

Abstract
Long noncoding RNAs (lncRNAs) have emerged as important regulators of many biological processes, including embryogenesis and development. To provide a systematic analysis of lncRNAs expressed during chicken embryogenesis, we used Iso-Seq and RNA-Seq to identify potential lncRNAs at embryonic stages from d 1 to d 8 of incubation: sequential stages covering gastrulation, somitogenesis, and organogenesis. The data characterized an expanded landscape of lncRNAs, yielding 45,410 distinct lncRNAs (31,282 genes). Amongst these, a set of 13,141 filtered intergenic lncRNAs (lincRNAs) transcribed from 9803 lincRNA gene loci, of which, 66.5% were novel, were further analyzed. These lincRNAs were found to share many characteristics with mammalian lincRNAs, including relatively short lengths, fewer exons, lower expression levels, and stage-specific expression patterns. Functional studies motivated by “guilt-by-association” associated individual lincRNAs with specific GO functions, providing an important resource for future studies of lincRNA function. Most importantly, a weighted gene co-expression network analysis suggested that genes of the brown module were specifically associated with the day 2 stage. LincRNAs within this module were co-expressed with proteins involved in hematopoiesis and lipid metabolism. This study presents the systematic identification of lincRNAs in developing chicken embryos and will serve as a powerful resource for the study of lincRNA functions.
Key words: lincRNA, RNA-Seq, Iso-Seq, embryonic development, chicken
INTRODUCTION
Genome-wide studies have discovered several transcripts that do not code for proteins (Eddy, 2001; Pauli, et al., 2012), such as small nuclear RNAs, microRNAs, small-interfering RNAs, Piwi-interacting RNAs, and long noncoding RNAs (lncRNAs) (Eddy, 2001). lncRNAs are RNAs >200 bp long and although they lack protein coding potential, they are capped, spliced, polyadenylated, and developmentally regulated in eukaryotes (Cabili, et al., 2011). Genome-wide studies using RNA-Seq technology have confirmed that mammalian genomes (Guttman, et al., 2009; Cabili, et al., 2011; Iyer, et al., 2015), chicken genome (Jehl, et al., 2020) and other livestock genomes (Kern, et al., 2018; Foissac, et al., 2019) are populated by large number of lncRNAs, which play critical roles in a variety of biological processes via a range of mechanisms including gene activation, repression, cis and trans gene expression regulation, and chromatin modification (Quinn and Chang, 2016; Kopp and Mendell, 2018; Ransohoff, et al., 2018). However, the vast majority of lncRNAs remain to be functionally characterized, and a global perspective of lncRNA is necessary to better understand this complex transcriptome.
The vertebrate embryo undergoes extensive morphological changes during gastrulation and organogenesis (Mitiku and Baker, 2007). Intricate spatiotemporal control of gene expression is undoubtedly critical for these morphological changes. Thus, the dynamic transcriptional landscape of the human embryo (Fang, et al., 2010; Xiang, et al., 2020), as well as many of the most popular model animal embryos, has been well-characterized, including that of the mouse (Mitiku and Baker, 2007; Cao, et al., 2019), zebrafish (Nepal, et al., 2013; White, et al., 2017), Drosophila (Graveley, et al., 2011; Schor, et al., 2018), and nematode (Levin, et al., 2012; West, et al., 2018). Despite these advances and enhanced understanding of expression patterns of protein coding gene during embryogenesis, progress has been hampered by limited information on lncRNA genes. Evidence has indicated that large-scale lncRNAs contribute greatly to the regulation of embryo development (Ulitsky, et al., 2011; Pauli, et al., 2012; Yan, et al., 2013; Bouckenheimer, et al., 2016; White, et al., 2017). For example, a study systematically identified thousands of lncRNAs in zebrafish embryogenesis, providing the first systematic identification of lncRNAs in a vertebrate embryo (Pauli, et al., 2012). The chicken embryo is one of the most valuable vertebrate model systems used for embryological studies due to its rapid in ovo development and high-quality reference genome (Brown, et al., 2003; Stern, 2005). Few studies have investigated the lncRNAs in chicken genome (Jehl, et al., 2020; Li, et al., 2020; Ning, et al., 2020). However, chicken embryonic lncRNAs remain poorly characterized, and therefore, a comprehensive powerful resource of lncRNA expression in developing chicken embryos is needed.
RNA-Seq has identified many potential lncRNAs, but there is still the technical challenge of accurately delineating full-length transcriptional lncRNAs, the expression of which is typically lower than mRNAs (Guttman, et al., 2009; Iyer, et al., 2015). Single-molecule long-read sequencing (Iso-Seq) technology has enabled a more accurate identification of spliced isoforms by sequencing full-length transcripts directly (Abdel-Ghany, et al., 2016; Bo, et al., 2016; Kuo, et al., 2017). Indeed, Iso-Seq has shown that there are > 20,000 lncRNAs in the chicken embryonic transcriptome, much more than the annotated lncRNAs in the chicken genome (Kuo, et al., 2017). Although lncRNA transcripts have been shown to be an important feature of the complex transcriptome, the dynamic expression patterns of lncRNAs in the chicken embryo remain unclear. Given the large number of lncRNAs whose functions have yet to be revealed and the wide range of regulatory mechanisms involved, the genome-wide screening of lncRNAs and construction of expression profiles during chick development will provide a range of novel insights. In particular, recent studies have focused on long intergenic noncoding RNAs (lincRNAs) (Ulitsky, et al., 2011; Ransohoff, et al., 2018), as their genomic location facilitates experimental manipulation and computational analysis.
Previously, Han et al. reported a whole transcriptomic analysis of pre-oviposited early chicken embryos (Hwang, et al., 2017; Hwang, et al., 2018), and we have recently analyzed the dynamic transcriptional landscape of protein coding genes in post-oviposited embryos (Ren, et al., 2019). Here we seek to add detail of global lncRNAs to these valuable resources for investigating early avian embryogenesis. Specifically, the sequencing data generated from embryos at eight stages (n = 3) of development, encompassing both gastrulation and early organogenesis, were reanalyzed to provide insight into the high-resolution dynamic of lncRNAs in chicken embryo.
MATERIALS AND METHODS
Embryo Collection
Commercial pureline White Leghorn fertilized eggs were collected and incubated in an automated egg incubator at 37.5°C and rotated every 6 h. Embryos were collected every day after 1 to 8 days (i.e., E1–E8) with three biological replicates. Embryos were washed with PBS and immediately processed for RNA isolation. Total RNA was isolated following standard TRIzol (Invitrogen, CA) protocol. All animal experiments were approved by the Animal Welfare Committee of China Agricultural University (permit number, XK622) and performed in accordance with the protocol outlined in the “Guide for Care and Use of Laboratory Animals.
Ribo-Zero RNA Sequencing
Whole transcriptome libraries were constructed using TruSeq Stranded Total RNA with Ribo-Zero Gold (Illumina, CA) following the manufacturer's instructions. The generated cDNA libraries were assessed and quantified using the Agilent Bioanalyzer 2100 system. The final set of 24 libraries was sequenced using the Illumina HiSeq 4000 platform (Illumina) and 150 bp paired-end sequencing strategy. The obtained raw reads were cleaned using the FASTX-Toolkit (v0.0.13). The clean reads with high quality were mapped to the chicken genome (Galgal 5) using TopHat2 (v2.0.12). Only reads with a perfect match or one mismatch were employed, and these reads were used to assemble transcripts using String Tie under the default parameters. The assembled transcripts were annotated using the gffcompare program. More details and parameters can be found in the previous publication (Ren, et al., 2019).
Differential Expression Analysis
The expression level for each transcript was normalized by Fragments Per Kilobase of exon model per Million mapped (FPKM) using String Tie. The FPKM of the protein-coding genes and lncRNA genes in each sample were computed by summing the FPKMs of the gene's all transcripts. Differential expression analysis between 2 groups was performed at gene level using the DESeq2 R package (v1.14.1). In our study, The Benjamini and Hochberg's approach were used to control the false discovery rate (FDR) of the P-value. Genes with FDR <0.01 and |log2fold change| >1 were identified as differentially expressed genes.
PacBio Library Construction and Iso-Seq
RNA samples extracted from E1, E3, E5, and E7 embryos were used to prepare the PacBio library. For each developmental stage, equal amounts of total RNA from three biological replicates were pooled together as one sample. For each sample, obtained cDNA were amplified by 16 cycles of PCR and then size selected into 1–2, 2–3, 3–6 and 1–6 kb libraries. The 12 short fragment libraries were sequenced on the Pacific Bioscience RS II platform with 13 SMRT cells and the 4 long fragment (1–6 kb) libraries were sequenced on a PacBio Sequel platform with 4 SMRT cells. PacBio data were processed and evaluated using the PacBio SMRT Analysis Package (v2.3.0) with default parameters. The obtained transcripts from all 16 PacBio libraries were then merged with Galgal 5 reference genome to create a PacBio GTF file. The new GTF file was used in the following analysis. The detailed RNA preparation, sequencing and data processing were performed as described previously (Ren, et al., 2019).
LincRNA Identification
All transcripts generated by the Illumina and PacBio data were employed to identify lncRNAs. Novel transcripts (<200 bp) were discarded, as these were most likely sequencing or assembly artifacts. Known lncRNAs were identified using a BLAST analysis against known chicken lncRNAs downloaded from the NONCODE v5 and Ensembl databases. According to the BLAST result, lncRNAs with >50% query coverage and >80% identity were considered as known lncRNAs. The protein coding potential of the remaining transcripts was tested using the CPC (Kong, et al., 2007), CNCI (Sun, et al., 2013), Pfam (Finn, et al., 2014), and CPAT methods (Wang, et al., 2013). Only transcripts with CPC score <0, CNCI score <0, Pfam E-value >0.001 and labeled with no coding probability by CPAT, were considered candidate lncRNAs. To reduce the interference of protein coding transcripts and transcriptional noise, lincRNAs that were expressed with an average FPKM <0.1 were discarded. Then, these lincRNAs cover with known pseudogenes, ribosomal RNA, miRNAs, and other types of known non coding RNAs (except lncRNAs) were further discarded.
Weighted Gene Co-expression Network Analysis
Weighted Gene Co-expression Network Analysis (WGCNA) construction and module detection were performed using the “WGCNA” R package (Langfelder and Horvath, 2008). Briefly, lincRNAs with an average FPKM > 0.1 and protein coding genes with an average FPKM > 1 were used for constructing the WGCNA. First, an unsigned weighted correlation network was constructed by creating a matrix of Pearson correlation coefficients between all pairs of genes, followed by establishing a weighted adjacency matrix with a soft thresholding power of 29. Then, the adjacency matrix was transformed into a topological matrix. Then each topological matrix was used as input for linkage hierarchical clustering analysis, and primary gene modules was detected by using a dynamic tree cutting algorithm (deep split = 2, cut height = 0.2). Primary modules with high correlation (module eigengene correlation >0.80) were then merged and the final gene modules were obtained. Then, module eigengenes were correlated with different developmental stages and searched against the most significant associations.
LincRNA Functional Investigation
“Guilt-by-association” analyses were applied to predict lncRNA functions (Guttman, et al., 2009). First, a correlation matrix between lincRNAs and protein coding genes was generated by computing the Pearson correlation coefficients for all pairwise combinations. Then, functional associations between lincRNAs and GO terms were computed using Gene Set Enrichment Analysis (GSEA) (Subramanian, et al., 2005; Guttman, et al., 2009; Pauli, et al., 2012). In brief, each lincRNA was used as a profile and a list of protein coding genes were ranked by their correlation coefficient with the lincRNA. Then, the protein coding genes list was subjected to GSEA. A total of 1,449 gene sets were constructed based on GO terms downloaded in DAVID (gene sets <8 genes were filtered). Gene sets were permuted 1,000 times to obtain corrected P-values. The lincRNA is defined as positively (Normalized enrichment score >0, P< 0.05), negatively (normalized enrichment score <0, P< 0.05), and not associated (P> 0.05) with each of the GO terms. Based on the GSEA results, an association matrix between lincRNAs and GO terms was constructed. Then, biclustering of the matrix was performed to identify the most susceptible GO terms.
GO and KEGG Pathway Enrichment
Functional enrichment of the genes module was analyzed using the web-based tools in DAVID (v6.8; https://david.ncifcrf.gov/) to identify enriched GO terms and KEGG pathways. Ensembl gene IDs were submitted to the Gene Functional Classification Tool, then biological process, cellular component, molecular function and KEGG pathway were selected to perform the enrichment. GO terms and KEGG pathways with P< 0.05 were defined as significantly enriched.
Quantitative Real-Time PCR
Total RNA was isolated using the standard TRIzol (Invitrogen) protocol. The purity and concentrations were determined using NanoDrop2000 (Thermo Scientific, DE). Qualified RNA was reverse transcribed into cDNA using a PrimeScript RT reagent kit (TaKaRa, Kyoto, Japan). Quantitative real-time PCR (qRT-PCR) was performed using the SYBR Green method in a Lightcycler 96 system (Roche Applied Science, IN). Each reaction contained 5 μL SYBR Green PCR Master Mix (TaKaRa), 3.5 μL RNase-free water, 0.5 μL each of the forward and reverse primers, and 0.5 μL extracted cDNA. The amplification protocol was as follows: 95°C for 5 min, 40 cycles at 95°C for 30 s, 60°C for 30 s, 72°C for 20 s, and 10 min extension at 72°C. All reactions were performed in triplicate. The housekeeping gene, β-actin, was used as an internal control for normalization. The relative expression of genes was calculated by the 2−∆∆Ct method. Primers were designed by the NCBI Primers-BLAST online program (Table S1).
Availability of the Sequencing Data
The datasets generated during the current study are available in the NCBI Sequence Read Archive under the accession number PRJNA488330.
RESULTS
Genome-Wide Identification of LincRNAs in Chicken Embryos
To systematically define the landscape of lncRNAs in chicken embryos, strand-specific Ribo-Zero RNA sequencing was performed, focusing on chicken embryos at 8 stages between 1 and 8 d of incubation (E1–E8). This time span covers Hamburger-Hamilton stages HH3–HH26 (Hamburger and Hamilton, 1951), and encompasses gastrulation, somitogenesis, and organogenesis. The RNA-seq generated 228.9 million clean reads from the 24 established libraries, and more than 83.6% reads were uniquely mapped to the chicken reference genome (details in Table S2). Accurately delineating full-length lncRNAs transcripts using ‘short reads’ technology remains a challenge, so in addition, Iso-Seq was also performed on embryos at E1, E3, E5, and E7. In brief, our Iso-Seq generated 69.89 Gb row data including 3.01 million reads of insert. Of these, 1.57 (52.1%) million reads were identified as full-length non-chimeric reads. After steps of polish, we finally obtained a total of 135,379 high-quality transcripts (details in Table S3). A detailed description of the transcriptomic data was outlined in a previous study (Ren, et al., 2019).
An integrative computational pipeline for the systematic identification of lincRNAs is shown in Figure 1. In brief, a total of 16 size-fractionated full length cDNA libraries were sequenced using the PacBio RS II and Sequel platform, which yielded 135,379 high-quality transcripts. After filtering out known protein coding transcripts and short length transcripts, the remaining transcripts (124,558) were subjected to the CPC, CNCI, CPAT, and Pfam programs to estimate their coding potential (detail in methods). In total, 25,935 potential full length lncRNAs were identified, similar to the number of lncRNAs described in a previous report (Kuo, et al., 2017). Subsequently, a total of 24 strand-specific sequencing libraries were sequenced. After removing the known transcripts, newly assembled transcripts were filtered and subjected to the CPC, CNCI, CPAT, and Pfam programs. Therefore, another 19,584 potential lncRNAs were identified. Thus, a total of 45,519 putative lncRNA transcripts (25,225 genes) were identified in the developing chicken embryo. After filtering out the low expressed lncRNAs, the remaining 31447 (20,869 genes) were blast against lncRNA database. Results showed that only ∼14.0% lncRNA transcripts were previously described and registered in the NONCODE and Ensembl databases (Figure 2a). In addition, these lncRNAs were further blast against a recently published lncRNA catalogue (Jehl, et al., 2020), which was built using four public databases and 364 RNA-seq data, resulting 5,327 (16.9%) common lncRNA transcripts. These results demonstrated that the chicken genome contains a large number of novel lncRNAs. Of all 45,519 lncRNA transcripts, most were lincRNAs (42%) and intronic lncRNAs (27%; Figure 2b). To better characterize the properties of lincRNAs, a set of 13,141 lincRNA transcripts (Figure 1) transcribed from 9,803 lincRNA genes were selected for subsequent analyses. The 13,141 lincRNAs transcripts were transcribed from 9,803 lincRNA gene loci and 33.5% (3,285) of the lincRNA genes had previously been registered in the recently published lncRNA catalogue (Jehl, et al., 2020).
Figure 1.

An integrative computational pipeline for the systematic identification of lincRNAs. Abbreviations: lincRNAs, Long noncoding RNAs.
Figure 2.

Overview of the identified lncRNAs: (a) Venn diagram illustrating the number of known lncRNAs recorded by the NONCODE and Ensembl databases. (b) Pie chart showing the classifications of lncRNAs. Abbreviations: lincRNAs, Long noncoding RNAs.
Genomic Characterization of Embryonic LincRNA Genes
The genomic features of lincRNAs were characterized and compared with that of protein coding genes where appropriate. Out of 13,141 lincRNAs transcripts transcribed from 9,803 lincRNA genes, only 1,679 lincRNA genes generated more than a single transcript, giving a multitranscript rate of 17.1%. In contrast, the multi-transcript rate was >40% for protein coding genes. Moreover, lincRNA transcripts (average length of 1,595 nucleotides) were significantly shorter than mRNAs (average length of 3,831 nucleotides) (Figure 3a; P< 2.2E-16, Kolmogorov-Smirnov test). Comparison of the intron/exon features of lncRNA genes and protein coding genes revealed that lincRNA genes (2.1 exons on average) span significantly fewer exons than protein coding genes (9 exons, on average) (Figure 3b; P< 2.2E-16, Kolmogorov-Smirnov test). Moreover, lincRNA transcripts were more A/U-rich than the coding sequences and 5’UTRs of protein coding genes, but were less A/U-rich than 3’UTRs (Figure 3c). We also calculated the Pearson correlation coefficients of these lincRNA genes and protein coding gene pairs. Results revealed that intronic lncRNA genes have relatively high correlation coefficients with their host genes, as expected. However, lincRNA genes and their neighboring coding genes (<100 kb) were significantly correlated with each other as opposed to random lincRNA-mRNA pairs (Figure 3d; P< 2.2E-16, Kolmogorov-Smirnov test). These properties are consistent with the lincRNAs of mammals (Bertone, et al., 2004; Ponjavic, et al., 2007; Dinger, et al., 2008; Jia, et al., 2010) and other model organisms (Nam and Bartel; Ulitsky, et al., 2011; Pauli, et al., 2012).
Figure 3.

Characterization of embryonic lincRNAs: (a) Transcript length distributions of lincRNAs and protein coding transcripts. (b) Exon number of lincRNA transcripts and protein coding transcripts. (c) A/U content of lncRNAs and the 5’ UTR, 3’ UTR, and CDS for protein coding transcripts. (d) Correlation between the expression levels of lincRNAs and adjacent protein coding genes. Spearman's correlation coefficient between the expression levels of each gene pair was calculated. A total of 10,000 lincRNAs and mRNA genes were randomly selected and paired as the control group. Abbreviations: lincRNAs, Long noncoding RNAs.
Temporal Expression Profiles of LincRNAs
To gain deeper insight into lincRNA expression patterns, individual lincRNA dynamic expression patterns across the embryonic development stages was generated using Illumina data from E1–E8. The mean expression level of the 9,803 lincRNA gene (median log2FPKM was −2.86) was significantly lower than protein coding genes (median log2FPKM was −1.75; Figure 4a; P< 2.2E-16, Kolmogorov-Smirnov test). A heatmap of lincRNA gene expression profiles across embryonic development stages was arranged in a manner dependent on their stage of maximum expression (Figure 4b). As contrast, the protein coding gene expression profiles across stages were showed in Figure s1. This arrangement clearly revealed a more stage-specific expression of lincRNA genes than protein coding genes. Pairwise comparisons between any 2 of the 8 stages revealed that a total of 2,811 lincRNA genes were differentially expressed (|log2fold change| >1, FDR <0.01). The number of differentially expressed lincRNA genes was not uniform across the entire time period of embryogenesis (Figure 4c). Specifically, differentially expressed lincRNA genes between distant stages were much more numerous than those identified between adjacent stages (Figure 4c). The dynamics of differentially expressed lincRNA genes between adjacent stages was examined to investigate whether expression turnover was continuous across E1–E8. Interestingly, the stages of somitogenesis (E1–E3) featured the most radical transcriptomic changes (Figure 4d), which matched the findings of previous studies on zebrafish and mouse embryos (Mitiku and Baker, 2007; White, et al., 2017). The high rate of differential expression in the first 3 d may reflect dramatic changes in genetic programs associated with pluripotency and the initial differentiation of embryonic stem cells. Next, qRT-PCR was performed to verify the RNA-Seq data. Results showed that the expression patterns of the random selected lincRNA genes were consistent with the RNA-Seq results (Figure 4e).
Figure 4.

Expression profile of lincRNAs: (a) Average expression levels of lincRNAs and protein coding transcripts. (b) Heatmap of lincRNA expression profiles across development, where lincRNAs were clustered based on the stage of maximum expression. (c) Visualization of the number of differentially expressed lincRNAs between each pair of stages. The size of the circles represents the number of differentially expressed lincRNAs. Blue and red represent the up- and downregulated lincRNAs; lincRNAs with |log2fold change| >1 and FDR <0.01 were considered differentially expressed lincRNAs. (d) Number of significantly up- and downregulated genes between stage transitions. (e) Validation of differentially expressed lincRNAs using qRT-PCR. The line graph represents the qRT-PCR data and the Heatmap in the bottom represents the RNA-Seq data. Abbreviations: FDR, false discovery rate; lincRNAs, Long noncoding RNAs. (Color version of figure is available online.)
Functional Annotation of LincRNAs
Few lincRNAs have been functionally annotated in the genome of all organisms, including humans. The lack of annotated features makes the functional assignment of lincRNAs a more challenging task than for proteins. To investigate the functions of lincRNAs, “guilt-by-association” analyses were employed to annotate the functions of lincRNAs (Dinger, et al., 2008; Guttman, et al., 2009; Pauli, et al., 2012). First, ∼1,449 gene sets were constructed using known GO terms (gene sets <8 genes were not accessible). Then, each lincRNA transcripts was used as a profile and Pearson correlation coefficients of expression abundance were calculated between each lincRNA and known protein coding genes using the 24 samples. The protein coding gene list was subjected to the GSEA (Subramanian, et al., 2005; Guttman, et al., 2009; Pauli, et al., 2012); genes were ranked by their correlation coefficient and tested with each GO term. Significantly enriched GO terms were identified with an FDR <0.05 and positive/negative associations were defined with the NES. Therefore, a matrix of the association of each lincRNA with each of the ∼1,449 GO terms was constructed (Figure 5a). For example, linc5006 was identified to be positively associated with 12 GO terms and negatively associated with 43 GO terms, including cilium assembly, neuron migration, gluconeogenesis, and structural constituent of ribosome (Figure 5b). This analysis associated each lincRNA with distinct and diverse biological processes, thereby achieving batch processing of lncRNA annotation. After biclustering, results revealed some lincRNAs associated with widespread GO terms, including embryonic skeletal system development, pigmentation, and BMP signaling pathway.
Figure 5.

Functional annotation of lincRNAs: (a) Expression-based association matrix of lincRNAs (rows) and GO terms (columns) derived from GSEA. lincRNAs and GO terms are positively (red; FDR <0.05; NES >0), negatively (blue; FDR <0.05; NES <0), or not associated (white; FDR >0.05). Black boxes highlight significant biclusters in the matrix. (b) GSEA enrichment plots of linc5006 associated with GO:0042384 (cilium assembly), GO:0001764 (neuron migration), GO:0006094 (gluconeogenesis), and GO:0003735 (structural constituent of ribosome). The green curve corresponds to the calculation of enrichment scores (ES). The horizontal bar (red to blue gradient) represents the known ranked genes ordered by Pearson correlation coefficients with linc5006. Vertical black lines represent the projection of individual genes within the tested GO term onto the ranked gene list. Abbreviations: FDR, false discovery rate; lincRNAs, Long noncoding RNAs; NES, normalized enrichment score. (Color version of figure is available online.)
WGCNA Analysis Revealed a Set of LincRNAs That Participate in Hematopoiesis
To better assess the interpretive potential of lincRNAs in the context of specific developmental stages, a WGCNA was conducted to investigate protein coding gene and lincRNA co-expression networks. The gene expression data of protein coding genes (average FPKM >1) and lincRNAs (average FPKM >0.1) in 24 samples were used as the input expression matrix. The analysis resulted in 13 distinct modules with module sizes ranging from 33 to 4,040 (Figure 6A). Module-trait correlation analyses suggested that 3 modules were significantly correlated (r > 0.7, P< 0.01) with specific developmental stages or with the entire developmental process (Figure 6b). In particular, genes that clustered in the brown module (612 protein coding genes, 242 lincRNAs) had the strongest correlation with stage E2 (Figure 6c; r = 0.98, P< 1e-15). GO and KEGG pathway enrichment analyses revealed that protein coding genes within the brown module were mainly enriched in hematopoiesis (i.e., extracellular exosome, blood microparticle, fibrinolysis, and chylomicron) and lipid metabolic (i.e., cholesterol homeostasis, PPAR signaling pathway, and lipid metabolic process) related functions (Figure 6d). Additionally, many lincRNAs in the brown module were annotated in hematopoiesis and metabolic-related functions, which were in line with the functional enrichment of the protein coding genes. Along with the morphological changes that occurred in the E2 stage, results uncovered the participation of these lincRNAs and known protein coding genes in hematopoiesis functions with which they were co-expressed or bound.
Figure 6.

WGCNA identified lincRNA and mRNA coexpression modules (a) Gene dendrogram obtained by average linkage hierarchical clustering. The color row (bottom) represents the assigned original module and merged module. (b) Heatmap of the correlations between modules and developmental stages. Each cell contains the correlation coefficient and corresponding P-value. The table is color coded by correlation according to the color legend. (c) Gene significance of genes in the brown module for E2 stage. (d) DAVID analysis of enriched GO terms of genes found in the brown module. Abbreviations: lincRNAs, Long noncoding RNAs; WGCNA, weighted gene co-expression network analysis. (Color version of figure is available online.)
DISCUSSION
The transcriptional landscape of the human embryo and of embryos of many model organisms has been well characterized. The chicken was the first genome-sequenced non-mammalian amniote, and possesses unique features that are valuable for developmental and evolutionary studies. Thus, transcriptome profiling of chicken embryos creates an opportunity to advance our understanding of molecular regulation in vertebrate development. Previously, Han et al. reported the whole transcriptomic characterization of pre-oviposited early chicken embryos (Hwang, et al., 2017; Hwang, et al., 2018), and more recently, using an integrated Iso-Seq and RNA-Seq approach, we have characterized an expanded landscape of transcription in the post-oviposited chicken embryo from E1 to E8 (Ren, et al., 2019). The integration of multilayered information across different stages of embryonic organogenesis provides insights into the dynamism of the transcriptome. In addition to studies focused on protein coding genes, some studies have investigated noncoding genes in the developing embryo. A microRNA catalog of the developing post-oviposited chicken embryo provided an important foundation for further investigations on miRNA gene regulation in the chicken embryo (Glazov, et al., 2008). Genome-wide identification of lncRNAs in embryonic skeletal muscle (Li, et al., 2012; Zhenhui, et al., 2017), liver (Ning, et al., 2020), and brain (Xu, et al., 2018), and embryonic stem cells has also been performed (Li, et al., 2017), suggesting lncRNA regulatory and functional roles in chicken embryonic development. These analyses show that noncoding RNAs constitute a substantial portion of the transcriptome and suggest that these play critical roles in animal embryogenesis. The present study provides a useful database resource of lincRNA and potential function during chicken embryogenesis.
To identify the lincRNAs involved in chicken embryo development, we used Iso-Seq and RNA-Seq data to define an expanded landscape of lincRNAs. This systematic analysis generated tens of thousands of potential lincRNAs for the chicken embryo, and revealed tremendous transcriptional complexity. The ∼13,141 identified lincRNAs were found to share many properties typical of previously described examples (Figure 3), including mammal and zebrafish lincRNAs (Cabili, et al., 2011; Ulitsky, et al., 2011; Pauli, et al., 2012). Specifically, chicken lincRNAs were expressed at lower levels compared to protein coding genes. Previously, the lower expression levels of lincRNAs compared to mRNAs has led to suggestions that lincRNAs represent transcriptional noise or that they lack biological significance (Clark, et al., 2011). However, there is evidence that lower expression levels may be due to their tissue-, stage-, and condition-specific expression patterns (Nam and Bartel, 2012; Schor, et al., 2018). As predicted, the data generated in the present study indicated that lincRNAs function in a stage-specific manner.
Because of locus variety and potential functional diversity, defining the function of individual lncRNA remains a challenge. For example, studies have suggested that some lincRNAs may act in cis and affect the gene expression of their chromosomal neighborhood (Orom, et al., 2010; Gil and Ulitsky, 2020), while others suggest that lincRNAs are trans-acting and regulate gene expression at independent loci (Rinn, et al., 2007; Qu, et al., 2019). Although certain specific lncRNAs have been functionally characterized, no unifying model exists that explains their function or mechanism of action. To inform this debate, we adopted a “guilt-by-association” approach (Guttman, et al., 2009) to assign putative functions to each lncRNA. In conjunction with the expression data, this analysis provides possible functional roles for identified lincRNAs. For example, the protein coding genes clustered in the brown module were most enriched in hematopoiesis and lipid metabolic process. After examining the annotation of lincRNAs co-expressed in the brown module, there are many examples of lincRNAs annotated in blood microparticle, PPAR signaling pathway, and cholesterol homeostasis GO terms, such as PB.22745, PB.23323 and PB.2902. Although functional annotations of embryonic lincRNAs are still far from perfect, the functional annotations provided here serve as the first genome-wide functional blueprint for studies on lincRNA biology in the chicken.
Comprehensive studies on the developing embryo reported here describe the dynamic expression of lincRNAs during embryogenesis in detail. Several developmentally synchronous lincRNAs were identified by examining a time series of 8 developmental stages. As suggested by the presence of several differentially expressed lincRNAs between stages (Figure 4c), many lincRNAs participate in embryonic development processes. Notably, the number of lincRNAs that exhibit differential expression was not uniform across the examined time period (Figure 4c, d). More than three times as many lincRNAs were differentially expressed during the first 2 d than at any subsequent time point (Figure 4d). This result was consistent with previous observations made on protein coding genes (Ren, et al., 2019). The similarity of these trends between lincRNAs and mRNAs suggests that they are subject to similar modes of regulation during embryogenesis. In particular, embryos in the E1–E2 window featured the most profound morphological changes; somitogenesis mainly occurs during this timeframe, suggesting that somitogenesis stage encompasses most of the transcriptomic changes. Consistent with this finding, somitogenesis stages in mouse embryos were also marked by most of the positive and negative changes occurring within the transcriptome (Mitiku and Baker, 2007). Uncovering the biological functional process will much helpful for understanding the dramatic changes during the E1–E2 time window. The WGCNA analysis revealed a set of genes clustered in the brown module had the strongest correlation with stage E2. We have employed GO enrichment for protein coding genes and applied "guilt-by-association" for lincRNAs to uncover the function of the gene set. Results revealed that the genes in brown module are most involved in hematopoiesis and lipid metabolic process, leading to hypothesize that the dramatic changes in gene expression aim to construct functional foundation for the entrance into organogenesis and differentiation. In conclusion, the high rate of differential expression during somitogenesis may reflect dramatic changes in genetic programs associated with pluripotency and initial specifications of differentiation trajectories.
Acknowledgments
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China under Grant number 31320103905 and Programs for Changjiang Scholars and Innovative Research in Universities under Grant number IRT15R62.
Disclosures
The authors have no conflicts of interest to disclose.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.psj.2021.101160.
Appendix. Supplementary materials
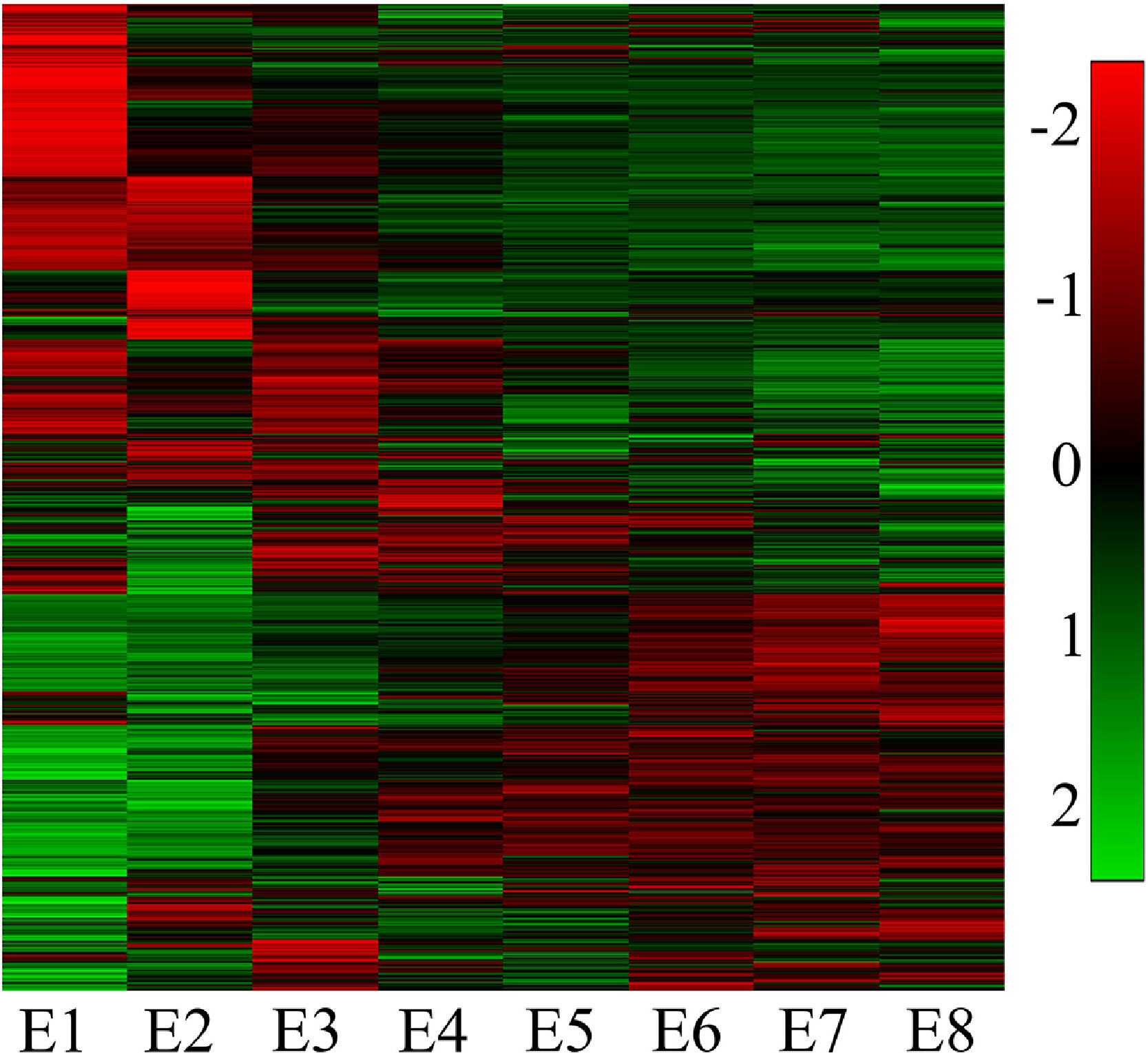
Figure s1. Heatmap of protein coding genes expression profiles across development.
{kind=link}
Table S1. The list of qRT-PCR primers.
Table S2. Summary of data generated by RNA-Seq.
Table S3. Summary of data generated by Iso-Seq.
REFERENCES
- Abdel-Ghany S.E., Michael H., Jacobi J.L., Peter N., Nicholas D., Faye S., Asa B.H., Reddy A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016;7:11706. doi: 10.1038/ncomms11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertone P., Stolc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- Bo W., Tseng E., Regulski M., Clark T.A., Hon T., Jiao Y., Lu Z., Olson A., Stein J.C., Ware D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016;7:11708. doi: 10.1038/ncomms11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckenheimer J., Assou S., Riquier S., Hou C., Philippe N., Sansac C., Lavabre-Bertrand T., Commes T., Lemaître J.M., Boureux A., De Vos J. Long non-coding RNAs in human early embryonic development and their potential in ART. Hum. Reprod. Update. 2016;23:19–40. doi: 10.1093/humupd/dmw035. [DOI] [PubMed] [Google Scholar]
- Brown W.R.A., Hubbard S.J., Tickle C., Wilson S.A. The chicken as a model for large-scale analysis of vertebrate gene function. Nat. Rev. Genet. 2003;4:87–98. doi: 10.1038/nrg998. [DOI] [PubMed] [Google Scholar]
- Cabili M.N., Trapnell C., Goff L., Koziol M., Tazonvega B., Regev A., Rinn J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J., Trapnell C., Shendure J. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M.B., Amaral P.P., Schlesinger F., Dinger M.E., Taft R.J., Rinn J.L., Ponting C.P., Stadler P.F., Morris K.V., Morillon A. The reality of pervasive transcription. PLoS Biol. 2011;9 doi: 10.1371/journal.pbio.1000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinger M.E., Amaral P.P., Mercer T.R., Pang K.C., Bruce S.J., Gardiner B.B., Askarianamiri M.E., Ru K., Soldà G., Simons C. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008;18:1433–1445. doi: 10.1101/gr.078378.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. Non–coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- Fang H., Yang Y., Li C., Fu S., Yang Z., Jin G., Wang K., Zhang J., Jin Y. Transcriptome analysis of early organogenesis in human embryos. Dev. Cell. 2010;19:174–184. doi: 10.1016/j.devcel.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Finn R.D., Bateman A., Clements J., Coggill P., Eberhardt R.Y., Eddy S.R., Heger A., Hetherington K., Holm L., Mistry J. Pfam: the protein families database. Nucleic Acids Res. 2014;42:222–230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foissac S., Djebali S., Munyard K., Vialaneix N., Giuffra E. Multi-species annotation of transcriptome and chromatin structure in domesticated animals. BMC Biol. 2019;17:108. doi: 10.1186/s12915-019-0726-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil N., Ulitsky I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020;21:102–117. doi: 10.1038/s41576-019-0184-5. [DOI] [PubMed] [Google Scholar]
- Glazov E.A., Cottee P.A., Barris W.C., Moore R.J., Dalrymple B.P., Tizard M.L. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res. 2008;18:957–964. doi: 10.1101/gr.074740.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley B.R., Brooks A.N., Carlson J.W., Duff M.O., Landolin J.M., Yang L., Artieri C.G., Baren M.J.V., Boley N., Booth B.W. The developmental transcriptome of drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V., Hamilton H.L. A series of normal stages in the development of the chick embryo. Dev. Dyn. 1951;195:231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- Hwang Y.S., Seo M., Bang S., Kim H., Han J.Y. Transcriptional and translational dynamics during maternal-to-zygotic transition in early chicken development. The FASEB J. 2017;32:2004–2011. doi: 10.1096/fj.201700955R. [DOI] [PubMed] [Google Scholar]
- Hwang Y.S., Seo M., Choi H.J., Kim S.K., Kim H., Han J.Y. The first whole transcriptomic exploration of pre-oviposited early chicken embryos using single and bulked embryonic RNA-sequencing. Gigascience. 2018;7:1–9. doi: 10.1093/gigascience/giy030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehl F., Muret K., Bernard M., Boutin M., Lagarrigue S. An integrative atlas of chicken long non-coding genes and their annotations across 25 tissues. Sci. Rep. 2020;10:20457. doi: 10.1038/s41598-020-77586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H., Osak M., Bogu G.K., Stanton L.W., Johnson R., Lipovich L. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA. 2010;16:1478–1487. doi: 10.1261/rna.1951310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern C., Wang Y., Chitwood J., Korf I., Zhou H. Genome-wide identification of tissue-specific long non-coding RNA in three farm animal species. BMC Genomics. 2018;19:684. doi: 10.1186/s12864-018-5037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Zhang Y., Ye Z.Q., Liu X.Q., Zhao S.Q., Wei L., Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F., Mendell J.T. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo R.I., Tseng E., Eory L., Paton I.R., Archibald A.L., Burt D.W. Normalized long read RNA sequencing in chicken reveals transcriptome complexity similar to human. BMC Genomics. 2017;18:323. doi: 10.1186/s12864-017-3691-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M., Hashimshony T., Wagner F., Yanai I. Developmental milestones punctuate gene expression in the Caenorhabditis embryo. Dev. Cell. 2012;22:1101–1108. doi: 10.1016/j.devcel.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Li D., Ji Y., Wang F., Wang Y., Wang M., Zhang C., Zhang W., Lu Z., Sun C., Ahmed M.F. Regulation of crucial lncRNAs in differentiation of chicken embryonic stem cells to spermatogonia stem cells. Anim. Genet. 2017;48:191–204. doi: 10.1111/age.12510. [DOI] [PubMed] [Google Scholar]
- Li T., Wang S., Wu R., Zhou X., Zhu D., Zhang Y. Identification of long non-protein coding RNAs in chicken skeletal muscle using next generation sequencing. Genomics. 2012;99:292–298. doi: 10.1016/j.ygeno.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Li W., Jing Z., Cheng Y., Wang X., Li D., Han R., Li W., Li G., Sun G., Tian Y., Liu X., Kang X., Li Z. Analysis of four complete linkage sequence variants within a novel lncRNA located in a growth QTL on chromosome 1 related to growth traits in chickens. J. Anim. Sci. 2020;98:1–11. doi: 10.1093/jas/skaa122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitiku N., Baker J.C. Genomic analysis of gastrulation and organogenesis in the mouse. Dev. Cell. 2007;13:897–907. doi: 10.1016/j.devcel.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Nam J.-W., Bartel D.P. Long noncoding RNAs in C. elegans. Genome Res. 2012;22:2529–2540. doi: 10.1101/gr.140475.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepal C., Hadzhiev Y., Previti C., Haberle V., Li N., Takahashi H., Suzuki A.M., Sheng Y., Abdelhamid R.F., Anand S. Dynamic regulation of the transcription initiation landscape at single nucleotide resolution during vertebrate embryogenesis. Genome Res. 2013;23:1938–1950. doi: 10.1101/gr.153692.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning C., Ma T., Hu S., Xu Z., Li D. Long Non-coding RNA and mRNA profile of liver tissue during four developmental stages in the chicken. Front. Genet. 2020;11:574. doi: 10.3389/fgene.2020.00574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orom U.A., Derrien T., Beringer M., Gumireddy K., Gardini A., Bussotti G., Lai F., Zytnicki M., Notredame C., Huang Q. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A., Valen E., Lin M.F., Garber M., Vastenhouw N.L., Levin J.Z., Fan L., Sandelin A., Rinn J.L., Regev A. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012;22:577–591. doi: 10.1101/gr.133009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponjavic J., Ponting C.P., Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007;17:556–565. doi: 10.1101/gr.6036807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X., Alsager S., Zhuo Y., Shan B. HOX transcript antisense RNA (HOTAIR) in cancer. Cancer Lett. 2019;454:90–97. doi: 10.1016/j.canlet.2019.04.016. [DOI] [PubMed] [Google Scholar]
- Quinn J.J., Chang H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- Ransohoff J.D., Wei Y., Khavari P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018;19:143–157. doi: 10.1038/nrm.2017.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Sun C., Clinton M., Yang N. Dynamic transcriptional landscape of the early chick embryo. Fron. Cell Dev. Bio. 2019;7:196. doi: 10.3389/fcell.2019.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn J.L., Kertesz M., Wang J.K., Squazzo S.L., Xu X.L., Brugmann S.A., Goodnough L.H., Helms J.A., Farnham P.J., Segal E. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schor I.E., Bussotti G., Maleš M., Forneris M., Viales R.R., Enright A.J., Furlong E.E.M. Non-coding RNA expression, function, and variation during drosophila embryogenesis. Curr. Biol. 2018;28:3547–3561. doi: 10.1016/j.cub.2018.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern C.D. The chick; a great model system becomes even greater. Dev. Cell. 2005;8:9–17. doi: 10.1016/j.devcel.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A.G., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., Liu Y., Chen R., Zhao Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013;41:e166. doi: 10.1093/nar/gkt646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I., Shkumatava A., Jan C.H., Sive H., Bartel D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Jung P.H., Surendra D., Wang S., Jean-Pierre K., Li W. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S.M., Mecenas D., Gutwein M., Aristizábalcorrales D., Piano F., Gunsalus K.C. Developmental dynamics of gene expression and alternative polyadenylation in the Caenorhabditis elegans germline. Genome Biol. 2018;19:8. doi: 10.1186/s13059-017-1369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.J., Collins J.E., Sealy I.M., Wali N., Dooley C.M., Digby Z., Stemple D.L., Murphy D.N., Billis K., Hourlier T. A high-resolution mRNA expression time course of embryonic development in zebrafish. Elife. 2017;6:e30860. doi: 10.7554/eLife.30860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang L., Yin Y., Zheng Y., Ma Y., Li Y., Zhao Z., Guo J., Ai Z., Niu Y., Duan K., He J., Ren S., Wu D., Bai Y., Shang Z., Dai X., Ji W., Li T. A developmental landscape of 3D-cultured human pre-gastrulation embryos. Nature. 2020;577:537–542. doi: 10.1038/s41586-019-1875-y. [DOI] [PubMed] [Google Scholar]
- Xu Z., Che T., Li F., Tian K., Zhu Q., Mishra S.K., Dai Y., Li M., Li D. The temporal expression patterns of brain transcriptome during chicken development and ageing. BMC Genomics. 2018;19:917. doi: 10.1186/s12864-018-5301-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L., Yang M., Guo H., Yang L., Wu J., Li R., Liu P., Lian Y., Zheng X., Yan J., Huang J., Li M., Wu X., Wen L., Lao K., Li R., Qiao J., Tang F. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- Zhenhui L., Hongjia O., Ming Z., Bolin C., Peigong H., A. A.B., Qinghua N., Xiquan Z. Integrated analysis of long non-coding RNAs (LncRNAs) and mRNA expression profiles reveals the potential role of LncRNAs in skeletal muscle development of the chicken. Fron. Physiol. 2017;7:687. doi: 10.3389/fphys.2016.00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure s1. Heatmap of protein coding genes expression profiles across development.
Table S1. The list of qRT-PCR primers.
Table S2. Summary of data generated by RNA-Seq.
Table S3. Summary of data generated by Iso-Seq.