

Abstract
Background
To identify genetic associations of quantitative metabolic syndrome (MetS) traits and characterize heterogeneity across ethnic groups.
Methods
Data was collected from GENetics of Noninsulin dependent Diabetes Mellitus (GENNID), a multiethnic resource of Type 2 diabetic families and included 1520 subjects in 259 African-American, European-American, Japanese-Americans, and Mexican-American families. We focused on eight MetS traits: weight, waist circumference, systolic and diastolic blood pressure, high-density lipoprotein, triglycerides, fasting glucose, and insulin. Using genotyped and imputed data from Illumina’s Multiethnic array, we conducted genome-wide association analyses with linear mixed models for all ethnicities, except for the smaller Japanese-American group, where we used additive genetic models with gene-dropping.
Results
Findings included ethnic-specific genetic associations and heterogeneity across ethnicities. Most significant associations were outside our candidate linkage regions and were coincident within a gene or intergenic region, with two exceptions in European-American families: (a) within previously identified linkage region on chromosome 2, two significant GLI2-TFCP2L1 associations with weight, and (b) one chromosome 11 variant near CADM1-LINC00900 with pleiotropic blood pressure effects.
Conclusions
This multiethnic family study found genetic heterogeneity and coincident associations (with one case of pleiotropy), highlighting the importance of including diverse populations in genetic research and illustrating the complex genetic architecture underlying MetS.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13098-021-00670-3.
Keywords: Metabolic syndrome, Genetic epidemiology, Family studies, Quantitative trait loci, Linkage
Background
Metabolic syndrome (MetS) is a common, complex condition characterized by hyperlipidemia, hypertension, hyperglycemia, and excess abdominal fat [1–3]. The National Cholesterol Education Program’s Adult Treatment Panel (NCEP ATP) III criteria [3], typically used in the United States for clinical diagnosis, defines MetS as the presence of at least three of five risk factors: elevated systolic and/or diastolic blood pressure (SBP, DBP), elevated triglycerides (TG), decreased high density lipoprotein (HDL)-cholesterol, elevated fasting glucose, and abdominal obesity [1, 3]. Due to the clustering of these characteristics [4, 5], individuals with MetS are at risk for cardiovascular and metabolic diseases such as stroke and diabetes [6–10]. Moreover, in several US-based studies of families [11–15], MetS quantitative and multivariate factor traits are highly heritable with about half of the variation between subjects explained by genetics in families of European descent [14, 15] and particularly for obesity and lipid-related traits in families of African Americans [12, 14], Mexican Americans [13] and Japanese Americans [11]. Family-based studies have been a primary approach for identifying genetic influences on a range of disease and still offer many advantages [16, 17] including being robust to confounding due to underlying population structure and phenotype model misspecifications, using pedigree structures and information on related individuals to detect genotyping errors [16], and having more power to detect rare variants [16, 17].
Candidate gene [18–23] and genome-wide association studies (GWAS) [18, 24–27] have already generated a number of candidate genes and variants possibly associated with MetS. However, the number of variants is still growing [8], particularly in the Asian population [28]. Nonetheless, many questions still remain about the underlying genetic architecture of MetS. For example, are the genetic influences the same regardless of which NCEP traits cluster within an individual? Accumulating evidence suggests that the specific combination of traits may matter and could explain the large number of variants associated with MetS [29, 30]. Several obesity-related loci have been shown to be associated with different MetS traits [8, 31]. For example, obesity, high TG, high fasting insulin, and low HDL are associated with MIP1, MC4R, and PRKD1, yet when these same traits are combined with hypertension, they are associated with FTO and TMEM18 [8].
Results from our previous studies suggest differences in the clustering due to the underlying genetics of MetS traits by ethnicity [32–34]. For example, while a significant genetic correlation between weight and waist is present in African American (AA), European American (EA), Japanese American (JA) and Mexican American (MA) families [32, 34], the genetic correlation between high systolic blood pressure (SBP) and diastolic blood pressure (DBP) is seen only in AA, EA, and MA families [34]. The significant genetic correlation of lipids (TG and HDL) has been shown to be characteristic among EA and JA families [32, 34]. These differences in clustering patterns may be driven by different sets of underlying genetic influences and could explain the large number of genetic variants and genes associated with MetS.
Previously, family-based genetic linkage analyses nominated chromosomal regions with putative causal variants for individual and multivariate MetS traits. Results indicated several high priority linkage regions, including a region on chromosome 2 for EA [32, 33] and AA families [35, 36] and a linkage region containing ADIPOQ on chromosome 3 among MA families [33]. These candidate linkage regions are large (between 150 and 540 Mbp), with multiple traits mapping to these regions and evidence for heterogeneity across ethnic groups [33]. A more in-depth evaluation of these regions to determine if linkage is due to pleiotropy or co-incident linkage/association, along with a broader focus on understanding if different trait clustering contributes to heterogeneity is needed. We used the GENetics of NonInsulin-dependent Diabetes mellitus (GENNID) resource [37], a multiethnic study of families with type 2 diabetes (T2D), and a GWAS approach to identify quantitative trait nucleotides (QTNs) with possible pleiotropic or coincident effects and to examine evidence for heterogeneity in genetic association findings for MetS traits across ethnic groups.
Methods
Study subjects
GENNID is an American Diabetes Association (ADA) resource of genetic, questionnaire, and laboratory data from multiplex, ethnically diverse AA, EA, JA and MA families with T2D, diagnosed using the National Diabetes Data Group criteria [38]. In this cross-sectional study from 1993 to 1997, T2D families were ascertained in two phases across multiple centers in the United States [37]. Phase 1 focused primarily on larger, multi-generational data collection of families with at least two T2D affected siblings in addition to at least three first-degree relatives. Phase 2 ascertained sibling pairs and nuclear families with at least two T2D affected siblings, and if at most one parent was ascertained, then data was collected on at least two additional siblings. AA, EA, and MA families were collected in both phases while JA families were only collected in Phase 1 [37, 39]. This study used all available data except for the Phase 2 EA data (N = 371 subjects) which were not yet genotyped. Self-identified race, family and medical histories, anthropometric and lab measurements were obtained from participants. Specifically, we focused on eight MetS-related, quantitative traits (i.e., HDL, TG, SBP, DBP, fasting insulin, fasting glucose, weight, and waist circumference) defined from anthropomorphic and lab measurements. Pedigree relationships, age, sex, and diabetes status were obtained from the data collection and questionnaires.
Genotying and imputation
Previously, using microsatellite markers, linkage analyses identified candidate regions for multivariate MetS traits as described in Edwards et al. [32]. For this study, the Northwest Genomics Center (NWGC) performed genome-wide genotyping using Illumina’s Infininium LCG genotyping assay on the Multiethnic Global beadchip (v1.0, genome build 37). DNA samples were normalized using a PerkinElmer Janus Workstation and then genotyped. We performed quality control (QC) of genotype data separately for each ethnic group [40]. Genetic imputation was then performed by first phasing each ethnic group’s QC’ed genotypes using Eagle2 software via the Sanger Imputation Service [41] with the corresponding reference panels for each ethnic group: 1000 Genomes Phase 3 [42] reference panel (for AAs, MAs, JAs) and the Haplotype Reference Consortium (HRC) [41] reference panel (for EAs). DuoHMM [43] was used to correct haplotype phasing switch errors based on pedigree relatedness. The final step for imputation was then performed using Minimac3 via the Michigan Imputation Server [44] with the HRC reference panel.
Statistical analysis
We performed genome-wide association testing across all imputed and genotyped QTNs. Quantitative traits with non-normal distributions were transformed in order to satisfy normality assumptions. The skewed distributions of HDL, TG, waist, and insulin measures were log-transformed, whereas a rank-based inverse normal transformation resulted in approximately normal distributions for weight, DBP, SBP, and fasting glucose. For AAs, EAs, and MAs, linear mixed models were used in Genome-wide Complex Trait Analysis (GCTA) software [45] with the kinship coefficient matrix empirically estimated by LD-adjusted kinships (LDAK) software [46]. However, due to asymptotic concerns with a smaller JA sample, association testing was performed using gene-dropping [40]. Univariate association analyses were adjusted by age, sex, and self-reported diabetes status. Association results with P ≤ 5 × 10–8 were genome-wide significant and with P between 5 × 10–8 and 10–6 (i.e., 5 × 10–8 < P ≤ 10–6) were suggestive of association. Additionally, after a Bonferroni correction for the testing of 8 traits, we also identified highly significant QTNs with P ≤ 6.25 × 10–9 [i.e., (5 × 10–8)/8]. Moreover, the I2 metric [47] was used to assess the degree of heterogeneity across ethnic groups. Using the METAL software [48], I2 was calculated as the percentage of variance that is due to heterogeneity of effect size (β) estimates across ethnic groups. I2 values of 0 indicate no heterogeneity of effect sizes across ethnic groups; values over 75% and up to 100% indicate considerable heterogeneity [49]. I2 values were not calculated for QTNs present in only one ethnic group (i.e., when the QTNs were monomorphic or were filtered out during QC in the other ethnic groups). The R program [50] was also used for statistical analysis, programming, and plotting. Circular Manhattan plots were made using the CMplot R package [51].
Functional and regulatory annotation
Finally, evidence of biological function was characterized by annotating significant QTNs and any QTNs in linkage disequilibrium (LD). Specifically, we used ANNOtate VARiation (ANNOVAR) software [52] to annotate significant QTNs with five different integrative annotations and their corresponding thresholds. In particular, based on support vector machine (SVM) supervised learning, Combined Annotation Dependent Depletion (CADD v1.3) [53] phred-scaled scores of at least 10 denoted deleterious variants belonging in the top 10%. Additionally, obtained from random forest methods, Genome Wide Annotation of VAriants (GWAVA) annotation tool [54], Training Stress Scores (TSS) of at least 0.40 defined variants with possible regulatory effects. Using spectral, unsupervised learning algorithms, EIGEN [55] scores greater than 0 indicated putative deleterious variants. Additional annotations for intronic variants included Functional Analysis Through Hidden Markov Models-Multiple Kernel Learning (FATHMM-MLK) [56] scores greater than 0.50 and RegSNPs-intron [57] disease-causing probabilities greater than 0.50. LDproxy [58] was used to identify potentially functional QTNs in LD (r2 > 0.80) with QTNs significantly associated with MetS traits. These QTNs in LD were considered to be functional if they were exonic or had a RegulomeDB [59] rank (which ranged from 1 to 7) of at most 3.
Results
Descriptives
Sample and family size, demographic characteristics, and phenotypic measures varied by ethnic group (Table 1). There were 281 subjects in 73 AA families, 516 subjects in 75 EA families, 125 subjects in 15 JA families, and 598 subjects in 96 MA families. In particular, EA and JA families were larger and at least three-generational with a median size of 6–7 members per family, respectively. AA families were typically smaller with a median of 4 members per family. Although the MA families had a median family size of 4 members, the mean family size was 6 members, and there were a few very large MA families.
Table 1.
GENNID genetic and phenotypic characteristics by ethnic group (mean ± SD)
| Characteristics | AA: African Americans | EA: European Americans | JA: Japanese Americans | MA: Mexican Americans |
|---|---|---|---|---|
| # Subjects (# families) | 281 (73 families) | 516 (75 families) | 125 (15 families) | 598 (96 families) |
|
Median (min, max) # Subjects per family |
4 (1, 7) | 6 (1, 29) | 7 (3, 17) | 4 (1, 112) |
| # QCa variants | 710,226 | 686,200 | 437,730 | 731,016 |
| # Variants genotyped and imputed (association analysis) | 13,042,663 | 7,681,619 | 5,455,666 | 7,907,815 |
| Age (years) | 52.4 ± 15.1 | 51.2 ± 16.8 | 56.1 ± 15.8 | 52.1 ± 15.8 |
| Sex (% male) | 31.3% | 43.8% | 52% | 36.0% |
| Self-reported diabetes status (% diabetes) | 51.2% | 37.2% | 27.20% | 47.7% |
| Systolic blood pressure (SBP) (mmHg) | 128.5 ± 19.8 | 127.2 ± 19.1 | 121.8 ± 18.1 | 126.2 ± 19.3 |
| Diastolic blood pressure (DBP) (mmHg) | 79.4 ± 10.8 | 78 ± 10.2 | 72.3 ± 11.5 | 74.6 ± 10.6 |
| High density lipoproteins (HDL) (mg/dL) | 47.5 ± 12.3 | 40.5 ± 11.2 | 45.6 ± 14.2 | 39.4 ± 10.2 |
| Triglycerides (TG), fasting (mg/dL) | 105.5 ± 110.3 | 143.8 ± 123.8 | 135.4 ± 104.5 | 160.9 ± 121.7 |
| Glucose, fasting (mg/dL) | 146.8 ± 75.3 | 128.3 ± 56.7 | 115 ± 29.6 | 152.2 ± 76.9 |
| Insulin, fasting (mg/dL) | 16.2 ± 21 | 11.5 ± 10.9 | 7.3 ± 6.3 | 17 ± 16.8 |
| Weight (kg) | 86.8 ± 21.1 | 84.1 ± 20.4 | 65.7 ± 13.4 | 79 ± 17.7 |
| Waist circumference (cm) | 98.5 ± 16 | 99.5 ± 16 | 88.7 ± 11.2 | 102.4 ± 14.3 |
SD standard deviation
aQC: quality control includes alignment to Haplotype Reference Consortium (HRC) panel (EA) or 1000 Genomes (1000G) panel (AA, MA, JA)
After QC, there was a similar number of QTNs for MA, AA, and EA families—about 731 K, 710 K, and 686 K QTNs, respectively. Among JA families, there were only ~ 437 K QTNs, which was ~ 40% less than the other ethnic groups. The lower number of QTNs among JA families was due to the removal of a large number of monomorphic markers, which may suggest a lower coverage for those of Asian descent on the multiethnic genotyping array [40].
Across ethnic groups, the mean age was similar and ranged between 51 and 56 years old (Table 1). About half the subjects in JA and EA families were men (52% and 43.8%, respectively), whereas AA and MA families had more females (68.7% and 64%, respectively). T2D was most frequent in AA and MA families with more than 51% and 47% of individuals self-reported as having T2D, respectively. About one-third of subjects (i.e., 37% and 27%) self-reported having T2D among EA and JA families, respectively. Although mean SBP and DBP measures were similar across ethnic groups, the mean blood pressure for this study population was elevated by 2017 clinical guidelines as defined by SBP > 120 mmHg and DBP > 80mmHg [60]. Furthermore, using NCEP ATP III guidelines [3], dyslipidemia, characterized by HDL measures < 40 mg/dL, was more evident in MA and EA families compared to AA and JA families (with the following means, respectively: 39.4 mg/dL and 40.5 mg/dL vs. 47.5 mg/dL and 45.6 mg/dL). Moreover, mean TG was most elevated among MAs with 160.9 mg/dL and lowest in AAs with 105.5 mg/dL. Hyperglycemia (when fasting glucose > 100 mg/dL) was present in all ethnic groups: MAs had the highest mean level (152.2 mg/dL) and JAs had the lowest mean level (115.0 mg/dL). Mean fasting insulin measures were elevated among MAs, AAs, and EAs (17.0 mg/dL, 16.2 mg/dL, and 16.5 mg/dL, respectively). Mean weight and mean waist circumference were both lowest among JAs (65.7 kg and 88.7 cm, respectively).
Genomewide association results
We evaluated genetic association results for traits with at least one significant QTN (P ≤ 5 × 10–8) using circular Manhattan plots in Fig. 1. Table 2 presents all significant (P ≤ 5 × 10–8) results for each ethnic group including two variants with suggestive evidence (5 × 10–8 < P ≤ 10–6) of an association with MetS traits for EA: (a) rs1260326, a nonsynonymous GCKR variant with possible functional importance and (b) rs186742063, a possible pleiotropic variant with evidence of association with SBP and DBP traits. Specifically, there was a suggestive association of log(TG) and a non-synonymous QTN (rs1260326, P = 1.4 × 10–7) in the GCKR gene among the EAs. Additionally, in the EA group, on chromosome 11 at 115,495,297 bp (hg19/GRCh37) between CADM1 and LINC00900, there was a pleiotropic QTN, rs186742063, with a significant association with DBP (P = 4.42 × 10–8) and a suggestive association with SBP (P = 9.92 × 10–8), respectively.
Fig. 1.

Family-based association results by Ethnic Group. Chromosomes are labeled 1 through 22 with QTN density denoted by right-hand legend. Each ring of the circular Manhattan plot indicates a selected quantitative trait that was analyzed (labeled at the top of each ring). Each dot represents the − log10(P) corresponding to test for association with the given quantitative trait. Genome-wide significant results are denoted by red box points at/above the red threshold line of − log10(5 × 10−8). Suggestive association is denoted by blue triangle points at/above the dotted blue line threshold at − log10(10−6). Only traits with significant results are shown for each ethnic group
Table 2.
Significant MetS Quantitative Trait Association results (P ≤ 5 × 10–8) by ethnic group
| Ethnic group | Traita | rsid | CHR | BP (hg19) | A1 | A2 | A1 Freq in studyb | A1 Freq in AFRc | A1 Freq in AMRc | A1 Freq in EASc | A1 Freq in NFEc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AA | log(TG) | rs75219957 | 1 | 231,296,755 | G | C | 0.020 | 0.012 | 0.133 | 0.278 | 0.0547 |
| log(TG) | rs568152609 | 4 | 31,060,113 | C | T | 0.010 | 0.007 | 0.002 | 0 | 0 | |
| Glucose | rs138724790 | 5 | 86,997,218 | C | T | 0.010 | 0.016 | 0 | 0 | 0 | |
| Glucose | rs78975723 | 6 | 39,338,990 | G | C | 0.010 | 0.033 | 0.002 | 0 | 6.5E−5 | |
| log(TG) | rs73123056 | 7 | 63,354,705 | A | C | 0.040 | 0.024 | 0.073 | 0 | 0.048 | |
| log(TG) | rs17137121 | 7 | 63,358,355 | T | C | 0.040 | 0.024 | 0.073 | 0 | 0.048 | |
| log(TG) | rs114606502 | 12 | 130,117,352 | C | T | 0 | 0.007 | 0 | 0 | 0 | |
| log(TG) | rs141776024 | 12 | 130,147,629 | A | G | 0 | 0.009 | 0 | 0 | 0 | |
| EA | log(TG) | rs1260326 | 2 | 27,730,940 | T | C | 0.400 | 0.135 | 0.314 | 0.501 | 0.400 |
| Weight | rs139940998 | 2 | 121,836,875 | A | G | 0.010 | 0.002 | 0.011 | 0 | 0.009 | |
| Weight | rs144756634 | 2 | 121,843,978 | A | T | 0.010 | 0.002 | 0.011 | 0 | 0.009 | |
| DBP | rs186742063f | 11 | 115,495,297 | A | G | 0.020 | 0.004 | 0.007 | 0 | 0.018 | |
| JA | Weight | rs2302308 | 4 | 122,258,149 | G | T | 0.012 | 0.025 | 0.062 | 0.071 | 0.106 |
| Weight | rs74398478 | 4 | 122,259,584 | A | C | 0.012 | 0.025 | 0.063 | 0.071 | 0.106 | |
| Weight | rs17051338 | 4 | 122,260,042 | G | T | 0.012 | 0.025 | 0.063 | 0.073 | 0.106 | |
| Weight | rs77438622 | 4 | 122,264,692 | G | C | 0.012 | 0.025 | 0.063 | 0.073 | 0.106 | |
| Weight | rs77006299 | 4 | 122,270,900 | C | A | 0.012 | 0.025 | 0.063 | 0.073 | 0.106 | |
| log(waist) | rs146792726 | 11 | 3,972,232 | A | G | 0.012 | 0 | 0 | 0.029 | 0 | |
| log(waist) | rs147605117 | 11 | 4,063,916 | G | C | 0.012 | 0 | 0 | 0.031 | 0 | |
| log(waist) | rs143126598 | 15 | 54,259,502 | A | G | 0.012 | 0.0002 | 0 | 0.009 | 0 | |
| MA | Glucose | rs17024841 | 4 | 149,556,752 | A | G | 0.030 | 0.010 | 0.020 | 0.144 | 0.051 |
| Glucose | rs111933650 | 4 | 149,558,682 | T | C | 0.030 | 0.010 | 0.020 | 0.144 | 0.050 | |
| Glucose | rs76209611 | 4 | 149,561,328 | G | T | 0.040 | 0.011 | 0.020 | 0.145 | 0.057 | |
| log(TG) | rs4522365 | 15 | 29,964,742 | T | C | 0.170 | 0.534 | 0.156 | 0.088 | 0.152 |
| Ethnic group | Traita | rsid | Gene | Functional info | I2d | Beta | SE | P | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AA | log(TG) | rs75219957 | FAM89A-TRIM67 | Intergenic | 80.4 | 1.04 | 0.19 | 3.0E−08 | |||
| log(TG) | rs568152609 | PCDH7 | Intronic | – | 1.36 | 0.23 | 5.3E−09 | ||||
| Glucose | rs138724790 | CCNH-TMEM161B | Intergenic | – | 1.93 | 0.35 | 3.7E−08 | ||||
| Glucose | rs78975723 | KIF6 | Intronic | – | 1.75 | 0.31 | 2.6E−08 | ||||
| log(TG) | rs73123056 | DQ599799-BC042811 | Intergenic | 84.5 | 0.81 | 0.15 | 2.4E−08 | ||||
| log(TG) | rs17137121 | DQ599799-BC042811 | Intergenic | 84.5 | 0.81 | 0.15 | 2.4E−08 | ||||
| log(TG) | rs114606502 | TMEM132D | Intronic | – | 2.32 | 0.40 | 7.9E−09 | ||||
| log(TG) | rs141776024 | TMEM132D | Intronic | – | 2.32 | 0.40 | 7.9E−09 | ||||
| EA | log(TG) | rs1260326 | GCKR | Nonsyn | 53.2 | 0.25 | 0.05 | 1.4E−07e | |||
| Weight | rs139940998 | GLI2-TFCP2L1 | Intergenic | – | 1.84 | 0.32 | 7.3E−09 | ||||
| Weight | rs144756634 | GLI2-TFCP2L1 | Intergenic | – | 1.84 | 0.32 | 7.3E−09 | ||||
| DBP | rs186742063f | CADM1-LINC00900 | Intergenic | 90.9 | 1.42 | 0.26 | 4.4E−08 | ||||
| JA | Weight | rs2302308 | QRFPR | Intronic | 0 | 2.13 | 0.59 | 5.0E−08 | |||
| Weight | rs74398478 | QRFPR | Intronic | 0 | 2.13 | 0.59 | 5.0E−08 | ||||
| Weight | rs17051338 | QRFPR | Intronic | 0 | 2.13 | 0.59 | 5.0E−08 | ||||
| Weight | rs77438622 | QRFPR | Intronic | 0 | 2.13 | 0.59 | 5.0E−08 | ||||
| Weight | rs77006299 | QRFPR | Intronic | 0 | 2.13 | 0.59 | 5.0E−08 | ||||
| log(waist) | rs146792726 | STIM1 | Intronic | – | − 0.39 | 0.08 | 5.0E−08 | ||||
| log(waist) | rs147605117 | STIM1 | Intronic | – | − 0.39 | 0.08 | 5.0E−08 | ||||
| log(waist) | rs143126598 | WDR72-UNC13C | Intergenic | – | − 0.39 | 0.08 | 5.0E−08 | ||||
| MA | Glucose | rs17024841 | 5S_rRNA-BC031092 | Intergenic | 80.6 | − 0.70 | 0.12 | 1.7E−08 | |||
| Glucose | rs111933650 | 5S_rRNA-BC031092 | Intergenic | 80.6 | − 0.70 | 0.12 | 1.7E−08 | ||||
| Glucose | rs76209611 | 5S_rRNA-BC031092 | Intergenic | 78.9 | − 0.67 | 0.12 | 4.2E−08 | ||||
| log(TG) | rs4522365 | FAM189A1-BC043570 | Intergenic | 74.8 | − 0.29 | 0.05 | 2.0E−09 |
AA African Americans, EA European Americans, JA Japanese Americans, MA Mexican Americans
aRank-based inverse normal transformation applied to weight, SBP (systolic blood pressure), DBP (diastolic blood pressure), and glucose traits
bFrequency of A1 allele in GENNID as estimated by GCTA [45]
cFrequency of A1 allele in gnomAD [76]: NFE (Non-Finnish Europeans) for EA, EAS (East Asians) for JA, AMR (Latinos) for MA, and AFR (African/African Americans) for AA
dStatistic summarizing heterogeneity across ethnic groups; the percentage of variation of beta estimates across ethnic groups due to heterogeneity; values of 0 indicate no heterogeneity of effect sizes across ethnic groups; values over 75% and up to 100% indicate high levels of heterogeneity; missing values indicate the QTN was only analyzed in one ethnic group (either due to the QTN being monomorphic in the other ethnic groups or being filtered out during quality control in the other ethnic groups)
ers1260326 nonsynonymous SNV was suggestive (5 × 10–8 > P ≥ 10–6) but was found to be significant in GENNID meta-analysis [40]
frs186742063 has evidence of pleiotropy with suggestive evidence (P = 9.9E−08) of being associated with systolic blood pressure (SBP) (see Additional file 1: Table S1)
Moreover, two variants were still significant after using a more stringent Bonferroni correction for analyzing 8 traits (i.e., P ≤ 6.25 × 10–9). Specifically, on chromosome 4, a rare, intronic PCDH7 variant (rs568152609 with MAF = 0.7% in the AFR population of gnomAD) was strongly associated with log(TG) in AA. Additionally, in MA on chromosome 15, an intergenic variant (rs4522365) between FAM189A1 and BC043570 was also significantly associated with log(TG) (P = 2.0 × 10–9). All suggestive results (5 × 10–8 < P ≤ 10–6) are included in Additional file 1 (Table S1).
The significant QTNs associated with MetS quantitative traits are also from intergenic or intronic chromosomal regions. Most of the significant QTNs (Table 2) were moderately rare with ~ 1–3% frequency in the corresponding ethnic-specific population based on data from gnomAD reference groups [61]. These variants were enriched in our GENNID families and had frequencies (estimated by GCTA) that were ~ 1–2% higher than in gnomAD. QTNs significantly associated with log(waist) in JA and variants associated with glucose and log(TG) in MA all had negative additive effects (β < 0) for each copy of the A1 minor allele. All other significant associations had a positive additive effect (β > 0).
Heterogeneity across ethnic groups
Although there were significant genetic associations with both fasting glucose and TG in both AA and MA (Fig. 1), the locations of the significant QTNs were found on different chromosomes (Table 2). For AA families, there were significant associations of log(TG) with multiple QTNs in the following genetic regions: 1q42.2, in PCDH7 on chromosome 4, intergenic region between DQ599799 and BC042811 on chromosome 7, and in TMEM132D on chromosome 12. In addition, significant QTNs associated with fasting glucose levels were found in intergenic regions on chromosome 5 (CCNH-TMEM161B) and within the KIF6 gene on chromosome 6. However, in MA families, log(TG) and fasting glucose levels were significantly associated with chromosomes 15q13.1 and 4q31, respectively. These regions were not associated with MetS quantitative traits in EA or JA groups (P > 10–6).
In addition, although in different physical locations, both EA and JA had significant genetic associations with weight (Fig. 1 and Table 2). For EA families, there was significant evidence of a genetic association for weight on chromosome 2q14.2, whereas in JA, QTNs were significantly associated with weight in the intronic region of QRFPR on chromosome 4. Moreover, in JA families, log-transformed waist circumference was significantly associated with QTNs on chromosomes 11 (STIM1) and 15 (at 54,259,502 bp between WDR72 and UNC13C), whereas in EA there were only suggestive associations with log(waist) (Additional file 1: Table S1) on chromosome 2 between 123,052,152 and 124,754,047 bp (hg19/GRCh37) near AX747402, 7SK, and TSN. These associations were unique to each ethnic group and were not found to be significant or suggestive in other ethnic groups (P > 10–6).
Thus, there was no overlap of significant or suggestive associations among ethnic groups. Moreover, there was evidence of heterogeneity between genetic effects across ethnic groups (I2 ≥ 75% in Table 2). Although chromosome 4 variants were significantly associated with weight only in JA families, these genetic effects were comparable across the other ethnic groups (i.e., I2 = 0). On the other hand, other variants besides the chromosome 4 QRFPR intronic variants associated with weight in JA families in Table 2, had effects that were either heterogeneous (I2 ≥ 75%) across ethnic groups or were unique to a single ethnic group (I2 not calculated) [61].
Association in candidate linkage regions
Some significant and suggestive associations overlapped or were near previously nominated candidate linkage regions found on chromosomes 1, 2, 3, 5, and 16 [32, 35, 36, 62–65] (Additional file 2: Table S2). In particular, unique only among EA families was a significant association between weight and QTNs (rs139940998 and rs144756634) on chromosome 2 at 121,836,875 bp and a second location at 121,843,978 bp (between GLI2 and TFC2L1) within 7.3 Mbp of the candidate linkage region (2q12.1–13) (Fig. 2). Also, in the MA families, the candidate linkage region on 3p26 harbored two suggestive QTN associations (i.e., rs17005939 with P = 8.02 × 10–8 at chr3:2004251; rs12631510 with P = 9.13 × 10–8 at chr3:2001175) between the genes CNTN6 and CNTN4 at 3p26.3–p26.2 with log(HDL); both QTNs had genetic effects that were heterogeneous and differed across ethnic groups (I2 of 80.7% and 79.2%, respectively). Suggestive evidence for association with log(TG) and a non-coding RNA intron within a pseudo-gene, AK126539, (nominated by QTN at chr16:11562798) was identified within our linkage candidate region of 16p13.2–16p12.1 and was unique to AA families. Additional suggestive associations in AA with log(TG), specifically, three QTNs unique to AA and one QTN, rs78637841 in WWOX, having a high level of heterogeneity with I2 = 85%, and weight (i.e., three QTNs being unique to AA) were also found on 16q13.13 and 16q23.1, respectively (Additional file 1: Table S1). There was no evidence for association within the previously candidate linkage region on chromosome 5 (5q33.1–5q34) in JA (nominated for log(waist)); however, there was a nearby QTN (at chr5:130581195 within 5q23.3–5q31.1) that had suggestive association with fasting glucose (P = 1.38 × 10–7). The genetic effect of this QTN is not unique to JA and was comparable across ethnic groups (I2 = 0).
Fig. 2.
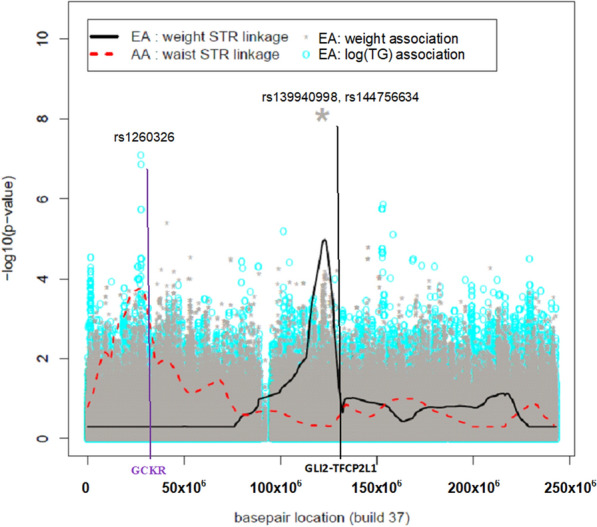
MetS Quantitative Trait Linkage and Association results: chromosome 2. Each color (black and red) and line type in the legend denotes ethnic group (European Americans (EA), African Americans (AA), respectively) univariate trait analysis for microsatellite (STR) linkage and SNP association testing. Genes of interest are denoted along the bottom of the x-axis in red and black font and colored vertical lines indicate suggestive and significant QTN locations. AA waist linkage had maxLOD = 2.78, and this location had prior evidence of association to GCKR
Potential functional roles and regulatory effects
Additionally, in Table 3, we used ANNOVAR to assign functional roles to QTNs from Table 2 that were significantly associated with MetS traits. In particular, although the majority of significant QTNs were intronic and intergenic, some of these non-exonic variants have evidence of functionality according to the various annotating scoring methods. In the AAs, two intergenic QTNs (rs75219957 and rs73123056) on chromosomes 1 and 7, respectively, that were both significantly associated with log(TG) had evidence of possible deleterious effects. In the EAs, the GCKR nonsynonymous variant rs1260326 had moderate evidence of function based on an EIGEN score of 0.29, and the intergenic GLI2-TFCP2L1 variant rs139940998 not only had a high CADD score of 19.05 but also evidence of possible deleterious effects from GWAVA, FATHMM, and EIGEN scores. Moreover, most of the significant QRFPR intronic variants associated with weight in JAs had moderate evidence of function based only on EIGEN scores; however, rs2302308 also had additional evidence of regulatory effects based on RegSNPs-intron with a 79% probability of being disease-causing. Also, in JAs, an STIM1 intronic variant (rs147605117) significantly associated with waist circumference also had potential damaging effects as indicated by both a CADD score of 10.86 and EIGEN score of 0.29. On the other hand, in MAs, only one intergenic variant (rs76209611) had weak evidence of being functional with an EIGEN score near zero at 0.08.
Table 3.
Significantly-associated MetS QTNs with functional annotation
| Ethnic group | Trait | Gene (distance in basepairs) from refGene | Function (refGene) | CHR | Basepair (hg19) | rsid | Alleles | Functional annotations with thresholdsa | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FATHMM-MLK (noncoding > 0.50) | GWAVA (TSS) > 0.40 | CADD v1.3 (phred-scaled) > 10 | EIGEN > 0 | ||||||||
| AA | log(TG) |
FAM89A(120,755) TRIM67(1919) |
Intergenic | 1 | 231,296,755 | rs75219957 | G/C | 0.20 | 0.62 | 4.48 | 0.38 |
| AA | log(TG) |
MIR4283-1(273,158) ZNF722P(104,479) |
Intergenic | 7 | 63,354,705 | rs73123056 | A/C | 0.97 | 0.21 | 3.38 | 0.61 |
| EA | log(TG) | GCKR | Exonic-NSVb | 2 | 27,730,940 | rs1260326 | T/C | 0.02c | 0.20 | 0.11 | 0.29 |
| EA | Weight |
GLI2(86,646) TFCP2L1(137,289) |
Intergenic | 2 | 121,836,875 | rs139940998 | A/G | 0.63 | 0.46 | 19.05 | 0.69 |
| JA | Weight | QRFPR | Intronic | 4 | 122,258,149 | rs2302308d | G/T | 0.12 | 0.34 | 3.83 | 0.02 |
| JA | Weight | QRFPR | Intronic | 4 | 122,260,042 | rs17051338 | G/T | 0.23 | 0.31 | 7.28 | 0.33 |
| JA | Weight | QRFPR | Intronic | 4 | 122,264,692 | rs77438622 | G/C | 0.12 | 0.21 | 6.40 | 0.09 |
| JA | Weight | QRFPR | Intronic | 4 | 122,270,900 | rs77006299 | C/A | 0.21 | 0.22 | 1.33 | 0.03 |
| JA | Waist | STIM1 | Intronic | 11 | 3,972,232 | rs146792726 | A/G | 0.14 | 0.12 | 4.48 | 0.08 |
| JA | Waist | STIM1 | Intronic | 11 | 4,063,916 | rs147605117 | G/C | 0.15 | 0.17 | 10.86 | 0.28 |
| MA | Glucose |
NR3C2(195,478) LOC105377480(300,770) |
Intergenic | 4 | 149,561,328 | rs76209611 | G/T | 0.18 | 0.2 | 6.35 | 0.08 |
AA African Americans, EA, European Americans, JA, Japanese Americans, MA, Mexican Americans
aBolded scores beyond recommended thresholds indicate QTNs with a deleterious or functional role
bExonic, nonsynonymous variant
cFATHMM-MLK coding score shown
dRegSNPs-intron probability of disease-causing was 0.79 and only annotated for variant rs2302308
Furthermore, we determined if the significant QTNs located in intronic and intergenic regions were in LD with nearby, functional QTNs. Table 4 summarizes these proxy QTNs in LD with the significant QTN, with possible deleterious or regulatory effects given for each ethnicity. From the LDproxy [58] analysis, we found three QTNs that had high regulatory potential according to their RegulomeDB ranks; however, all of the proxy QTNs were in the same gene as the significant QTNs. Specifically, in the AAs, one TMEM132D intronic variant (rs14606502) was in LD with QTNs (rs142863227 and rs116163662) that had annotations of regulatory effects based on: transcription factor (TF) binding, having a DNase peak (indicating DNase I hypersensitivity sites), and/or being in any motif that could be functional. Notably, in the EAs, rs1260326 was in LD with another GCKR intronic variant, rs780094, that had evidence of regulatory effects including: TF binding, matched TF motif, and having a DNase peak. The five intronic QRFPR variants significantly associated with weight among the JAs were all in LD with rs55975435, an exonic but synonymous variant.
Table 4.
Proxy QTNs in linkage disequilibrium (LD) with significantly-associated MetS QTNs
| Ethnic group | Trait | QTN significantly associated with trait in Table 2 | Proxy QTN (rsid) | CHR | bp (GRCh37) | Distance (bp) | MAF (%)a | r2 (LD) | Correlated alleles (significant QTN = proxy QTN) | RegulomeDBb | Gene: function | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Significant QTN (rsid) | Gene: function | |||||||||||
| AA | log(TG) | rs114606502 | TMEM132D: intronic | rs142863227 | 12 | 130,094,067 | 23,285 | 0.4 | 1 |
T = G C = C |
4 | TMEM132D: intronic |
| rs116163662 | 12 | 130,114,869 | 2483 | 0.5 | 0.83 |
T = G C = A |
3a | TMEM132D: intronic | ||||
| EA | log(TG) | rs1260326 | GCKR: exonic, Nonsynonymous | rs780094 | 2 | 27,741,237 | 10,297 | 41 | 0.91 |
T = T C = C |
2c | GCKR: intronic |
| JA | Weight | rs2302308, rs74398478, rs17051338, rs77438622, rs77006299 | QRFPR: intronic | rs55975435 | 4 | 122,254,014 | 6028 | 12 | 0.98 |
T = A G = G |
7 | QRFPR: exonic, synonymous |
AA African Americans, EA European Americans, JA Japanese Americans, MA Mexican Americans
aMinor allele frequency (MAF) from 1000G corresponding reference group: AFR (for AA GENNID), EAS (for JA GENNID), EUR (for EA GENNID)
bRegulomeDB rank is defined by the following supporting data for evidence of regulatory effects: 2c: TF binding + matched TF motif + DNase peak; 3a: TF binding + any motif + DNase peak; 4: TF binding + DNase peak; 7: other; see https://www.regulomedb.org/regulome-help/
Discussion
This study identified several suggestive and significant associations within previously defined candidate linkage regions. Multiple significant associations were also identified outside candidate regions, nominating other putative genes associated with MetS traits. We showed substantial heterogeneity as evidenced by trait-to-genotype associations that were unique to each ethnic group, a lack of sharing significant genetic associations between ethnic groups, and differences in genetic effects across ethnic groups. Interestingly, only one of these QTN associations (rs186742063) appeared to have pleiotropic effects only in the EA families. A large percentage of our findings were in intronic and intergenic regions, which are consistent with results of the ENCODE project [66].
There were several associations that were not within candidate linkage regions/regions. The only significant association findings on chromosome 1 was within 1q42.2, outside our candidate linkage region of 1q12–1q21.1 [32]. Among the EAs only and on chromosome 2, there were two intergenic GLI2-TFCP2L1 variants (rs139940998 and rs144756634) that were associated with weight but did not lie within the previously identified candidate linkage region. In addition, the suggestively associated, non-synonymous QTN (rs1260326) within the GCKR gene in EA was found to be significant in both our trans-ethnic meta-analysis [40] as well as other studies [67, 68] and with evidence of nearby regulatory effects (Table 4). Moreover, the GCKR gene is located within a previous microsatellite linkage region nominated for harboring QTNs influencing the variation of waist circumference among GENNID AAs. Two suggestive associations on chromosome 3q26–27 region near the CNTN4 gene were found to be unique to the MA sample. The CNTN4 gene has been implicated with MetS traits [62, 63] and serum uric acid levels, and positively associated with increased risk for chronic kidney disease and cardiovascular disease [65]. The ADIPOQ gene was nominated as a candidate gene in our linkage region on 3p26 [32]; however, in the current study there was no evidence of associated QTNs in the ADIPOQ region with any of the univariate MetS traits, despite previous reports of ADIPOQ being associated with multiple underlying MetS conditions [8] and being associated with MetS in Han Chinese [69] and North Indian Punjabi [70]. In the GENNID JA families, there were two intronic STIM1 variants (rs146792726 and rs147605117) on chromosome 11 that were significantly associated with waist circumference (with the latter variant having evidence of function based on CADD and EIGEN scores). However, both variants were in linkage equilibrium (uncorrelated) with another STIM1 variant rs12290747 (r2 = 0.004) that was found to be significantly associated with urolithiasis in a recent, large-scale Japanese population GWAS [71].
Together the linkage and association results indicated differences in genetic and phenotypic architecture that are unique to each ethnic group. Furthermore, there was no overlap in the significant genes nominated among the four ethnicities. For example, we found significant evidence of a genetic association on chromosome 2q14.2 with weight and waist circumference in EAs; these two QTNs (rs139940998, rs144756634) are located in between GLI2 and TFCP2L1, which is expressed in the kidneys [72] and may act as a transcriptional suppressor of UBP1-mediated transcription activation [73]. Moreover, rs139940998 was identified as being functional across multiple annotations (Table 3). However, among JA, weight was significantly associated with the intronic region of QRFPR on chromosome 4, and there were associations between waist circumference and QTNs on chromosomes 11 (STIM1) and 15 (between WDR72 and UNC13C). These two regions were not nominated in the MA and AA groups. Similarly, in MA families, a significant genetic association was demonstrated between log(TG) and 15q13.1, but in AA families, log(TG) was significantly associated with multiple QTNs in 1q42.2, PCDH7 on chromosome 4, the intergenic region between DG599799 and BC04811 on chromosome 7, and in TMEM132D with possible regulatory effects (Table 4) on chromosome 12. These findings are consistent with our previous studies [32, 33] which have shown that the clustering of MetS traits in the GENNID differs by ethnicity. The Multi-Ethnic Study of Atherosclerosis (MESA), also found heterogeneity of effects across ethnic groups and ethnic-specific results while investigating genetic associations of body mass index (BMI); in particular the intronic rs6435678 in ERBB4 on chromosome 2 was significantly associated with BMI only in AA; however, these results were uncorrelated with our significant chromosome 2 associations with weight in the GENNID EA families [74]. This phenotypic heterogeneity could be driven by different sets of underlying genes [34] that could in turn explain variation in risk for MetS-related conditions. LD analysis and annotation of our top candidate QTNs revealed possible regulatory roles in several of these genes. However, additional functional validation studies are needed.
Furthermore, the use of family-based analyses enabled us to use a sample size smaller than what is needed for a traditional case–control GWAS to detect rare QTN associations [75]. Although the JAs had the fewest number of families, these families were multi-generational (at least three generations deep) and had more members per family. Nonetheless, for the JAs, we used genedropping to calculate p-values using empirical distributions for statistics when large-sample, asymptotic approximations may not have been valid. In addition, we used conservative genome-wide significance thresholds to assess associations in our candidate linkage regions. However, we may have been too conservative in our univariate association testing approach. Further multivariate trait analyses accounting for between trait correlations could increase power to detect genetic associations and pleiotropy.
Conclusions
Our study associations of MetS traits across a diverse group of multiethnic Americans. We built on our previous linkage analysis using highly informative microsatellites and utilized the power of genome-wide QTN association testing in families to refine and extend our previous work examining evidence for heterogeneity and pleiotropy. In summary, heterogeneity across ethnic groups was evident in not only the genetic location of the QTN, but with different associated traits and genetic effects. There was some evidence of intergenic and intronic variants having functional properties based on annotation scoring. Most significant associations were outside our candidate linkage regions and were coincident. However, in EA families and within the chromosome 2 candidate region, two significant GLI2-TFCP2L1 associations with weight were found; also, there was one chromosome 11 variant (rs186742063) with pleiotropic blood pressure effects found in the EAs. The results of this project provide new insights into the complexity and genetic architecture of MetS and highlight the utility of family-based studies and the importance of including diverse populations in genomic research.
Supplementary Information
Additional file 1. Table S1: Suggestive MetS association results (5 × 10−8 < p < 10−6) by ethnic group. Summary Table of suggestive results.
Additional file 2. Table S2: Association within or nearby linkage regiona (P< 10−6 and LOD ≤ 1.9). Summary Table of association results within or nearby candidate linkage regions.
Acknowledgements
This work was supported by the American Diabetes Association. We would like to thank the following: the ADA for providing access to the GENNID resource, Harwood Garland and Lewis A. Simon for their review and discussion of the manuscript, and Brian Fish for computing support. Genetic material collected by, and families characterized by, the American Diabetes Association GENNID Study Group, which includes Eric Boerwinkle, Ph.D., University of Texas Health Science Center; John Buse, MD, Ph.D., University of North Carolina; Ralph DeFronzo, MD, University of Texas Health Science Center; David Ehrmann, MD, University of Chicago; Steven C. Elbein, MD, University of Utah/University of Arkansas; Wilfred Fujimoto, MD, and Steven E. Kahn, MB, ChB, University of Washington; Craig L. Hanis, Ph.D., University of Texas Health Science Center; Richard A. Mulivor, Ph.D., and Jeanne C. Beck, Ph.D., Coriell Cell Repositories; Jill Norris, Ph.D., University of Colorado School of Medicine; M. Alan Permutt, MD, and Philip Behn, MD, Washington University School of Medicine; Leslie Raffel, MD, Cedars-Sinai Medical Center; and David C. Robbins, MD, Medlantic Research Institute, USA.
Abbreviations
- GENNID
GENetics of Noninsulin dependent Diabetes Mellitus
- MetS
Metabolic syndrome
- NCEP ATP III
National Cholesterol Education Program’s Adult Treatment Panel III
- HDL
High-density lipoproteins
- TG
Triglycerides
- SBP
Systolic blood pressure
- DBP
Diastolic blood pressure
- GWAS
Genome-wide association studies
- EA
European American
- MA
Mexican American
- AA
African American
- JA
Japanese American
- T2D
Type 2 diabetes
- QTN
Quantitative trait nucleotide
- ADA
American Diabetes Association
- NWGC
Northwest Genomics Center
- QC
Quality control
- HRC
Haplotype Reference Consortium
- GCTA
Genome-wide Complex Trait Analysis
- LDAK
LD-adjusted kinships
- LD
Linkage disequilibrium
- ANNOVAR
ANNOtate VARiation
- SVM
Support vector machine
- CADD
Combined Annotation Dependent Depletion
- GWAVA
Genome Wide Annotation of VAriants
- FATHMM-MLK
Functional Analysis Through Hidden Markov Models-Multiple Kernel Learning
- MESA
Multi-Ethnic Study of Atherosclerosis
Authors’ contributions
JYW analyzed the data, wrote, reviewed/revised the manuscript. DLG contributed to writing the manuscript, reviewed/edited the manuscript. ELW analyzed data, reviewed/edited the manuscript, contributed to discussion. ARF contributed to discussion/interpretation and reviewed/edited the manuscript. TMN-K contributed to discussion/interpretation and to writing the manuscript, reviewed/edited the manuscript. SAS contributed to discussion/interpretation and reviewed/edited the manuscript. KLE designed the analyses, researched the data, wrote, and reviewed/edited the manuscript. ADA GENNID Study group performed the data collection and provided a repository for the research of the data. All authors read and approved the final manuscript.
Funding
The design of the study and collection, analysis, interpretation of data and the writing the manuscript was funded by NHLBI (1R01HL113189, Edwards KL, PI): Life After Linkage Consortium.
Availability of data and materials
The data that support the findings of this study are available from American Diabetes Association but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of American Diabetes Association.
Declarations
Ethics approval and consent to participate
The study was classified as non-human subjects research, and the need for approval was waived.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Karen L. Edwards, Email: kedward1@hs.uci.edu
American Diabetes GENNID Study Group:
Eric Boerwinkle, John Buse, Ralph DeFronzo, David Ehrmann, Steven C. Elbein, Wilfred Fujimoto, Steven E. Kahn, Craig L. Hanis, Richard A. Mulivor, Jeanne C. Beck, Jill Norris, M. Alan Permutt, Philip Behn, Leslie Raffel, and David C. Robbins
References
- 1.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 2.Grundy SM. Metabolic syndrome update. Trends Cardiovasc Med. 2016;26(4):364–373. doi: 10.1016/j.tcm.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 3.National Cholesterol Education Program Expert Panel on Detection E, Treatment of High Blood Cholesterol in A Third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) final report. Circulation. 2002;106(25):3143–3421. doi: 10.1161/circ.106.25.3143. [DOI] [PubMed] [Google Scholar]
- 4.Kraja AT, Rao DC, Weder AB, Cooper R, Curb JD, Hanis CL, et al. Two major QTLs and several others relate to factors of metabolic syndrome in the family blood pressure program. Hypertension. 2005;46(4):751–757. doi: 10.1161/01.HYP.0000184249.20016.bb. [DOI] [PubMed] [Google Scholar]
- 5.Carmelli D, Cardon LR, Fabsitz R. Clustering of hypertension, diabetes, and obesity in adult male twins: same genes or same environments? Am J Hum Genet. 1994;55(3):566–573. [PMC free article] [PubMed] [Google Scholar]
- 6.Bell DSH, Goncalves E. Stroke in the patient with diabetes (part 1)—epidemiology, etiology, therapy and prognosis. Diabetes Res Clin Pract. 2020;164:108193. doi: 10.1016/j.diabres.2020.108193. [DOI] [PubMed] [Google Scholar]
- 7.DeBoer MD, Filipp SL, Sims M, Musani SK, Gurka MJ. Risk of ischemic stroke increases over the spectrum of metabolic syndrome severity. Stroke. 2020;51(8):2548–2552. doi: 10.1161/STROKEAHA.120.028944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziki MDA, Mani A. Metabolic syndrome: genetic insights into disease pathogenesis. Curr Opin Lipidol. 2016;27(2):162–171. doi: 10.1097/MOL.0000000000000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Decker JJ, Norby FL, Rooney MR, Soliman EZ, Lutsey PL, Pankow JS, et al. Metabolic syndrome and risk of ischemic stroke in atrial fibrillation: ARIC study. Stroke. 2019;50(11):3045–3050. doi: 10.1161/STROKEAHA.119.025376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogers AJ, Kaplan I, Chung A, McFarlane SI, Jean-Louis G. Obstructive sleep apnea risk and stroke among blacks with metabolic syndrome: results from metabolic syndrome outcome (MetSO) registry. Int J Clin Res Trials. 2020;5(1):143. doi: 10.15344/2456-8007/2020/143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Austin MA, Edwards KL, McNeely MJ, Chandler WL, Leonetti DL, Talmud PJ, et al. Heritability of multivariate factors of the metabolic syndrome in nondiabetic Japanese Americans. Diabetes. 2004;53(4):1166–1169. doi: 10.2337/diabetes.53.4.1166. [DOI] [PubMed] [Google Scholar]
- 12.Khan RJ, Gebreab SY, Sims M, Riestra P, Xu R, Davis SK. Prevalence, associated factors and heritabilities of metabolic syndrome and its individual components in African Americans: the Jackson heart study. BMJ Open. 2015;5(10):e008675. doi: 10.1136/bmjopen-2015-008675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mamtani M, Kulkarni H, Dyer TD, Goring HH, Neary JL, Cole SA, et al. Genome- and epigenome-wide association study of hypertriglyceridemic waist in Mexican American families. Clin Epigenet. 2016;8:6. doi: 10.1186/s13148-016-0173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musani SK, Martin LJ, Woo JG, Olivier M, Gurka MJ, DeBoer MD. Heritability of the severity of the metabolic syndrome in Whites and Blacks in 3 large cohorts. Circ Cardiovasc Genet. 2017;10(2):e001621. doi: 10.1161/CIRCGENETICS.116.001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panizzon MS, Hauger RL, Sailors M, Lyons MJ, Jacobson KC, Murray McKenzie R, et al. A new look at the genetic and environmental coherence of metabolic syndrome components. Obesity. 2015;23(12):2499–2507. doi: 10.1002/oby.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laird NM, Lange C. The role of family-based designs in genome-wide association studies. Stat Sci. 2009;24(4):388–97, 10.
- 17.Morris N, Elston RC, Barnholtz-Sloan JS, Sun X. Novel approaches to the analysis of family data in genetic epidemiology. Front Genet. 2015;6:27. doi: 10.3389/fgene.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown AE, Walker M. Genetics of insulin resistance and the metabolic syndrome. Curr Cardiol Rep. 2016;18(8):75. doi: 10.1007/s11886-016-0755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chuluun-Erdene A, Sengeragchaa O, Altangerel TA, Sanjmyatav P, Dagdan B, Battulga S, et al. Association of candidate gene polymorphism with metabolic syndrome among Mongolian subjects: a case–control study. Med Sci. 2020;8(3):38. doi: 10.3390/medsci8030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munoz AM, Velasquez CM, Agudelo GM, Uscategui RM, Estrada A, Patino FA, et al. Examining for an association between candidate gene polymorphisms in the metabolic syndrome components on excess weight and adiposity measures in youth: a cross-sectional study. Genes Nutr. 2017;12:19. doi: 10.1186/s12263-017-0567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagrani R, Foraita R, Gianfagna F, Iacoviello L, Marild S, Michels N, et al. Common genetic variation in obesity, lipid transfer genes and risk of metabolic syndrome: results from IDEFICS/I. Family study and meta-analysis. Sci Rep. 2020;10(1):7189. doi: 10.1038/s41598-020-64031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada Y, Ichihara S, Kato K, Yoshida T, Yokoi K, Matsuo H, et al. Genetic risk for metabolic syndrome: examination of candidate gene polymorphisms related to lipid metabolism in Japanese people. J Med Genet. 2008;45(1):22–28. doi: 10.1136/jmg.2007.052415. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Liu J, Liu J, Li W, Li X, He Y, et al. Genetic association study with metabolic syndrome and metabolic-related traits in a cross-sectional sample and a 10-year longitudinal sample of Chinese elderly population. PLoS ONE. 2014;9(6):e100548. doi: 10.1371/journal.pone.0100548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraja AT, Vaidya D, Pankow JS, Goodarzi MO, Assimes TL, Kullo IJ, et al. A bivariate genome-wide approach to metabolic syndrome: STAMPEED consortium. Diabetes. 2011;60(4):1329–1339. doi: 10.2337/db10-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lind L. Genome-wide association study of the metabolic syndrome in UK Biobank. Metab Syndr Relat Disord. 2019;17(10):505–511. doi: 10.1089/met.2019.0070. [DOI] [PubMed] [Google Scholar]
- 26.Ridker PM, Pare G, Parker A, Zee RY, Danik JS, Buring JE, et al. Loci related to metabolic-syndrome pathways including LEPR, HNF1A, IL6R, and GCKR associate with plasma C-reactive protein: the women’s genome health study. Am J Hum Genet. 2008;82(5):1185–1192. doi: 10.1016/j.ajhg.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Setoh K, Terao C, Muro S, Kawaguchi T, Tabara Y, Takahashi M, et al. Three missense variants of metabolic syndrome-related genes are associated with alpha-1 antitrypsin levels. Nat Commun. 2015;6:7754. doi: 10.1038/ncomms8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamada Y, Kato K, Oguri M, Horibe H, Fujimaki T, Yasukochi Y, et al. Identification of four genes as novel susceptibility loci for early-onset type 2 diabetes mellitus, metabolic syndrome, or hyperuricemia. Biomed Rep. 2018;9(1):21–36. doi: 10.3892/br.2018.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lusis AJ, Attie AD, Reue K. Metabolic syndrome: from epidemiology to systems biology. Nat Rev Genet. 2008;9(11):819–830. doi: 10.1038/nrg2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monda KL, North KE, Hunt SC, Rao DC, Province MA, Kraja AT. The genetics of obesity and the metabolic syndrome. Endocr Metab Immune Disord Drug Targets. 2010;10(2):86–108. doi: 10.2174/187153010791213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards KL, Hutter CM, Wan JY, Kim H, Monks SA. Genome-wide linkage scan for the metabolic syndrome: the GENNID study. Obesity. 2008;16(7):1596–1601. doi: 10.1038/oby.2008.236. [DOI] [PubMed] [Google Scholar]
- 33.Edwards KL, Wan JY, Hutter CM, Fong PY, Santorico SA. Multivariate linkage scan for metabolic syndrome traits in families with type 2 diabetes. Obesity. 2011;19(6):1235–1243. doi: 10.1038/oby.2010.299. [DOI] [PubMed] [Google Scholar]
- 34.Wan J, Edwards K, Santorico S. Investigating genetic and environmental correlations between traits of metabolic syndrome in the multi-ethnic GENNID study. JP J Biostat. 2001;6(2):77–96. [Google Scholar]
- 35.Elbein SC, Das SK, Hallman DM, Hanis CL, Hasstedt SJ. Genome-wide linkage and admixture mapping of type 2 diabetes in African American families from the American Diabetes Association GENNID (genetics of NIDDM) study cohort. Diabetes. 2009;58(1):268–274. doi: 10.2337/db08-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasstedt SJ, Highland HM, Elbein SC, Hanis CL, Das SK, American Diabetes Association GSG Five linkage regions each harbor multiple type 2 diabetes genes in the African American subset of the GENNID study. J Hum Genet. 2013;58(6):378–383. doi: 10.1038/jhg.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raffel LJ, Robbins DC, Norris JM, Boerwinkle E, DeFronzo RA, Elbein SC, et al. The GENNID study. A resource for mapping the genes that cause NIDDM. Diabetes Care. 1996;19(8):864–872. doi: 10.2337/diacare.19.8.864. [DOI] [PubMed] [Google Scholar]
- 38.National Diabetes Data Group Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes. 1979;28(12):1039–1057. doi: 10.2337/diab.28.12.1039. [DOI] [PubMed] [Google Scholar]
- 39.Ehm MG, Karnoub MC, Sakul H, Gottschalk K, Holt DC, Weber JL, et al. Genomewide search for type 2 diabetes susceptibility genes in four American populations. Am J Hum Genet. 2000;66(6):1871–1881. doi: 10.1086/302950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willems EL, Wan JY, Norden-Krichmar TM, Edwards KL, Santorico SA. Trans-ethnic meta-analysis of metabolic syndrome in a multi-ethnic study. Genet Epidemiol. 2018;42(7):741–742. doi: 10.1002/gepi.22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Connell J, Gurdasani D, Delaneau O, Pirastu N, Ulivi S, Cocca M, et al. A general approach for haplotype phasing across the full spectrum of relatedness. Plos Genet. 2014;10(4):e1004234. doi: 10.1371/journal.pgen.1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang JA, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Speed D, Cai N, Johnson MR, Nejentsev S, Balding DJ, Consortium U Reevaluation of SNP heritability in complex human traits. Nat Genet. 2017;49(7):986. doi: 10.1038/ng.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21(11):1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 48.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deeks JJ, Higgins JPT, Altman DG, Group CSM. Chapter 10: Analysing data and undertaking meta-analyses. In: Cochrane handbook for systematic reviews of interventions. Cochrane. Version 6. 2019. https://training.cochrane.org/handbook/current/chapter-10.
- 50.R Core Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2020. [Google Scholar]
- 51.Yin L. CMplot: circle Manhattan plot. R package version 3.6.2 ed. https://CRAN.R-project.org/package=CMplot2020.
- 52.Wang K, Li MY, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11(3):294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48(2):214–220. doi: 10.1038/ng.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31(10):1536–1543. doi: 10.1093/bioinformatics/btv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin H, Hargreaves KA, Li R, Reiter JL, Wang Y, Mort M, et al. RegSNPs-intron: a computational framework for predicting pathogenic impact of intronic single nucleotide variants. Genome Biol. 2019;20(1):254. doi: 10.1186/s13059-019-1847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22(9):1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whelton. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines (vol 71, pg 1269, 2018). Hypertension. 2018;71(6):E136–9. [DOI] [PubMed]
- 61.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kissebah AH, Sonnenberg GE, Myklebust J, Goldstein M, Broman K, James RG, et al. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc Natl Acad Sci USA. 2000;97(26):14478–14483. doi: 10.1073/pnas.97.26.14478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rich SS, Bowden DW, Haffner SM, Norris JM, Saad MF, Mitchell BD, et al. Identification of quantitative trait loci for glucose homeostasis: the insulin resistance atherosclerosis study (IRAS) family study. Diabetes. 2004;53(7):1866–1875. doi: 10.2337/diabetes.53.7.1866. [DOI] [PubMed] [Google Scholar]
- 64.Cai G, Cole SA, Freeland-Graves JH, MacCluer JW, Blangero J, Comuzzie AG. Principal component for metabolic syndrome risk maps to chromosome 4p in Mexican Americans: the San Antonio family heart study. Hum Biol. 2004;76(5):651–665. doi: 10.1353/hub.2005.0001. [DOI] [PubMed] [Google Scholar]
- 65.Chittoor G, Kent JW, Jr, Almeida M, Puppala S, Farook VS, Cole SA, et al. GWAS and transcriptional analysis prioritize ITPR1 and CNTN4 for a serum uric acid 3p26 QTL in Mexican Americans. BMC Genom. 2016;17:276. doi: 10.1186/s12864-016-2594-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Consortium EP An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bi M, Kao WH, Boerwinkle E, Hoogeveen RC, Rasmussen-Torvik LJ, Astor BC, et al. Association of rs780094 in GCKR with metabolic traits and incident diabetes and cardiovascular disease: the ARIC study. PLoS ONE. 2010;5(7):e11690. doi: 10.1371/journal.pone.0011690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mohas M, Kisfali P, Jaromi L, Maasz A, Feher E, Csongei V, et al. GCKR gene functional variants in type 2 diabetes and metabolic syndrome: do the rare variants associate with increased carotid intima-media thickness? Cardiovasc Diabetol. 2010;9:79. doi: 10.1186/1475-2840-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li XT, Wei DY, He HJ, Zhang JH, Wang CX, Ma MJ, et al. Association of the adiponectin gene (ADIPOQ) +45T>G polymorphism with the metabolic syndrome among Han Chinese in Sichuan province of China. Asia Pac J Clin Nutr. 2012;21(2):296–301. [PubMed] [Google Scholar]
- 70.Kaur H, Badaruddoza B, Bains V, Kaur A. Genetic association of ADIPOQ gene variants (−3971A>G and +276G>T) with obesity and metabolic syndrome in North Indian Punjabi population. PLoS ONE. 2018;13(9):e0204502. doi: 10.1371/journal.pone.0204502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ishigaki K, Akiyama M, Kanai M, Takahashi A, Kawakami E, Sugishita H, et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat Genet. 2020;52(7):669–679. doi: 10.1038/s41588-020-0640-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.TFCP2L1 transcription factor CP2 like 1 [Homo sapiens (human)]. NCBI. 2021. https://www.ncbi.nlm.nih.gov/gene/29842.
- 73.Taracha A, Kotarba G, Wilanowski T. Neglected functions of TFCP2/TFCP2L1/UBP1 transcription factors may offer valuable insights into their mechanisms of action. Int J Mol Sci. 2018;19(10):2852. doi: 10.3390/ijms19102852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Salinas YD, Wang L, DeWan AT. Multiethnic genome-wide association study identifies ethnic-specific associations with body mass index in Hispanics and African Americans. BMC Genet. 2016;17(1):78. doi: 10.1186/s12863-016-0387-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sul JH, Cade BE, Cho MH, Qiao D, Silverman EK, Redline S, et al. Increasing generality and power of rare-variant tests by utilizing extended pedigrees. Am J Hum Genet. 2016;99(4):846–859. doi: 10.1016/j.ajhg.2016.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv. 2020 doi: 10.1101/531210v4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Table S1: Suggestive MetS association results (5 × 10−8 < p < 10−6) by ethnic group. Summary Table of suggestive results.
Additional file 2. Table S2: Association within or nearby linkage regiona (P< 10−6 and LOD ≤ 1.9). Summary Table of association results within or nearby candidate linkage regions.
Data Availability Statement
The data that support the findings of this study are available from American Diabetes Association but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of American Diabetes Association.