigated the anti-inflammatory activity of 1a in a different model, i.e., the RAW264.7 murine macrophage cell line.23 As expected, 4a did not influence the expression of all the tested inflammatory mediators.
The anti-inflammatory activity of 5 and 6 has been also confirmed by ELISA (Figure 5). The pretreatment with 5 and 6 was able to significantly decrease the release of IL-1β in the culture medium when compared to LPS-treated cells.
Figure 5.

Secretion of IL1-β in activated BV-2 cells treated with 5 and 6. BV-2 cells were treated with 5 and 6 (0.1 μM) for 24 h and exposed to 100 ng/mL LPS for further 24 h, and ELISA was performed to detect IL1-β concentration in the culture medium. Each bar represents means ± SEM of three independent experiments. Data were analyzed with a one-way ANOVA followed by Tukey’s test. *p < 0.05 compared to LPS.
The increased expression of proinflammatory enzymes and cytokines is mediated by the migration of transcription factor NF-κB to the nucleus. NF-κB is usually located in the cytoplasm in association with IκBα. Upon IκBα phosphorylation and degradation, NF-κB is isolated and translocated to the nucleus.78,79 Therefore, we further aimed to determine whether 5 and 6 could modulate LPS-induced nuclear translocation of NF-κB. BV-2 cells were treated with 0.1 μM 5 and 6 for 24 h and exposed to LPS for further 24 h, and the localization of the transcription factor NF-κB was evaluated by confocal immunofluorescence (Figure 6). 1a and 4a were used as reference compounds. LPS induced a strong increase in NF-κB levels both in the cytoplasm and in the nucleus. Consistent with the RT-PCR results, LPS-mediated nuclear translocation of NF-κB was considerably blocked by pretreatment with 5 and 6 and, at a lower extent, with 1a. This suggests that 5 and 6 counteract neuroinflammation by inhibiting the transcriptional activation of NF-κB. This finding also confirms their higher neuroinflammatory activity with respect to 1a.
Figure 6.

Nuclear translocation of NF-κB in LPS-induced BV-2 cells treated with 1a, 4a, 5, and 6. Cells were treated with compounds 5 and 6 (0.1 μM) and 1a and 4a as reference compounds (0.1 μM) for 24 h and then exposed to 100 ng/mL LPS for further 24 h. BV-2 cells were immunostained with a primary antibody against NF-κB p65 followed by secondary Alexa Fluor 488-conjugated antirabbit IgG antibody (green), and cell nuclei (blue) were visualized with DAPI. Scale bars: 10 μm.
Blood–Brain Barrier Permeability Prediction
A key feature for AD drugs is their effective delivery into the brain at therapeutic concentrations, mainly because of the BBB presence. PAMPA-BBB is an in vitro tool developed to rapidly predict passive BBB permeation. BBB permeability of the most promising 5 and 6 was estimated using the PAMPA-BBB model in comparison with standard drugs, including AChEI tacrine (4a) and donepezil. The measurement predicted that both 5 and 6 have the potential to cross the BBB. Particularly, Pe values matched those of two standard AD drugs (donepezil and 4a), known for effective BBB penetration (Table 2).
Table 2. In Vitro Permeability (Pe) Values with Related Predictive Penetrations into the CNS of Commercial Drugs, 5 and 6.
| BBB penetration estimation | ||
|---|---|---|
| compound | Pe ± SEM (× 10–6 cm/s)a | CNS (+/−) |
| 5 | 6.99 ± 1.04 | CNS + |
| 6 | 17.70 ± 4.63 | CNS + |
| furosemide | 0.19 ± 0.07 | CNS – |
| ranitidine | 0.35 ± 0.31 | CNS – |
| donepezil | 21.93 ± 2.06 | CNS – |
| tacrine (4a) | 5.96 ± 0.59 | CNS – |
Pe ± SEM (n = 3). Each compound was assessed in quadruplicate.
Plasma Stability Assay
Considering that both 5 and 6 carry a labile ester functionality, we preliminary performed a human plasma stability assay. As determined by HPLC–MS analysis (Figure 7), 5 showed no decomposition over a 6 h range. This suggests that 5 is stable and not eventually transformed into the less active demethylated 14 in this time frame.
Figure 7.

Human plasma stability of compound 5 upon incubation at 37 °C. Concentration was assessed by means of HPLC–MS. Analyses were performed in duplicate.
Conclusions
After decades of massive research efforts and continuous clinical failures, the reason to target more than one pathway in AD seems rather clear. Inhibition of BChE is a promising target to increase the acetylcholine level and attenuate the cognitive decline, especially at a late disease stage. It could be also beneficial for modulating other AD hallmarks as a link between BChE and amyloid, and tau pathologies have been proposed. Furthermore, there is much evidence indicating that neuroinflammation and microglia are major contributors to AD. As such, molecules inhibiting proinflammatory microglia and neuronal death have been intensively investigated as possible disease-modifying drugs, although not exempt from complications.
In this study, we have developed a new series of hybrids by combining the cholinesterase activity of tacrine derivatives 4a–4c with the anti-inflammatory properties of CNSL-derived 1. Enzymatic studies have disclosed potent and selective inhibitors of both AChE and BChE. Compounds 5, 6, and 12, endowed with subnanomolar activities, can be listed among the most effective BChE inhibitors so far developed. Specifically, to the best of our knowledge, 5 stands as the one with the lowest IC50 value (0.0352 nM). Our investigation in BV-2 microglial cells has revealed a protective activity against neurotoxic insults for 5 and 6 already at the remarkably low concentration of 0.01 μM. Both suppress LPS-induced IL-1β, COX-2, and iNOS (TNF-α only for 6) overexpression. Particularly, they are capable of counteracting neuroinflammation by inhibiting the transcriptional activation of NF-κB, without causing cytotoxicity in microglial, neuronal, and hepatic cell lines.
On the basis of the obtained biological and PAMPA-BBB data, we can speculate that 5 and 6 may access the brain at their active nanomolar concentration.
Finally, as an important remark, the utilization of components from CNSL, a cheap and highly available food waste, offers an invaluable resource for developing new MTDLs for combating AD. It could be of importance and economic feasibility in low- and middle-income countries that are among the cashew top producers and, at the same time, areas with high current and future disease prevalence. In principle, the new molecules, in addition to the peculiar advantages of MTDLs, could offer accessible drugs to patients living in those countries, who might otherwise be excluded from access to therapy. Clearly, this is a long-term aspirational goal; however, in our opinion, it deserves attention by the medicinal chemistry community for the potential benefits to the global patient population and the environment.
Experimental Section
Chemistry
All the commercially available reagents and solvents were purchased from Sigma-Aldrich, Alfa Aesar, VWR, and TCI and used without further purification. Reactions were followed by analytical thin-layer chromatography (TLC) on precoated TLC plates (layer: 0.20 mm silica gel 60 with a fluorescent indicator UV254, from Sigma-Aldrich). Developed plates were air-dried and analyzed under a UV lamp (UV 254/365 nm). A CEM Discover SP focused microwave reactor was used for microwave-assisted reactions. Nuclear magnetic resonance (NMR) experiments were run on a Varian VXR 400 (400 MHz for 1H and 100 MHz for 13C). 1H and 13C NMR spectra were acquired at 300 K using deuterated chloroform (CDCl3) and methanol (CD3OD) as solvents. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane (TMS) as the internal reference, and coupling constants (J) are reported in hertz (Hz). The spin multiplicities are reported as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were recorded on a Waters ZQ4000, XevoG2-XSQTof, Acquity arc-QDA LC–MS apparatus with electrospray ionization (ESI) in positive mode. Catalytic hydrogenation was performed on an H-Cube continuous-flow hydrogenation reactor (H-Cube, ThalesNano Nanotechnology, Budapest, Hungary). Compounds were named following IUPAC rules as applied by ChemBioDraw Ultra (version 16.0). The purity of compounds was determined using a Kinetex 5 μM EVO C18 100 Å, LC column 150 × 4.6 mm and an HPLC JASCO Corporation (Tokyo, Japan) instrument (PU-1585 UV equipped with a 20 μL loop valve). All the tested compounds (except compound 9, whose purity is 94%) showed ≥95% purity by analytical HPLC.
Extraction of Anacardic Acid Mixtures (1) from Natural CNSL
A solution of 15 g of calcium hydroxide in methanol/water (6:1, 210 mL) was added to 30 g of natural CNSL. The system was stirred at 60 °C for 3 h. After this period, the mixture was concentrated under vacuum and filtered. The solid was transferred to a 1 L Erlenmeyer in which were added ethyl acetate (150 mL), distilled water (50 mL), and 50% HCl solution to reach pH = 1.0. The resulting solution was washed with saturated sodium chloride solution (50 mL) and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the mixture was purified by silica gel column chromatography (hexane/ethyl acetate: 0–30%), giving 16.5 g of the anacardic acid mixture (1), corresponding to approximately 55% of the mass of natural CNSL used.
Extraction of Mixtures of Cardanols (2) and Cardols (3) from Technical CNSL
Twenty grams of technical CNSL donated from the company Resibras was purified by silica gel column chromatography (hexane/ethyl acetate: 5–35%) to provide 14 g of the mixture of cardanols (2, 70% of the applied mass) and 4.8 g of the mixture of cardols (3, 24% of the applied mass).
General Procedure I (for Compounds 18a–18c)
Mixtures of 1 (12.5 g, 36.5 mmol) or 2 (10 g, 33.5 mmol) or 3 (6 g, 18.7 mmol) were stirred with potassium carbonate (3.0 equiv) in acetone (300 mL). After 2 h, methyl iodide (4.0 or 6.0 equiv) was added, and the reaction was refluxed at 110 °C with a cooling system at −8 °C for 24 h. After cooling, the solvent was evaporated in vacuo. Distilled water (40 mL) was added, and the residue was extracted with dichloromethane (3 × 30 mL). The combined organic phases were washed with 10% HCl solution (10 mL) and saturated sodium chloride solution (10 mL), dried anhydrous Na2SO4, filtered, and concentrated under vacuum. The crude product was purified by column chromatography (hexane/ethyl acetate: 5–20%), providing the unchanged mixture of saturated derivatives and the mixtures of methyl O-methylanacardates (18a, 74%), O-methylcardanols (18b, 80%), and O,O-dimethylcardols (18c, 66%) with different unsaturation degrees as brown oils. The mixtures have been used for the further step without characterization.
General Procedure II (for Compounds 19a–19c)
To an ozonation flask was added a solution (5.0 mmol, 1.0 equiv) of the O-methylated methylanacardate mixture 18a or O-methylated cardanol 18b or O,O-dimethylated cardol 18c in dichloromethane/methanol (1:1, 60 mL). The reaction was cooled at 0 °C with an ice–water bath, and the solution was treated with ozone for approximately 3 h. After purging the reaction mixture with nitrogen, NaBH4 (20.0 mmol, 4.0 equiv) was added, and the mixture was left warming to room temperature, under vigorous stirring for 24 h. The reaction was quenched with water and 10% aqueous HCl (10 mL) and extracted with dichloromethane (3 × 30 mL). The combined organic layers were washed with brine solution (10 mL), dried over anhydrous Na2SO4, and evaporated. The residue was purified by chromatography (hexane/ethyl acetate: 20–35%), providing the corresponding alcohol.
General Procedure III (for Compounds 21a–21d)
Compounds 19a–19c or 20 (0.68 mmol, 1.0 equiv) and NEt3 (0.88 mmol, 1.3 equiv) were stirred in dichloromethane (0.35 mL) at 0 °C for 15 min. Methanesulfonyl chloride (0.88 mmol, 1.3 equiv) was added dropwise to the mixture and stirred overnight at room temperature. The reaction was quenched with water and extracted with dichloromethane (3 ×10 mL). The organic layers were collected, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The crude products were purified by column chromatography.
General Procedure IV (for Compounds 5–11, 22, and 23)
The appropriate compounds 4a–4c (0.15 mmol, 1.0 equiv) were solubilized in dry DMSO (1.3 mL), together with KOH (0.24 mmol, 1.6 equiv) and 4 Å molecular sieves. The solution was stirred at room temperature for 1 h under N2. The derivatives 21a–21d (0.18 mmol, 1.2 equiv) were then added to the mixture, and the reaction was carried out under microwave irradiation at 120 °C for 12 min. The mixture was poured into water and extracted with dichloromethane (3 × 10 mL). The organic layers were collected, dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The crude product was purified by column chromatography to afford the corresponding intermediate.
General Procedure V (for Compounds 12 and 13)
BBr3 (1 mmol, 4.0 equiv) was added at 0 °C to a stirred solution of compound 5 or 6 (0.25 mmol, 1.0 equiv) in dichloromethane (3 mL). The resulting mixture was stirred at room temperature for 40 min. When completed, the reaction was quenched with a saturated solution of aqueous NaHCO3 and 20 mL of water. The mixture was extracted with dichloromethane (3 × 10 mL), and the organic layers were collected, dried over anhydrous Na2SO4, filtered, and evaporated to dryness under vacuum. The crude product was purified by column chromatography to afford the corresponding intermediate 12 or 13.
General Procedure VI (for Compounds 14 and 15)
Compound 12 or 13 (0.07 mmol, 1.0 equiv) was solubilized in 2 mL of 3.5 M KOH solution (water/methanol, 2:1), and the reaction was performed under microwave irradiation at 100 °C for 10 min. The mixture was cooled down at 0 °C, and a solution of 2 N HCl was added dropwise until pH = 2. After 20 min, the obtained white precipitate was collected by filtration. The filtrate was suspended in methanol, and the inorganic insoluble precipitate was filtered off. The organic phase was dried under vacuum to give the corresponding hydrochloride derivative 14 or 15.
General Procedure VII (for Compounds 16 and 17)
A ThalesNano H-Cube Mini flow reactor was equipped with a Pd/C 10% catalyst cartridge. The reactor was programmed to run at 25 °C and 5 bar. A solution of 0.01 M compounds 22 and 23 (0.07 mmol, 1.0 equiv) (ethyl acetate/methanol, 1:1) was pumped through the reactor at a flow rate of 1 mL/min. The crude product coming out of the exit port was collected, concentrated under vacuum, and purified by flash chromatography.
Methyl 2-Methoxy-6-(8-((1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)benzoate (5)
The title compound was obtained according to general procedure IV using 4a and 21a. The crude product was purified by column chromatography (6.5:3:0.5 petroleum ether/ethyl acetate/NEt3). Compound 5 was obtained as a sticky yellow-brown oil. Yield: 20%. HPLC purity: 95%. 1H NMR (CDCl3, 400 MHz): δ 1.30–1.39 (m, 8H), 1.55–1.66 (m, 4H), 1.92 (m, 4H), 2.53 (t, 2H, J = 8.0 Hz), 2.70 (m, 2H), 3.06 (m, 2H), 3.47 (t, 2H, J = 7.6 Hz), 3.81 (s, 3H), 3.89 (s, 3H), 6.75 (d, 1H, J = 8.4 Hz), 6.80 (d, 1H, J = 7.2 Hz), 7.26 (t, 1H, J = 7.6 Hz), 7.33 (t, 1H, J = 7.2 Hz), 7.56 (t, 1H, J = 7.2 Hz), 7.90–7.96 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.3, 22.8, 24.5, 26.8, 29.1, 29.2, 29.2, 29.3, 30.9, 31.0, 31.6, 32.8, 31.7, 33.3, 33.4, 49.3, 51.9, 55.8, 108.4, 113.4, 121.4, 121.4, 122.9, 123.5, 123.8, 127.3, 128.9, 130.1, 141.1, 156.2, 168.8. HRMS (ESI+) m/z: [M + H]+ calcd for C30H38N2O3, 475.29552; found, 475.29597.
Methyl 2-(8-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)-6-methoxybenzoate (6)
The title compound was obtained according to general procedure IV using 4b and 21a. The crude product was purified by column chromatography (7.5:2:0.5 petroleum ether/ethyl acetate/NEt3). Compound 6 was obtained as a sticky yellow-brown oil. Yield: 34%. HPLC purity: 98%. 1H NMR (CDCl3, 400 MHz): δ 1.28–1.35 (m, 8H), 1.53–1.66 (m, 4H), 1.89 (s, 4H), 2.51 (t, 2H, J = 7.6 Hz), 2.63 (m, 2H), 3.04 (m, 2H), 3.50 (t, 2H, J = 7.2 Hz), 3.79 (s, 3H), 3.87 (s, 3H), 6.72–6.79 (m, 2H), 7.24 (m, 2H), 7.89–7.93 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.3, 22.7, 24.4, 26.7, 29.1, 29.1, 29.2, 30.9, 31.6, 33.1, 33.3, 49.4, 51.9, 55.8, 108.4, 115.0, 117.8, 121.4, 123.5, 124.4, 124.6, 126.5, 130.2, 141.1, 151.4, 156.3, 168.8. HRMS (ESI+) m/z: [M + H]+ calcd for C30H37ClN2O3, 509.256547; found, 509.25651.
Methyl 2-Methoxy-6-(8-((7-methoxy-1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)benzoate (7)
The title compound was obtained according to general procedure IV using 4c and 21a. The crude product was purified by column chromatography (7.5:2:0.5 petroleum ether/ethyl acetate/NEt3). Compound 7 was obtained as a sticky yellow-brown oil. Yield: 23%. HPLC purity: 99%. 1H NMR (CDCl3, 400 MHz): δ 1.29–1.37 (m, 8H), 1.55–1.66 (m, 4H), 1.90 (m, 4H), 2.51 (t, 2H, J = 7.6 Hz), 2.69 (m, 2H), 3.05 (m, 2H), 3.42 (t, 2H, J = 6.8 Hz), 3.79 (s, 3H), 3.87 (s, 3H), 3.89 (s, 3H), 6.73–6.80 (m, 2H), 7.24 (m, 4H), 7.99 (d, 1H, J = 7.2 Hz). 13C NMR (CDCl3, 100 MHz): δ 20.9, 22.1, 24.2, 26.6, 28.8, 29.0, 29.1, 29.2, 30.9, 31.4, 33.3, 48.1, 52.1, 55.0 103.3, 108.4, 117.8, 121.4, 122.9, 123.2, 130.2, 141.0, 154.0, 156.2, 156.6, 168.3. MS (ESI+) m/z: [M + H]+ calcd for C31H40N2O4, 504; found, 505.
N-(8-(3-Methoxyphenyl)octyl)-1,2,3,4-tetrahydroacridin-9-amine (8)
The title compound was obtained according to general procedure IV using 4a and 21b. The crude product was purified by column chromatography (7:2.5:0.5 petroleum ether/ethyl acetate/NEt3). Compound 8 was obtained as a sticky yellow-brown oil. Yield: 24%. HPLC purity: 95%. 1H NMR (CDCl3, 400 MHz): δ 1.30–1.39 (m, 8H), 1.55–1.66 (m, 4H), 1.92 (m, 4H), 2.53 (t, 2H, J = 8.0 Hz), 2.70 (m, 2H), 3.06 (m, 2H), 3.47 (t, 2H, J = 7.6 Hz), 3.77 (s, 3H), 6.70–6.75 (m, 3H), 7.17 (t, 1H, J = 7.6 Hz), 7.35 (t, 1H, J = 7.2 Hz), 7.58 (t, 1H, J = 7.2 Hz), 7.99 (d, 1H, J = 8.8 Hz), 8.09 (d, 1H, J = 7.6 Hz). 13C NMR (CDCl3, 100 MHz): δ 21.5, 22.3, 22.6, 22.8, 24.0, 26.7, 29.1, 29.1, 29.3, 29.6, 31.2, 31.4, 35.9, 49.0, 55.0, 110.6, 114.2, 117.6, 120.8, 123.5, 124.4, 129.1, 130.6, 144.3, 153.4, 154.4, 159.5. MS (ESI+) m/z: [M + H]+ calcd for C28H36N2O, 416; found, 417.
6-Chloro-N-(8-(3-methoxyphenyl)octyl)-1,2,3,4-tetrahydroacridin-9-amine (9)
The title compound was obtained according to general procedure IV using 4b and 21b. The crude product was purified by column chromatography (8:1.5:0.5 petroleum ether/ethyl acetate/NEt3). Compound 9 was obtained as a sticky yellow-brown oil. Yield: 29%. HPLC purity: 94%. 1H NMR (CDCl3, 400 MHz): δ 1.32–1.38 (m, 8H), 1.60–1.68 (m, 4H), 1.91 (m, 4H), 2.57 (t, 2H, J = 7.6 Hz), 2.65 (m, 2H), 3.05 (m, 2H), 3.51 (t, 2H, J = 7.6 Hz), 3.79 (s, 3H), 6.72–6.77 (m, 3H), 7.19 (t, 1H, J = 8.4 Hz), 7.27 (m, 1H), 7.90–7.93 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.4, 22.8, 24.4, 26.8, 29.1, 29.2, 29.3, 31.2, 31.7, 33.5, 35.9, 49.5, 55.1, 110.7, 114.2, 115.2, 118.0, 120.8, 124.3, 124.67, 126.9, 129.1, 134.3, 144.3, 151.1, 159.5. MS (ESI+) m/z: [M + H]+ calcd for C28H35ClN2O, 450; found, 451.
N-(8-(3,5-Dimethoxyphenyl)octyl)-1,2,3,4-tetrahydroacridin-9-amine (10)
The title compound was obtained according to general procedure IV using 4a and 21c. The crude product was purified by column chromatography (7.5:2:0.5:1:0.1 petroleum ether/ethyl acetate/dichloromethane/methanol/aqueous 32% ammonia). Compound 10 was obtained as a sticky yellow-brown oil. Yield: 25%. HPLC purity: 95%. 1H NMR (CDCl3, 400 MHz): δ 1.46–1.13 (m, 8H), 1.73–1.46 (m, 4H), 1.91 (t, 4H, J = 6.6 Hz), 2.58–2.39 (m, 2H), 2.68 (s, 2H), 3.08 (s, 2H), 3.49 (t, 2H, J = 7.1 Hz), 3.61 (t, 2H, J = 6.6 Hz), 3.75 (s, 6H), 6.46–5.79 (m, 2H), 6.76 (m, 1H), 7.28 (m, 1H), 7.54 (t, 1H), 7.95 (d, 2H, J = 8.6 Hz). 13C NMR (CDCl3, 100 MHz): δ 22.6, 24.4, 25.2, 26.8, 29.1, 29.3, 31.6, 33.4, 39.6, 49.9, 52.2, 54.8, 55.1, 57.6, 97.4, 106.5, 107.0, 108.3, 119.9, 121.4, 123.0, 123.8, 130.1, 141.0, 141.2, 145.1, 159.2, 160.6. MS (ESI+) m/z: [M + H]+ calcd for C29H38N2O2, 446; found, 447.
6-Chloro-N-(8-(3,5-dimethoxyphenyl)octyl)-1,2,3,4-tetrahydroacridin-9-amine (11)
The title compound was obtained according to general procedure IV using 4b and 21c. The crude product was purified by column chromatography (7.5:2:0.5:1:0.1 petroleum ether/ethyl acetate/dichloromethane/methanol/aqueous 32% ammonia). Compound 11 was obtained as a sticky yellow-brown oil. Yield: 25%. HPLC purity: 95%. 1H NMR (CDCl3, 400 MHz): δ 1.40–1.10 (m, 8H), 1.57–1.62 (m, 4H), 1.91 (t, 4H, J = 2.9 Hz), 2.58–2.47 (m, 2H), 2.65 (s, 2H), 3.02 (s, 2H), 3.45 (s, 2H), 3.77 (s, 6H), 6.46–6.16 (m, 3H), 7.57–6.97 (m, 2H), 7.8–7.90 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.6, 22.8, 24.5, 26.8, 29.1, 29.2, 29.3, 31.1, 31.7, 33.9, 36.2, 49.5, 55.2, 97.4, 106.4, 115.5, 118.3, 124.1, 124.6, 127.4, 133.9, 145.1, 148.0, 150.8, 159.3, 160.6. MS (ESI+) m/z: [M + H]+ calcd for C29H37ClN2O2, 480; found, 481.
Methyl 2-Hydroxy-6-(8-((1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)benzoate (12)
The title compound was obtained according to general procedure V starting from 5. The crude product was purified by column chromatography (7:2.5:0.5:0.05 petroleum ether/dichloromethane/methanol/aqueous 32% ammonia). Compound 12 was obtained as a sticky colorless oil. Yield: 25%. HPLC purity: 99%. 1H NMR (CDCl3, 400 MHz): δ 1.25–1.38 (m, 8H), 1.50–1.52 (m, 2H), 1.64 (t, 2H, J1 = 8 Hz), 1.91 (s, 4H), 2.69 (s, 2H), 2.85 (t, 2H, J1 = 8 Hz), 3.06 (s, 2H), 3.48 (t, 2H, J = 7.1 Hz), 3.92 (s, 4H), 6.69 (d, 1H, J = 7.5 Hz), 6.83 (d, 1H, J = 8.2 Hz), 7.29 (dt, 2H, J = 15.8, 7.7 Hz), 7.54 (t, 1H, J = 7.5 Hz), 7.80–8.05 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.7, 23.0, 24.7, 26.9, 29.3, 29.3, 29.6, 31.7, 31.9, 33.8, 36.4, 49.4, 52.0, 112.1, 115.5, 115.6, 120.0, 122.2, 122.8, 123.5, 128.3, 128.4, 134.0, 145.8, 147.1, 150.9, 158.2, 162.2, 171.7. MS (ESI+) m/z: [M + H]+ calcd for C29H36N2O3, 460; found, 461.
Methyl 2-(8-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)-6-hydroxybenzoate (13)
The title compound was obtained according to general procedure V starting from 6. The crude product was purified by column chromatography (7:2.5:0.5:0.05 petroleum ether/dichloromethane/methanol/aqueous 32% ammonia). Compound 13 was obtained as a sticky colorless oil. Yield: 25%. HPLC purity: 97%. 1H NMR (CDCl3, 400 MHz): δ 1.23–1.40 (m, 8H), 1.47–1.54 (m, 2H), 1.67 (dt, 2H, J = 8 Hz), 1.88–1.89 (m, 4H), 2.63 (s, 2H), 2.79–2.92 (m, 2H), 3.04 (s, 2H), 3.51 (t, 2H, J = 6.8 Hz), 3.92 (s, 3H), 4.19 (s, 1H), 6.69 (d, 1H, J = 7.5 Hz), 6.75–6.98 (m, 1H), 7.11–7.33 (m, 2H), 7.92 (dd, 2H, J1 = 16.0, J2 = 5.2 Hz). 13C NMR (CDCl3, 100 MHz): δ 22.3, 22.7, 24.4, 26.8, 29.2, 29.3, 29.6, 31.7, 31.9, 33.2, 36.4, 49.4, 52.1, 111.0, 114.9, 115.6, 117.8, 122.3, 124.4, 124.7, 126.5, 134.1, 134.6, 145.8, 151.3, 158.5, 162.4, 171.8. MS (ESI+) m/z: [M + H]+ calcd for C29H35ClN2O3, 495; found, 496.
2-Hydroxy-6-(8-((1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)benzoic Acid (14)
The title compound was obtained according to general procedure VI starting from 12. The crude product was purified by filtration. Compound 14 was obtained as a white solid. Yield: 84%. HPLC purity: 95%. 1H NMR (CD3OD, 400 MHz): δ 1.21–1.43 (m, 8H), 1.52 (s, 2H), 1.76–1.83 (m, 2H), 1.93 (s, 4H), 2.67 (s, 2H), 2.88–2.94 (m, 2H), 2.99 (s, 2H), 3.92 (t, 2H, J = 6.4 Hz), 6.62 (dd, 2H, J1 = 18.5, J2 = 7.7 Hz), 7.11 (t, 1H, J = 7.8 Hz), 7.55 (t, 1H, J = 7.6 Hz), 7.74 (d, 1H, J = 8.4 Hz), 7.81 (t, 1H, J = 7.2 Hz), 8.35 (d, 1H, J = 8.7 Hz). 13C NMR (CD3OD, 100 MHz): δ 20.4, 21.5, 23.4, 26.0, 27.8, 28.5, 28.7, 29.0, 29.9, 31.2, 31.5, 34.9, 111.4, 113.9, 118.6, 121.0, 124.8, 125.0, 132.6, 137.7, 148.2, 154.8, 159.8. HRMS (ESI+) m/z: [M + H]+ calcd for C28H34N2O3, 447.26422; found, 447.26436.
2-(8-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)-6-hydroxybenzoic Acid (15)
The title compound was obtained according to general procedure VI starting from 13. The crude product was purified by filtration. Compound 15 was obtained as a white solid. Yield: 54%. HPLC purity: 95%. 1H NMR (CDCl3, 400 MHz): δ 1.41–1.25 (m, 5H), 1.53 (s, 1H), 1.79 (dd, 1H, J1 = 14.1, J2 = 6.8 Hz), 1.92 (d, 3H, J = 2.8 Hz), 2.64 (s, 1H), 2.89–2.77 (m, 1H), 2.97 (s, 1H), 3.31–3.24 (m, 1H), 3.91 (t, 2H, J = 7.3 Hz), 6.66 (dd, 2H, J1 = 13.1, J2 = 8.0 Hz), 7.17 (t, 1H, J = 7.8 Hz), 7.52 (dd, 1H, J = 9.3, J2 = 1.9 Hz), 7.75 (d, 1H, J = 1.9 Hz), 8.35 (d, 1H, J = 9.3 Hz). 13C NMR (CDCl3, 100 MHz): δ 20.4, 21.9, 24.1, 25.9, 27.8, 27.8, 28.4, 28.6, 29.6, 30.7, 31.8, 36.2, 48.3, 110.6, 111.7, 115.3, 118.5, 122.2, 125.5, 126.6, 134.0, 138.3, 138.4, 147.1, 151.6, 155.3, 163.1, 173.2. HRMS (ESI+) m/z: [M + H]+ calcd for C28H33ClN2O33, 481.22525; found, 481.20619.
3-(8-((1,2,3,4-Tetrahydroacridin-9-yl)amino)octyl)phenol (16)
The title compound was obtained according to general procedure VII starting from 22. The crude product was purified by column chromatography (9.5:0.5:0.1 dichloromethane/methanol/aqueous 32% ammonia). Compound 16 was obtained as a sticky yellow-brown oil. Yield: 41%. HPLC purity: 95%. 1H NMR (CDCl3, 400 MHz): δ 1.22–1.45 (m, 10H), 1.67 (q, 2H, J = 6.8 Hz), 1.87 (m, 4H), 2.44 (t, 2H, J = 7.6 Hz), 2.66 (m, 2H), 3.10 (m, 2H), 3.62 (t, 2H, J = 6.8 Hz), 4.44 (br, NH), 6.62 (m, 2H), 6.73 (d, 1H, J = 8.0 Hz), 7.07 (t, 1H, J = 8.0 Hz), 7.34 (t, 1H, J = 8.0 Hz), 7.53 (t, 1H, J = 8.0 Hz), 8.00–8.06 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.3, 22.7, 24.4, 26.4, 28.5, 28.6, 29.0, 29.6, 30.6, 31.3, 32.3, 35.4, 48.9, 113.0, 114.4, 115.3, 119.1, 119.5, 123.1, 123.8, 126.8, 129.1, 129.1, 144.1, 152.0, 157.1, 157.1. MS (ESI+) m/z: [M + H]+ calcd for C27H34N2O, 402; found, 403.
3-(8-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)phenol (17)
The title compound was obtained according to general procedure VII starting from 23. The crude product was purified by column chromatography (7:2.5:0.5:0.05 petroleum ether/ethyl acetate/methanol/aqueous 32% ammonia). Compound 17 was obtained as a sticky yellow-brown oil. Yield: 45%. HPLC purity: 97%. 1H NMR (CDCl3, 400 MHz): δ 1.40–1.13 (m, 8H), 1.51 (m, 2H), 1.69 (m, 4H), 2.00–1.84 (m, 2H), 2.55–2.43 (m, 2H), 2.73–2.63 (m, 2H), 3.17–3.02 (m, 2H), 3.64–3.51 (m, 2H), 4.36–4.00 (m, 1H), 6.70 (m, 3H), 7.12 (m, 1H), 7.28 (m, 1H), 7.97 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.3, 22.7, 24.3, 26.4, 28.6, 28.7, 29.1, 30.7, 31.4, 32.8, 35.5, 49.1, 112.9, 114.7, 115.4, 117.6, 119.7, 124.3, 124.8, 126.1, 129.2, 134.7, 144.3, 146.8, 151.7, 157.1, 158.6.
Methyl 2-(8-Hydroxyoctyl)-6-methoxybenzoate (19a)
The title compound was obtained according to general procedure II starting from 18a. The crude product was purified by column chromatography (from 8:2 to 6.5:3.5 petroleum ether/ethyl acetate). Compound 19a was obtained as a light brown oil. Yield: 60%. 1H NMR (CDCl3, 500 MHz): δ 1.30 (m, 8H, 3–6), 1.51–1.56 (m, 4H), 1.86 (s, 1H), 2.52 (t, 2H, J = 7.0 Hz), 2.61 (t, 2H, J = 6.0 Hz), 3.80 (s, 3H), 3.90 (s, 3H), 6.75 (d, 1H, J = 8.2 Hz), 6.81 (d, 1H, J = 7.6 Hz), 7.26 (t, 1H, J = 8.0 Hz). 13C NMR (CDCl3, 125 MHz): δ 25.8, 29.4–29.5, 31.2, 32.8, 33.6, 52.3, 56.0, 63.1, 108.5, 121.6, 123.5, 130.4, 141.4, 156.4, 169.1.
3-Methoxyphenyloctan-1-ol (19b)
The title compound was obtained according to general procedure II starting from 18b. The crude product was purified by column chromatography (from 8:2 to 6.5:3.5 petroleum ether/ethyl acetate). Compound 19b was obtained as a light yellow oil. Yield: 70%. 1H NMR (CDCl3, 300 MHz): δ 1.33 (m, 8H), 1.54–1.58 (m, 2H), 1.60–1.63 (m, 2H), 2.59 (t, 2H, J = 6.0 Hz), 3.64 (t, 2H, J = 6.0 Hz), 3.81 (s, 3H), 6.72–6.79 (m, 3H), 7.18–7.21 (m, 1H). 13C NMR (CDCl3, 75 MHz): δ 25.9, 29.4–29.6, 31.5, 32.9, 36.2, 55.3, 63.2, 110.9, 114.4, 121.1, 129.3, 144.7, 159.7.
8-(3,5-Dimethoxyphenyl)octan-1-ol (19c)
The title compound was obtained according to general procedure II starting from 18c. The crude product was purified by column chromatography (from 8:2 to 6.5:3.5 petroleum ether/ethyl acetate). Compound 19c was obtained as a light brown oil. Yield: 60%. 1H NMR (CDCl3, 300 MHz): δ 1.32 (s, 8H), 1.53–1.60 (m, 4H), 2.07 (s, 1H), 2.54 (t, 2H, J = 7.7 Hz), 3.62 (t, 2H, J = 6.6 Hz), 3.78 (s, 6H), 6.30 (t, 1H, J = 2.2 Hz), 6.35 (d, 2H, J = 2.2 Hz). 13C NMR (CDCl3, 75 MHz): δ 25.8, 29.3–29.6, 31.3, 32.8, 36.4, 55.3, 63.1, 97.7, 106.6, 145.4, 160.8.
Synthesis of 8-(3-Hydroxyphenyl)octan-1-ol (19d)
Mixture of cardanol 2 (2 g, ∼6.6 mmol), distilled acetic anhydride (1.25 mL, 13.1 mmol), and phosphoric acid (4 drops) were heated in a conventional microwave oven for 3 min (3 × 1 min) at a power of 450 W (50%). Then, the mixture was extracted with ethyl acetate (3 × 10 mL) and the combined organic layers were washed with 5% sodium bicarbonate solution (10 mL), 10% hydrochloric acid solution (10 mL), and saturated saline (10 mL) and dried over anhydrous Na2SO4. After evaporating the solvent under reduced pressure, the reaction mixture was purified by silica gel column chromatography (dichloromethane, 100%), providing the acetylated mixture intermediates in 90% yield. The mixture of acetylated cardanols (2 g, ∼9.4 mmol) was solubilized in dichloromethane/methanol (1:1, 60 mL). The reaction system was cooled at 0 °C, and the solution was treated with ozone for approximately 3 h. After that, the excess of ozone was purged with the nitrogen flow and the solution was transferred to another flask. The mixture was dissolved in methanol/ethanol (1:1, 60 mL) and cooled at 0 °C under an ice–water bath, and sodium borohydride (2 g, 8.0 equiv) was added. The reaction was left warming to rt under vigorous stirring for 16 h. Then, the mixture was acidified to pH 3.0 with concentrated hydrochloric acid solution and the residue was extracted with ethyl acetate (3 × 20 mL). The combined organic fractions were washed with saturated sodium chloride solution (10 mL), dried over anhydrous Na2SO4, and evaporated. The product was purified by silica gel column chromatography (dichloromethane/ethanol, 0–5%), giving 19d as a light yellow oil. Yield: 67%. 1H NMR (CDCl3, 500 MHz): δ 1.30 (s, 8H), 1.56–1.58 (m, 4H), 2.53 (t, 2H, J = 7.5 Hz), 3.65 (t, 2H, J = 6.5 Hz), 4.11 (s, 2H), 6.66 (d, 1H, J = 8.3 Hz), 6.67 (s, 1H), 6.72 (d, 1H, J = 7.4 Hz), 7.12 (t, 1H, J = 7.6 Hz). 13C NMR (CDCl3, 125 MHz): δ 25.8, 29.2, 29.3, 29.4, 31.3, 32.7, 35.9, 63.3, 112.8, 115.6, 120.8, 129.5, 144.9, 156.0.
Synthesis of 8-(3-(Benzyloxy)phenyl)octan-1-ol (20)
Compound 19d (0.74 mmol, 1.0 equiv) and K2CO3 (1.11 mmol, 1.5 equiv) were stirred in acetone (7.40 mL). Benzyl bromide (0.74 mmol, 1.0 equiv) was added, and the reaction was stirred at reflux temperature for 12 h. The solvent was then removed in vacuo, and the residue was dissolved in ethyl acetate and washed with water. The organic layers were combined, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography (7.5:2:0.5 petroleum ether/ethyl acetate/methanol). Compound 20 was obtained as a colorless oil. Yield: 92%. 1H NMR (CDCl3, 400 MHz): δ 1.36 (s, 8H), 1.56–1.66 (m, 4H), 1.94 (s, OH), 2.62 (t, 2H, J = 7.6 Hz), 3.63 (t, 2H, J = 6.8 Hz), 5.07 (s, 2H), 6.87–6.82 (m, 3H), 7.22 (s, 1H, J = 8 Hz), 7.35–7.48 (m, 5H). 13C NMR (CDCl3, 100 MHz): δ 25.7, 29.2, 29.4, 29.5, 31.3, 32.7, 36.0, 62.9, 69.9, 111.7, 115.2, 121.2, 127.5, 127.9, 128.5, 129.2, 137.2, 144.6, 158.8.
Methyl 2-Methoxy-6-(8-((methylsulfonyl)oxy)octyl)benzoate (21a)
The title compound was obtained according to general procedure III starting from 19a. The crude product was purified by column chromatography (100% dichloromethane). Compound 21a was obtained as a colorless oil. Yield: 85%. 1H NMR (CDCl3, 500 MHz): δ 1.30 (m, 6H), 1.38 (m, 2H), 1.57 (m, 2H), 1.70–1.76 (m, 2H), 2.53 (t, 2H, J = 7.8 Hz), 3.00 (s, 3H), 3.81 (s, 3H), 3.90 (s, 3H), 4.21 (t, 2H, J = 6.0 Hz), 6.76 (d, 1H, J = 8.3 Hz), 6.81 (d, 1H, J = 7.7 Hz), 7.27 (t, 1H, J = 8.0 Hz). 13C NMR (CDCl3, 125 MHz): δ 25.5, 29.0, 29.2–29.4, 31.2, 33.6, 37.5, 52.3, 56.0, 70.3, 108.5, 121.6, 123.6, 130.4, 141.3, 156.4, 169.1.
8-(3-Methoxyphenyl)octyl Methanesulfonate (21b)
The title compound was obtained according to general procedure III starting from 19b. The crude product was purified by column chromatography (100% dichloromethane). Compound 21b was obtained as a light brown oil. Yield: 60%. 1H NMR (CDCl3, 300 MHz): δ 1.32–1.42 (m, 8H), 1.56–1.63 (m, 2H), 1.79–1.89 (m, 2H), 2.57 (t, 2H, J = 6.0 Hz), 2.98 (s, 3H), 3.40 (t, 2H, J = 6.0 Hz), 3.79 (s, 3H), 6.71–6.78 (m, 3H), 7.20 (dd, 1H, J = 9.0 Hz). 13C NMR (CDCl3, 75 MHz): δ 28.2, 28.7–29.3, 31.3, 32.8, 34.1, 36.0, 37.5, 55.1, 110.8, 114.2, 120.8, 129.2, 144.5, 159.5.
8-(3,5-Dimethoxyphenyl)octyl Methanesulfonate (21c)
The title compound was obtained according to general procedure III starting from 19c. The crude product was purified by column chromatography (100% dichloromethane). Compound 21c was obtained as a light brown oil. Yield: 80%. 1H NMR (CDCl3, 300 MHz): δ 1.33–1.44 (s, 8H), 1.60 (t, 2H, J = 6.5 Hz), 1.69–1.78 (m, 2H), 2.54 (t, 2H, J = 7.6 Hz), 2.99 (s, 3H), 3.78 (s, 6H), 4.21 (t, 2H, J = 6.5 Hz), 6.30 (t, 1H, J = 2.2 Hz), 6.34 (d, 2H, J = 2.2 Hz). 13C NMR (CDCl3, 75 MHz): δ 25.5, 29.0, 29.2–29.4, 31.3, 36.3, 37.4, 55.3, 70.3, 97.7, 106.6, 145.3, 160.8.
8-(3-(Benzyloxy)phenyl)octyl Methyl Sulfate (21d)
The title compound was obtained according to general procedure III starting from 20. The crude product was purified by column chromatography (6.5:3.5 dichloromethane/petroleum ether). Compound 21d was obtained as a colorless oil. Yield: 87%. 1H NMR (CDCl3, 400 MHz): δ 1.35–1.41 (m, 8H), 1.64–1.76 (m, 4H), 2.61 (t, 2H, J = 7.6 Hz), 2.96 (s, 3H), 4.21 (t, 2H, J = 6.0 Hz), 5.06 (s, 2H), 6.81–6.85 (m, 3H), 7.22 (t, 1H, J = 8.0 Hz), 7.34–7.47 (m, 5H). 13C NMR (CDCl3, 100 MHz): δ 25.4, 28.9, 29.1, 29.1, 29.29, 31.2, 35.9, 37.2, 69.8, 70.2, 111.7, 115.2, 121.1, 127.5, 127.9, 128.5, 129.2, 137.2, 144.5, 158.8.
N-(8-(3-(Benzyloxy)phenyl)octyl)-1,2,3,4-tetrahydroacridin-9-amine (22)
The title compound was obtained according to general procedure IV using 4a and 21d. The crude product was purified by column chromatography (6.5:3:0.5 petroleum ether/ethyl acetate/NEt3). Compound 22 was obtained as a sticky yellow-brown oil. Yield: 32%. 1H NMR (CDCl3, 400 MHz): δ 1.30–1.39 (m, 8H), 1.56–1.89 (m, 4H), 1.89 (m, 4H), 2.55 (t, 2H, J = 7.6 Hz), 2.62 (m, 2H), 3.14 (m, 2H), 3.61 (t, 2H, J = 7.2 Hz), 5.02 (s, 2H), 6.75–6.79 (m, 3H), 7.17 (t, 1H, J = 7.6 Hz), 7.30–7.42 (m, 6H), 7.53 (t, 1H, J = 7.2 Hz), 8.00 (d, 1H, J = 8.4 Hz), 8.12 (d, 1H, J = 8.00 Hz). 13C NMR (CDCl3, 100 MHz): δ 22.7, 23.0, 24.7, 26.9, 29.1, 29.2, 29.3, 31.2, 31.7, 33.8, 35.9, 49.5, 69.8, 111.6, 115.1, 115.6, 120.0, 121.1, 122.9, 123.5, 127.5, 127.8, 128.3, 128.5, 129.1, 137.1, 144.4, 147.2, 150.9, 158.2, 158.8.
N-(8-(3-(Benzyloxy)phenyl)octyl)-6-chloro-1,2,3,4-tetrahydroacridin-9-amine (23)
The title compound was obtained according to general procedure IV using 4b and 21d. The crude product was purified by column chromatography (7:2:0.5 petroleum ether/ethyl acetate/NEt3). Compound 23 was obtained as a sticky yellow-brown oil. Yield: 24%. HPLC purity: 95%. 1H NMR (CDCl3, 400 MHz): δ 1.36–1.24 (m, 8H), 1.64–1.59 (m, 4H), 1.91–1.88 (t, 4H, J = 8 Hz), 2.56 (t, 2H, J = 8 Hz), 2.63 (t, 2H, J = 8 Hz), 3.02 (t, 2H, J = 8 Hz), 3.47 (t, 2H, J = 8164 Hz), 5.03 (s, 2H), 6.81–6.76 (m, 3H), 7.43–7.15 (m, 8H), 7.90–7.87 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 22.5, 22.8, 24.4, 26.8, 29.1, 29.2, 29.3, 31.2, 31.7, 33.8, 35.9, 49.5, 69.8, 111.6, 115.2, 121.1, 124.2, 124.6, 127.4, 127.8, 128.5, 129.1, 137.1, 144.4, 151.1, 158.8.
Crystallization of Human BChE in Complex with 5
Recombinant 4sugOff/L530Stop human BChE (hBChE) was produced in Chinese hamster ovary cells80 and purified by hBChE specific affinity chromatography (Hupresin; CHEMFORASE, Rouen, France) and size exclusion chromatography (Superdex 200, GE Healthcare) as previously described.81 The stock solution of compound 5 (100 mM) was prepared in MeOH. Crystallization was carried out at 293 K with the hanging drop vapor diffusion method using a 6.5 mg/mL hBChE solution and a mother liquor containing 1 mM ligand, of composition 1% MeOH, and 2.15 M (NH4)2SO4 in 100 mM 2-(N-morpholino)ethanesulfonic acid (MES) (pH 6.5) buffer. Large crystals (200 to 400 μM) grew in about a week. Crystals were cryoprotected in a solution of 0.1 M MES (pH 6.5), 2.15 M (NH4)2SO4, 20% glycerol, 1 mM ligand, and 1% MeOH before flash cooling into liquid nitrogen.
Structure Determination of Human BChE in Complex with 5
X-ray diffraction data were collected at synchrotron SOLEIL (Saint Aubin, France) at the PROXIMA-2 beamline at 100 K. Images from two isomorphous crystals of equal quality recorded on an EIGER 16M detector were processed with the XDS suite of software.82 The structure resolution and refinement were realized using the PHENIX software suite.83 An initial model was obtained by molecular replacement using Phaser-MR84 included in PHENIX and the hBChE structure (PDB entry: 1p0i) devoid of any ligands, glycans, or water molecules. Extra electron density was observed close to the active-site gorge that allowed modeling of compound 5. Ligand geometry restraints were calculated using PHENIX eLBOW85 included in PHENIX and the semiempirical quantum mechanical method (AM1). The model was refined by iterative cycles of phenix.refine and model building using Coot.86 Coordinates and structure factors of the hBChE–5 complex are deposited into the Protein Data Bank under accession code 7bgc. Data collection and refinement statistics, as calculated using PHENIX, are shown in Table S2. The protein structures were illustrated using the program PyMOL (Schrodinger LLC).
Biology
Inhibition of Human AChE and BChE Activities
The method of Ellman et al. was followed.38 AChE stock solution was prepared by dissolving human recombinant AChE lyophilized powder (Sigma, Italy) in 100 mM phosphate buffer (pH = 8.0) containing 0.1% Triton X-100. Stock solution of BChE from human serum (Sigma, Italy) was prepared by dissolving the lyophilized powder in an aqueous solution of 0.1% gelatine. Stock solutions of tested compounds (1 mM) were prepared in methanol and diluted in methanol. The assay solution consisted of a 0.1 M phosphate buffer (pH 8.0), with the addition of 340 μM 5,5′-dithio-bis(2-nitrobenzoic acid), 0.02 units of hAChE or hBChE, and 550 μM substrate (acetylthiocoline iodide or butyrylthiocholine iodide, respectively). Assays were done with a blank containing all components except the enzyme to account for nonenzymatic substrate hydrolysis. Tested tacrine hybrids were added to the assay solution and preincubated with the enzyme for 20 min before the addition of the substrate. Initial rate assays were performed at 37 °C with a JASCO V-530 double-beam spectrophotometer (JASCO Europe, Italy) equipped with a thermostated cuvette holder (37 °C). The absorbance value at 412 nm was recorded for 240 s, and enzyme activity was calculated from the slope of the obtained linear trend. The reaction rates obtained in the presence and in the absence of the tested compound were compared, and the percent inhibition was calculated. Five different concentrations of each compound were used to obtain inhibition of enzyme activity between 20 and 80%. IC50 values were determined graphically from log concentration–inhibition curves (GraphPad Prism 4.03 software, GraphPad Software Inc.). Each final value is the mean of at least two independent experiments each performed in triplicate.
Determination of Hepatotoxicity of 5–17 on HepG2 Cells
HepG2 cells (human hepatocytes from liver carcinoma, American Type Culture Collection, ATCC) were grown in the DMEM supplemented with 10% FBS and 50 units/mL penicillin/streptomycin (Life Technologies Italia, Monza, MB, Italy) at 37 °C in a humidified atmosphere containing 5% CO2. For the experiments, cells (0.3 × 105 cells/well) were seeded in a 96-well plate in a complete medium; after 24 h, the medium was removed, and cells were exposed to the increasing concentrations of compounds 5–17, reference compounds (0.1, 1, and 10 μM), or vehicle and dissolved in the complete DMEM for 24 h. Cell viability was measured by the MTT assay.
Determination of Toxicity and Activity Profiles of Selected Compounds on SH-SY5Y and BV-2 Cells
Chemicals and Reagents
High-glucose Dulbecco’s modified Eagle’s medium (DMEM), l-glutamine solution, penicillin/streptomycin, trypsin–EDTA solution, phosphate-buffered saline (PBS), LPS from Escherichia coli serotype O127:B8, all-trans-retinoic acid (RA), dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), and primers for RT-PCR were purchased from Sigma-Aldrich–Merck (Milan, Italy). Fetal bovine serum (FBS) and low-endotoxin FBS were purchased from Euroclone (Milan, Italy).
Cell Cultures and Treatments
SH-SY5Y cell line was purchased from Sigma-Aldrich–Merck (ECACC 94030304) (Milan, Italy) and was grown in a high-glucose DMEM supplemented with 10% (v/v) FBS, 2 mM l-glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin, as previously reported.87 Before starting the experiments, cells were differentiated with all-trans-retinoic acid (10 μM) for 7 days. Differentiated SH-SY5Y cells were treated for 24 h with various concentrations of the tested compounds. BV-2 murine microglial cells were kindly provided by Prof. Elisabetta Blasi (University of Modena and Reggio Emilia, Modena, Italy) and were grown in a high-glucose DMEM supplemented with 10% (v/v) low-endotoxin FBS, 2 mM l-glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin. BV-2 cells were treated for 24 h with various concentrations of the tested compounds and then exposed to LPS (100 ng/mL) for further 24 h.
MTT Viability Assay
Cell viability was evaluated by measuring MTT reduction as reported in ref (88). The cells were seeded in 96-well tissue culture plates, and at the end of treatments, cells were incubated with 0.5 mg/mL MTT solution for 30 min (BV-2 cells) or 90 min (SH-SY5Y cells). At the end, the MTT solution was replaced with DMSO in order to solubilize the formed formazan crystals. Finally, formazan formation was measured spectrophotometrically at 595 nm using a microplate spectrophotometer (VICTOR3 V Multilabel Counter; PerkinElmer, Wellesley, MA, USA). Data are expressed as a percentage of control cells, which are considered as 100% cell viability.
RNA Extraction
The extraction of total RNA was conducted using an RNeasy Mini Kit (QIAGEN GmbH, Hilden, Germany), following the manufacturer’s protocol. The yield and purity of the RNA were measured using a NanoVue spectrophotometer (GE Healthcare, Milan, Italy).
Real-Time Polymerase Chain Reaction (PCR)
One microgram of total RNA was reverse-transcribed to cDNA using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA), following the manufacturer’s instructions. The real-time PCR was carried out in a total volume of 10 μL containing 2.5 μL (12.5 ng) of cDNA, 5 μL of SsoAdvanced Universal SYBR Green Supermix (Bio-Rad), and 0.5 μL (500 nM) of each primer. IL-1β, TNF-α, iNOS, and COX-2 (Sigma-Aldrich–Merck, Milan, Italy) expression levels were evaluated, while as the reference gene for BV-2 cells was used GAPDH. The primer sequences are reported in Table S3. The following protocol was followed to amplify the cDNA: 30 s at 95 °C (to activate the polymerase) followed by 5 s at 95 °C and 30 s at 60 °C for 40 cycles. Normalized expression levels were evaluated in respect to control cells according to the 2–ΔΔCT method.
Immunofluorescence Confocal Microscopy
BV-2 cells were seeded directly on glass coverslips in 6-well plates. At the end of treatments, cells were fixed at room temperature with paraformaldehyde 2% for 15 min and then permeabilized with Triton X-100 0.1% for 10 min. Subsequently, BV-2 cells were incubated overnight with a polyclonal antibody (1:500) against NF-κB p65. After PBS extensive washing, cells were exposed, for 1 h at room temperature, to a secondary Alexa Fluor 488-conjugated antirabbit IgG antibody (1:1000). Nuclei were stained with 1 μg/mL 4′-6-diamidino-2-phenylindole (DAPI). Slides were analyzed with a C2 Plus confocal laser scanning microscope (Nikon Instruments, Firenze, Italy). Images were processed using NIS-Elements imaging software (Nikon Instruments, Firenze, Italy).
IL-1β Quantification
BV-2 cells were seeded in 6-well plates, and at the end of treatments, the culture media were taken to detect IL-1β concentration. IL-1β quantification was performed using an IL-1β ELISA Kit following the manufacturer’s instructions (Sigma-Aldrich–Merck). Absorbance (450 nm) was measured using a microplate spectrophotometer (VICTOR3 V Multilabel Counter).
Statistical Analysis
All the analyses were carried out at least in triplicate, and data were expressed as mean ± standard error. To compare differences among groups, one-way ANOVA followed by Dunnett’s or Tukey’s test (Prism 7; GraphPad Software, San Diego, CA) was used. Differences at the level of p < 0.05 were considered statistically significant.
PAMPA Assay
The filter membrane of the donor plate was coated with PBL (polar brain lipid, Avanti, AL, USA) in dodecane (4 μL of 20 mg/mL PBL in dodecane), and the acceptor well was filled with 300 μL of PBS (pH 7.4) buffer (VA). Tested compounds were dissolved first in DMSO and diluted with PBS (pH 7.4) to reach the final concentration in the donor well (40–100 μM). Concentration of DMSO did not exceed 0.5% (v/v) in the donor solution. Three hundred microliters of the donor solution was added to the donor wells (VD), and the donor filter plate was carefully put on the acceptor plate so that the coated membrane was “in touch” with both donor solution and acceptor buffer. The test compound diffused from the donor well through the lipid membrane (area = 0.28 cm2) to the acceptor well. The concentrations of the drug in both donor and acceptor wells were assessed after 3, 4, 5, and 6 h of incubation in quadruplicate using the UV plate reader Synergy HT (Biotek, Winooski, VT, USA) at the maximum absorption wavelength of each compound. In addition to that, solution of theoretical compound concentration, simulating the equilibrium state established if the membrane was ideally permeable, was prepared and assessed as well. Concentrations of the compounds in the donor and acceptor wells and equilibrium concentration were calculated from the standard curve and expressed as the permeability (Pe) according to the equation89
where 
Plasma Stability Assay
Stability of 5 in plasma was assessed by incubating the compound over a 6 h time frame at 37 °C and analyzing its concentration by high-performance liquid chromatography (HPLC) coupled with mass spectrometry (MS). Pooled human plasma was purchased from VWR (Stříbrná Skalice, Czech Republic). Stock solution of 5 in DMSO was diluted with plasma to reach a 10 μM final concentration of 5. Two hundred microliters of 10 μM plasma solution was pipetted to mictrotubes. The first sample was taken to be extracted, while the others were incubated at 37 °C. Every hour, one sample was taken and extracted. Ten microliters of internal standard (IS; 200 μM tacrine-trolox hybrid described by Nepovimova et al. in methanol)90 was added to every sample, vortexed, and supplemented by 700 μL of LC–MS grade acetonitrile (VWR, Stříbrná Skalice, Czech Republic). Then, the sample was shaken (3 min, level 7, VM-10 Witeg, Wertheim, Germany) and centrifuged (10,000 RPM, 2 min, Roth Gusto, ROTILABO, Illinois, USA) and 700 μL of supernatant was transferred to the vial and analyzed by HPLC–MS. Calibration samples were prepared by adding 10 μL of 5 (20–400 μM in DMSO) to 190 μL of blank plasma; final concentrations were in a range of 1–20 μM. Then, 10 μL of IS was added and samples were extracted as above and analyzed by HPLC–MS.
HPLC–MS analysis was performed using Dionex UltiMate 3000 UHPLC consisting of an RS LPG quaternary pump, RS column compartment, RS autosampler, and diode array detector controlled by Chromeleon (version 7.2.9 build 11323) software (Thermo Fisher Scientific, Germering, Germany) with a Q Exactive Plus Orbitrap mass spectrometer with Thermo Xcalibur (version 3.1.66.10.) software (Thermo Fisher Scientific, Bremen, Germany). Detection was performed by mass spectrometry in positive mode. Settings of the heated electrospray source were as follows: spray voltage, 3.5 kV; capillary temperature, 300 °C; sheath gas, 55 arbitrary units; auxiliary gas, 15 arbitrary units; spare gas, 3 arbitrary units; probe heater temperature, 250 °C; max spray current, 100 μA; and S-lens RF level: 50. The concentration assessment of 5 was performed in reverse-phase gradient mode using a Kinetex EVO C18 column (2.1 × 50 mm, 1.7 μm, Phenomenex, Torrance, California, USA) with a Kinetex SecurityGuard Ultra C18 guard column (2.1 mm, Phenomenex, Torrance, California, USA). Mobile phase A was ultrapure water of ASTM I type (resistance, 18.2 MΩ cm at 25 °C) prepared by Barnstead Smart2Pure 3 UV/UF apparatus (Thermo Fisher Scientific, Bremen, Germany) with 0.1% (v/v) formic acid (LC–MS grade, VWR, Stříbrná Skalice, Czech Republic); mobile phase B was acetonitrile (LC–MS grade, VWR, Stříbrná Skalice, Czech Republic) with 0.1% (v/v) of formic acid. The column was tempered to 35 °C, mobile phase flow was set to 0.5 mL/min, and injection volume was 5 μL. The method started with 5% B and was steady for 0.3 min, and then the gradient went from 5 to 100% B in 3 min, was kept at 100% B for 0.5 min, then went back to 5% B, and equilibrated for 2.2 min. Total runtime of the method was 6 min. The compound and IS were detected with a mass spectrometer in the total ion current scan in a range of 105–700 m/z in positive mode. Retention time for 5 was 3.32 min with mass searched 475.2952, and retention time for IS was 3.29 min with mass searched 592.3297. Calibration had five points (1, 5, 10, 15, and 20 μM) and was linear along the entire range.
Acknowledgments
The authors acknowledge “Multi-target paradigm for innovative ligand identification in the drug discovery process (MuTaLig)” COST Action CM15135. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001 (L.C.N. Ph. D. fellow 2018–2021). L.A.S.R. is a fellow holder for Productivity in Technological Development and Innovative Extension - DT (CNPq 312709/2017-0). The authors would like to thank the SOLEIL synchrotron for long-term beamtime access 20181022 (IBS BAG). The authors also thank Stefano Perna and Carlo Faggiotto for the technical assistance with synthesis of the compounds.
Glossary
Abbreviations
- Aβ
amyloid β
- AChE
acetylcholinesterase
- AChEI
acetylcholinesterase inhibitor
- AD
Alzheimer’s disease
- BChE
butyrylcholinesterase
- CNS
central nervous system
- CNSL
cashew nutshell liquid
- hBChE
human butyrylcholinesterase
- HDAC
histone deacetylase
- IL-1β
interleukin-1β
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MTDL
multi-target-directed ligand
- MW
microwave
- NF-κB
nuclear factor kappa B
- PAMPA
parallel artificial membrane permeability assay
- Pe
effective permeability
- RT-PCR
real-time polymerase chain reaction
- SAR
structure–activity relationships
- TLR4
Toll-like receptor 4
- TNF-α
tumor necrosis factor-α
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00048.
Author Contributions
○ M.R., M.F., and L.d.C.N. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the University of Bologna (grant RFO 2017 and 2018), by National Council for Scientific and Technological Development (grant CNPq 401864/2013-8), and by Czech Science Foundation (project no. 20-12047S and student project no. SV/FVZ 202010). M.R. acknowledges the “Piano triennale Alte competenze per la ricerca, il trasferimento tecnologico e l’imprenditorialità” Emilia Romagna regional Ph.D. program (Ph.D. fellow 2016–2019). F.N. was funded by the Direction Générale de l’Armement (DGA) and Service de Santé des Armées (SSA) of the French Ministry of Armed Forces, under grant number NBC-5-C-4210.
The authors declare no competing financial interest.
Notes
The structural data of the hBChE–5 complex were deposited in the Protein Data Bank under accession number 7bgc. Authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Alzheimer’s Disease International . https://www.alzint.org/about/dementia-facts-figures/dementia-statistics (accessed Jan 8, 2021).
- Cummings J.; Aisen P. S.; DuBois B.; Frölich L.; Jack C. R. Jr.; Jones R. W.; Morris J. C.; Raskin J.; Dowsett S. A.; Scheltens P. Drug development in Alzheimer’s disease: the path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. 10.1186/s13195-016-0207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J.; Lee G.; Ritter A.; Sabbagh M.; Zhong K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dementia 2020, 6, e12050 10.1002/trc2.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J. L. Translational scoring of candidate treatments for Alzheimer’s Disease: a systematic approach. Dementia Geriatr. Cognit. Disord. 2020, 49, 22–37. 10.1159/000507569. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Barabási A.-L.; Gulbahce N.; Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith C. T.; Borisy A. A.; Stockwell B. R. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discovery 2005, 4, 71–78. 10.1038/nrd1609. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- Cavalli A.; Bolognesi M. L.; Minarini A.; Rosini M.; Tumiatti V.; Recanatini M.; Melchiorre C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- Danon J. J.; Reekie T. A.; Kassiou M. Challenges and Opportunities in Central Nervous System Drug Discovery. Trends Chem. 2019, 1, 612–624. 10.1016/j.trechm.2019.04.009. [DOI] [Google Scholar]
- Zhou J.; Jiang X.; He S.; Jiang H.; Feng F.; Liu W.; Qu W.; Sun H. Rational design of multitarget-directed ligands: strategies and emerging paradigms. J. Med. Chem. 2019, 62, 8881–8914. 10.1021/acs.jmedchem.9b00017. [DOI] [PubMed] [Google Scholar]
- Alarcón-Espósito J.; Mallea M.; Rodríguez-Lavado J. From hybrids to new scaffolds: the latest medicinal chemistry goals in multi-target directed ligands for Alzheimer’s disease. Curr. Neuropharmacol. 2020, 10.2174/1570159X18666200914155951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemes L. F. N.; de Andrade Ramos G.; de Oliveira A. S.; da Silva F. M. R.; de Castro Couto G.; da Silva Boni M.; Guimarães M. J. R.; Souza I. N. O.; Bartolini M.; Andrisano V.; do Nascimento Nogueira P. C.; Silveira E. R.; Brand G. D.; Soukup O.; Korábečný J.; Romeiro N. C.; Castro N. G.; Bolognesi M. L.; Romeiro L. A. S. Cardanol-derived AChE inhibitors: towards the development of dual binding derivatives for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 108, 687–700. 10.1016/j.ejmech.2015.12.024. [DOI] [PubMed] [Google Scholar]
- Soares Romeiro L. A.; da Costa Nunes J. L.; de Oliveira Miranda C.; Simões Heyn Roth Cardoso G.; de Oliveira A. S.; Gandini A.; Kobrlova T.; Soukup O.; Rossi M.; Senger J.; Jung M.; Gervasoni S.; Vistoli G.; Petralla S.; Massenzio F.; Monti B.; Bolognesi M. L. Novel sustainable-by-design HDAC inhibitors for the treatment of Alzheimer’s Disease. ACS Med. Chem. Lett. 2019, 10, 671–676. 10.1021/acsmedchemlett.9b00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Das J.; Braje W. M.; Dash A. K.; Handa S. A Glimpse into Green Chemistry Practices in the Pharmaceutical Industry. ChemSusChem 2020, 13, 2806–2806. 10.1002/cssc.202001204. [DOI] [PubMed] [Google Scholar]
- Brun N.; Hesemann P.; Esposito D. Expanding the biomass derived chemical space. Chem. Sci. 2017, 8, 4724–4738. 10.1039/C7SC00936D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi M. L.Chapter Six - Sustainable anti-trypanosomatid drugs: An aspirational goal for medicinal chemistry. In Annual Reports in Medicinal Chemistry, Chibale K., Ed., Academic Press; 2019; Vol. 52, pp. 153–176.
- Decker M.Design of hybrid molecules for drug development, Elsevier; 2017. [Google Scholar]
- Carlier P. R.; Chow E. S.; Han Y.; Liu J.; El Yazal J.; Pang Y. P. Heterodimeric tacrine-based acetylcholinesterase inhibitors: investigating ligand-peripheral site interactions. J. Med. Chem. 1999, 42, 4225–4231. 10.1021/jm990224w. [DOI] [PubMed] [Google Scholar]
- Milelli A.; De Simone A.; Ticchi N.; Chen H. H.; Betari N.; Andrisano V.; Tumiatti V. Tacrine-based multifunctional agents in Alzheimer’s disease: an old story in continuous development. Curr. Med. Chem. 2017, 24, 3522–3546. 10.2174/0929867324666170309123920. [DOI] [PubMed] [Google Scholar]
- Hemshekhar M.; Sebastin Santhosh M.; Kemparaju K.; Girish K. S. Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin. Pharmacol. Toxicol. 2012, 110, 122–132. 10.1111/j.1742-7843.2011.00833.x. [DOI] [PubMed] [Google Scholar]
- Lomonaco D.; Pinheiro Santiago G. M.; Ferreira Y. S.; Campos Arriaga Â. M.; Mazzetto S. E.; Mele G.; Vasapollo G. Study of technical CNSL and its main components as new green larvicides. Green Chem. 2009, 11, 31–33. 10.1039/B811504D. [DOI] [Google Scholar]
- de Souza M. Q.; Teotônio I. M. S. N.; de Almeida F. C.; Heyn G. S.; Alves P. S.; Romeiro L. A. S.; Pratesi R.; de Medeiros Nóbrega Y. K.; Pratesi C. B. Molecular evaluation of anti-inflammatory activity of phenolic lipid extracted from cashew nut shell liquid (CNSL). BMC Complementary Altern. Med. 2018, 18, 181. 10.1186/s12906-018-2247-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T.; Carson M. J.; El Khoury J.; Landreth G. E.; Brosseron F.; Feinstein D. L.; Jacobs A. H.; Wyss-Coray T.; Vitorica J.; Ransohoff R. M.; Herrup K.; Frautschy S. A.; Finsen B.; Brown G. C.; Verkhratsky A.; Yamanaka K.; Koistinaho J.; Latz E.; Halle A.; Petzold G. C.; Town T.; Morgan D.; Shinohara M. L.; Perry V. H.; Holmes C.; Bazan N. G.; Brooks D. J.; Hunot S.; Joseph B.; Deigendesch N.; Garaschuk O.; Boddeke E.; Dinarello C. A.; Breitner J. C.; Cole G. M.; Golenbock D. T.; Kummer M. P. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 2015, 14, 388–405. 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W.-Y.; Wang X.; Ip N. Y. Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: mechanisms, drug candidates, and new opportunities. ACS Chem. Neurosci. 2019, 10, 872–879. 10.1021/acschemneuro.8b00402. [DOI] [PubMed] [Google Scholar]
- Reale M.; Di Nicola M.; Velluto L.; D’Angelo C.; Costantini E.; Lahiri D. K.; Kamal M. A.; Yu Q.-s.; Greig N. H. Selective acetyl- and butyrylcholinesterase inhibitors reduce amyloid-β ex vivo activation of peripheral chemo-cytokines from Alzheimer’s disease subjects: exploring the cholinergic anti-inflammatory pathway. Curr. Alzheimer Res. 2014, 11, 608–622. 10.2174/1567205010666131212113218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi M. L. Harnessing polypharmacology with medicinal chemistry. ACS Med. Chem. Lett. 2019, 10, 273–275. 10.1021/acsmedchemlett.9b00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero A.; Cacabelos R.; Oset-Gasque M. J.; Samadi A.; Marco-Contelles J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013, 23, 1916–1922. 10.1016/j.bmcl.2013.02.017. [DOI] [PubMed] [Google Scholar]
- Sameem B.; Saeedi M.; Mahdavi M.; Shafiee A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 128, 332–345. 10.1016/j.ejmech.2016.10.060. [DOI] [PubMed] [Google Scholar]
- Mesiti F.; Chavarria D.; Gaspar A.; Alcaro S.; Borges F. The chemistry toolbox of multitarget-directed ligands for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 181, 111572. 10.1016/j.ejmech.2019.111572. [DOI] [PubMed] [Google Scholar]
- Lane R. M.; Potkin S. G.; Enz A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. 10.1017/S1461145705005833. [DOI] [PubMed] [Google Scholar]
- Lin H.; Li Q.; Gu K.; Zhu J.; Jiang X.; Chen Y.; Sun H. Therapeutic agents in Alzheimer’s disease through a multi-target-directed ligands strategy: recent progress based on tacrine core. Curr. Top. Med. Chem. 2017, 17, 3000–3016. 10.2174/1568026617666170717114944. [DOI] [PubMed] [Google Scholar]
- Mollabagher H.; Taheri S.; Mojtahedi M. M.; Seyedmousavi S. Cu-metal organic frameworks (Cu-MOF) as an environment-friendly and economical catalyst for one pot synthesis of tacrine derivatives. RSC Adv. 2020, 10, 1995–2003. 10.1039/C9RA10111J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajouhesh H.; Lenz G. R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S.-S.; Wang X.-B.; Li J.-Y.; Yang L.; Kong L.-Y. Design, synthesis and evaluation of novel tacrine-coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer’s disease. Eur. J. Med. Chem. 2013, 64, 540–553. 10.1016/j.ejmech.2013.03.051. [DOI] [PubMed] [Google Scholar]
- Prat D.; Wells A.; Hayler J.; Sneddon H.; McElroy C. R.; Abou-Shehada S.; Dunn P. J. CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 2016, 18, 288–296. 10.1039/C5GC01008J. [DOI] [Google Scholar]
- Fanelli F.; Parisi G.; Degennaro L.; Luisi R. Contribution of microreactor technology and flow chemistry to the development of green and sustainable synthesis. Beilstein J. Org. Chem. 2017, 13, 520–542. 10.3762/bjoc.13.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman G. L.; Courtney K. D.; Andres V.; Featherstone R. M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–90. 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Savini L.; Gaeta A.; Fattorusso C.; Catalanotti B.; Campiani G.; Chiasserini L.; Pellerano C.; Novellino E.; McKissic D.; Saxena A. Specific targeting of acetylcholinesterase and butyrylcholinesterase recognition sites. Rational design of novel, selective, and highly potent cholinesterase inhibitors. J. Med. Chem. 2003, 46, 1–4. 10.1021/jm0255668. [DOI] [PubMed] [Google Scholar]
- Soukup O.; Jun D.; Zdarova-Karasova J.; Patocka J.; Musilek K.; Korabecny J.; Krusek J.; Kaniakova M.; Sepsova V.; Mandikova J.; Trejtnar F.; Pohanka M.; Drtinova L.; Pavlik M.; Tobin G.; Kuca K. A resurrection of 7-MEOTA: a comparison with tacrine. Curr. Alzheimer Res. 2013, 10, 893–906. 10.2174/1567205011310080011. [DOI] [PubMed] [Google Scholar]
- Elsinghorst P. W.; González Tanarro C. M.; Gütschow M. Novel heterobivalent tacrine derivatives as cholinesterase inhibitors with notable selectivity toward butyrylcholinesterase. J. Med. Chem. 2006, 49, 7540–7544. 10.1021/jm060742o. [DOI] [PubMed] [Google Scholar]
- Greig N. H.; Lahiri D. K.; Sambamurti K. Butyrylcholinesterase: an important new target in Alzheimer’s disease therapy. Int Psychogeriatr 2002, 14, 77–91. 10.1017/S1041610203008676. [DOI] [PubMed] [Google Scholar]
- Darreh-Shori T.; Vijayaraghavan S.; Aeinehband S.; Piehl F.; Lindblom R. P. F.; Nilsson B.; Ekdahl K. N.; Långström B.; Almkvist O.; Nordberg A. Functional variability in butyrylcholinesterase activity regulates intrathecal cytokine and astroglial biomarker profiles in patients with Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2465–2481. 10.1016/j.neurobiolaging.2013.04.027. [DOI] [PubMed] [Google Scholar]
- Xing S.; Li Q.; Xiong B.; Chen Y.; Feng F.; Liu W.; Sun H. Structure and therapeutic uses of butyrylcholinesterase: Application in detoxification, Alzheimer’s disease, and fat metabolism. Med. Res. Rev. 2021, 41, 858–901. 10.1002/med.21745. [DOI] [PubMed] [Google Scholar]
- Dighe S. N.; Deora G. S.; De la Mora E.; Nachon F.; Chan S.; Parat M.-O.; Brazzolotto X.; Ross B. P. Discovery and Structure–Activity Relationships of a Highly Selective Butyrylcholinesterase Inhibitor by Structure-Based Virtual Screening. J. Med. Chem. 2016, 59, 7683–7689. 10.1021/acs.jmedchem.6b00356. [DOI] [PubMed] [Google Scholar]
- Košak U.; Brus B.; Knez D.; Žakelj S.; Trontelj J.; Pišlar A.; Šink R.; Jukič M.; Živin M.; Podkowa A.; Nachon F.; Brazzolotto X.; Stojan J.; Kos J.; Coquelle N.; Sałat K.; Colletier J.-P.; Gobec S. The magic of crystal structure-based inhibitor optimization: development of a butyrylcholinesterase inhibitor with picomolar affinity and in vivo activity. J. Med. Chem. 2018, 61, 119–139. 10.1021/acs.jmedchem.7b01086. [DOI] [PubMed] [Google Scholar]
- Hoffmann M.; Stiller C.; Endres E.; Scheiner M.; Gunesch S.; Sotriffer C.; Maurice T.; Decker M. Highly selective butyrylcholinesterase inhibitors with tunable duration of action by chemical modification of transferable carbamate units exhibit pronounced neuroprotective effect in an Alzheimer’s disease mouse model. J. Med. Chem. 2019, 62, 9116–9140. 10.1021/acs.jmedchem.9b01012. [DOI] [PubMed] [Google Scholar]
- Li Q.; Xing S.; Chen Y.; Liao Q.; Xiong B.; He S.; Lu W.; Liu Y.; Yang H.; Li Q.; Feng F.; Liu W.; Chen Y.; Sun H. Discovery and biological evaluation of a novel highly potent selective butyrylcholinsterase inhibitor. J. Med. Chem. 2020, 63, 10030–10044. 10.1021/acs.jmedchem.0c01129. [DOI] [PubMed] [Google Scholar]
- Panek D.; Wiȩckowska A.; Jończyk J.; Godyń J.; Bajda M.; Wichur T.; Pasieka A.; Knez D.; Pišlar A.; Korabecny J.; Soukup O.; Sepsova V.; Sabaté R.; Kos J.; Gobec S.; Malawska B. Design, synthesis, and biological evaluation of 1-benzylamino-2-hydroxyalkyl derivatives as new potential disease-modifying multifunctional anti-Alzheimer’s agents. ACS Chem. Neurosci. 2018, 9, 1074–1094. 10.1021/acschemneuro.7b00461. [DOI] [PubMed] [Google Scholar]
- Viayna E.; Coquelle N.; Cieslikiewicz-Bouet M.; Cisternas P.; Oliva C. A.; Sánchez-López E.; Ettcheto M.; Bartolini M.; De Simone A.; Ricchini M.; Rendina M.; Pons M.; Firuzi O.; Pérez B.; Saso L.; Andrisano V.; Nachon F.; Brazzolotto X.; García M. L.; Camins A.; Silman I.; Jean L.; Inestrosa N. C.; Colletier J.-P.; Renard P.-Y.; Muñoz-Torrero D. Discovery of a potent dual inhibitor of acetylcholinesterase and butyrylcholinesterase with antioxidant activity that alleviates Alzheimer-like pathology in old APP/PS1 mice. J. Med. Chem. 2021, 64, 812–839. 10.1021/acs.jmedchem.0c01775. [DOI] [PubMed] [Google Scholar]
- Butini S.; Brindisi M.; Brogi S.; Maramai S.; Guarino E.; Panico A.; Saxena A.; Chauhan V.; Colombo R.; Verga L.; de Lorenzi E.; Bartolini M.; Andrisano V.; Novellino E.; Campiani G.; Gemma S. Multifunctional cholinesterase and amyloid beta fibrillization modulators. Synthesis and biological investigation. ACS Med. Chem. Lett. 2013, 4, 1178–1182. 10.1021/ml4002908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachon F.; Carletti E.; Ronco C.; Trovaslet M.; Nicolet Y.; Jean L.; Renard P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. 10.1042/BJ20130013. [DOI] [PubMed] [Google Scholar]
- Rydberg E. H.; Brumshtein B.; Greenblatt H. M.; Wong D. M.; Shaya D.; Williams L. D.; Carlier P. R.; Pang Y. P.; Silman I.; Sussman J. L. Complexes of alkylene-linked tacrine dimers with Torpedo californica acetylcholinesterase: Binding of Bis(5)-tacrine produces a dramatic rearrangement in the active-site gorge. J. Med. Chem. 2006, 49, 5491–5500. 10.1021/jm060164b. [DOI] [PubMed] [Google Scholar]
- Rosenberry T. L.; Brazzolotto X.; Macdonald I. R.; Wandhammer M.; Trovaslet-Leroy M.; Darvesh S.; Nachon F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: a crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. 10.3390/molecules22122098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung J.; Rudolph M. J.; Burshteyn F.; Cassidy M. S.; Gary E. N.; Love J.; Franklin M. C.; Height J. J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- Masson P.; Froment M.-T.; Gillon E.; Nachon F.; Lockridge O.; Schopfer L. M. Hydrolysis of oxo- and thio-esters by human butyrylcholinesterase. Biochim. Biophys. Acta 2007, 1774, 16–34. 10.1016/j.bbapap.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Moriarty N. W.; Poon B. K.; Sobolev O. V.; Terwilliger T. C.; Adams P. D. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Cryst. 2017, 73, 148–157. 10.1107/S2059798316018210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Lockridge O.; Masson P.; Fontecilla-Camps J. C.; Nachon F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- Cummings J. L.; Morstorf T.; Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L.; Appenroth D.; Decker M.; Kiehntopf M.; Roegler C.; Deufel T.; Fleck C.; Peng S.; Zhang Y.; Lehmann J. Synthesis and biological evaluation of NO-donor-tacrine hybrids as hepatoprotective anti-Alzheimer drug candidates. J. Med. Chem. 2008, 51, 713–716. 10.1021/jm701491k. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zenger K.; Lupp A.; Kling B.; Heilmann J.; Fleck C.; Kraus B.; Decker M. Tacrine-silibinin codrug shows neuro- and hepatoprotective effects in vitro and pro-cognitive and hepatoprotective effects in vivo. J. Med. Chem. 2012, 55, 5231–5242. 10.1021/jm300246n. [DOI] [PubMed] [Google Scholar]
- Nepovimova E.; Uliassi E.; Korabecny J.; Peña-Altamira L. E.; Samez S.; Pesaresi A.; Garcia G. E.; Bartolini M.; Andrisano V.; Bergamini C.; Fato R.; Lamba D.; Roberti M.; Kuca K.; Monti B.; Bolognesi M. L. Multitarget drug design strategy: quinone-tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J. Med. Chem. 2014, 57, 8576–8589. 10.1021/jm5010804. [DOI] [PubMed] [Google Scholar]
- Leng F.; Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here?. Nat. Rev. Neurol. 2021, 17, 157–172. 10.1038/s41582-020-00435-y. [DOI] [PubMed] [Google Scholar]
- Streit W. J. Microglial response to brain injury: a brief synopsis. Toxicol. Pathol. 2000, 28, 28–30. 10.1177/019262330002800104. [DOI] [PubMed] [Google Scholar]
- Kim S. U.; de Vellis J. Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313. 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Gomez-Nicola D.; Perry V. H. Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist 2015, 21, 169–184. 10.1177/1073858414530512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S. A.; Boddeke H. W. G. M.; Kettenmann H. Microglia in physiology and disease. Annu. Rev. Physiol. 2017, 79, 619–643. 10.1146/annurev-physiol-022516-034406. [DOI] [PubMed] [Google Scholar]
- Cherry J. D.; Olschowka J. A.; O’Banion M. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 98. 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch U.-K.; Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Huang J.; Huang N.; Xu S.; Luo Y.; Li Y.; Jin H.; Yu C.; Shi J.; Jin F. Signaling mechanisms underlying inhibition of neuroinflammation by resveratrol in neurodegenerative diseases. J. Nutr. Biochem. 2021, 88, 108552. 10.1016/j.jnutbio.2020.108552. [DOI] [PubMed] [Google Scholar]
- Martinez F. O.; Helming L.; Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- de Oliveira A. C. P.; Candelario-Jalil E.; Bhatia H. S.; Lieb K.; Hüll M.; Fiebich B. L. Regulation of prostaglandin E2 synthase expression in activated primary rat microglia: evidence for uncoupled regulation of mPGES-1 and COX-2. Glia 2008, 56, 844–855. 10.1002/glia.20658. [DOI] [PubMed] [Google Scholar]
- Azrad M.; Zeineh N.; Weizman A.; Veenman L.; Gavish M. The TSPO ligands 2-Cl-MGV-1, MGV-1, and PK11195 differentially suppress the inflammatory response of BV-2 microglial cell to LPS. Int. J. Mol. Sci. 2019, 20, 594. 10.3390/ijms20030594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olajide O. A.; Kumar A.; Velagapudi R.; Okorji U. P.; Fiebich B. L. Punicalagin inhibits neuroinflammation in LPS-activated rat primary microglia. Mol. Nutr. Food Res. 2014, 58, 1843–1851. 10.1002/mnfr.201400163. [DOI] [PubMed] [Google Scholar]
- Henn A.; Lund S.; Hedtjärn M.; Schrattenholz A.; Pörzgen P.; Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 2009, 26, 83–94. 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- Rubio-Perez J. M.; Morillas-Ruiz J. M. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.-H.; Aid S.; Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol. Sci. 2009, 30, 174–181. 10.1016/j.tips.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Verma I. M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Kopitar-Jerala N. Innate Immune Response in Brain, NF-Kappa B Signaling and Cystatins. Front Mol Neurosci 2015, 8, 73. 10.3389/fnmol.2015.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachon F.; Nicolet Y.; Viguié N.; Masson P.; Fontecilla-Camps J. C.; Lockridge O. Engineering of a monomeric and low-glycosylated form of human butyrylcholinesterase: expression, purification, characterization and crystallization. Eur. J. Biochem. 2002, 269, 630–637. 10.1046/j.0014-2956.2001.02692.x. [DOI] [PubMed] [Google Scholar]
- Brazzolotto X.; Wandhammer M.; Ronco C.; Trovaslet M.; Jean L.; Lockridge O.; Renard P.-Y.; Nachon F. Human butyrylcholinesterase produced in insect cells: huprine-based affinity purification and crystal structure. FEBS J. 2012, 279, 2905–2916. 10.1111/j.1742-4658.2012.08672.x. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Cryst. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty N. W.; Grosse-Kunstleve R. W.; Adams P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Cryst. 2009, 65, 1074–1080. 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Cryst. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nzekoue F. K.; Angeloni S.; Navarini L.; Angeloni C.; Freschi M.; Hrelia S.; Vitali L. A.; Sagratini G.; Vittori S.; Caprioli G. Coffee silverskin extracts: Quantification of 30 bioactive compounds by a new HPLC-MS/MS method and evaluation of their antioxidant and antibacterial activities. Food Res. Int. 2020, 133, 109128. 10.1016/j.foodres.2020.109128. [DOI] [PubMed] [Google Scholar]
- Antognoni F.; Potente G.; Mandrioli R.; Angeloni C.; Freschi M.; Malaguti M.; Hrelia S.; Lugli S.; Gennari F.; Muzzi E.; Tartarini S. Fruit quality characterization of new sweet cherry cultivars as a good source of bioactive phenolic compounds with antioxidant and neuroprotective potential. Antioxidants 2020, 9, 677. 10.3390/antiox9080677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Fan K.; McConnell O. J.; Carter G. T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. 10.1016/S0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- Nepovimova E.; Korabecny J.; Dolezal R.; Babkova K.; Ondrejicek A.; Jun D.; Sepsova V.; Horova A.; Hrabinova M.; Soukup O.; Bukum N.; Jost P.; Muckova L.; Kassa J.; Malinak D.; Andrs M.; Kuca K. Tacrine-Trolox Hybrids: A Novel Class of Centrally Active, Nonhepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low In Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. 10.1021/acs.jmedchem.5b01325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.