

Abstract
Background
Classical Ehlers‐Danlos syndrome (cEDS) is a heterogeneous connective tissue disorder that mainly results from the germline mutation of COL5A1 and COL5A2. The majority of the COL5A2 mutations reported to date represent structural mutations, including missense or in‐frame exon‐skipping splice mutations. The only reported synonymous mutation was expected to affect on splicing of exon 29 by prediction programs which should be further confirmed.
Methods
Whole exome sequencing was performed to identify the genetic variants of a Chinese boy who was characterized by skin hyperextensibility, abnormal scarring, hypermobile joints and scoliosis. Sanger sequencing was used to validate the variants in his parents. Reverse transcription polymerase chain reaction (RT‐PCR) was performed to analyze the functional effects of the variant.
Results
A de novo heterozygous synonymous variant (NM_000393.5:c.1977 G>A) of COL5A2 gene was identified in the patient. The results of RT‐PCR revealed that the synonymous variant led to skipping of exon 29 in the RNA transcript.
Conclusions
Our study supplies further supporting evidence that the synonymous COL5A2 mutation c.1977 G>A can cause skipping of exon 29 in the RNA transcript, thus resulting in the production of mutant α2(V)‐chains and clinical phenotype of cEDS. This result highlights the need to include splicing‐altering synonymous mutations into the screening for cEDS.
Keywords: classic Ehlers Danlos syndrome, COL5A2, splicing, synonymous mutation, whole‐exome sequencing
Synonymousmutation can cause skipping of exon in the RNA transcript, thus resulting in the production of mutant α2(V)‐chains and clinical phenotype of classical Ehlers‐Danlos syndrome. Our results highlights the need to include splicing‐altering synonymous mutations into the screening for cEDS.

1. INTRODUCTION
Ehlers‐Danlos syndrome (EDS) is a clinically and genetically heterogeneous group of heritable connective tissue disorder characterized mainly by skin hyperextensibility, abnormal scarring, hypermobile joints, and other systemic involvement (Mitakides & Tinkle, 2017). The international EDS Consortium revised EDS classification into 13 subtypes in 2017 and proposed the definite diagnosis of all EDS subtypes, except for the hypermobile type, relying on molecular confirmation with identification of (a) causative genetic variant(s) (Malfait et al., 2017). Classical EDS (cEDS, MIM#130000 and MIM#130010) is an autosomal dominant disorder, which is characterized by skin hyperextensibility, widened atrophic (cigarette paper) scars and generalized joint hypermobility (Malfait et al., 2010). The prevalence of cEDS has been estimated to be 1:20,000 (Malfait et al., 2010). More than 90% of cEDS patients harbor a heterozygous mutation in the COL5A1 (MIM:120215) or COL5A2 (MIM:120190) gene, which encode the proα1(V) chain and proα2(V) chain of type V collagen respectively (Ritelli et al., 2013; Symoens et al., 2012). Rarely, specific mutations in the gene encoding type I collagen (COL1A1, MIM:120150) can be associated with a cEDS‐phenotype (Takahara et al., 2002). Type V collagen is a quantitatively minor fibrillar collagen with a wide tissue distribution. It represents mainly as [α1(V)]2α2(V) heterotrimers in skin, bone, cornea and tendon. It forms heterotypic type I/V collagen fibrils with type I collagen and regulates the diameter of these fibrils by retention its very large amino‐terminal propeptide (Birk, 2001).
Comprehensive molecular analysis demonstrated that only 14% of individuals with classical EDS were caused by structural pathogenic variants in COL5A2 (Ritelli et al., 2013; Symoens et al., 2012). These structural variants are most point mutations that result in exon skipping or substitution for glycine in the triple‐helical region of the collagen molecule (Michalickova et al., 1998) and a few nucleotide deletion/insertion variants result in reading frame shifts and downstream premature termination codons (Colombi et al., 2017; Michalickova et al., 1998; Symoens et al., 2012). The only pathogenic synonymous mutation in exon 29 of COL5A2 (c.1977 G>A p.Gly642_Pro659del) was first reported by Ritelli et al., (2013), and the effect on splicing of exon 29 predicted by using of four prediction programs (SpliceSite‐Finder‐like, MaxEntScan, NNSPLICE and Human Splicing Finder) should be further confirmed. Here, we identified the same synonymous mutation in a Chinese boy who suffered from cEDS, characterized by skin hyperextensibility, easy bruising, joint hypermobility and scoliosis. The effect of reported c.1977 G>A of COL5A2 identified by whole exome sequencing was confirmed by sequence analysis of COL5A2 mRNA and electron microscopy in skin.
2. MATERIALS AND METHODS
2.1. Study subjects and ethical compliance
A Chinese boy was recruited to identify the genetic etiology of skin hyperextensibility at the Reproductive and Genetic Hospital of CITIC Xiangya (Changsha). Written informed consent was obtained from patient's parents. This study was approved by the institutional ethics committees of the Reproductive and Genetic Hospital of CITIC Xiangya of Central South University (approved number LL‐SC‐2019‐033).
2.2. Molecular genetic study
Genomic DNA was extracted from peripheral blood lymphocytes of the patient and his parents using a QIAamp® DNA blood midi kit (Qiagen) according to standard protocols. Whole exome sequencing (WES) was performed on the Illumina HiSeq2000 platform (Illumina) using the IDT xGen exome capture panel v1.0 (Integrated DNA Technologies). The analysis of the high‐throughput sequencing data was performed using the GATK software package (Broad Institute). Sanger sequencing was performed for candidate variants.
The patient's skin of left knee was obtained from accidental fall. The skin tissue was divided into three parts: hematoxylin and eosin stain (H&E), transmission electron microscopy and RT‐PCR. Total RNA was extracted from the patient's skin using the RNeasy Midi Kit (Qiagen). Subsequently, cDNA synthesis was performed according to standard procedures using random primers. The part of COL5A2 cDNA (NM_000393.5) carrying the mutation was amplified using forward primer 5′‐GCTCCATAGGAATCAGAGGGC‐3′ (located in exon 27) and reverse primer 5′‐TTCTCACCAGGGAGTCCAGTTA‐3′ (located in exon 33) with a total number of 35 PCR cycles. PCR products were analyzed using 3.0% agarose gel electrophoresis, followed by excised from gel, purified and sequenced as described previously (Zhang et al., 2020).
2.3. Pathology and electron microscopy
Formalin‐fixed paraffin‐embedded material was cut into 4‐μm‐thick sections and stained with hematoxylin and eosin (H&E); Transmission electron microscopy was performed as described previously (Malfait et al., 2005).
3. RESULTS
3.1. Case presentation
This 5‐year‐old Chinese boy is the only child of healthy non‐consanguineous parents who were 38 (mother) and 42 (father) at the time of conception. The pregnancy was complicated by severe gestational hypertension. He was a premature baby with low birth weight (1,180 g (< 3rd centile), 32 gestational weeks). Psychomotor development was delayed: he achieved head control at the age of 6 months, started to sit independently at the age of 1 year and began walking independently at the age of 20 months. He was diagnosed with scoliosis and underwent surgical correction at the age of 28 months (Figure 1a). The patient first visited our clinic when he was 5 years old. At that time, his weight was 19 kg (50th centile), height 110 cm (50th centile). Physical examination revealed that he had widen atrophic scars on forehead, elbows, knees and pretibial area that had developed since childhood, as well as skin hyperextensibility, joint hypermobility (Beightonscore 9/9; Figure 1b,c,e), papyraceous scar (Figure 1d) and easy bruising. No vascular abnormalities were found, and the family history of EDS was negative.
FIGURE 1.
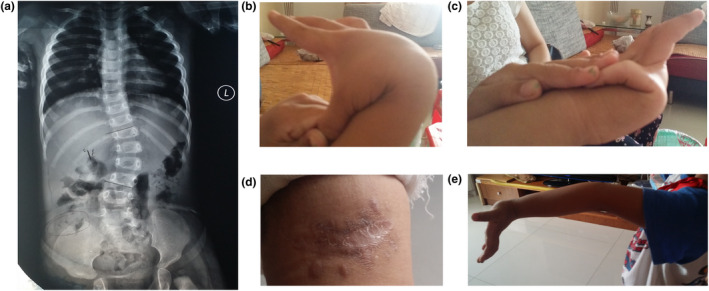
Clinical pictures of the patient. (a) X‐ray photographs of the thoracoabdominal of the patient. The patient presented marked scoliosis at age of 28 months. (b) Extreme joint hypermobility: the thumb can be passively moved to touch the ipsilateral forearm. (c) Extreme joint hypermobility: metacarpal‐phalangeal joint of the fifth finger can be hyperextended more than 90° with respect to the dorsum of the hand. (d) Atrophic scars of the knee. (e) Extreme joint hypermobility: elbow extends more than 10°
3.2. Molecular genetic findings
Whole exome sequencing revealed that the patient carried a heterozygous G‐to‐A transition in exon 29 of COL5A2 (NM_000393.5: c.1977G>A), which was confirmed by Sanger sequencing (Figure 2a). This mutation was absent in the unaffected parents (Figure 2a).
FIGURE 2.
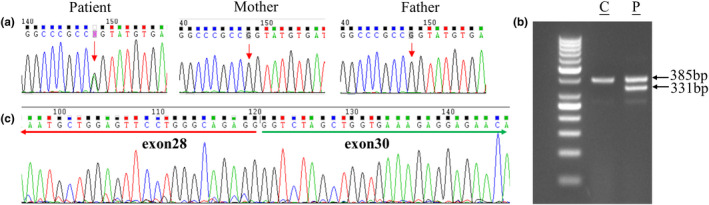
Effects of COL5A2 mutations on splicing. (a) Sanger sequencing of the c.1977 G>A (pointed by red arrows) mutation of COL5A2 in this family. Parents are free of the mutation. (b) RT‐PCR of RNA from the patient. The cDNA was amplified with primers in exons 27 and 33 and the expected 385 bp product is indicated. The smaller band of patient results from skipping of exon 29. There is an additional heteroduplex in the subject sample. (c) The sequence analysis of the 331‐bp PCR product presented in (a). As indicated by the cDNA sequences of exons 28 and 30 of COL5A2, exon 29 is clearly absent from this transcript. C, control individual; P, subject
The c. 1977G>A substitution abolished the COL5A2 exon 29 consensus donor splice site, as predicted by four prediction programs in Alamut software, which was published by Ritelli et al in 2013. According to the prediction, this mutation could cause the in‐frame deletion of the 18 amino acids that are encoded by exon 29 (p.Gly642_Pro659del). To determine whether the synonymous change affects splicing of COL5A2 mRNA, cDNA was synthesized from total RNA derived from the skin of patient's knee. Amplification of the COL5A2 cDNA fragments produced a faster‐migrating band in addition to the expected band (Figure 2b). Sequencing of the faster‐migrating cDNA led to identification of exon 29 skipping (p.Gly642_Pro659del; Figure 2c).
3.3. Pathology and ultrastructural collagen studies
Microscopic examination of the H&E stained section showed reticular areas are filled with deep staining “cloudy” masses of collagen and the connective tissue just above the subcutaneous fat tissue is not as closely packed as normal, but seems to disintegrate (Figure 3a,b). Ultrastructural studies revealed collagen bundles contained fibrils of variable diameter and loosely‐packed (Figure 3d) and rope‐like longitudinal sections (Figure 3e).
FIGURE 3.
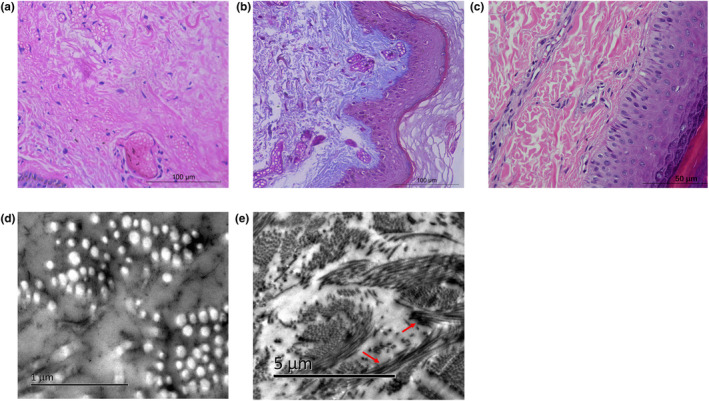
Pathology and Ultrastructural Collagen of patient and normal human. (a) Irregularly organized collagen bundles are intensely stained and have a cloudy appearance. (b) The overall aspect of the connective tissue is loose. (c) Regularly and tightly organized collagen bundles are intensely stained in normal human skin. (d) Cross‐sectioned collagen fibrils of variable diameter and loosely‐packed. (e) Longitudinal section of a collagen bundle with several thickened rope‐like and twisted fibrils (arrows)
4. DISCUSSION
Synonymous mutations are still rarely reported in human diseases despite the growing awareness that they can cause changes in protein expression, conformation, and function, by affecting posttranscriptional process and regulation of RNA expression (Sauna & Kimchi‐Sarfaty, 2011). This underreporting is likely to be caused by the perception in many earlier investigations that synonymous variants were “silent” and by the absence of functional studies of many of these variants (Sauna & Kimchi‐Sarfaty, 2011). In cEDS, despite over 90% patients harbor a type V collagen defect and more than 200 distinct mutations have been described in gene COL5A1 and COL5A2, only one pathogenic synonymous mutation of COL5A2 (c.1977G>A) has been described (Ritelli et al., 2013) and the functional change of this mutation was characterized in our study. This indicates the importance of investigating apparently silent variants when searching for causative gene mutations.
Until now, only 44 and 68 distinct mutations of COL5A2 are reported in the HGMD professional 2019.4 and LOVD EDS Variant database, respectively. The only reported pathogenic synonymous mutation was firstly predicted to be intolerant by four prediction programs (Ritelli et al., 2013) and it is further confirmed that this synonymous mutation caused the in‐frame deletion of the 18 amino acids that are encoded by exon 29 (p.Gly642_Pro659del) by sequencing of the expressed transcript of mutated COL5A2 in our study. This synonymous mutation might abolish the consensus donor splice site by disrupting an exonic splicing regulatory sequence. These short nucleotide sequences within exons are important for binding splicing regulatory proteins that are involved in the correct removal of introns (splicing) in the RNA transcript (Sauna & Kimchi‐Sarfaty, 2011). Efficient splicing has limited tolerance of mutations in the exonic splicing enhancers, even if they have no effect on protein coding, which explains the abnormal RNA processing observed in our study. The skipping of exon 29 in the RNA transcript is expected to lead to a in‐frame deletion of 18 amino acids and a truncated production of COL5A2 with shorted triple‐helix domain. Studies (Chanut‐Delalande et al., 2004; Mak et al., 2016) have shown that functional mutation impairs the assembly and/or secretion of the α1(V)2α2(V) isoform of COLV and leads to reduced thickness of the basement membranes underlying the epidermis and increased apoptosis of the stromal fibroblasts. These data suggests that in‐frame exon‐skipping splice mutation of our patient produces a mutant α2(V)‐chains that probably incorporate into collagen molecules and interferes the formation of heterotrimers, thus resulting in the growth of collagen fibrils into thicker ones as seen by the electron microscope.
According to the 2017 revised nosology of EDS (Malfait et al., 2017), our patient with a heterozygous COL5A2 mutation does not differ significantly from that of other reported cEDS patients with COL5A2 mutations. The patient meets the main diagnostic criteria for cEDS: skin hyperextensibility, widened atrophic scarring, joint hypermobility, associated with scoliosis, but no structural cardiac malformations which are uncommon in the cEDS and can be caused by homozygous COL5A2 mutations (Jin et al., 2017). By the comparison of our case with patient described by Ritelli et al., (2013), apart from the main diagnostic criteria for cEDS, both of them showed molluscoid pseudotumor and few distinctive facial features: epicanthus, strabismus, anteverted nostrils. In addition to scoliosis, our case didn't show other skeletal abnormalities complained in the previously reported 21‐year‐old male, such as mild pectus excavatum, ulnar deviation, asymmetric legs, hands and feet and deformity, hallux valgus, pes planus, enthesopathies (recurrent), dislocations (recurrent), chronic articular pain, chronic fatigue syndrome. The possible reason is that our case is younger that no more serious symptoms have appeared.
Although there appear to be no genotype‐phenotype correlations, it has been suggested that pathogenic variants in COL5A2 are thought to result in a phenotype at the more severe end of the classical EDS spectrum and the triple helix region appears to be related with the highest phenotypic variability, with a hypermobility Beighton score ranging from 0/9 to 9/9 (Paladin et al., 2015; Ritelli et al., 2013).
In conclusion, our study further demonstrates the functional changes of an unusual synonymous mutation of the COL5A2 gene and highlights the importance of including splicing‐altering synonymous mutations into the screening for EDS disease.
CONFLICT OF INTEREST
The authors declare that they have no conflicting interests.
AUTHOR CONTRIBUTIONS
NM, ZZ, JL and JD had major roles in the design of the study. NM and JD drafted the manuscript. NM, XZ, WT and BG were involved in the laboratory experiments and data interpretation. ZZ, JL, YP, ZJ, HX, and JD analyzed the clinical data. ZZ, JL and HW critically revised the manuscript for important intellectual content. HW and JD are corresponding authors of this manuscript. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
We thank the family members for participation in this study. We also thank laboratory staff at Prenatal Diagnosis Center of Hunan provincial Maternal and Child Health Care Hospital. And, we are grateful for the experimental support provided by laboratory staff at Institute of Reproductive and Stem Cell Engineering, School of Basic Medical Science, Central South University. This study was supported by grants from the Major Scientific and Technological Projects for Collaborative Prevention and Control of Birth Defects in Hunan Province (2019SK1010, 2019SK1014), the Natural Science Foundation of Hunan Province (No. 2018JJ3274, 2018JJ3275).
Funding Information
This study was supported by Major Scientific and Technological Projects for collaborative prevention and control of birth defects in Hunan Province, grant/award number: 2019SK1010 and 2019SK1014; Natural Science Foundation of Hunan Province, grant/award number: 2018JJ3274, 2018JJ3275.
Contributor Information
Hua Wang, Email: wanghua213@aliyun.com.
Juan Du, Email: tandujuan@csu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Birk, D. E. (2001). Type V collagen: heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron, 32, 223–237. 10.1016/S0968-4328(00)00043-3 [DOI] [PubMed] [Google Scholar]
- Chanut‐Delalande, Hélène , Bonod‐Bidaud, C. , Cogne, S. , Malbouyres, M. , Ramirez, F. , Fichard, Agnès , & Ruggiero, F. (2004). Development of a functional skin matrix requires deposition of collagen V heterotrimers. Molecular and Cellular Biology, 24, 6049–6057. 10.1128/MCB.24.13.6049-6057.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombi, M. , Dordoni, C. , Venturini, M. , Ciaccio, C. , Morlino, S. , Chiarelli, N. , Zanca, A. , Calzavara‐Pinton, P. , Zoppi, N. , Castori, M. , & Ritelli, M. (2017). Spectrum of mucocutaneous, ocular and facial features and delineation of novel presentations in 62 classical Ehlers‐Danlos syndrome patients. Clinical Genetics, 92, 624–631. 10.1111/cge.13052. [DOI] [PubMed] [Google Scholar]
- Jin, S. C. , Homsy, J. , Zaidi, S. , Lu, Q. , Morton, S. , DePalma, S. R. , Zeng, X. , Qi, H. , Chang, W. , Sierant, M. C. , Hung, W.‐C. , Haider, S. , Zhang, J. , Knight, J. , Bjornson, R. D. , Castaldi, C. , Tikhonoa, I. R. , Bilguvar, K. , Mane, S. M. , … Brueckner, M. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nature Genetics, 49, 1593–1601. 10.1038/ng.3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, K. M. , Png, C. Y. , & Lee, D. J. (2016). Type V collagen in health, disease, and fibrosis. The Anatomical Record, 299(5), 613–629. 10.1002/ar.23330 [DOI] [PubMed] [Google Scholar]
- Malfait, F. , Coucke, P. , Symoens, S. , Loeys, B. , Nuytinck, L. , & De Paepe, A. (2005). The molecular basis of classic Ehlers‐Danlos syndrome: a comprehensive study of biochemical and molecular findings in 48 unrelated patients. Human Mutation, 25(1), 28–37. 10.1002/humu.20107 [DOI] [PubMed] [Google Scholar]
- Malfait, F. , Francomano, C. , Byers, P. , Belmont, J. , Berglund, B. , Black, J. et al (2017). The 2017 international classification of the Ehlers‐Danlos syndromes. American Journal of Medical Genetics, 175, 8–26. 10.1002/ajmg.c.31552 [DOI] [PubMed] [Google Scholar]
- Malfait, F. , Wenstrup, R. J. , & De Paepe, A. (2010). Clinical and genetic aspects of Ehlers‐Danlos syndrome, classic type. Genetics in Medicine, 12, 597–605. 10.1097/GIM.0b013e3181eed412 [DOI] [PubMed] [Google Scholar]
- Michalickova, K. , Susic, M. , Willing, M. C. , Wenstrup, R. J. , & Cole, W. G. (1998). Mutations of the alpha2(V) chain of type V collagen impair matrix assembly and produce ehlers‐danlos syndrome type I. Human Molecular Genetics, 7, 249–255. 10.1093/hmg/7.2.249 [DOI] [PubMed] [Google Scholar]
- Mitakides, J. , & Tinkle, B. T. (2017). Oral and mandibular manifestations in the Ehlers‐Danlos syndromes. American Journal of Medical Genetics, 175, 220–225. 10.1002/ajmg.c.31541 [DOI] [PubMed] [Google Scholar]
- Paladin, L. , Tosatto, S. C. , & Minervini, G. (2015). Structural in silico dissection of the collagen V interactome to identify genotype‐phenotype correlations in classic Ehlers‐Danlos Syndrome (EDS). FEBS Letters, 589, 3871–3878. 10.1016/j.febslet.2015.11.022 [DOI] [PubMed] [Google Scholar]
- Ritelli, M. , Dordoni, C. , Venturini, M. , Chiarelli, N. , Quinzani, S. , Traversa, M. , Zoppi, N. , Vascellaro, A. , Wischmeijer, A. , Manfredini, E. , Garavelli, L. , Calzavara‐Pinton, P. , & Colombi, M. (2013). Clinical and molecular characterization of 40 patients with classic Ehlers‐Danlos syndrome: identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet Journal of Rare Diseases, 8, 58. 10.1186/1750-1172-8-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauna, Z. E. , & Kimchi‐Sarfaty, C. (2011). Understanding the contribution of synonymous mutations to human disease. Nature Reviews. Genetics, 12, 683–691. 10.1038/nrg3051 [DOI] [PubMed] [Google Scholar]
- Symoens, S. , Syx, D. , Malfait, F. , Callewaert, B. , De Backer, J. , Vanakker, O. , Coucke, P. , & De Paepe, A. (2012). Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Human Mutation, 33, 1485–1493. 10.1002/humu.22137 [DOI] [PubMed] [Google Scholar]
- Takahara, K. , Schwarze, U. , Imamura, Y. , Hoffman, G. G. , Toriello, H. , Smith, L. T. et al (2002). Order of intron removal influences multiple splice outcomes, including a two‐exon skip, in a COL5A1 acceptor‐site mutation that results in abnormal pro‐alpha1(V) N‐propeptides and Ehlers‐Danlos syndrome type I. American Journal of Human Genetics, 71, 451–465. 10.1086/342099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. X. , He, W. B. , Xiao, W. J. , Meng, L. L. , Tan, C. , Du, J. et al (2020). Novel loss‐of‐function mutation in MCM8 causes premature ovarian insufficiency. Molecular Genetics & Genomic Medicine, 8, e1165. 10.1002/mgg3.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.