
Figure 8.
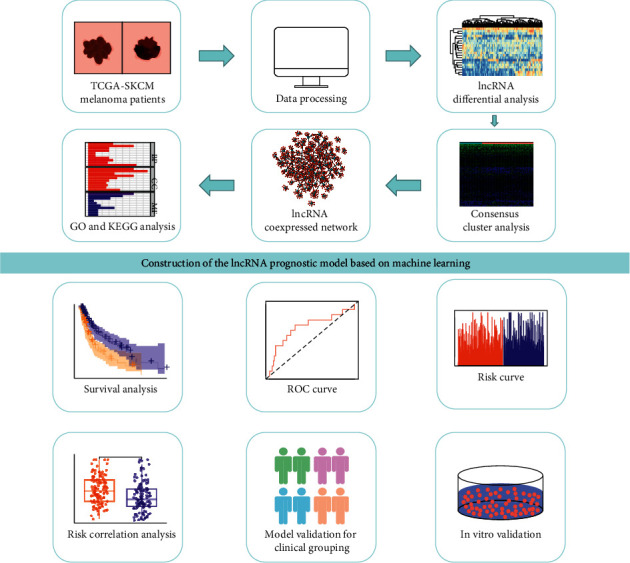
Flow chart of this study. First, RNA expression data, clinical follow-up data, and gene mutation data of melanoma patients were downloaded from the TCGA database. After data processing, two groups of samples were screened out according to the number of somatic cell mutations, and the differences were analyzed. According to the results of the difference analysis, the consensus cluster analysis was carried out on the total samples, and the samples were divided into the gene stable group and the gene unstable group. Then, a coexpression network of lncRNA-mRNA was constructed, and GO analysis and KEGG analysis were performed for this network. The machine learning method, Lasso regression analysis, and SVM-RFE method were combined to screen out key lncRNAs. Cox proportional hazards regression model was established, and key lncRNAs were selected. For this model, survival analysis, clinical subgroup analysis, risk correlation analysis, and in vitro validation were performed.