

Abstract
To identify novel risk genes and better understand the molecular pathway underlying Alzheimer’s disease (AD), whole-exome sequencing was performed in 215 early-onset AD (EOAD) patients and 255 unrelated healthy controls of Han Chinese ethnicity. Subsequent validation, computational annotation and in vitro functional studies were performed to evaluate the role of candidate variants in EOAD. We identified two rare missense variants in the phosphodiesterase 11A (PDE11A) gene in individuals with EOAD. Both variants are located in evolutionarily highly conserved amino acids, are predicted to alter the protein conformation and are classified as pathogenic. Furthermore, we found significantly decreased protein levels of PDE11A in brain samples of AD patients. Expression of PDE11A variants and knockdown experiments with specific short hairpin RNA (shRNA) for PDE11A both resulted in an increase of AD-associated Tau hyperphosphorylation at multiple epitopes in vitro. PDE11A variants or PDE11A shRNA also caused increased cyclic adenosine monophosphate (cAMP) levels, protein kinase A (PKA) activation and cAMP response element-binding protein phosphorylation. In addition, pretreatment with a PKA inhibitor (H89) suppressed PDE11A variant-induced Tau phosphorylation formation. This study offers insight into the involvement of Tau phosphorylation via the cAMP/PKA pathway in EOAD pathogenesis and provides a potential new target for intervention.
Introduction
Alzheimer’s disease (AD) is a leading cause of dementia, affecting between 23 and 35 million people worldwide (1). AD is one of the major contributors to disability and causes increases in the burdens of patients and the families of patients, as well as of the health care systems (2). The neuropathologic hallmarks of AD brains are extracellular accumulation of diffuse and neuritic amyloid plaques, composed of amyloid-β (Aβ) peptide, and the intraneuronal accumulation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated protein Tau (p-Tau). Based on age at onset (AAO), AD is classified as either early onset (AAO <65 years) or late onset (AAO ≥65 years) (3). Of all AD patients, around 10% are diagnosed with early-onset AD (EOAD) (4). Late-onset AD (LOAD) is a complex disorder with a heterogeneous etiology and a heritability of 58–79% (3,5). Genome-wide association studies (GWAS) have identified over 50 risk loci associated with AD (5). However, most GWAS loci are non-coding common variants of uncertain function. Indeed, GWAS hits explain a relatively small proportion of the phenotype in populations, and it is estimated that the ‘missing heritability’ may be explained by rare but functionally important variants. EOAD is an almost entirely genetically determined disease with a heritability ranging from 92 to 100% (5). High-penetrant variants in amyloid precursor protein (APP) (3), presenilins 1 (PSEN1) and presenilins 2 (PSEN2) are the main genetic risk factors underlying EOAD, but are only found in ~11% of all EOAD patients (6). Exome sequencing has recently identified rare missense variants in TREM2, ABCA7, SORL1, granulin precursor (GRN) and other genes that increase the risk for developing AD (7–9). However, a large number of EOAD patients remain genetically unexplained. EOAD patients are more likely to carry pathogenic variants (5) and suitable for identifying novel AD risk genes. The number of patients with dementia in China accounts for ~25% of the entire population with dementia worldwide (1). However, few studies on risk genes in EOAD cohorts have been carried out in China.
In this study, exome sequencing was performed in 215 EOAD patients and 255 unrelated healthy controls of Han Chinese ethnicity to identify novel AD risk genes. We identified two rare non-synonymous variants (p.Arg202His and p.Leu756Gln) in phosphodiesterase 11A (PDE11A) in individuals with EOAD. Functional analyses in vitro revealed that both variants enhanced Tau phosphorylation. We also investigated the mechanisms through which PDE11A may be relevant in AD.
Results
Identification of PDE11A variants
We sequenced and analyzed the whole exome of 470 individuals (the pipeline is shown in Fig. 1). The cohort consisted of 215 EOAD cases and 255 unrelated control samples (Table 1). About 80 000 variants per sample passed our quality control filters. For investigation of novel genes, patients carrying known EOAD risk genes (TREM2, VPS35, SORL1, MARK4, RUFY and TCIRG1) or genes associated with LOAD (ABCA7, ADAM17, IGHG3, PLD3, UNC5C, BIN1, CD2AP, CLU, CR1, EPHA1, MS4A4A, PLCG2, ABI3, AKAP9 and ZNF655) were excluded (number of genes =13) (Supplementary Material, Table S1). We then selected only variants rare in the population (<0.01% minor allele frequency (MAF)) and coding variants, lowering the count to 317. Next, we excluded synonymous variants and used in silico analysis to restrict our findings to those predicted as damaging for the protein, revealing 32 variants (Supplementary Material, Table S2). To further narrow the search for variants of interest, variant pathogenicity was evaluated using sorting intolerant from tolerant (SIFT), Polyphen-2, MutationTaster, Mendelian Clinically Applicable Pathogenicity (M-CAP), Combined Annotation Dependent Depletion (CADD), Likelihood Ratio Test (LRT), Protein Variation Effect Analyzer (PROVEAN), deleterious annotation of genetic variants using neural networks (DANN), Variant Effect Scoring Tool 3 (VEST3), fathmm-MKL, Genomic Evolutionary Rate Profiling (GERP), SiPhy, phastCons and phyloP tools. The variants that all the tools predicted pathogenic were from ADRA2B, AHSA2, PDE11A, MYO5A, COL6A6 and ETFDH (Supplementary Material, Table S3). Then, we used data from Online Mendelian Inheritance in Man (OMIM), Mouse Genome Informatics (MGI), Gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), The American College of Medical Genetics and Genomics (ACMG) and UKBiobank PheWeb to perform a systems-level analysis of the six genes.
Figure 1 .
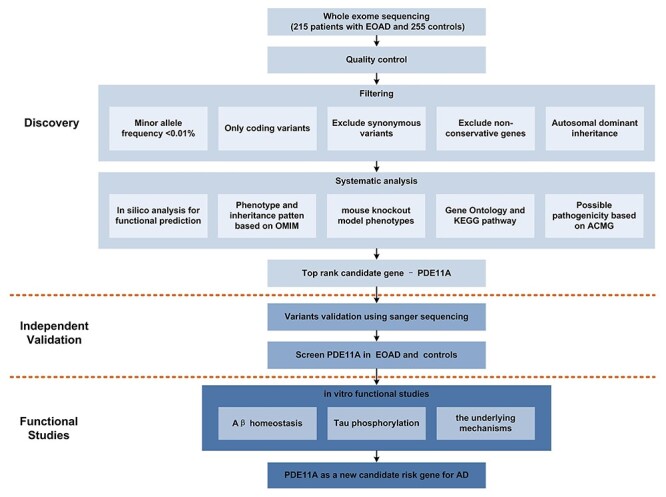
Workflow of the current study. Exome sequencing was performed in 215 Chinese individuals with EOAD and 255 controls. Subsequent direct sequencing was performed in an independent cohort to validate the selected rare variants. Then in vitro functional studies were performed to evaluate the role of candidate variants in EOAD and the underlying mechanisms.
Table 1.
Demographics
| Characteristic | WES cohort | Direct sequencing cohort | ||
|---|---|---|---|---|
| Controls (n = 255) | Cases (n = 215) | Controls (n = 4489) | Cases (n = 535) | |
| Male, n | 130 | 101 | 2145 | 261 |
| Female, n | 125 | 114 | 2344 | 274 |
| Age-at-study, mean (SD), years | 67.3 (5.3) | 51.2 (3.5) | 63.8 (6.5) | 53.2 (4.2) |
| Age at onset, mean (SD), years | NA | 48.5 (3.8) | NA | 49.8 (4.4) |
| Education (years) | 11 (6.3) | 11 (4.6) | 12 (3.8) | 9 (5.3) |
| MMSE score, mean (SD) | 27.8 (1.7) | 12.1 (2.5) | 27.5 (3.4) | 11.7 (3.4) |
| CDR score, mean (SD) | 0 | 2.7 (0.8) | 0 | 2.6 (1.3) |
| No. of APOE ε4 alleles, No. (%) | ||||
| 0 | 198 (77.6) | 121 (56.3) | 3668 (81.7) | 318 (59.4) |
| 1 | 57 (22.4) | 86 (40.0) | 806 (18.0) | 198 (37.0) |
| 2 | 0 | 8 (3.7) | 15 (0.3) | 19 (3.6) |
APOE, apolipoprotein E; CDR, Clinical Dementia Rating Scale; MMSE, Mini-Mental State Examination.
Among them, PDE11A met all criteria. We identified two variants in the PDE11A gene (NM_016953.4: rs752822096: c.605G>A: p.Arg202His and NM_016953.4: rs201572288: c.2267 T>A: p.Leu756Gln). One p.Arg202His heterozygote and one p.Leu756Gln heterozygote was found in EOAD patients, but none in normal controls. Confirmation by Sanger sequencing is shown in Figure 2A. PDE11A is a gene located on an autosome. A significant association was shown (P-value = 1.0 × 10−13) between PDE11A and AD using the UKBiobank PheWeb tool (https://pheweb.org/UKB-Neale/gene/PDE11A). Based on genetic databases, these two variants are rare in the East Asian population (p.Arg202His, gnomAD exomes_EAS: allele frequency = 0.0000; p.Leu756Gln, gnomAD exomes_EAS: allele frequency = 0.00431). The two variants are also rare in other populations. The p.Arg202His and p.Leu756Gln variants are predicted by 11 bioinformatics tools, including Polyphen2 HDIV, Polyphen2 HVAR, SIFT, LRT, PROVEAN, MutationTaster, DANN, VEST3, fathmm-MKL, CADD and M-CAP, to be damaging to the protein and by three algorithms (GERP, phastCons, phyloP) to be conserved (Table 2). The p.Arg202His and p.Leu756Gln variants are likely pathogenic and of uncertain significance, respectively, according to the American College of Medical Genetics and Genomics guidelines (10). Subsequent Sanger sequencing analysis was performed in an expanded cohort of individuals (N = 5024: 535 EOAD, 4489 controls). We identified two p.Arg202His heterozygotes in EOAD patients but none in normal controls, and six p.Leu756Gln heterozygotes in EOAD patients and 16 heterozygotes in normal controls. Meta-analysis for the two variants with two-stage sequencing data revealed a significant association with AD (p.Arg202His, P = 0.04; p.Leu756Gln, P = 0.01).
Figure 2 .
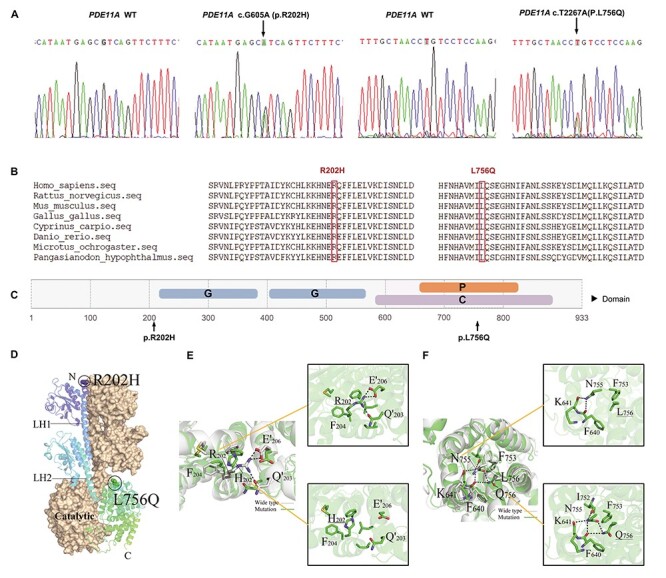
Functional annotation of PDE11A variants. (A) Sanger sequencing confirmation of PDE11A variants. (B) Protein homologs in different species were aligned. The p.Arg202His and p.Leu756Gln are conserved in all PDE11A orthologs. The variants are highlighted in the red boxes. (C) Predicted domain maps of PDE11A protein with the identified missense variants marked: G: GAF domain (amino acids 217–380 and amino acids 402–568), P: HD/PDEase domain (amino acids 663–839), C: 3′5′-cyclic nucleotide phosphodiesterase, catalytic domain (amino acids 588–912). The p.Arg202His variant is located near the GAF domain, and p.Leu756Gln variant in the 3′5′-cyclic nucleotide phosphodiesterase, catalytic domain. (D) Predicted three-dimensional structure of wide type (WT) and mutant PDE11A protein. (E) Structure comparison between wild-type PDE11A and R202H variant. Critical hydrogen bonds with surrounding amino acids were predicted to be eliminated. Dashed lines indicate hydrogen bonds. (F) Structure comparison between wild-type PDE11A and L756Q variant. Q756 mutant was predicted to induce more hydrogen bonds and then affect helix structure.
Table 2.
Pathogenicity prediction of PDE11A variants
| Variant | SIFT | PolyPhen2 HDIV | Polyphen2 HVAR | LRT | Mutation Taster | DANN | CADD | M -CAP | PROVEAN | VEST3 | fathmm-MKL | GERP++ | SiPhy | phast Cons100way vertebrates | phylo P100way vertebrates |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.G605A (p.Arg202His) | Damaging (0.028) | Probably damaging (0.997) | Possibly damaging (0.792) | Deleterious (0.000071) | Disease causing (1) | 0.998 | 6.96 | Damaging (0.027) | Damaging (−3.92) | 0.821 | Deleterious (0.986) | Conserved (4.34) | 14.443 | Highly conserved (1) | Conserved (5.622) |
| c.T2267A (p.Leu756Gln) | Damaging (0.001) | Probably damaging (0.996) | Probably damaging (0.943) | Deleterious (0) | Disease causing (1) | 0.995 | 6.12 | Damaging (0.174) | Damaging (−4.71) | 0.952 | Deleterious (0.945) | Conserved (5.57) | 15.739 | Highly conserved (1) | Conserved (8.857) |
PDE11A variants and clinical features
The PDE11A p.Arg202His variant was detected in a 54-year-old female patient who had visited our hospital complaining of progressive memory decline over the past 4 years. She presented with amnesia as well as executive function and orientation deficits. She scored 9/30 on the Mini-Mental State Examination (MMSE) and 8/30 on the Montreal Cognitive Assessment, which was below the recommended cutoff values of 22 and 24, respectively. She had a Clinical Dementia Rating (CDR) score of 3 and only remembered one word from the World Health Organization and University of California, Los Angeles (WHO-UCLA) Delayed Recall Memory Test. Magnetic resonance imaging (MRI) revealed moderate cerebral atrophy, especially in the hippocampus (medial temporal lobe atrophy (MTA) = 4). Her apolipoprotein E (APOE) genotype was ε3/ε3. Her parents and siblings were cognitively normal without complaints. Both her parents were deceased, and no DNA was available.
PDE11A p.Leu756Gln was found in a male patient who presented episodic memory decline at the age of 52 years. He had progressive difficulties in understanding and orientation, and he developed motor aphasia and personality changes in subsequent years. MRI showed atrophy of the temporoparietal lobe. The patient’s APOE genotype was ε4/ε3. He denied a family history of dementia.
Functional annotation of rare PDE11A variants
PDE11A p.Arg202His is uniquely present in the PDE11A4 isoform. The PDE11A protein sequence in which the two rare variants are located contains highly conserved amino acids across different species (Fig. 2B), and their GERP scores are 4.34 and 5.57, respectively, implicating potential interference of important protein biological functions by the variants. PDE11A p.Arg202His and p.Leu756Gln are predicted by Poly-Phen2 and SIFT to be damaging or possibly damaging (Table 1). As depicted in the schematic diagram of full-length PDE11A in Figure 2C, the two variants are located near or in functional domains, including the cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase (PDE), adenylyl cyclase and FhlA (GAF) and catalytic domains, suggesting a potential functional impact of these variants on the PDE11A protein.
Global conformations of the p.Arg202His and p.Leu756Gln variants changed significantly from wild-type human PDE11A in three-dimensional (3D) homology models (Fig. 2D–F). Specifically, the 3D model predicts that p.Arg202His abolishes critical hydrogen bonds with surrounding amino acids; in contrast, p.Leu756Gln leads to a new hydrogen-bonded network, which affects the helical structure. Taken together, the model predicts that both p.Arg202His and p.Leu756Gln variants identified in patients with AD may impair PDE11A function.
PDE11A expression in AD brain tissues
The PDE11A gene is expressed in several regions of the mouse hippocampus, including the CA1, the subiculum and the amygdalohippocampal area (11). Moreover, Pde11a-knockout mice exhibit enlarged lateral ventricles and abnormal social investigation (11). PDE11A is also reported to be involved in the regulation of cGMP-mediated signaling. These results suggest that PDE11A has an important role in the brain, with a possible role in central nervous system disorders.
We assessed PDE11A expression in a publicly available single-nuclei RNA sequencing (RNA-seq) study from AD cases and control brain samples (12) and found the PDE11A gene to be expressed in almost all types of cells, including neurons, astrocytes and microglia (Fig. 3A and B).
Figure 3 .
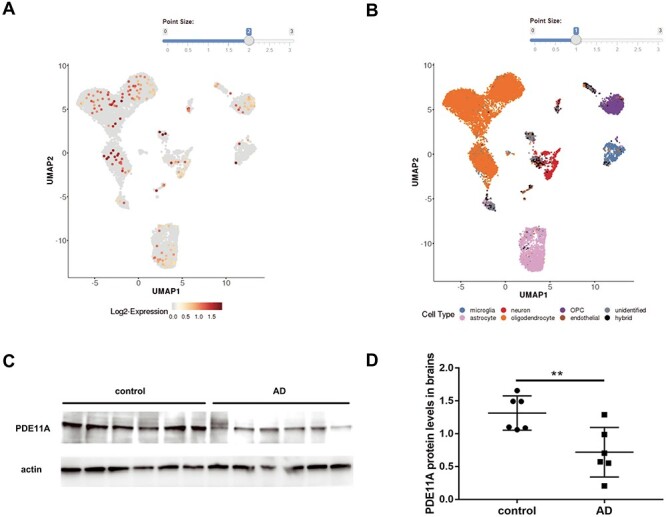
The PDE11A expression in brain tissues of AD. (A and B) Based on single-nucleus RNA sequencing data, PDE11A gene was expressed almost in all kinds of cells. (C) Representative western blots illustrate the expression of PDE11A in postmortem brain tissues. (D) The histogram shows the quantification of PDE11A detected by immunoblot relative to control levels. The data are represented as the mean ± SEM, based on three unrelated measurements. **P < 0.01 by Student’s t-test.
To characterize the involvement of PDE11A in AD, we analyzed the PDE11A protein in fresh frozen postmortem brain tissues from cognitively normal healthy controls (n = 6) and patients with AD (n = 6). All participants were matched for age and sex. This analysis revealed significantly decreased levels of PDE11A in those with AD relative to healthy controls (Fig. 3C and D).
Effects of PDE11A variants on Aβ homeostasis
To further confirm the pathogenesis of PDE11A variants, we performed in vitro studies to test the effect on Aβ homeostasis. Lower levels of the PDE11A protein was observed in AD patients. Therefore, we used PDE11A short hairpin RNA (shRNA) to knockdown (KD) PDE11A levels in cell models. A markedly lower PDE11A messenger RNA (mRNA) and protein levels were obtained in SH-SY5Y cells (Supplementary Material, Fig. S1A–C).
SH-SY5Y cells were transduced with APP695 lentiviruses and PDE11A lentiviruses (PDE11A wide type (WT), p.Arg202His, p.Leu756Gln, scramble or shRNA). PDE11A variants did not significantly change PDE11A levels. And it showed no significant differences in secreted Aβ40 or Aβ42 levels or the Aβ42/Aβ40 ratio (Supplementary Material, Fig. S2A–C). PDE11A variants or KD did not change the levels of APP or Beta-Secretase 1 (BACE-1) (Supplementary Material, Fig. S3A–C). These results suggest that PDE11A may not affect Aβ homeostasis.
PDE11A variants affect Tau phosphorylation
To understand the influence of PDE11A variants on Tau phosphorylation, PDE11A lentiviruses (WT PDE11A, PDE11A p.Arg202His, PDE11A p.Leu756Gln, shRNA or scramble) were used to transduce SH-SY5Y. Because of the relative low levels of endogenous phosphorylation Tau in cells, human Microtubule Associated Protein Tau (MAPT) lentiviruses were transduced cells with PDE11A lentiviruses simultaneously. Effects on Tau phosphorylation levels in transduced cells were assessed by immunoblotting. A significant reduction in Tau phosphorylation was detected at multiple sites, including T181, S404, S202/T205, S416, S214 and S396, in WT PDE11A-expressing cells compared with cells infected with mock lentivirus (Fig. 4). Moreover, both variants notably increased Tau phosphorylation at multiple sites compared with the WT (Fig. 4A and B). PDE11A shRNA treatment reduced PDE11A mRNA and protein levels by 60% and significantly increased Tau phosphorylation at multiple sites compared with scramble shRNA treatment (Fig. 4A–C). These results suggest that both variants could be loss-of-function.
Figure 4 .
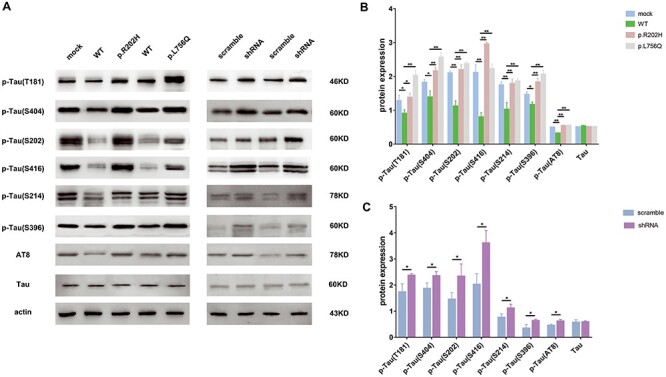
The PDE11A variants led to significantly high Tau phosphorylation levels. The cells were co-infected with MAPT and PDE11A lentivirus (PDE11A WT, mutants, scramble or shRNA). A total of 72 h after infection, cell lysates were used to detect levels of p-Tau(T181), p-Tau(S404), p-Tau(S202), p-Tau(S416), p-Tau(S214), p-Tau(S396), p-Tau(AT8) and Tau. (A) Representative western blots illustrate the expressions of phosphorylated Tau. (B and C) The histograms show the quantification of phosphorylated Tau levels detected by immunoblot relative to control levels. The data are represented as the mean ± SEM, based on three unrelated measurements. *P < 0.05, **P < 0.01 by one-way ANOVA and Dunnett test or Student’s t-test.
The protein level of glycogen synthase kinase-3β (GSK-3β), a major kinase involved in Tau phosphorylation, and its inactive form p-Ser9-GSK3β was not significantly altered in any of the groups (Supplementary Material, Fig. S4A–C). These data suggest that PDE11A variants likely affect Tau phosphorylation independent of GSK-3β signaling.
PDE11A variants exhibit alterations in cAMP/protein kinase A (PKA) signaling
The cAMP/PKA/cAMP response element-binding protein (CREB) signaling plays an important role in AD. However, the link with PDE11A is unknown. To further clarify the underlying mechanisms, we tested the effects of PDE11A on cAMP/PKA/CREB signaling. Transduction of WT PDE11A (with MAPT) in cells decreased cAMP levels compared with the mock group (with MAPT). The p.Arg202His and p.Leu756Gln variants increased cAMP levels relative to WT PDE11A (Fig. 5A and B). These results suggest that the p.Arg202His and p.Leu756Gln variants reduced the ability of PDE11A to degrade cAMP. Total PKA and p(Thr197)-PKA levels, as well as the ratio of phosphorylated CREB (p-CREB) to CREB, were also increased in variants PDE11A-expressing cells compared with WT PDE11A-expressing cells (Fig. 5C and D). Similar results were obtained in PDE11A shRNA-treated cells compared with scramble shRNA-treated cells (Fig. 5C and E).
Figure 5 .
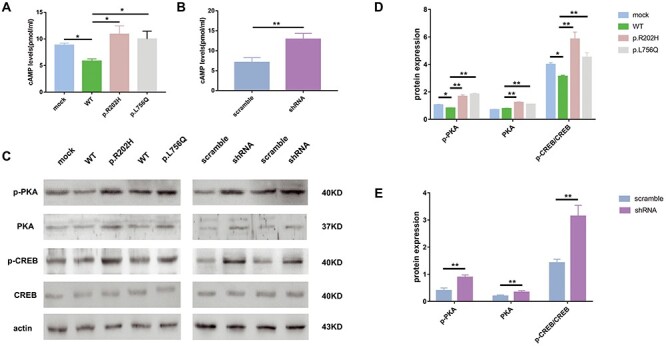
The PDE11A variants affect cAMP/PKA signaling. ELISA analysis showed cAMP levels in cells after infection with WT PDE11A (with MAPT), decreased as expected; they increased following infection with PDE11A p.Arg202His, PDE11A p.Leu756Gln (A) or shRNA (with MAPT) (B). (C) Western blot of PKA signaling. The histograms show the quantification of p-PKA, PKA, p-CREB/CREB levels detected by immunoblot relative to control levels in cells infected with PDE11A mutants (D) or shRNA (with MAPT) (E). The data are represented as the mean ± SEM. *P < 0.05, **P < 0.01 by one-way ANOVA and Dunnett test or Student’s t-test.
In addition, pretreating the cells with the PKA inhibitor (H89) reduced PDE11A p.Arg202His and p.Leu756Gln variant-induced Tau phosphorylation (Fig. 6A–C). H89 pretreatment also decrease phosphorylated PKA (p-PKA) and p-CREB/CREB levels (Supplementary Material, Fig. S5A–E). Furthermore, these results were recapitulated using PDE11A shRNA. Therefore, these data demonstrate that the PDE11A variants affect the cAMP/PKA pathway, which is associated with increased Tau phosphorylation through a loss-of-function mechanism.
Figure 6 .
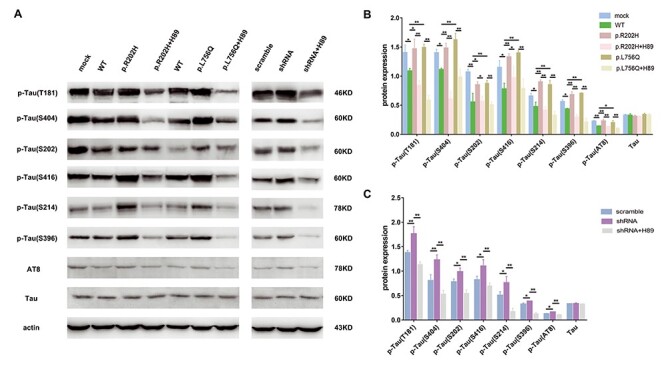
The effects of PDE11A mutants on phosphorylation of Tau after treatment with PKA inhibitor. SH-SY5Y cells were treated with or without the PKA inhibitor H89 (10M). Then, cells were co-transfected with MAPT and PDE11A mutant or shRNA plasmids. (A) Cell lysates were collected to examine Tau phosphorylation levels using western blot. (B and C) The histograms show the quantification of phosphorylated Tau levels detected by immunoblot relative to control levels. The data are represented as the mean ± SEM. *P < 0.05, **P < 0.01 by one-way ANOVA and Dunnett test.
Discussion
In the present study, we report PDE11A as a novel candidate risk gene for EOAD. Our results showed that PDE11A variants did not affect amyloid production but cause dysregulation of cAMP/PKA signaling and increase Tau phosphorylation. These findings potentially improve our understanding of the molecular mechanisms of EOAD and reveal new AD target pathways.
We selected 215 cases of very EOAD (with age of onset before 55 years) to detect new AD risk genes. EOAD has a higher heritability than LOAD and absence of known causal or risk AD gene variants, sequencing EOAD increases the chances of discovering new AD genes. Using a systematic analysis of exome sequencing data, we identified PDE11A as a novel candidate risk gene for EOAD. Combined with the subsequent direct sequencing of the two variants in 4962 individuals, it revealed the two variants were significantly enriched in AD. The human PDE11A gene is located at 2q31.2 and consists of 23 exons (13). Four transcript isoforms designated PDE11A1–4, which result from different transcription initiation sites and alternative splicing, encode different PDE11A isoforms with unique N-terminal domains. Germline mutations, expression changes and functional alterations in PDE11A have been linked to brain function, tumorigenesis and inflammation (11,14–16). Moreover, PDE11A negatively regulates the efficacy of lithium in treating bipolar disorder (17). Expression of PDE11A4 mRNA in the brain tends to occur in CA1 neurons and in the subiculum in ventral hippocampal (VHIPP) formation. Atrophy of VHIPP has been observed in patients with AD (18), and the deficits in the VHIPP appear to correlate with the severity of clinical impairment in patients with AD (19). In addition, PDE11A4 can directly influence synaptic plasticity (20). PDE11A4 also plays a role in systems consolidation, which has relevance for cognitive deficits (21). PDEs have been linked to mental processes of emotions, learning and memory, with implications in mood and cognitive disorders, including Huntington disease, Parkinson’s disease and AD (22–25). This evidence suggests that PDE11A plays a role in brain function and may also be involved in AD.
In our study, we identified two rare non-synonymous variants p.Arg202His and p.Leu756Gln in PDE11A in EOAD patients. The p.Arg202His variant is only present in the PDE11A4 isoform, whereas the p.Leu756Gln variant is present in all PDE11A isoforms. PDE11A isoforms are expressed in a tissue-specific manner (26). In a previous study, Kelly (27) found that PDE11A4 is highly expressed in hippocampal areas but not in any of the other tissues examined. PDE11A4 is the only PDE with restricted expression in hippocampal areas, which are affected in AD. Both variants identified in this study affect PDE11A4 isoforms through a loss-of-function mechanism. Both variants are highly conserved evolutionarily and located near or in an important PDE11A4 domain. In addition, both mutations disrupt hydrogen bonding and dramatically alter protein conformation. Furthermore, both variants are predicted to be damaging by 11 in silico tools and are considered conserved by all four algorithms applied. Taken together, these findings suggest that important biological functions can be attributed to these variants. Moreover, using bulk RNA-seq data derived from 1536 individuals (28), we found that the PDE11A gene is significantly associated with neuronal proportion in cortical tissues. Neuronal loss in the cerebral cortex is one of the main characteristic pathological changes of AD. We also detected significantly decreased protein levels of PDE11A in brain tissues from AD patients. It indicates that these variants may affect PDE11A function in the brain and facilitate disease onset.
We conducted cellular and biochemical studies and found that the PDE11A variants increase Tau phosphorylation at multiple sites. PDE11A shRNA also induces significant Tau hyperphosphorylation, whereas overexpression of PDE11A markedly decreases Tau phosphorylation. These findings suggest a putative role for PDE11A and variants in Tau phosphorylation. In contrast, the conditioned medium of cells transduced with lentivirus carrying the p.Arg202His or p.Leu756Gln variant or PDE11A shRNA showed no significant differences in Aβ42 or Aβ40 levels or Aβ42 to Aβ40 ratio, indicating that the variants may not affect Aβ peptides homeostasis. GSK-3β signaling affects Tau hyperphosphorylation. However, PDE11A variants or shRNA groups did not affect GSK-3β levels, indicating that the GSK-3β pathway is unaffected by the PDE11A variants. It is likely that other signaling pathways contribute to Tau hyperphosphorylation. The underlying mechanisms connecting PDE11A and Tau needs to be further evaluated.
Our data demonstrate that PDE11A affects Tau phosphorylation through the cAMP/PKA pathway. PDEs hydrolyze cAMP and are key enzymes regulating intracellular cAMP levels. cAMP is known as a crucial second messenger that participates in transducing signals in many types of cells. cAMP activates PKA, which phosphorylates and activates CREB. Activated CREB binds to cAMP-response elements (CREs) in target genes to regulate their expression. cAMP/PKA signaling pathway dysregulation has been reported in AD patients, and the pathway plays a crucial role in AD pathogenesis (29). Therefore, we reasoned that PDE11A variants might affect AD-relevant phenotypes through cAMP/PKA signaling. Our results showed higher cAMP levels in lysates from cells transduced with PDE11A p.Arg202His, p.Leu756Gln or shRNA compared with those expressing WT PDE11A or scramble shRNA. We also found that PDE11A p.Arg202His or p.Leu756Gln variant led to elevated PKA, p-PKA and p-CREB levels. Increases in cAMP and PKA levels have been observed in the cerebrospinal fluid (CSF) or cortex in AD patients (30,31). Expression of PKA and p-CREB is decreased in AD patients and in vitro models (32,33), suggesting that PKA and p-CREB levels may vary on different tissues at different stages of disease progression.
PKA phosphorylates Tau, and it is important for the formation of NFTs (34,35). Martinez et al. (30) showed that cAMP levels correlate with levels of Tau protein in the CSF. In the present study, H89 (a specific PKA inhibitor) pretreatment suppressed PDE11A mutation-associated p-Tau formation, which further confirmed the role of the cAMP/PKA/CREB signaling pathway in PDE11A-mediated regulation of Tau phosphorylation. Carlyle et al. (36) reported an age-related increase in cAMP-dependent PKA-mediated phosphorylation of Tau as a risk factor for aged cortex degeneration. PKA activation induces Tau hyperphosphorylation and spatial memory deficits, which are reversed by the PKA inhibitor H89 inhibits (35). Ikezu et al. (37) identified two rare variants in AKAP9 in African American AD patients. AKAP9 variants had no effect on Aβ, but significantly increased Tau phosphorylation in cells treated with a PDE4 inhibitor (38). PKA enhances Tau phosphorylation by other Tau kinases (39). Thus, the effect of PKA on AD-associated Tau phosphorylation may be either direct or indirect, and the exact mechanism has yet to be elucidated.
There are some limitations in our study. First, an independent replication of the association of PDE11A variants with EOAD in other Han Chinese population and other different populations would have been supportive of our genetic findings. Second, hopefully future studies into the pathogenesis of EOAD will be able to use our findings regarding PDE11A. Third, some other candidate risk genes for AD were missing using our selection strategies. Further analysis and functional study on other risk genes need to be investigated in future study. Fourth, frozen postmortem AD brain samples were not from EOAD patients. When the adequate EOAD postmortem brains are available, the PDE11A protein levels should be detected in these samples.
Conclusions
In summary, we report for the first time two novel rare PDE11A variants in individuals with EOAD. These PDE11A variants enhance both cAMP/PKA signaling and Tau phosphorylation. PDE11A could be a novel candidate predisposing genetic factor for AD.
Materials and Methods
Study design and subjects
Data from 215 individuals with EOAD from Xuanwu Hospital were evaluated in this observational study and compared with healthy cognitively normal controls (Table 1). Each individual underwent a neuropsychological examination, MRI; CSF analysis was performed for a subset of individuals (n = 115) in which levels of Aβ42, Aβ40, Tau and p-Tau181 were assessed. The criteria for the recruited EOAD patients were set as follows: (1) met the National Institute of Neurological and Communicative Disorders and the Stroke and the Alzheimer Disease and Related Disorders Association (40) or the National Institute on Aging-Alzheimer’s Association diagnostic criteria (41); (2) the onset age of affected individuals was below 55 years; and (3) no known pathogenic mutations in PSEN1, PSEN2, APP, MAPT or GRN genes. Healthy controls were cognitively normal; amnesia was not present, MMSE scores were higher than the appropriate cutoff for dementia, Clinical Memory Scale scores ≥90 and global CDR scores were equal to 0. Additional 535 DNA samples from patients with EOAD and 4489 healthy controls were collected at the Xuanwu Hospital from 2015 to 2020 and used for Sanger sequencing. Signed informed consent was provided by all the patients and control subjects. The study protocol was approved and monitored by the ethics committee of Xuanwu Hospital.
Relative PDE11A protein levels were quantified using immunoblot in samples from fresh frozen postmortem parietal lobe tissue of six AD patients and six cognitively healthy controls, recruited at Washington University in St. Louis Charles F. and Joanne Knight Alzheimer’s Disease Research Center Brain Bank and approved by the institutional review board of Washington University. Written informed consent for brain autopsy was obtained from all participants or their legal representatives.
Exome sequencing and data analysis
Genomic DNA was obtained from the peripheral blood of all participants. Briefly, the exome was enriched using Agilent SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA, USA) and analyzed using an Illumina HiSeq 2500 (150 bp paired-end, Illumina, San Diego, CA, USA). After conducting quality control, high-quality paired-end reads were mapped to the human genome build GRCh37 using Burrows–Wheeler Aligner software. Verita Trekker was employed to identify variants. Enliven® and Annotate Variation (ANNOVAR) were utilized to perform annotation for Variant Call Format, and variants were selected according to an autosomal dominant model. Non-synonymous exonic or close-to splice-site variants with minor allele frequency <0.01% were prioritized for analysis. Variant pathogenicity was evaluated using SIFT, Polyphen-2, MutationTaster, M-CAP, CADD, LRT, PROVEAN, DANN, VEST3, fathmm-MKL, GERP, SiPhy, phastCons and phyloP tools. Candidate variants were prioritized based on OMIM, MGI, GO, KEGG, ACGM and the published literature.
Annotation of PDE11A variants
InterPro was used to predict domain maps of the PDE11A protein. DNAMAN software (Lynnon Corporation, Quebec, Canada) was applied for multiple sequence alignment. A homology model of the wild-type PDE11A structure was built in MODELLER software using the crystal structure of the PDE template (PDB: 3IBJ). Based on the homology modeling structure of wild-type PDE11A, Arg202His- and Leu756Gln-mutated proteins were generated with Pymol software. AMBER software was used for molecular dynamics simulations for wild-type PDE11A and the Arg202His and Leu756Gln variants. To investigate in which major cell type the PDE11A gene is expressed, we queried the gene in a publicly available single-cell/nuclei RNA-seq dataset of AD cohorts of human brain tissues (http://adsn.ddnetbio.com/) (12).
Generation of PDE11A variants
Human wild-type PDE11A complementary DNA (cDNA) (NM_016953) was cloned into the pCDNA3.1-enhanced green fluorescent protein (EGFP) vector. Site-directed PDE11A mutagenesis (p.Arg202His and p.Leu756Gln) was performed using a KOD-Plus-Mutagenesis kit (Stratagene, La Jolla, CA, USA). For shRNA, three optimal targeting human PDE11A and one scrambled control were designed (Supplementary Material, Table S4). Lentiviral particles were generated by co-transfecting the transfer vector (psh2-u6-egfp-puro containing shRNA or scramble, pCDH-CMV-MCS-EF1-GFP-T2A-Puro containing WT PDE11A, PDE11A p.Arg202His or PDE11A p.Leu756Gln) and the packaging vectors into HEK293T cells.
Cell culture
SH-SY5Y cells were maintained in 60 mm culture dishes with Eagle’s Minimal Essential Medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and antibiotics (1% penicillin–streptomycin; Invitrogen). Cells (105/cm2) were transduced with lentiviruses.
Real-time quantitative polymerase chain reaction (PCR)
Total RNA was purified from freshly harvested cells using an RNAeasy Mini Kit (Thermo Fisher Scientific) and TRIzol (Thermo Fisher Scientific). Reverse transcription was carried out using a SuperScript First-Strand Synthesis Kit (Thermo Fisher Scientific), and total mRNA was measured by real-time quantitative PCR. Diluted cDNA templates, upstream and downstream primers, and SYBR Green I Master mix (Thermo Fisher Scientific) were mixed in a 20 μl volume, and reactions were performed using the StepOne Plus real-time PCR platform. Human Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) (forward, 5′-ACAGCCTCAAGATCATCAGCAAT-3′; reverse, 5′-GATGGCATGGACTGTGGTCAT-3′) was used as the internal reference gene, and relative expression levels of the PDE11A gene were calculated using the 2−ΔΔCt method. PDE11A quantitative PCR (q-PCR) primers were as follows: forward, 5′-CTGGAGTGGATTGATAGCATCTG-3′; reverse, 5′-CAGTCGTTTTTGGTGTAGCTCTT-3′.
Western blotting
Briefly, cells were lysed in radio immunoprecipitation assay (RIPA) buffer containing protease inhibitor cocktails and phosphatase inhibitors. The cell lysate was pelleted by centrifugation, and the protein concentration was measured by the Bicinchoninic Acid (BCA) assay. Samples were separated by 10% Tris-glycine sodium dodecyl sulfate polyacrylamide gel electrophoresis and then electroblotted onto a polyvinylidene fluoride (PVDF) membrane. After blocking in 5% non-fat dry milk, the membrane was probed with appropriate primary antibodies (listed in Supplementary Material, Table S5) overnight at 4°C. The next day, the membrane was washed with Tris-buffered saline containing Tween-20 and incubated with horseradish peroxidase-linked secondary antibodies (1:5000; Santa Cruz Biotechnology) for 1 h at room temperature. After washing, the proteins were visualized by chemiluminescence. Western blots were normalized using the housekeeping protein β-actin.
Enzyme-linked immunosorbent assay (ELISA)
The concentrations of secreted Aβ40 and Aβ42 in conditioned medium were quantified using commercial ELISA kits (IBL International, Hamburg, Germany). Levels of cAMP in cell lysates were determined using a commercially available assay kit (R&D Systems, Minneapolis, MN, USA), according to the protocol provided by the manufacturer. Briefly, after washing cells with phosphate-buffered saline, the cells were resuspended in cell lysis buffer. A total of 300 μl of cell lysates were used to perform the assay in duplicate. The optical density of each well was determined with a microplate reader which is set to 450 nm. The concentrations of cAMP were calculated corresponding to the mean absorbance from the standard curve.
Statistical analysis
Quantification data were obtained from three independent repeats. Two-tailed unpaired Student’s t-test or one-way analysis of variance (ANOVA) was performed using GraphPad Prism, version 8.00 (San Diego, CA, USA). Dunnett test was performed for multiple comparisons after one-way ANOVA. To determine if the candidate risk gene is enriched in AD, a Fisher’s exact test on the variant count data was performed. Meta-analysis of the two-stage sequencing data was done using the Review Manager 5.3 (The Cochrane Collaboration, Oxford, UK). A P-value <0.05 was used to indicate a statistically significant difference.
Supplementary Material
Acknowledgement
The authors thank Chengran Yang and Zeran Li for bioinformatics assistance. The authors sincerely thank the patients and their relatives for participation.
Conflict of Interest statement. All authors except C.C. report no disclosures. C.C. receives research support from Biogen, EISAI, Alector and Parabon. The funders of the study had no role in the collection, analysis or interpretation of data, in the writing of the report, or in the decision to submit the paper for publication. C.C. is a member of the advisory board of Vivid Genetics, Halia Therapeutics and ADx Healthcare.
Funding
Beijing Natural Science Foundation (7192077); National Key R&D Program of China (2017YFC1310100, 2016YFC1306305); the Key Project of the National Natural Science Foundation of China (81530036, 31627803, U20A20354); Beijing Municipal Science & Technology Commission (Z181100001718110); Beijing Scholars Program; Beijing Municipal Science & Technology Commission (Z201100005520016, Z201100005520017); Beijing Municipal Commission of Health and Family Planning (PXM2019_026283_000003); National Institutes of Health (R01AG044546, P01AG003991, RF1AG053303, R01AG058501, U01AG058922, RF1AG058501, R01AG064614).
References
- 1. GBD Dementia Collaborators (2019) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol., 18, 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brookmeyer, R., Johnson, E., Ziegler-Graham, K. and Arrighi, H.M. (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement., 3, 186–191. [DOI] [PubMed] [Google Scholar]
- 3. Barber, I.S., Braae, A., Clement, N., Patel, T., Guetta-Baranes, T., Brookes, K., Medway, C., Chappell, S., Guerreiro, R., Bras, J. et al. (2017) Mutation analysis of sporadic early-onset Alzheimer’s disease using the NeuroX array. Neurobiol. Aging, 49, 215.e211–215.e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cacace, R., Sleegers, K. and Van Broeckhoven, C. (2016) Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement., 12, 733–748. [DOI] [PubMed] [Google Scholar]
- 5. Sims, R., Hill, M. and Williams, J. (2020) The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci., 23, 311–322. [DOI] [PubMed] [Google Scholar]
- 6. Lanoiselee, H.M., Nicolas, G., Wallon, D., Rovelet-Lecrux, A., Lacour, M., Rousseau, S., Richard, A.C., Pasquier, F., Rollin-Sillaire, A., Martinaud, O. et al. (2017) APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med., 14, e1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Roeck, A., Van Broeckhoven, C. and Sleegers, K. (2019) The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol., 138, 201–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma, Y., Jun, G.R., Zhang, X., Chung, J., Naj, A.C., Chen, Y., Bellenguez, C., Hamilton-Nelson, K., Martin, E.R., Kunkle, B.W. et al. (2019) Analysis of whole-exome sequencing data for Alzheimer disease stratified by APOE genotype. JAMA Neurol., 76, 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jin, S.C., Pastor, P., Cooper, B., Cervantes, S., Benitez, B.A., Razquin, C., Goate, A., Ibero-American Alzheimer Disease Genetics Group, R and Cruchaga, C. (2012) Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res. Ther., 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W.W., Hegde, M., Lyon, E., Spector, E. et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kelly, M.P., Logue, S.F., Brennan, J., Day, J.P., Lakkaraju, S., Jiang, L., Zhong, X., Tam, M., Sukoff Rizzo, S.J., Platt, B.J. et al. (2010) Phosphodiesterase 11A in brain is enriched in ventral hippocampus and deletion causes psychiatric disease-related phenotypes. Proc. Natl. Acad. Sci. U. S. A., 107, 8457–8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grubman, A., Chew, G., Ouyang, J.F., Sun, G., Choo, X.Y., McLean, C., Simmons, R.K., Buckberry, S., Vargas-Landin, D.B., Poppe, D. et al. (2019) A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci., 22, 2087–2097. [DOI] [PubMed] [Google Scholar]
- 13. Fawcett, L., Baxendale, R., Stacey, P., McGrouther, C., Harrow, I., Soderling, S., Hetman, J., Beavo, J.A. and Phillips, S.C. (2000) Molecular cloning and characterization of a distinct human phosphodiesterase gene family: PDE11A. Proc. Natl. Acad. Sci. U. S. A., 97, 3702–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelly, M.P., Adamowicz, W., Bove, S., Hartman, A.J., Mariga, A., Pathak, G., Reinhart, V., Romegialli, A. and Kleiman, R.J. (2014) Select 3′,5′-cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cell. Signal., 26, 383–397. [DOI] [PubMed] [Google Scholar]
- 15. Wong, M.L., Whelan, F., Deloukas, P., Whittaker, P., Delgado, M., Cantor, R.M., McCann, S.M. and Licinio, J. (2006) Phosphodiesterase genes are associated with susceptibility to major depression and antidepressant treatment response. Proc. Natl. Acad. Sci. U. S. A., 103, 15124–15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coon, H., Darlington, T., Pimentel, R., Smith, K.R., Huff, C.D., Hu, H., Jerominski, L., Hansen, J., Klein, M., Callor, W.B. et al. (2013) Genetic risk factors in two Utah pedigrees at high risk for suicide. Transl. Psychiatry, 3, e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pathak, G., Agostino, M.J., Bishara, K., Capell, W.R., Fisher, J.L., Hegde, S., Ibrahim, B.A., Pilarzyk, K., Sabin, C., Tuczkewycz, T. et al. (2017) PDE11A negatively regulates lithium responsivity. Mol. Psychiatry, 22, 1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee, P., Ryoo, H., Park, J., Jeong, Y. and Alzheimer’s Disease Neuroimaging Initiative (2017) Morphological and microstructural changes of the hippocampus in early MCI: a study utilizing the Alzheimer’s Disease Neuroimaging Initiative database. J. Clin. Neurol., 13, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nie, X., Sun, Y., Wan, S., Zhao, H., Liu, R., Li, X., Wu, S., Nedelska, Z., Hort, J., Qing, Z. et al. (2017) Subregional structural alterations in hippocampus and nucleus accumbens correlate with the clinical impairment in patients with Alzheimer’s disease clinical spectrum: parallel combining volume and vertex-based approach. Front. Neurol., 8, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hegde, S., Capell, W.R., Ibrahim, B.A., Klett, J., Patel, N.S., Sougiannis, A.T. and Kelly, M.P. (2016) Phosphodiesterase 11A (PDE11A), enriched in ventral hippocampus neurons, is required for consolidation of social but not nonsocial memories in mice. Neuropsychopharmacology, 41, 2920–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pilarzyk, K., Klett, J., Pena, E.A., Porcher, L., Smith, A.J. and Kelly, M.P. (2019) Loss of function of phosphodiesterase 11A4 shows that recent and remote long-term memories can be uncoupled. Curr. Biol., 29, 2307–2321.e2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tibbo, A.J., Tejeda, G.S. and Baillie, G.S. (2019) Understanding PDE4’s function in Alzheimer's disease; a target for novel therapeutic approaches. Biochem. Soc. Trans., 47, 1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheng, J., Liao, Y., Dong, Y., Hu, H., Yang, N., Kong, X., Li, S., Li, X., Guo, J., Qin, L. et al. (2020) Microglial autophagy defect causes Parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy, 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fazio, P., Fitzer-Attas, C.J., Mrzljak, L., Bronzova, J., Nag, S., Warner, J.H., Landwehrmeyer, B., Al-Tawil, N., Halldin, C., Forsberg, A. et al. (2020) PET molecular imaging of phosphodiesterase 10A: an early biomarker of Huntington's disease progression. Mov. Disord., 35, 606–615. [DOI] [PubMed] [Google Scholar]
- 25. Farmer, R., Burbano, S.D., Patel, N.S., Sarmiento, A., Smith, A.J. and Kelly, M.P. (2020) Phosphodiesterases PDE2A and PDE10A both change mRNA expression in the human brain with age, but only PDE2A changes in a region-specific manner with psychiatric disease. Cell. Signal., 70, 109592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelly, M.P. (2015) Does phosphodiesterase 11A (PDE11A) hold promise as a future therapeutic target? Curr. Pharm. Des., 21, 389–416. [DOI] [PubMed] [Google Scholar]
- 27. Kelly, M.P. (2017) A role for phosphodiesterase 11A (PDE11A) in the formation of social memories and the stabilization of mood. Adv. Neurobiol., 17, 201–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li, Z., Farias, F.H.G., Dube, U., Del-Aguila, J.L., Mihindukulasuriya, K.A., Fernandez, M.V., Ibanez, L., Budde, J.P., Wang, F., Lake, A.M. et al. (2020) The TMEM106B FTLD-protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol., 139, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masutomi, H., Kawashima, S., Kondo, Y., Uchida, Y., Jang, B., Choi, E.K., Kim, Y.S., Shimokado, K. and Ishigami, A. (2017) Induction of peptidylarginine deiminase 2 and 3 by dibutyryl cAMP via cAMP-PKA signaling in human astrocytoma U-251MG cells. J. Neurosci. Res., 95, 1503–1512. [DOI] [PubMed] [Google Scholar]
- 30. Martinez, M., Fernandez, E., Frank, A., Guaza, C., de la Fuente, M. and Hernanz, A. (1999) Increased cerebrospinal fluid cAMP levels in Alzheimer’s disease. Brain Res., 846, 265–267. [DOI] [PubMed] [Google Scholar]
- 31. van der Harg, J.M., Eggels, L., Bangel, F.N., Ruigrok, S.R., Zwart, R., Hoozemans, J.J.M., la Fleur, S.E. and Scheper, W. (2017) Insulin deficiency results in reversible protein kinase A activation and tau phosphorylation. Neurobiol. Dis., 103, 163–173. [DOI] [PubMed] [Google Scholar]
- 32. Bonkale, W.L., Cowburn, R.F., Ohm, T.G., Bogdanovic, N. and Fastbom, J. (1999) A quantitative autoradiographic study of [3H]cAMP binding to cytosolic and particulate protein kinase A in post-mortem brain staged for Alzheimer’s disease neurofibrillary changes and amyloid deposits. Brain Res., 818, 383–396. [DOI] [PubMed] [Google Scholar]
- 33. Du, H., Guo, L., Wu, X., Sosunov, A.A., McKhann, G.M., Chen, J.X. and Yan, S.S. (2014) Cyclophilin D deficiency rescues Abeta-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim. Biophys. Acta, 1842, 2517–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kelly, M.P. (2018) Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal., 42, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang, H.H., Li, Y., Li, A., Yan, F., Li, Z.L., Liu, Z.Y., Zhang, L., Zhang, J., Dong, W.R. and Zhang, L. (2018) Forskolin induces hyperphosphorylation of tau accompanied by cell cycle reactivation in primary hippocampal neurons. Mol. Neurobiol., 55, 696–706. [DOI] [PubMed] [Google Scholar]
- 36. Carlyle, B.C., Nairn, A.C., Wang, M., Yang, Y., Jin, L.E., Simen, A.A., Ramos, B.P., Bordner, K.A., Craft, G.E., Davies, P. et al. (2014) cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc. Natl. Acad. Sci. U. S. A., 111, 5036–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Logue, M.W., Schu, M., Vardarajan, B.N., Farrell, J., Bennett, D.A., Buxbaum, J.D., Byrd, G.S., Ertekin-Taner, N., Evans, D., Foroud, T. et al. (2014) Two rare AKAP9 variants are associated with Alzheimer’s disease in African Americans. Alzheimers Dement., 10, 609–618.e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ikezu, T., Chen, C., DeLeo, A.M., Zeldich, E., Fallin, M.D., Kanaan, N.M., Lunetta, K.L., Abraham, C.R., Logue, M.W. and Farrer, L.A. (2018) Tau phosphorylation is impacted by rare AKAP9 mutations associated with Alzheimer disease in African Americans. J. Neuroimmune Pharmacol., 13, 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu, F., Liang, Z., Shi, J., Yin, D., El-Akkad, E., Grundke-Iqbal, I., Iqbal, K. and Gong, C.X. (2006) PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners. FEBS Lett., 580, 6269–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D. and Stadlan, E.M. (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology, 34, 939–944. [DOI] [PubMed] [Google Scholar]
- 41. McKhann, G.M., Knopman, D.S., Chertkow, H., Hyman, B.T., Jack, C.R., Jr., Kawas, C.H., Klunk, W.E., Koroshetz, W.J., Manly, J.J., Mayeux, R. et al. (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement., 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.