

Abstract
Background
Mycobacterium tuberculosis (Mtb), the bacterium that causes tuberculosis, causes 10 million infections and 1.5 million deaths per year worldwide. The success of Mtb as a human pathogen is directly related to its ability to suppress host responses, which are critical for clearing intracellular pathogens. Emerging evidence suggests that key response pathways may be regulated by a novel class of small noncoding RNA, called transfer RNA (tRNA)–derived fragments (tRFs). tRFs can complex with Argonaute proteins to target and degrade messenger RNA targets, similarly to micro RNAs, but have thus far been overlooked in the context of bacterial infections.
Methods
We generated a novel miRge2.0-based tRF-analysis tool, tRFcluster, and used it to analyze independently generated and publicly available RNA-sequencing datasets to assess tRF dysregulation in host cells following infection with Mtb and other intracellular bacterial pathogens.
Results
We found that Mtb and Listeria monocytogenes drive dramatic tRF dysregulation, whereas other bacterial pathogens do not. Interestingly, Mtb infection uniquely increased the expression of mitochondria-derived tRFs rather than genomic-derived tRFs, suggesting an association with mitochondrial damage in Mtb infection.
Conclusions
tRFs are dysregulated in some, but not all, bacterial infections. Biased dysregulation of mitochondria-derived tRFs in Mtb infection suggests a link between mitochondrial distress and tRF production.
Keywords: Mycobacterium tuberculosis, small RNAs, tRNA-derived fragments, tRFs, mitochondria, mitochondrial dysfunction
tRNA-derived fragments (tRFs) are dysregulated by intracellular bacterial infection. While tRFs can derive from the nuclear or mitochondrial genomes of infected cells, only Mycobacterium tuberculosis biases mitochondria-derived tRF production, suggesting unique mitochondrial distress during M. tuberculosis infection.
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is the leading cause of death due to a single infectious agent [1]. Its success as a human pathogen is due to its ability to evade host immune responses [2]. Mtb inhibits key antibacterial immune pathways, such as apoptosis, autophagy, phagosome-lysosome maturation, and cytokine signaling [3, 4], through various mechanisms, including altered expression of host microRNAs (miRNAs). miRNAs regulate expression of proteins in these pathways, thereby altering the outcome of infection [5–7].
Like miRNAs, transfer RNA (tRNA)–derived fragments (variably termed tRFs, tDRs, tiRNAs, or tRNA halves), are a diverse class of small noncoding RNAs, which are generated in response to host cell stress across a variety of disease states [8–10]. The term “tRFs” encompasses various subtypes of tRNA-derived molecules, including tRF-1, tRF-3, tRF-5, i-tRF, and tiRNAs. These tRFs may derive from the cell’s nuclear or mitochondrial genome. The human nuclear genome encodes 433 tRNAs and the mitochondrial genome encodes 22 mitochondrial tRNAs (mtRNAs), each of which serves as a source of tRFs [2]. tRFs block translation by interfering with the eIF4F complex and polysomes [11, 12]. As in the case of miRNAs, they may also complex with Argonaute machinery to regulate protein expression via translational repression and messenger RNA target degradation [13–16]. Several studies have reported increased tRF formation in cancer [17, 18] and viral infection [19]. However, the potential role of tRFs has not been studied during infection with Mtb or other bacterial pathogens.
Because it is known that Mtb dysregulates miRNAs, we hypothesized that Mtb may also significantly dysregulate the production of tRFs. We also predicted that patterns of tRF dysregulation in Mtb infection may differ from patterns observed during infection with other bacterial pathogens, such as Mycobacterium bovis, Salmonella enterica subspecies enterica serovar Typhimurium (S. Typhimurium), Listeria monocytogenes, and Yersinia pseudotuberculosis. Here, we tested these hypotheses by comparing tRF dysregulation in Mtb-infected primary human macrophages with dysregulation in infections with other intracellular bacterial pathogens from publicly available RNA-sequencing (RNA-seq) datasets. To determine tRF abundance, we developed and validated a new tRF-finding tool, tRFcluster, which was incorporated into our miRNA alignment tool, miRge2.0 [20].
MATERIALS AND METHODS
Ethical Approvals
This study was approved by the Johns Hopkins University Institutional Review Board.
Isolating Human Macrophages
Human platelet–depleted whole blood was obtained from healthy platelet donors at Anne Arundel Medical Center, Maryland. Primary human peripheral blood mononuclear cells were isolated using Ficoll-Paque (GE Healthcare, catalog number 17144003) density gradient centrifugation. Monocytes were isolated via plastic adherence in serum-free media for 4 hours. Following incubation, nonadherent lymphocytes and residual red blood cells were removed by 5 washes in 1× phosphate-buffered saline. Adherent monocytes were allowed to differentiate into monocyte-derived macrophages (MDMs) for 1 week in complete media (RPMI 1640 + 4 mM L-glutamine + 10% fetal bovine serum).
Mtb Infection
Primary human MDMs from each donor were infected with H37Rv-lux [21] for 24 or 48 hours (multiplicity of infection [MOI] = 5 or 10). To avoid altering cellular transcriptional responses, extracellular bacteria were not removed by washes or antibiotic treatment. Infected cells were incubated at 37°C with 5% carbon dioxide. At each time point, matched wells were used for assessment of MDM viability, bacterial burden, and collection of RNA for small RNA-seq (sRNA-seq) library preparation.
Assessment of Bacterial Burden
H37Rv-lux contains an integrated bacterial luciferase operon (LuxAB), including genes encoding the enzyme luciferase and its substrate luciferin under control of a constitutive promoter [21]. Therefore, relative luminescent units can be used as a real-time readout for bacterial burden, which is directly proportional to colony-forming units. Triplicate MDMs for each donor were lysed in 0.05% sodium dodecyl sulfate and immediately read in a single tube luminometer (Promega GloMax 20/20) (Supplementary Figure 2).
Assessment of MDM Viability
MDM viability was determined by MTS assay (Promega, CellTiter 96 AQueous One Solution Cell Proliferation Assay). At 24 and 48 hours postinfection, triplicate wells were incubated with MTS reagent for 4 hours, following manufacturer protocol. Reactions were read in a BMG Labtech Optima Fluorescence Microplate Reader (Supplementary Figure 3).
RNA Extraction and sRNA-seq Library Preparation
RNA was extracted with the miRNeasy Mini Kit (Qiagen, catalog number 217004), following manufacturer instructions. RNA was converted into sRNA-seq libraries using the QIAseq miRNA Library Kit (Qiagen) following kit protocol. RNA and library quantity and quality were assessed by fragment analyzer at the Johns Hopkins DNA Services Core Facility.
sRNA Sequencing of Mtb-Infected Human MDMs
Libraries were pooled to a concentration of 1 ng/μL for sRNA-seq. Sequencing was performed on an Illumina NextSeq 500 at the Transcriptomics and Deep Sequencing Core Facility at Johns Hopkins University. Samples were sequenced with sufficient depth to achieve at least 1 million reads per sample. Sequencing run quality, including total reads, trimmed reads, and number of reads per type of small RNA, was assessed using miRge2.0. All samples were sequenced within 1 run to avoid batch effects.
Analyzing tRF Reads Through miRge2.0
FASTQ files were analyzed in miRge 2.0 for tRFs calling the –trf function. tRF-related output comprises 1 tRF folder showing all exact alignments and 3 tRF files: tRF.Counts.csv, tRF.RP100K.csv, and tRFs.potential.report.tsv reporting counts.
Exclusion of Unwanted Variation Normalization of tRF Samples
The remove-unwanted-variation algorithm [22] using replicate samples (RUVSeq R/BioC package) was used to estimate 20 latent factors separately for adjustment of batch effects in the 345 samples described in the Supplementary Extended Methods (Supplementary Table 1).
t-Distributed Stochastic Neighbor Embedding
The t-distributed stochastic neighbor embedding (t-SNE) was performed using Rtsne package in R on RUV-normalized data with perplexity set at 15 after evaluations of perplexity values of 5–100 [23]. All RUV data were log2-transformed.
Analysis of Differential Expression of tRFs
DESeq2 was used for differential analysis of tRFs [24]. Benjamini–Hochberg correction was performed. Significantly differentially expressed tRFs were applied to hierarchical clustering, where pairwise distance was based on Euclidean distance. Average linkage clustering was used as agglomeration criteria.
Use of Comparison Tools
tDRmapper and MINTmap [25, 26] were installed locally and used to detect tRFs for the samples in Supplementary Table 1. The parameters were set as default.
Generating a New miRge2.0-Based tRF Detection Method, tRFcluster
Four hundred thirty-three mature tRNA sequences were downloaded from the GtRNAdb database (release 2.0) [2]. The 22 human mtRNA sequences were obtained from MINTmap [26].
The sRNA-seq FASTQ files from various primary human cell types were obtained from Bioprojects PRJNA358331, PRJNA391912, and PRJNA385925 [27–29]. Samples with <10 000 tRNA-mapped reads were excluded, resulting in 711 samples for further analysis (Supplementary Table 2).
Input FASTQ file(s) were analyzed using the standard miRge2.0 workflow modified only to have separate alignment to mature tRNA and precursor tRNA 3′-trailer [20, 30]. Due to known RNA editing of tRNAs, the search strategy employed allowed 1 mismatch to mature tRNA sequences. For tRF-1 alignments to the pre-tRNA tail, reads are required to have poly-T (≥ 3 “Ts”) tails and no mismatch in the read sequence region where the poly-Ts are excluded. Since some sRNA-seq reads can be mapped equally to several tRNA families, all best alignments to mature tRNA and precursor tRNA 3′-trailers were reported (Supplementary Figure 1).
Clustering Potential tRFs by Means of Density Peak Clustering
tRFcluster uses an unsupervised clustering algorithm to detect centers with local maxima in the density as a basis for assigning clusters. The cluster centers are empirically determined when local density ρ i is ≥ rhomin 5.0 and ≥ deltamin 8.0.
The most abundant read in any given cluster is determined as the representative tRF, and the tRF read count and RP100K values are the sum of the read counts and RP100K values of all reads in the cluster core.
Refining tRF Entities From tRF Clusters
The 711 sRNA-seq samples generated 36 901 possible tRF clusters across the tRNA libraries. These were secondarily collapsed together using the same clustering method, as described above, resulting in 2463 tRFs. The sequence and genomic loci information of these tRF clusters is listed in Supplementary Table 3. Many of the tRFs had high similarity, with equal sequences but of different lengths (for example, having sequence lengths of 24 vs 25 base pairs), or differing by a single nucleotide. Using a pairwise distance strategy, 540 tRF entities were generated. The entities and their tRF members are listed in Supplementary Table 4. The i-tRF, tRF-1, -3, and -5, 5′ half and 3′ half assignments were made on these 540 tRFs and all halves were ≥31 nt (further details are shown in the Supplementary Extended Methods).
Biologic sRNA-seq Datasets
To validate this tRF tool, FASTQ files of 9 cell types (macrophage, B lymphocyte, T lymphocyte, fibroblast, endothelial cell, retinal pigment epithelial cell, smooth muscle cell, monocyte, and neural stem cell) were chosen because they were represented in 2 or more of BioProjects PRJNA358331, PRJNA391912, or PRJNA385925 [27–29] (n = 345) (Supplementary Table 1).
Raw data from human sRNA-seq studies in cell culture models involving bacteria or hypoxia were mined from public National Center for Biotechnology Information BioProjects PRJNA206504, PRJNA480576, PRJNA298741, PRJNA297139, and PRJNA270244 (Supplementary Table 5). FASTQ files were analyzed using tRFcluster in miRge2.0. Samples were compared across studies with DESeq2 (R).
RESULTS
Creation and Validation of tRFcluster
Due to the complexity of tRNA structure, we currently lack a widely accepted nomenclature for cataloging tRFs. Our new miRge2.0-based tool, tRFcluster, which uses a clustering approach optimized to account for tRF sequence complexities, was validated using 711 samples (Table 1 and Supplementary Figure 1). After alignment to 455 human tRNA sequences, reads were clustered together. Clusters of 2461 unique tRFs were collapsed into 540 tRF entities comprising tRFs and tRNA halves (Table 1). Each entity has between 1 and 33 clusters (average 4.5) assigned to it, with approximately 50% having only 1 specific sequence. Eight entities had sequences that could be aligned to >1 tRNA type. This new clustering method balances the total number of tRFs to be counted with appropriate alignment that accounts for sequences mapping to >1 genetic locus.
Table 1.
Comparison of tRFcluster, MINTmap, tDRmapper, tRFfinder, and tRFdb
| Characteristic | tRFcluster | MINTmap | tDRmapper | tRFfinder | tRFdb |
|---|---|---|---|---|---|
| Runs locally | Yes | Yes | Yes | No | No |
| Catalogs tRFs and miRNAs | Yes | No | No | No | No |
| No. of human tRFs | 2461a/540b | 594 972 | NA | NA | 552 |
| tRF-5 | Yes | Yes | Yes | Yes | Yes |
| tRF-3 | Yes | Yes | Yes | Yes | Yes |
| i-tRF | Yes | Yes | Yes | No | Yes |
| tRF-1 | Yes | No | Yes | Yes | Yes |
Abbreviations: miRNA, micro RNA; NA, not applicable; tRF, transfer RNA–derived fragment.
aPrecollapsed unique tRFs.
bPostcollapsed tRF combinations.
We then validated tRFcluster against MINTmap and tDRmapper to test our ability to generate robust and meaningful analyses of sRNA-seq data. We determined the distributions of tRFs in 345 human primary cell samples, comprising 9 cell types, across tRFcluster, MINTmap, and tDRmapper (Table 2). We found that the most abundant tRF subtype was 5′ tRNA halves (31%) (Figure 1A). The most abundant tRFs came from valine, glycine, glutamine, and lysine tRNAs (Figure 1B). Finally, using a t-SNE, we found that tRFcluster demonstrated good clustering agreement of similar cell types that was comparable to MINTmap and superior to tDRmapper (Supplementary Figure 4). These data suggest that our goal of developing a robust new tRF analysis tool that captures a high number of tRF reads in a reasonable number of entities was achieved.
Table 2.
Depth of Transfer RNA–Derived Fragment Discovery Across 3 Platforms Over 345 Samples
| Tool | Alignment Strategy | Total Read Counts of tRFs | Total Read Counts of tRFs After Filter | Proportion of tRF Reads After Filter | Total tRF Counts | Total tRF Counts After Filter |
|---|---|---|---|---|---|---|
| tRFcluster | 1 mismatch | 176 584 695 | 175 683 293 | 0.99 | 540 | 324 |
| MINTmap | 0 mismatch | 143 792 494 | 133 141 746 | 0.93 | 79 404 | 858 |
| tDRmapper | 2 mismatches and 3 deletions | 193 952 012 | 187 599 715 | 0.97 | 1469 | 374 |
Abbreviation: tRF, transfer RNA–derived fragment.
Figure 1.

Features of transfer RNA (tRNA)–derived fragments (tRFs) from 345 cell samples. A, Distribution of tRNA fragments (tRFs and tRNA halves) showing that 5′ halves and tRF-3 segments are the most abundant. B, Distribution of tRF entities by tRNA family. Abundance of tRF reads does not fully correlate with number of tRF entities by tRNA family.
tRFs Show Pathogen-Specific Patterns of Alteration During Infection
Using tRFcluster, we examined how tRFs differed in infection with Mtb compared to other intracellular bacterial pathogens. We infected primary human MDMs ex vivo with virulent Mtb strain H37Rv-lux [21]. Small RNAs from uninfected MDMs and MDMs infected at MOIs of 5 or 10 were sequenced and analyzed at 24 and 48 hours postinfection. We found that tRFs were significantly dysregulated by infection with Mtb at all MOIs and time points (Figure 2A). To validate these findings, we performed the same tRF analysis using a publicly available sRNA-seq dataset of dendritic cells that had been infected with virulent Mtb strain H37Rv (lineage 4) or Mtb Beijing strain GC1237 (lineage 2) [31]. We found strikingly similar patterns of tRF dysregulation between our own dataset and the publicly available Mtb dataset (Figure 2B). Additionally, H37Rv and Beijing strains did not cluster separately, suggesting that Mtb infection drives significant lineage-independent changes in tRF production.
Figure 2.

Virulent strains of Mycobacterium tuberculosis (Mtb), but not Mycobacterium bovis, significantly dysregulate transfer RNA (tRNA)–derived fragment (tRF) production. A, tRF changes between uninfected and primary human monocyte–derived macrophages. Top bar indicates infection type. Uninfected (black), or infected with Mtb strain H37Rv-lux at a multiplicity of infection (MOI) of 5 (green) or 10 (red). Identity of each of the 4 biological replicates (donors) is color coded by the second horizontal bar to show interdonor variability. B, Assessment of tRFs in infection with virulent Mtb strains from publicly available dataset (SRP051119). tRFs were assessed in uninfected dendritic cells (DCs) (black) vs DCs infected with Mtb strain H37Rv (green) or Beijing GC1237 (red) for 48 hours. C, Assessment of tRFs in infection with nontuberculous mycobacterial M. bovis strain BCG from publicly available dataset (SRP051119). tRFs were compared between uninfected DCs and DCs infected with BCG for 48 hours.
Unlike other intracellular pathogens, Mtb possesses unique mechanisms of immune evasion, allowing it to survive within host cells [32–34]. Therefore, we hypothesized that Mtb-induced patterns of tRF dysregulation may be distinct from those of other, less virulent intracellular bacterial pathogens. To test this, we analyzed publicly available datasets containing sRNA-seq data of cells infected with M. bovis, S. Typhimurium, L. monocytogenes, and Y. pseudotuberculosis [31, 35]. We then used tRFcluster to examine tRF dysregulation during each of these infection conditions.
Interestingly, compared to virulent strains of Mtb, M. bovis strain BCG, a live-attenuated strain of M. bovis used as a vaccine for TB, did not cluster based on infection status (Figure 2C). This suggests that tRFs are not significantly dysregulated by infection with BCG and implies that there may be a connection between degree of virulence and tRF dysregulation. We next examined tRF dysregulation in nonmycobacterial infections. We found that L. monocytogenes drove the strongest changes in tRF dysregulation. In contrast, neither S. Typhimurium nor Y. pseudotuberculosis showed significant tRF dysregulation (Figure 3). Interestingly, all species examined here are known to alter miRNA expression [31, 35]. However, our data show that only some dysregulate tRFs. This suggests that tRF dysregulation may be species-specific and independent of miRNA dysregulation.
Figure 3.

Listeria monocytogenes shows stark transfer RNA–derived fragment (tRF) dysregulation compared to other nonmycobacterial intracellular bacterial pathogens. A, tRF production was compared for uninfected macrophages (black) vs macrophages infected with Salmonella Typhimurium (green) or Listeria monocytogenes (red) for 24 hours. Raw data from publicly available dataset (SRP064235). B, tRFs were compared between uninfected macrophages (black) and macrophages infected with S. Typhimurium (green) or Yersinia pseudotuberculosis (red) for 48 hours. Raw data from publicly available dataset (SRP051119).
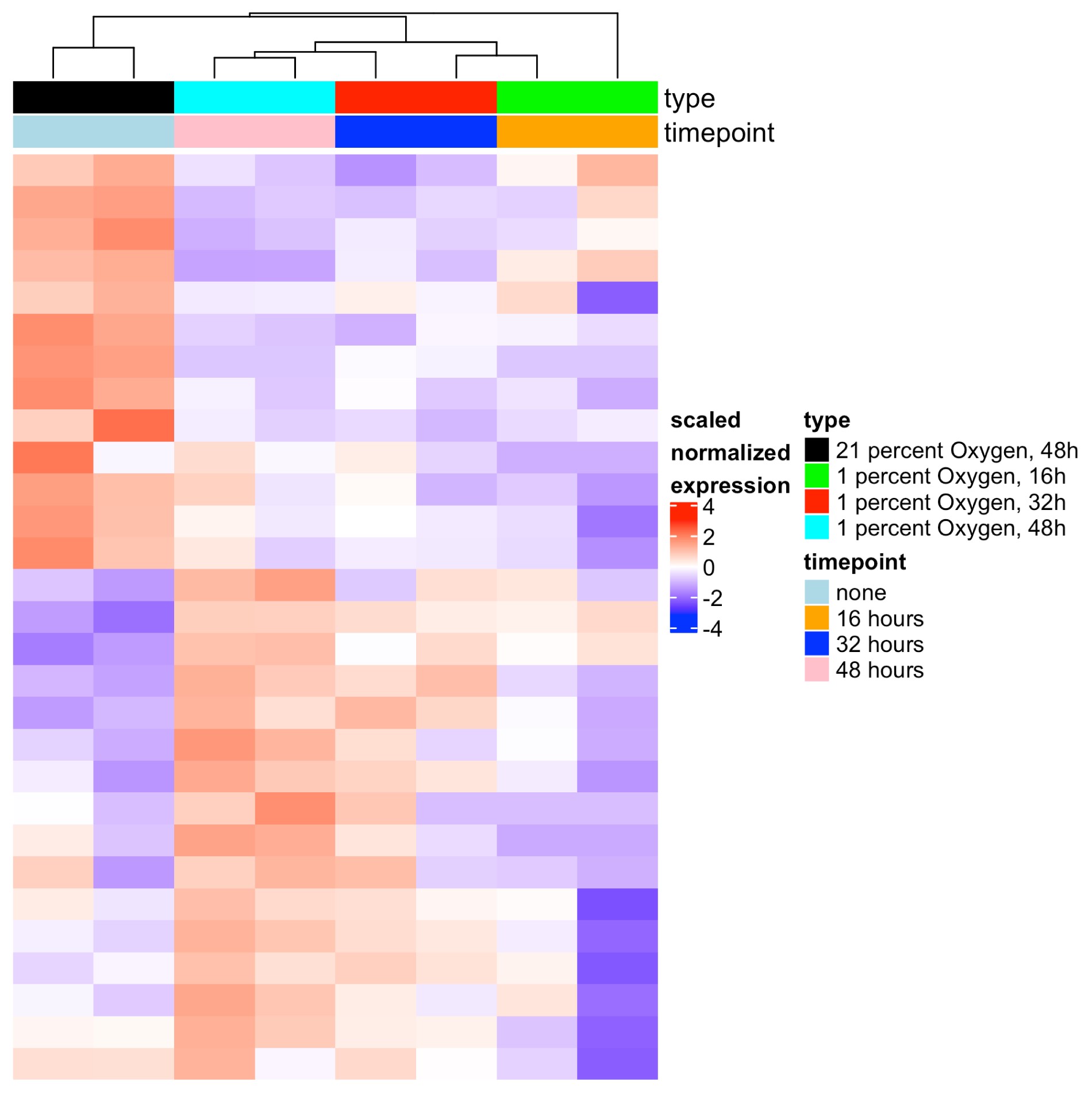
Finally, we sought to compare Mtb infection–mediated tRF dysregulation with that of other TB pathology–related cell stresses. As hypoxia is known to be an important stress encountered by Mtb within necrotic lung granulomas and is known to drive tRF dysregulation [36, 37], we selected this as our noninfectious comparison. We found that hypoxic stress led to alterations in tRF production that increased with degree of hypoxic condition (Supplementary Figures 5 and 6). While this is unlikely to be relevant to the tRF dysregulation we observe in ex vivo Mtb infections in primary human MDMs, it may be important to understand in the context of how cells respond to disease within a human host.
tRF Dysregulation Increases With Severity of Mtb Infection
Next, we sought to determine whether there was a relationship between tRF production and intracellular bacillary burden and/or duration of Mtb infection. To test this, we assessed tRF production for all Mtb-infected samples and for each individual condition, and compared them to matched uninfected controls. When considering all samples without distinguishing between MOI or time postinfection, we found 66 tRFs that met our thresholds for significant dysregulation (log2 [fold-change] > 1; adjusted P < .01) (Figure 4A and Supplementary Table 6).
Figure 4.

Transfer RNA–derived fragment (tRF) dysregulation increases with severity of infection with Mycobacterium tuberculosis (Mtb). A, Volcano plot showing differentially produced tRFs in Mtb-infected primary human monocyte–derived macrophages. tRFs from 4 infection conditions (multiplicity of infection [MOI] of 5–24 hours, MOI of 5–48 hours, MOI of 10–24 hours, and MOI of 10–48 hours) are compared to tRFs from uninfected cells at matched time points. Red lines indicate significance cutoffs (log2 [fold change] > 1; adjusted P < .01). Red points indicate tRFs that are significantly dysregulated between infected and uninfected conditions. Red points indicate tRFs that are significantly dysregulated between infected and uninfected conditions. Red points indicate tRFs that are significantly dysregulated between infected and uninfected conditions (66 total). B, Volcano plots showing differential production of tRFs in each independent infection condition separated by MOI and time postinfection. Significance indicators are the same as in (A). Red bolded numbers in upper left corner of each volcano plot represent the total number of significantly dysregulated tRFs. C, Venn diagram showing the number of significantly dysregulated tRFs shared in each infection condition. Bolded numbers outside each oval represent the MOI and time point (in parentheses).
The number of dysregulated tRFs increased within each MOI between 24 and 48 hours postinfection. A similar increase was observed within each time point with increasing MOI (Figure 4B). Importantly, many of the tRFs dysregulated by less severe infection conditions were also significantly dysregulated in the more severe conditions. Five of 6 tRFs that were significantly different at an MOI of 5 at 24 hours were also observed at 48 hours. All of the tRFs that were significantly different at an MOI of 10 at 24 hours were also significantly dysregulated at 48 hours. Cells infected at MOIs of 5 and 10 shared approximately 50% of their significantly different tRFs at 48 hours (Figure 4C, Supplementary Tables 7–10). Importantly, all tRFs that were significantly dysregulated in >1 condition were dysregulated in the same direction (Supplementary Figure 7). The consistent dysregulation of these tRFs suggests that their altered abundance likely reflects a true biological phenomenon.
Mtb Infection Is Associated With a Bias Toward Dysregulation of Mitochondria-Derived tRFs
Given the stark differences in tRF dysregulation among various bacterial species, and the propensity for Mtb to induce mitochondrial distress, we next sought to assess the origin of significantly dysregulated tRFs by determining whether the sequence originated from the host nuclear or mitochondrial genome. We then compared the percentage of significantly dysregulated mitochondrial-derived tRFs (mtRFs) from our own dataset to that from publicly available datasets generated for host cells infected by Mtb, L. monocytogenes, Y. pseudotuberculosis, and S. Typhimurium. Hypoxia in MCF7 cells was again used as a noninfectious control. In addition to its relevance to TB disease, hypoxia also alters mitochondrial function [38], allowing us to determine whether mtRF dysregulation is specific to Mtb infection or simply due to general stress-induced mitochondrial dysfunction. Interestingly, we found that Mtb infection was associated with a strong bias toward mtRFs, whereas other intracellular bacterial pathogens and hypoxia were not. Of the significantly dysregulated tRFs in Mtb infection, 59%–76% originated from mitochondria. For datasets that showed clear clustering of tRFs based on infection status (Mtb and L. monocytogenes), we examined mtRF dysregulation among all time points, as well as at the last time point only. The last time points were analyzed independently, as tRF dysregulation increases with time postinfection. For datasets that did not show clustering, we included all time points, as time postinfection did not significantly alter tRF dysregulation. Analysis of our independently generated dataset showed that mtRFs accounted for 66.7% of all dysregulated tRFs (χ 2 = 57.20; P = 3.94 × 10–14) across all time points and 59% at 48 hours postinfection (χ 2 = 24.20, P = 8.60 × 10–7). Similarly, the analysis of the publicly available Mtb dataset showed that mtRFs accounted for 76% of all dysregulated tRFs (χ 2 = 104.00, P = 2.20 × 10–16) across all time points and 69% at 48 hours postinfection (χ 2 = 80.86, P = 2.20 × 10–16). In contrast, mtRFs were less common in other intracellular bacteria and hypoxia. Listeria monocytogenes was the only other organism that achieved statistical significance, though the percentage of mtRFs was much lower than in Mtb infections (39%; P = 2.05 × 10–3; Table 3). This suggests that Mtb infections bias toward tRF production from mtRNAs.
Table 3.
Transfer RNA–Derived Fragments Dysregulated in Mycobacterial Infections Are Primarily of Mitochondrial Origin
| Characteristic | Time, h | mtRF | Non-mtRF | % mtRF | χ 2 | P Value |
|---|---|---|---|---|---|---|
| Mtb | All | 57 | 18 | 76.0 | 1.04 × 102 | 2.20 × 10–16 |
| Mtb | 48 | 54 | 24 | 69.2 | 8.09 × 101 | 2.20 × 10–16 |
| Mtb (Current study) | All | 44 | 22 | 66.7 | 5.72 × 101 | 3.94 × 10–14 |
| Mtb (Current study) | 48 | 27 | 19 | 58.7 | 2.42 × 101 | 8.60 × 10–7 |
| Listeria monocytogenes | 24 | 45 | 72 | 38.5 | 9.51 × 100 | 2.05 × 10–3 |
| Hypoxia MCF7 cells | All | 29 | 66 | 30.5 | 5.80 × 10–1 | 4.46 × 10–1 |
| L. monocytogenes | All | 17 | 44 | 27.9 | 1.40 × 10–3 | 9.70 × 10–1 |
| Yersinia pseudotuberculosis | All | 3 | 21 | 12.5 | 1.92 × 100 | 1.65 × 10–1 |
| Salmonella Typhimuriuma | All | 1 | 11 | 0.1 | NA | NA |
| BCGa | All | 0 | 1 | 0 | NA | NA |
Genomic origins of significantly dysregulated transfer RNA–derived fragments (tRFs) for each dataset were analyzed using miRge2.0 (Supplementary Table 11). Percentages of significantly dysregulated tRFs for each dataset that originated from the mitochondrial genome (% mtRF) are reported.
Abbreviations: Mtb, Mycobacterium tuberculosis; mtRF, mitochondrial transfer RNA–derived fragment; NA, not applicable.
aχ 2 analysis was excluded due to insufficient data points.
DISCUSSION
Until now, tRFs have not been assessed in the context of bacterial infections. We found that Mtb drives significant dysregulation of tRFs and that tRF dysregulation increased with severity of infection. Listeria monocytogenes induced a pattern of tRF dysregulation that was similar to that seen in Mtb infection. However, other bacteria tested here did not significantly alter tRF abundance.
Only Mtb infection drove strongly biased dysregulation of mtRFs. This may reflect the severity of mitochondrial distress during Mtb infection, manifested by disruption of membrane potential and a shift toward necrosis, which is associated with mitochondrial swelling, mitochondrial outer membrane permeabilization, and bacterial release and survival [39–43]. These changes to mitochondrial membrane architecture can allow movement of certain protein and nucleic acid factors across the membrane [44, 45]. We predict that angiogenin (ANG), one of the RNAses that cleaves tRFs, and/or mtRNAs may be among these factors. Under normal conditions, ANG is sequestered to the nucleus. However, during cell stress, ANG is translocated to the cytoplasm [46]. Mtb infection induces cell stress and may drive translocation of ANG out of the nucleus. Given this, we have developed 2 hypotheses that may help explain the overabundance of mtRFs produced in Mtb infection: (1) mtRNAs are cleaved within the mitochondria after ANG (or other tRF-cleaving RNAses) is translocated into the mitochondria; or (2) intact mtRNAs are released from mitochondria into the cytoplasm, allowing them to be cleaved by ANG.
Reasons for the lack of mtRF-biased dysregulation in other infections remain unclear, but may be related to the duration of mitochondrial dysfunction in each infection. Host cells infected with Mtb exhibit robust, stable signs of mitochondrial distress at least through 24 hours [40, 41]. This may allow for sustained interaction between mtRNAs and ANG and could result in increased production of mtRFs. In contrast, although L. monocytogenes and S. Typhimurium induce mitochondrial fragmentation, this effect is transient and mitochondria recover within 24 hours [47, 48]. There is no clearly defined link between Y. pseudotuberculosis infection and mitochondrial distress. It is, therefore, possible that the degree of mtRF dysregulation is proportional to the duration and extent of mitochondrial distress. However, additional research is required to elucidate this mechanism.
Future studies will investigate the biological mechanisms underlying these findings, including the localization of mtRFs, ANG, and other relevant RNases during infection. Specifically, we will determine if these entities colocalize and interact with markers of mitochondrial distress. We will also assess whether blocking mitochondrial damage and necrosis may prevent or alter the production of mtRFs and potentially modulate the overall outcome of infection.
In this study, we also designed tRFcluster as a module to miRge2.0. This tool allows simultaneous identification of miRNAs and tRFs for downstream analyses. Herein, we validated our clustering approach for these complicated reads, successfully identified tRF clusters, and had performance at least equivalent to stand-alone tRF tools. Our approach is intermediate between MINTmap/MINTbase version 2.0, which take a maximalist approach in uniquely identifying and naming each sequence aligning to tRNA, resulting in 23 413 human tRFs, and the minimalist approach of tRFdb, which lists only 552 human tRF sequences [26, 49, 50]. Despite our bacterial focus, this tool should have generalizable utility for analysis of tRF production and regulation in virtually any biological condition or disease state.
Limitations of this study include the inherent variability in experimental conditions between datasets. Factors that may have impacted comparisons are different cell types, library preparation protocols, and equipment for sRNA-seq. However, despite these methodological differences, we observed good consistency between our Mtb dataset and the publicly available Mtb dataset, suggesting that our results are reproducible and biologically relevant.
Overall, our results suggest that host cell tRFs are dysregulated by infection with certain bacterial pathogens, but the particular pattern is dependent on the infecting bacterial species. Additionally, our tRF data suggest that Mtb infection may alter mitochondrial physiology in such a way that creates a unique opportunity for interaction between mtRNAs and tRF-cleaving enzymes.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
Acknowledgments. We thank Laura Kasch-Semenza of the Johns Hopkins DNA Services Core Facility and Haiping Hao of the Johns Hopkins Transcriptomics and Deep Sequencing Core Facility for their assistance on this project.
Financial support. This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (grant numbers UH3AI122309 and K24AI143447 to P. C. K.).
Potential conflicts of interest. All authors: No reported conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Presented in part: Tuberculosis: Immunity and Immune Evasion Keystone Meeting, Santa Fe, New Mexico, January 2020. Abstract 2034.
References
- 1. World Health Organization. Global tuberculosis report. Geneva, Switzerland: WHO, 2019. [Google Scholar]
- 2. The Lowe Lab. GtRNAdb tRNAscan-SE analysis of complete genomes. Vol. 18.1. Santa Cruz, CA: The Lowe Lab, 2019. [Google Scholar]
- 3. Pahari S, Kaur G, Negi S, et al. . Reinforcing the functionality of mononuclear phagocyte system to control tuberculosis. Front Immunol 2018; 9:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Martino M, Lodi L, Galli L, Chiappini E. Immune response to Mycobacterium tuberculosis: a narrative review. Front Pediatr 2019; 7:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Das K, Garnica O, Dhandayuthapani S. Modulation of host miRNAs by intracellular bacterial pathogens. Front Cell Infect Microbiol 2016; 6:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ryan B, Joilin G, Williams JM. Plasticity-related microRNA and their potential contribution to the maintenance of long-term potentiation. Front Mol Neurosci 2015; 8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harapan H, Fitra F, Ichsan I, et al. . The roles of microRNAs on tuberculosis infection: meaning or myth? Tuberculosis (Edinb) 2013; 93:596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mesitov MV, Soldatov RA, Zaichenko DM, et al. . Differential processing of small RNAs during endoplasmic reticulum stress. Sci Rep 2017; 7:46080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fu H, Feng J, Liu Q, et al. . Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett 2009; 583:437–42. [DOI] [PubMed] [Google Scholar]
- 10. Anderson P, Ivanov P. tRNA fragments in human health and disease. FEBS Lett 2014; 588:4297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamasaki S, Ivanov P, Hu GF, Anderson P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol 2009; 185:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sobala A, Hutvagner G. Small RNAs derived from the 5′ end of tRNA can inhibit protein translation in human cells. RNA Biol 2013; 10:553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010; 16:673–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Z, Ender C, Meister G, Moore PS, Chang Y, John B. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res 2012; 40:6787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuscu C, Kumar P, Kiran M, Su Z, Malik A, Dutta A. tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. RNA 2018; 24:1093–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martinez G, Choudury SG, Slotkin RK. tRNA-derived small RNAs target transposable element transcripts. Nucleic Acids Res 2017; 45:5142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 2016; 165:1416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brubaker PE, Moran JP, Bridbord K, Hueter FG. Noble metals: a toxicological appraisal of potential new environmental contaminants. Environ Health Perspect 1975; 10:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruggero K, Guffanti A, Corradin A, et al. . Small noncoding RNAs in cells transformed by human T-cell leukemia virus type 1: a role for a tRNA fragment as a primer for reverse transcriptase. J Virol 2014; 88:3612–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu Y, Baras AS, Halushka MK. miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinformatics 2018; 19:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andreu N, Zelmer A, Fletcher T, et al. . Optimisation of bioluminescent reporters for use with mycobacteria. PLoS One 2010; 5:e10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 2014; 32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laurens van der Maaten GH. Visualizing data using t-SNE. J Mach Learn Res 2008; 9:2579–605. [Google Scholar]
- 24. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Selitsky SR, Sethupathy P. tDRmapper: challenges and solutions to mapping, naming, and quantifying tRNA-derived RNAs from human small RNA-sequencing data. BMC Bioinformatics 2015; 16:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loher P, Telonis AG, Rigoutsos I. MINTmap: fast and exhaustive profiling of nuclear and mitochondrial tRNA fragments from short RNA-seq data. Sci Rep 2017; 7:41184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Juzenas S, Venkatesh G, Hübenthal M, et al. . A comprehensive, cell specific microRNA catalogue of human peripheral blood. Nucleic Acids Res 2017; 45:9290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Rie D, Abugessaisa I, Alam T, et al. . FANTOM Consortium . An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat Biotechnol 2017; 35:872–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCall MN, Kim MS, Adil M, et al. . Toward the human cellular microRNAome. Genome Res 2017; 27:1769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baras AS, Mitchell CJ, Myers JR, et al. . miRge—a multiplexed method of processing small RNA-seq data to determine microRNA entropy. PLoS One 2015; 10:e0143066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siddle KJ, Tailleux L, Deschamps M, et al. . Bacterial infection drives the expression dynamics of microRNAs and their isomiRs. PLoS Genet 2015; 11:e1005064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu CH, Liu H, Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol 2017; 14:963–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhai W, Wu F, Zhang Y, Fu Y, Liu Z. The immune escape mechanisms of Mycobacterium tuberculosis. Int J Mol Sci 2019; 20:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 2006; 124:767–82. [DOI] [PubMed] [Google Scholar]
- 35. Pai AA, Baharian G, Pagé Sabourin A, et al. . Widespread shortening of 3’ untranslated regions and increased exon inclusion are evolutionarily conserved features of innate immune responses to infection. PLoS Genet 2016; 12:e1006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cui Y, Huang Y, Wu X, et al. . Hypoxia-induced tRNA-derived fragments, novel regulatory factor for doxorubicin resistance in triple-negative breast cancer. J Cell Physiol 2019; 234:8740–51. [DOI] [PubMed] [Google Scholar]
- 37. Prosser G, Brandenburg J, Reiling N, Barry CE 3rd, Wilkinson RJ, Wilkinson KA. The bacillary and macrophage response to hypoxia in tuberculosis and the consequences for T cell antigen recognition. Microbes Infect 2017; 19:177–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fuhrmann DC, Brüne B. Mitochondrial composition and function under the control of hypoxia. Redox Biol 2017; 12:208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dubey RK. Assuming the role of mitochondria in mycobacterial infection. Int J Mycobacteriol 2016; 5:379–83. [DOI] [PubMed] [Google Scholar]
- 40. Jamwal S, Midha MK, Verma HN, Basu A, Rao KV, Manivel V. Characterizing virulence-specific perturbations in the mitochondrial function of macrophages infected with Mycobacterium tuberculosis. Sci Rep 2013; 3:1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abarca-Rojano E, Rosas-Medina P, Zamudio-Cortéz P, Mondragón-Flores R, Sánchez-García FJ. Mycobacterium tuberculosis virulence correlates with mitochondrial cytochrome c release in infected macrophages. Scand J Immunol 2003; 58:419–27. [DOI] [PubMed] [Google Scholar]
- 42. Dallenga T, Repnik U, Corleis B, et al. . M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe 2017; 22:519–30.e3. [DOI] [PubMed] [Google Scholar]
- 43. Divangahi M, Behar SM, Remold H. Dying to live: how the death modality of the infected macrophage affects immunity to tuberculosis. Adv Exp Med Biol 2013; 783:103–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zorova LD, Popkov VA, Plotnikov EY, et al. . Mitochondrial membrane potential. Anal Biochem 2018; 552:50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Samali A, Fulda S, Gorman AM, Hori O, Srinivasula SM. Cell stress and cell death. Int J Cell Biol 2010; 2010:245803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu W, Goncalves KA, Li S, et al. . Plexin-B2 mediates physiologic and pathologic functions of angiogenin. Cell 2017; 171:849–64.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci U S A 2011; 108:3612–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hos NJ, Ganesan R, Gutiérrez S, et al. . Type I interferon enhances necroptosis of Salmonella Typhimurium–infected macrophages by impairing antioxidative stress responses. J Cell Biol 2017; 216:4107–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pliatsika V, Loher P, Magee R, et al. . MINTbase v2.0: a comprehensive database for tRNA-derived fragments that includes nuclear and mitochondrial fragments from all the Cancer Genome Atlas projects. Nucleic Acids Res 2018; 46:D152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kumar P, Mudunuri SB, Anaya J, Dutta A. tRFdb: a database for transfer RNA fragments. Nucleic Acids Res 2015; 43:D141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.