

Abstract
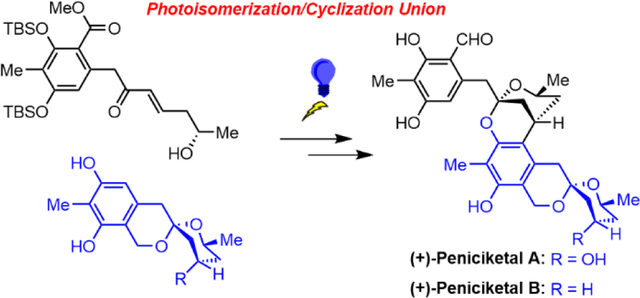
The enantioselective total syntheses of (+)-peniciketals A and B, two members of a family of architecturally complex spiroketals, has been achieved. Key synthetic transformations comprise Type I Anion Relay Chemistry (ARC) to construct the benzannulated [6,6]-spiroketal skeleton, a Negishi cross-coupling/olefin cross-metathesis reaction sequence to generate the trans-enone structure, and a late-stage large fragment union exploiting our recently developed photoisomerization/cyclization tactic.
Graphical Abstract
The peniciketals A-C (1–3; Scheme 1), architecturally complex spiroketals, isolated in 2014 from the fungus Penicillium raistrickii found in saline soil samples isolated from Bohai Bay (China), display cytotoxicity against HL-60 cells with IC50 values of 3.2, 6.7 and 4.5 μM, respectively.1 Peniciketal A (1), in particular, proved to be cytotoxic, with time-dependent inhibition/proliferation of the human non-small lung cancer cell line A549 (IC50 = 22.33 μM in 72 hr), as well as inhibition of both migration and invasion of A549 cells by reducing the levels of the MMP-2 and MMP-9 protein.2 More recently, peniciketal A (1) was also revealed to reduce cell proliferation in three leukaemia cell lines,3,4 as well as high selectivity for cancer cells with lower toxicity towards normal cells (L02, MRC5 and MEFs).2,3 This high level of antitumor activity recently led to more mechanistic studies including a global proteomic profile of peniciketal A (1),4 that suggests this natural product may possess additional bioactivities and as such constitutes a promising drug lead candidate.
Scheme 1.

(+)-Peniciketal A-C
The structures of the peniciketals are unprecedented in the literature, comprising one phenyl ring fused not only to a [6,6]- or [5,6]-spiroketal but also to a 2,8-dioxabicyclo-[3.3.1]nonane moiety. Such benzannulated spiroketals, especially those possessing a [6,6]-spiro-ring, are relative rare in nature, and as such have led to significant synthetic efforts in past two decades.5 From our perspective, we envisioned our Type I Anion Relay Chemistry (ARC), a multicomponent union protocol developed for the elaboration of structurally complex scaffolds,6 could be an effective tactic to access rapidly such aryl spiroketals. Equally significant, the unique benzo-fused 2,8-dioxabicy-clo[3.3.1]nonane framework, is also a challenging framework for current synthetic methods. For example, transition-metal-catalyzed asymmetric Michael addition,7 followed by acid-mediated ketalization to construct such structures, is particularly difficult in the presence of free hydroxy, carbonyl, spiroketal and/or phenol groups. Attracted by the challenging architecture and extensive biological activities, we initiated a synthetic program toward peniciketals A (1) and B (2), exploiting Type I ARC and our newly developed photochemical protocol (vide infra) to construct the complex benzo-fused [3.3.1]nonane core, the latter in a single step.
Having a long-standing interest in developing novel photochemical protocols for total synthesis, see the paniculides,8 hibiscone C,9 echinosporin,10 and recently the danshenspiroketal-lactones,11 etc., we disclosed in 2015 a tandem photoisomerization/cyclization tactic to construct cyclic and spirocyclic ketals (Scheme 2a).12 Pleasingly, this mild photochemical protocol proceeds with high diastereoselectivity (dr >20:1), and as such holds great potential for the construction of complex polycyclic structures, when extended to an intermolecular version. For example (Scheme 2b), irradiation (355 nm) of trans-enone A in mild acid was envisioned to give rise to a mixture of olefin isomers with the cis-isomer B, and then undergo cyclization to deliver diene C. Under the acidic conditions, C could then protonate to generate oxonium D. This electrophilic intermediate is structurally rigid and as such could then lead to a stereoselective [3+3] cyclization with electron-rich aromatic nucleophiles to access the unique benzo-fused 2,8-dioxabicyclo[3.3.1] nonane structure E in a one-pot fashion.
Scheme 2.

Tandem Photoisomerization/Cyclization
With this overview, the [3.3.1] nonane core of peniciketal A would be disconnected to yield the northern hemisphere (4) and benzannulated [6,6]-spiroketal (5) as the southern hemisphere (Scheme 3), exploiting the above proposed late-stage photoisomerization/cyclization union tactic. The northern hemisphere (4), a highly functionalized trans-enone was anticipated to be constructed via a Negishi cross-coupling/olefin cross-metathesis reaction sequence, involving ester 6, acryloyl chloride 7 and commercially available homoallylic alcohol (+)-8. The southern hemisphere (5) in turn would derive from linear precursor 9, possessing the 1,3,5-triol functionality, well suited for our Type I Anion Relay Chemistry (ARC), employing linchpin 11 and two different electrophiles 10 and (+)-12.13
Scheme 3.

Retrosynthetic Analysis
We first conducted a model reaction (Table 1) to validate the intermolecular photochemical protocol with enone (+)-13 (see Supporting Information) and commercial resorcinol 14. Pleasingly, the desired bicyclo[3.3.1]nonane (+)-15 was obtained in 45% yield with high diastereoselectivity (entry 1, dr > 20:1). The key intermediate diene (+)-16 was isolated, which can be resubjected into acidic condition with 14 to yield (+)-15 without UV-A light. Further optimization indicated that with camphor sulfonic acid (CSA) as the Bronsted acid, in conjunction with more concentrated solutions, the yield increased to 80% (entry 3). A control experiment demonstrated the essential role of UV-A light (entry 5). Neither isomerization nor cyclization occurred when the reaction was conducted in the dark.
Table 1.
Model Studies of the Photochemical Protocol
 | |||
|---|---|---|---|
| entry | Bronsted Acid | concentration | yielda (%) of 15 |
| 1 | PTSA (20 mol%) | 0.1 M | 45 |
| 2 | PTSA (20 mol%) | 0.2 M | 71 |
| 3 | CSA (20 mol%) | 0.2 M | 80 |
| 4 | PPTS (50 mol%) | 0.2 M | 49 |
| 5b | CSA (20 mol%) | 0.2M | NR |
Reaction conditions: (+)-13 (0.2 mmol), 14 (0.2 mmol), Bronsted acid (cat.), in THF, rt, UV-A light (λ=355 nm), 24 h. The isolated yields of products (+)-15 are obtained by flash chromatography. The dr was measured by 1H NMR.
no light. NR = no reaction, and both starting materials remained.
With the success of the model study, we initiated the synthesis of peniciketal A (1), with construction of the southern hemisphere (5), employing Type I ARC as outlined in Scheme 4. The known compound 18 was prepared from 17 in 47% yield by a 5-step sequence.14 Benzyl bromide 10 was then prepared from 18 in 3 steps. The ARC process was then initiated by deprotonation of the dithiane linchpin 11 with n-BuLi, followed by the addition of epoxide (+)-12. The resulting alkoxide 19 then underwent a [1,4]-Brook rearrangement,15 triggered by the addition of HMPA to relay the negative charge via silyl group migration from the dithiane carbon in 19 to oxygen in 20. Carbanion 20 was then captured with benzyl bromide 10 to deliver the desired three-component adduct 21, which was subjected to dithiane removal with simultaneous hydrolysis of the TMS ether to provide linear precursor (+)-9 in a total yield of 64% for the 2 steps on a 2-gram scale. At this stage, a variety of conditions for global removal of TBS groups, including TBAF, TBAF/HOAc buffer, HF/pyridine, PTSA/MeOH and other acidic conditions, were explored, but only poor results were obtained. Pleasingly however, the gold-catalyzed deprotection protocol of Zhang et al.16 enabled the selective deprotection of two aliphatic TBS ethers with simultaneous cyclization to deliver spiroketal (−)-22 as a single diastereomer! We envision the high diastereoselectivity to be due to the anomeric effect17 and presumably a chelation effect of the gold catalyst. Final removal of remaining phenolic TBS group with TBAF completed the synthesis of the southern hemisphere (−)-5.
Scheme 4.

Synthesis of the Southern Hemisphere
Turning to construct the northern hemisphere, we began with the naturally abundant/commercially available atraric acid 23, which was first protected with two TBS groups to deliver 6 in 90% yield. Deprotonation of 6 with freshly prepared LiTMP formed the ortho-ester benzylic anion 24. Pleasingly, anion 24, with the OTBS group adjacent to the ester, that does not undergo self-condensation,18 is sufficiently stable for further transmetallation with ZnCl2, and in turn undergoes the desired Negishi coupling19 with acryloyl chloride 7 to furnish enone 25 in 80% yield. Olefin cross-metathesis between 25 and homoallylic alcohol (+)-8 in the presence of Hoveyda-Grubbs II catalyst then produced the northern hemisphere (+)-4 in 84% yield, with the enone in the E configuration. It is particularly noteworthy that the highly functionalized northern hemisphere (+)-4, bearing an enone, an ester and a free hydroxy group could be constructed in only 3 steps on gram scale.
Having arrived at the requisite northern and southern hemisphere (+)-4 and (−)-5, we next explored the key large-fragment union employing our photoisomerization/cyclization tactic (Scheme 6). Pleasingly under UV-A light, the enone (+)-4 underwent photoisomerization to form the key cyclic oxonium diene intermediate 26 in situ. The hindrance of the methyl group in 26 then led to a stereoselective [3+3]-cyclization with the bulky nucleophile (−)-5 to furnish the desired [3.3.1] nonane adduct (−)-27 in 72% isolated yield (88% based on recovered 5) with a 13:1 dr. The endgame to complete the total synthesis of peniciketal A now only required reduction of the ester to aldehyde. Unfortunately, various attempts to reduce (−)-27 to the corresponding aldehyde in a single step proved unsuccessful. Alternatively, TBS protection of (−)-27 followed by DIBAL-H reduction furnished alcohol (−)-29, which under oxidation with Dess-Martin periodinane (DMP) and silyl group removal with TBAF completed the total synthesis of (+)-peniciketal A (1). It is also noteworthy that in the endgame of this synthesis, (−)-27 and (−)-29, once fully deprotected, could serve as the ester and alcohol analogues of peniciketal A, which are potentially important for possible structure-activity relationship studies to improve the bioactivities.
Scheme 6.

Total Synthesis of (+)-Peniciketal A
The NMR spectra of the synthetic (+)-1 are identical to the natural sample. The absolute configuration of (+)-1 has been confirmed by a single-crystal X-ray anomalous dispersion, which is also identical to the natural product.1 The optical rotation obtained from crystalized synthetic sample is (c 1.0, acetone), compared with natural (c 0.53, acetone). See Supporting Information for details.
With the success of (+)-peniciketal A, we were encouraged to continue our synthetic drive to access (+)-peniciketal B (2), which structurally only differs at the C(3)-OH site of (+)-peniciketal A. The synthesis began with a Barton-McCombie deoxygenation20 of (−)-22 (Scheme 7), which furnished the de oxygenated benzannulated [6,6]-spiroketal (−)-30 in 80% yield. Treatment of (−)-30 with TBAF generated the southern hemisphere (−)-31, required for (+)-peniciketal B (2). Toward the latter end, the photoisomerization/cyclization tactic again pleasingly achieved the large fragment union of (+)-4 and (−)-31 to furnish the desired adduct (−)-32 in 73% yield, with dr = 12 :1. Further elaboration of (−)-32 with the same reduction/oxidation sequence as for (+)-peniciketal A (1), then completed the total synthesis of (+)-peniciketal B (2), the spectral properties of which proved to be identical in all respects to those reported for the natural product. The overall yield for the 8-step sequence from (−)-22 was 24%.
Scheme 7.

Total Synthesis of (+)-Peniciketal B
We further evaluated the cytotoxicity of synthetic (+)-peniciketals A and B against human lung cancer cell lines. (+)-Peniciketal A (1) showed cytotoxic against A549 cells with IC50 values of 16.4 μM, close to the literature report.2 It also dis-played activity against H1975 (IC50 = 14.3 μM). Pleasingly, we found (+)-peniciketal B (2) is more potent against both A549 and H1975 cell lines, with IC50 values of 6.8 and 7.3 μM respectively. Synthetic peniciketals A (1) and B (2) showed less toxicity to human normal lung fibroblast IMR90 cells with IC50 > 50 μM and IC50 = 38 μM respectively.
In summary, the first total synthesis of (+)-peniciketal A (1) and (+)-peniciketal B (2) has been achieved. The total synthesis of (+)-peniciketal A (1) was achieved with the longest linear sequence of 17 steps from 17. The central features of this synthetic venture entailed the further development and application of a novel photoisomerization/cyclization union protocol to construct the complex benzo-fused 2,8-dioxabicyclo[3.3.1]nonane skeleton, in conjunction with a three-component Type I ARC tactic to construct the rare benzannulated [6,6]-spiroketal. Studies toward the synthesis of other members of the peniciketal family, as well as the development of analogues for biological evaluations, continue in our laboratory.
Supplementary Material
Scheme 5.

Synthesis of the Northern Hemisphere
ACKNOWLEDGMENT
Financial support was provided by the NIH through Grant No. CA-19033 and the Bader fellowship. We thank Dr. C. Ross, III for HRMS analysis. We thank Dr. Patrick J. Carroll and Michael Gau for X-ray crystallographic. We thank Dr. David C. Schultz at the University of Pennsylvania High-Throughput Screening Core for supporting the in vitro cell viability studies on IMR90 cells.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Liu W-Z; Ma L-Y; Liu D-S; Huang Y-L; Wang C-H; Shi S-S; Pan X-H; Song X-D; Zhu R-X Peniciketals A-C, New Spiroketals from Saline Soil Derived Penicillium raistrichii. Org. Lett 2014, 16 (1), 90–93. [DOI] [PubMed] [Google Scholar]
- (2).Gao X; Zhou Y; Zheng X; Sun H; Zhang J; Liu W; Pan X Peniciketal A, A Novel Spiroketal Compound, Exerts Anticancer Effects by Inhibiting Cell Proliferation, Migration and Invasion of A549 Lung Cancer Cells. Anticancer Agents Med. Chem 2018, 18 (11), 1573–1581. [DOI] [PubMed] [Google Scholar]
- (3).Gao X; Zhou Y; Sun H; Liu D; Zhang J; Zhang J; Liu W; Pan X Effects of a Spiroketal Compound Peniciketal A and Its Molecular Mechanisms on Growth Inhibition in Human Leukemia. Toxicol. Appl. Pharmacol 2019, 366, 1–9. [DOI] [PubMed] [Google Scholar]
- (4).Gao X; Zhou Y; Sun H; Liu D; Zhang J; Zhang J; Liu W; Pan X Analysis of Comparative Proteomeic and Potent Targets of Peniciketal A in Human Acute Monocytic Leukemia. Anicancer Agents Med. Chem 2019, 19 (4), 515–527. [DOI] [PubMed] [Google Scholar]
- (5).Gillard RM; Brimble MA Benzannulated Spiroketal Natural Products: Isolation, Biological Activity, Biosynthesis, and Total Synthesis. Org. Biomol. Chem 2019, 17 (36), 8272–8307. [DOI] [PubMed] [Google Scholar]
- (6).Deng Y; Smith AB III. Evolution of Anion Relay Chemistry: Construction of Architecturally Complex Natural Products. Acc. Chem. Res 2020, 53 (4), 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For reviews:; (a) Heravi MM; Hajabbasi P; Hamidi H Recent Development in the Asymmetric Michael Addition for Carbon-Carbon Bond Formation. Curr. Org. Chem 2014, 18 (4), 489–511. [Google Scholar]; (b) Hui C; Pu F; Xu J Metal-Catalyzed Asymmetric Michael Addition in Natural Product Synthesis. Chem Eur. J 2017, 23 (17), 4023–4036. [DOI] [PubMed] [Google Scholar]
- (8).Smith AB III; Richmond RE A Common Strategy for Construction of the Paniculides (A-C); Total Synthesis of Paniculide A. J. Org. Chem 1981, 46 (23), 4814–4816. [Google Scholar]
- (9).Koft ER; Smith AB III. Total Synthesis of Hibiscone C (Gmelofuran). J. Am. Chem. Soc 1982, 104 (20), 5568–5570. [Google Scholar]
- (10).Smith AB III.; Sulikowski GA; Fujimoto K Total Synthesis of Natural (−)-Echinosporin. Determination of the Absolute Configuration. J. Am. Chem. Soc 1989, 111 (20), 8039–8041. [Google Scholar]
- (11).Deng Y; Nguyen MD; Zou Y; Houk KN; Smith AB III.; Generation of Dithianyl and Dioxolanyl Radicals Using Photoredox Catalysis: Application in the Total Synthesis of the Danshenspiroketal-lactones via Radical Relay Chemistry. Org. Lett 2019, 21 (6), 1708–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Li B; Williams BD; Smith AB III. A Mild, Diastereoselective Construction of Cyclic and Spirocyclic Ketals Employing a Tandem Photoisomerization/Cyclization Tactic. Org. Lett 2015, 17 (1), 3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(+)-12 is prepared from (+)-8 in 4 steps following literature report:; Mohapatra DK; Pulluri K; Gajula S; Yadav JS 13-Step Total Synthesis of Dendrodolide K Following Iterative Barlett-Smith Iodo-carbonate Cyclization. Tetrahedron Lett. 2015, 56 (46), 6377–6380. [Google Scholar]
- (14).Elix JA; Jayanthi WK Synthetic Confirmation of the Structure of the Lichen Benzyl Esters Alectorialic and Barbatolic Acids. Aust. J. Chem 1987, 40 (11), 1841–1850. [Google Scholar]
- (15).Brook AG Molecular Rearrangement of Organosilicon Compounds. Acc. Chem. Res 1974, 7 (3), 77–84. [Google Scholar]
- (16).Zhang Q; Kang X; Long L; Zhu L; Chai Y Mild and Selective Deprotection of tert-Butyl(dimethyl)silyl Ethers with Catalytic Amounts of Sodium Tetrachloroaurate(III) Dihydrate. Synthesis. 2015, 47 (1), 55–64. [Google Scholar]
- (17).Perron F; Albizati KF Chemistry of Spiroketals. Chem. Rev 1989, 89 (7), 1617–1661. [Google Scholar]
- (18).Lewis CN; Spargo PL; Staunton J A Convenient Synthesis of 3-Substituted 8-Methoxy- and 6,8-Dimethoxyisocoumarins. Synthesis. 1986, 11, 944–946. [Google Scholar]
- (19).Negishi E-I; Bagheri V; Chatterjee S; Luo F-T; Miller JA; Stoll AT Palladium-Catalyzed Acylation of Organozincs and Other Organometallics as A Convenient Route to Ketones. Tetrahedron Lett. 1983, 24 (47), 5181–5184. [Google Scholar]
- (20).Barton DHR; McCombie SWJ A New Method for the De-oxygenation of Secondary Alcohols. J. Chem. Soc. Perkin Trans. 1 1975, 16, 1574–1585. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.