

ABSTRACT
The phylogenetic and functional diversities of microbial communities in tropical rainforests and how these differ from those of temperate communities remain poorly described but are directly related to the increased fluxes of greenhouse gases such as nitrous oxide (N2O) from the tropics. Toward closing these knowledge gaps, we analyzed replicated shotgun metagenomes representing distinct life zones and an elevation gradient from four locations in the Luquillo Experimental Forest (LEF), Puerto Rico. These soils had a distinct microbial community composition and lower species diversity compared to those of temperate grasslands or agricultural soils. In contrast to the overall distinct community composition, the relative abundances and nucleotide sequences of N2O reductases (nosZ) were highly similar between tropical forest and temperate soils. However, respiratory NO reductase (norB) was 2-fold more abundant in the tropical soils, which might be relatable to their greater N2O emissions. Nitrogen fixation (nifH) also showed higher relative abundance in rainforest than in temperate soils, i.e., 20% versus 0.1 to 0.3% of bacterial genomes in each soil type harbored the gene, respectively. Finally, unlike temperate soils, LEF soils showed little stratification with depth in the first 0 to 30 cm, with ∼45% of community composition differences explained solely by location. Collectively, these results advance our understanding of spatial diversity and metabolic repertoire of tropical rainforest soil communities and should facilitate future ecological studies of these ecosystems.
IMPORTANCE Tropical rainforests are the largest terrestrial sinks of atmospheric CO2 and the largest natural source of N2O emissions, two greenhouse gases that are critical for the climate. The microbial communities of rainforest soils that directly or indirectly, through affecting plant growth, contribute to these fluxes remain poorly described by cultured-independent methods. To close this knowledge gap, the present study applied shotgun metagenomics to samples selected from three distinct life zones within the Puerto Rico rainforest. The results advance our understanding of microbial community diversity in rainforest soils and should facilitate future studies of natural or manipulated perturbations of these critical ecosystems.
KEYWORDS: Puerto Rico, diversity, nitrous oxide, nosZ, soil microbial communities
INTRODUCTION
Soil microbiomes are some of the most complex ecosystems owing to microenvironments and steep physicochemical gradients, which can change on a micrometer or millimeter scale (1–3). Tropical rainforests (“forests” hereafter) are characterized by humid and wet climate patterns and account for a large portion of the world’s total forest cover (4). These forests have high levels of primary productivity (∼30% of the total global production) due to large amounts of precipitation coupled with year-long warm temperatures and high levels of light (5). Consequently, high levels of biodiversity are observed in these forests, above- and belowground. The soil microbial communities of these forests, in particular, harbor taxa and genes that are exclusive to these habitats/locations, along with only a few cosmopolitan taxa that are shared with other (nontropical forest) habitats (6, 7). Although tropical forest soils are critical ecosystems that host a plethora of distinct ecological niches, little is known about the metabolic potential of tropical soils, especially across elevation and depth gradients. Describing this metabolic diversity is important for studying and monitoring the microbial activities related to greenhouse gas fluxes, namely, nitrous oxide (N2O) and carbon dioxide (CO2), from the tropical soils (8).
Notably, tropical forests represent the largest terrestrial sinks of atmospheric CO2 and the largest natural source of N2O emissions (9–12). Natural soils have been reported to contribute over 43% of the total global N2O emissions, with tropical ecosystems being the highest contributors, having 2 to 4 times higher contributions than natural temperate ecosystems (13–16). These soils are also responsible for about 70% of terrestrial nitrogen fixation, which underlies, at least in part, their high rates of net primary productivity (8, 17).
Microbially mediated nitrification and denitrification are the biotic processes contributing the most to global soil N2O emissions (60 to 70%) (16, 18, 19), although chemodenitrification, i.e., ferrous iron generated by ferric iron-reducing bacteria reacting with nitrite to produce N2O abiotically, is also likely high in iron-rich tropical soils (20). In soils, N2O is biologically produced as a result of DNRA (dissimilatory nitrite reduction to ammonium) or denitrification respiratory pathways (19, 21, 22). Respiratory nitric oxide reductase (nor) is a key contributor to the microbial production of N2O and is commonly found in the genomes of denitrifying bacteria as well as in that of some ammonia-oxidizing organisms (19, 23–27).
While both biotic and abiotic processes contribute to N2O production, consumption of N2O is exclusively mediated by microbial N2O reductase (NosZ) activity (22, 28, 29). Yet, whether the denitrifying microorganisms in the tropical forest soils differ from their counterparts in temperate soils and whether their functional genes present in the community reflect the high nitrogen fluxes remain unanswered questions despite their apparent importance for better management and modeling of tropical soil ecosystems. It has also been demonstrated that tropical forests have significantly higher rates of nitrogen fixation (∼70% of total terrestrial nitrogen fixation) than other ecosystems, significantly affecting the nitrogen budgets in these ecosystems (3, 30–32). How these ecosystem rates translate to the nitrogen-fixing microbial (sub)community diversity and gene relative abundance remains unclear.
The Luquillo Experimental Forest (LEF), also known as the El Yunque National Forest in Puerto Rico (PR), has been a long-term ecological research (LTER) site since 1988. The site is dedicated to the assessment of the effects of climate drivers on the biota and biogeochemistry. The forest has been subjected to several disturbance regimes over the last few decades, mostly natural and, to a smaller extent, anthropogenic, such as tourism and experimental manipulations (33, 34). This site encompasses distinct “life zones” characterized by sharp environmental gradients even across small spatial scales (33, 35, 36). The broad life zones based on the Holdridge classification system include the rain forest, wet forest, lower montane wet forest, and lower montane rain forest. These life zones are distinguished by elevation and temperature and rainfall patterns, in addition to other edaphic factors (37–40). The elevation and rainfall patterns also tend to influence oxygen availability, redox potential, nutrient uptake, and organic decomposition rates (37, 40, 41). The dynamic interplay of existing physicochemical gradients and climatic factors gives rise to a complex mosaic of biodiversity patterns observed in this forest (38). Hence, LEF represents an ideal environment to study tropical microbial community diversity patterns and their effect on carbon and nitrogen cycling. The four sampling sites of this study (see below) were chosen to represent the distinct vegetation and life zones within the LEF.
Previous studies in the LEF, and in similar forest regions, have mostly focused on the effects of redox dynamics, litter decomposition, nitrogen (N), and other nutrient fertilization on microbial community activity through enzyme assays and biochemical process rate measurements. Few studies have examined microbial diversity patterns across an elevation gradient, and those were based only on low-resolution techniques such as terminal restriction fragment length polymorphism analysis (4, 10, 11, 42–44). Furthermore, studies linking marker-gene abundances (related to nitrogen cycling) with in situ flux measurements have been met with mixed success in the forest soils, despite the high N2O fluxes often measured in these ecosystems (45). A possible underlying reason for this mixed success could be that the commonly used nosZ primers target only the clade I (typical) nosZ sequences and miss the often numerically more dominant clade II (atypical) sequences (46), a bias that can be circumvented by employing metagenomic analyses (47).
With recent developments in next-generation DNA sequencing and associated bioinformatics genome binning algorithms, nearly complete metagenome-assembled genomes (MAGs) can be recovered without cultivation (48, 49), opening new windows into studying soil microbial communities. Here, shotgun metagenomes originating from soils from the four different locations/life zones and three different depths in the LEF were analyzed to describe the microbial community diversity, biogeographical patterns, and metabolic potential differences across samples. Furthermore, the metagenomic data obtained from these soils were compared to similar data from temperate grasslands in Oklahoma (OK) (50) and agricultural soils from Illinois (IL), USA (49) previously obtained by our team. By analyzing nearly complete MAGs, we show that the most abundant microbial populations (based on number of reads recruited) at each of the sampling locations represent sequence-discrete populations, similar to those observed in other habitats (45). Using such sequence-discrete populations as the fundamental unit of microbial communities, we subsequently assess the population distribution at high resolution across the sampling sites (biogeography) and the gene content they carry, with a focus on nitrogen metabolism.
RESULTS
Sampling sites.
Soil samples were collected on February 2016 from four locations/sites across the LEF (18.3′ N, 65.80′ W). The four sites, namely, Sabana, El Verde field station, Palm Nido, and Pico del Este, each located at different elevations from the mean sea level, i.e., 265, 434, 634, and 953 m, respectively, were chosen due to their unique landscapes and rainfall patterns, thereby creating distinct ecological niches (Fig. 1A).
FIG 1.
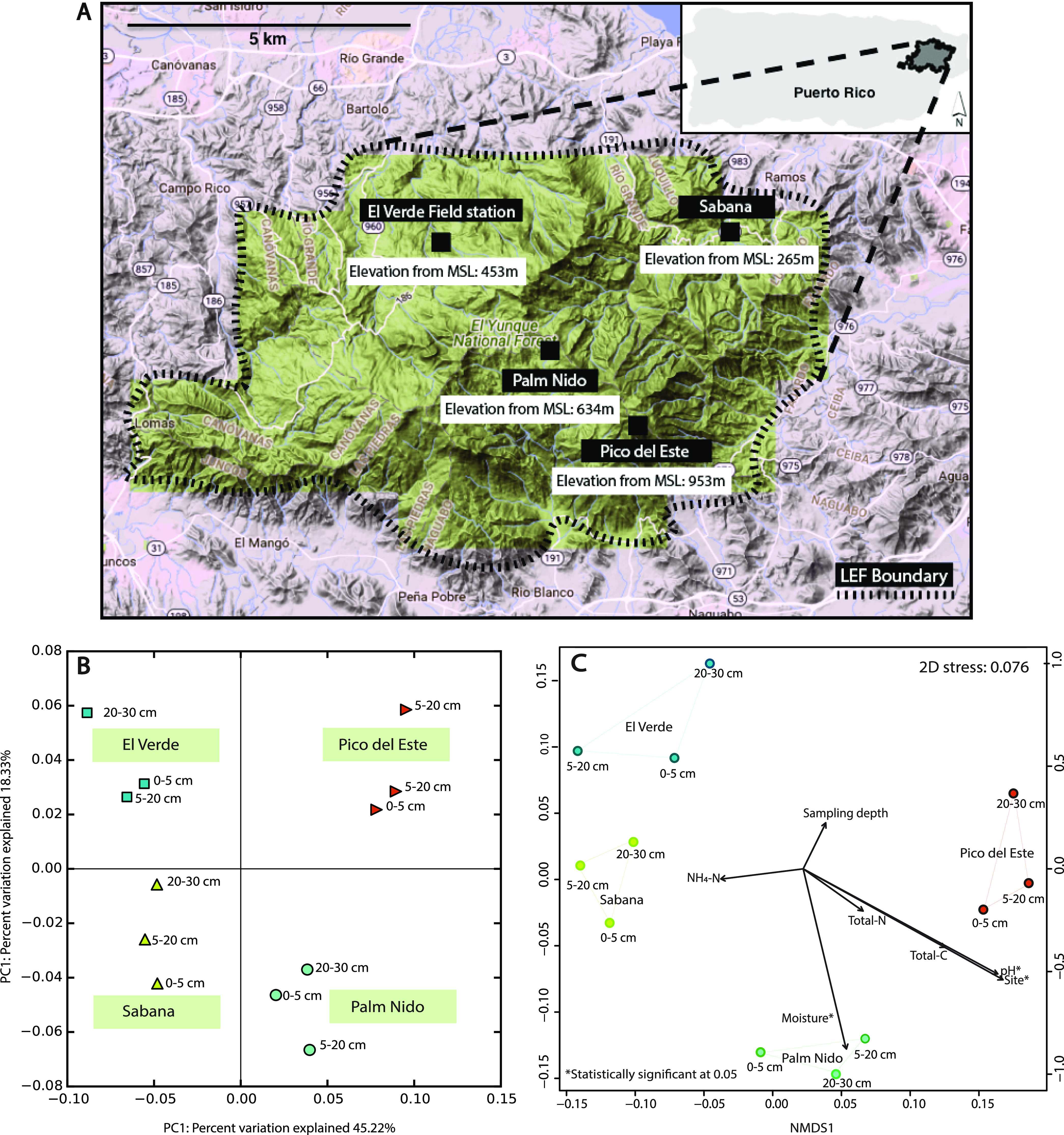
Sampling location map and microbial community diversity among the study sites. (A) Map of the four sampling sites within the Luquillo Experimental Forest (LEF). (B) Principal coordinate analysis (PCoA) plots based on Mash distances, colored by sampling site. (C) Nonmetric multidimensional scaling (NMDS) plot with the soil physicochemical parameters incorporated. The arrow lengths are proportional to the strength of the correlations obtained between measured soil physicochemical parameters and each ordination axis. The coordinates for the Luquillo Experimental Forest (LEF) were obtained from DEIMS-SDR (Dynamic Ecological Information Management System, site and dataset registry; https://deims.org/bd0b5bcf-4f2e-4038-8275-629ffa5bf2aa) and visualized using the terrain feature in Google Maps. The map was refined and annotated in Adobe Illustrator.
The El Yunque forest is categorized into four distinct vegetation zones, namely, the Tabonuco, Palo Colorado, Sierra Palm, and Dwarf/Elfin forests. Sites Sabana and El Verde, which are located at the lowest elevation among the four sites within the LEF, fall under the Tabonuco forest category in terms of vegetation, dominated by the tree species Dacryodes excelsa (native to Puerto Rico). They are characterized by canopy cover and low light intensities at the ground level which account for the sparsely vegetated forest floor. However, these sites still harbor the richest flora of all sites (51). Palm Nido is characterized by unstable, wetter soils, steeper slopes, and vegetation that is dominated by the Sierra Palm (Prestoea montana). The site at the highest elevation, Pico del Este (dwarf forest ecosystem or “elfin woodlands”) is characterized by higher winds, lower temperatures, and vegetation that is enveloped by clouds (34, 52), and its main vegetation is comprised of moss and epiphytes. Furthermore, highly acidic soil and continuously water-saturated soils deficient in oxygen are some major characteristics of this ecosystem, with most mineral inputs for plants being dissolved in the rain and fog.
Three adjacent soil profiles were taken from each of the four LEF sites (4 sites encompassing 3 life zones; Palo Colorado was not sampled). For each profile, individual soil cores were taken at each depth (0 to 5 cm, 5 to 20 cm, 20 to 30 cm) using a 3-cm diameter by 15-cm length soil corer (AMS Inc, ID) that was decontaminated between samplings by washing with 70% ethanol. The three cores at each sampling depth were pooled for community DNA extraction, producing a total of 12 samples across the four sites.
Diversity of forest microbial communities.
The LEF soil communities were compared to those of intensively studied ecosystems, namely, the Oklahoma temperate grassland (OK) (1, 50) and Illinois agricultural soils (IL) (49), which were previously characterized with similar shotgun metagenomics approaches. Shotgun metagenomic sequencing recovered a total of 370 million reads across the 4 sites (see Table S2 in the supplemental material). Nonpareil 2.0 (53) was used to estimate sequence coverage, i.e., what fraction of the total extracted community DNA was sequenced. Nonpareil analysis of community diversity (Fig. S1) showed that the agricultural Urbana (IL) site had the highest diversity of all the soils compared (Nd diversity 24.02; note that Nd values are given in log scale) and consequently the lowest sequence coverage at (only) 37.23%. El Verde and Pico del Este (20 to 30 cm) were the least diverse or most completely sequenced with 87.1% and 73.4% coverage, respectively (Nonpareil diversity of 19.6 and 20.6, respectively, or about 2 to 3 orders of magnitude less diverse). Overall, OK and IL soils appear to be more diverse than the PR soils by about 2 orders of magnitude, on average, with an average Nonpareil value of 22.75 ± 0.37. Nearly complete coverage for El Verde and Pico del Este (20 to 30 cm samples) would require 2.402e+09 bp and 8.735e+09 bp, respectively, and for the same level of coverage, the more complex communities in Urbana (IL) would require a substantially higher sequencing effort of 1.282e+12 bp. The OK soils had an estimated sequencing depth of 2.063e+11 ± 1.436e+11 bp.
Community composition variation across the forest sites based on 16S rRNA gene sequences.
The number of total 16S rRNA gene-based OTUs (operational taxonomic units) observed in each metagenome and the Chao1 estimate of total OTUs present reflected the degree of undersampling at each site (Fig. S1 and S2) and were also consistent with the Nonpareil coverage estimates (Fig. 1). When Puerto Rico (PR) tropical soils were compared with the agricultural and grassland soils from the United States at the phylum level, Proteobacteria, Acidobacteria, and Actinobacteria were the most abundant taxa across all ecosystems. However, in the forest soils, a few highly abundant OTUs dominated the entire soil community, whereas in the OK and IL soils, OTUs were more evenly distributed (Fig. S2), consistent with the Nonpareil diversity results. Only 1.28% of the total detected OTUs (out of a total 8,019, non-singleton OTUs) were shared among all PR samples, while 49.95% of OTUs were exclusive to a particular sampling site in PR, partly reflecting the undersampling of the extant diversity by sequencing. Only 0.37% of the OTUs (out of a total 13,760, non-singleton OTUs) were shared among all the sites across all 3 ecosystems, all of which were assignable to Alphaproteobacteria, Acidobacteria, Verrucomicrobia, and Actinobacteria.
It is important to note that the above-mentioned data sets were obtained based on different DNA extraction methods and at different time points and laboratories. To test for the possible effect of the DNA extraction method on the derived results, we applied four additional DNA extraction methods on a selected subset of our PR samples, including two manual phenol-chloroform-based methods that are often advantageous for iron-rich soils like those in tropical forests. Our results revealed comparable levels of diversity among the different methods tested, showing that the method used here for extraction performed comparably to if not better than non-kit-based (manual) extraction methods. For example, Nd values for soils from the four sampling sites ranged between 19 and 22, and the MP Bio FastDNA SPIN kit method used for the samples reported herein provided among the highest diversity values, especially for mid- and high-altitude samples (Fig. S3). Hence, the diversity patterns reported here are presumably independent of the DNA extraction method used. We would also like to note that given the differences in the soil compositions across the different sampling regions in PR soils alone, a single/uniform extraction protocol would have some inherent biases (i.e., some soils had higher clay/iron content than others in the same forest).
Factors driving community diversity in the forest soils: multidimensional scaling analysis of beta diversity.
The principal coordinate analysis (PCoA) plots, constructed based on the Mash distances among whole metagenomes, showed a clustering pattern that was primarily governed by site/location. Accordingly, site explained 45.22% of the total diversity (Fig. 1B). The nonmetric multidimensional scaling (NMDS) analysis of the data revealed only site, pH, and soil moisture to be statistically significant physicochemical parameters in explaining the observed community diversity (Fig. 1C; Table S3). Analysis of similarity (ANOSIM) values also indicated site to be a more important factor than depth, with P values of 0.001 and 0.94, respectively. Based on the distance-based redundancy analysis (dbRDA), site was the most significant factor, even when the interplay between site and sampling depth was accounted for (Table S4). Table 1 shows the partitioning of the variance between the proportion that is explained by constrained axes (i.e., environmental variables measured) and the proportion explained by unconstrained axes (i.e., variance not explained by environmental variables measured). The total variance explained by all (measured) environmental variables was 80.2% (Table 1), which is remarkably high for a soil ecosystem (54).
TABLE 1.
Proportion of total microbial community diversity explained by measured soil environmental factorsa
| Axis type | Inertia | Proportion | Rank |
|---|---|---|---|
| Constrained | 0.0876 | 0.8021 | 6 |
| Unconstrained | 0.02161 | 0.1978 | 5 |
| Total | 0.1092 | 1 |
Site, sampling depth, pH, total nitrogen, total carbon, and moisture data were considered in the analysis.
Major N-cycling pathways.
Genes encoding proteins involved in denitrification and nitrogen fixation were the most abundant nitrogen (N) cycling pathway genes detected at different sites. Overall, the forest soils harbored an ∼2- to 3-fold higher abundance of denitrification genes, i.e., narG, nirK, and norB (catalyzing the reduction of nitrate, nitrite, and nitric oxide, respectively), than the grassland and agricultural soils (Fig. 2A). For instance, the norB gene abundance was found to be at the highest abundance among the denitrification genes, with ∼37% (standard deviation [SD] 9.5%) of the genomes in the PR soils predicted to carry a norB gene, compared to ∼17% (SD 4%) and ∼14% (SD 1.3%) at IL and OK, respectively. Similarly, narG showed a 3-fold higher abundance in the PR soils than in IL and OK soils (Fig. 2B). While denitrification gene abundances appeared higher in the tropical soils, the relative abundance of nosZ gene (i.e., 11.6% [SD 3%] of the total genomes across the four locations in the LEF were predicted to carry nosZ) was similar to nosZ relative abundance in IL and OK soils, i.e., 11.75% (SD 5%) and 11.08% (SD 3%), respectively (not statistically significant at P = 0.05). Similar to those of nosZ, DNRA gene abundances (namely, nrfA) were comparable across all sites studied herein (9%, SD 1.9%).
FIG 2.

Abundance of N-cycling genes and their distribution across soil ecosystems. (A) Abundance of hallmark genes for denitrification, DNRA, and nitrogen fixation pathways, represented as genome equivalents (% of total bacterial genomes sampled that carry the gene) in the metagenomes studied (see figure key). (B) Frequency of genomes carrying the respective denitrifying gene across the three ecosystems studied. Genes denoted by the same letter are not statistically significantly different between ecosystems (analysis of variance [ANOVA] Tukey test). Statistical significance reported at P < 0.05. Note that nitrification genes were not detected in any of the Puerto Rico sites.
Predominant nosZ clades are shared among soil ecosystems.
Placing nosZ-carrying reads to a reference nosZ phylogenetic tree revealed that clade II nosZ, affiliated predominantly with Opitutus, Anaeromyxobacter, and other closely related genera, dominated the nosZ gene pool in the tropical forests (Fig. 3; Figs. S4 to S7). In contrast, a very small fraction of reads (<10% of total nosZ reads) were recruited to clade I nosZ. Members belonging to the clade II nosZ dominated the nosZ gene pool in OK and IL soils as well, with IL agricultural soils showing the greatest nosZ sequence diversity among the three regions. Notably, Opitutus terrae-affiliated sequences represented the most abundant subclade (nosZ OTUs/subclades were defined at the 95% nucleotide sequence identity level) in all regions. Furthermore, most of the O. terrae-affiliated reads in the forest soil data set appeared to be assigned to a single subclade, while their counterparts in the OK and IL soils appeared to be more evenly distributed among several closely related nosZ subclades, i.e., showing higher sequence diversity (Fig. 3; Fig. S4 to S7). O. terrae (strain DSM 11246/PB90-1) nosZ reads at >95% identity made up between 20% and 60% of the total nosZ reads recovered from the El Verde site and, together with the second most abundant subclade from Anaeromyxobacter sp., contributed over 30% of the total nosZ reads across all four PR locations (Fig. 3). Despite the significant taxonomic diversity observed in these soils (Fig. S2), the soils from PR shared several abundant nosZ gene sequences/subclades at >95% nucleotide identity with soils in OK and IL (Fig. 3). Furthermore, in order to compare the predominant nosZ sequence variants across the samples shown here, a new phylogenetic reference tree was constructed based on almost-full-length sequences obtained from the assemblies/MAGs obtained from the metagenomes studied here (namely, PR, OK, IL). The short reads identified as nosZ from the PR soils were placed on this tree and show that the majority of these reads are recruited by the nosZ sequences obtained from these assemblies/MAGs, indicating that the nosZ sequences across the ecosystems studied here are similar (Fig. S8.)
FIG 3.

Phylogenetic diversity of nosZ-carrying metagenomic reads recovered in each soil ecosystem. nosZ sequences were identified by the ROCker pipeline and placed in a reference nosZ phylogeny as described in Materials and Methods. The radii of the pie charts are proportional to the number of reads assigned to each subclade and the colors represent the sampling sites from each ecosystem (see figure key). Subclades highlighted in gray indicate the most abundant subclades across all three ecosystems, whereas the ones highlighted in blue were abundant only in agricultural soils (IL). (A) nosZ reads from every sampling site recruiting to clade II clades. (B) nosZ reads recruiting to clade I clades. Inset shows the most abundant subclade (Opitutus terrae) from panel A and its distribution across all sites. Note that in all three ecosystems most of the reads recruit to clade II subclades. Fig. S7 shows the distribution of the reads among the most abundant subclades in detail.
Nitrogen fixation potential.
The nitrogen fixation genes (mainly nifH) were present at a much lower abundance in the lower altitude forest samples. For instance, only 1 to 3% of all genomes in the lower altitude samples were predicted to carry nifH, compared to ∼20% of the genomes in the higher-elevation samples (Pico del Este) (Fig. 2A), and almost none of the reads from IL and OK metagenomes appeared to carry nifH (<0.1%). Therefore, nitrogen fixation gene abundance patterns indicated a much stronger selection for nitrogen fixation in the tropical forest relative to that in temperate agricultural or natural prairie soils, especially at higher elevations. Furthermore, no ammonia-oxidizing genes (amoA) were detected in any of the soils except for Urbana soils (IL), which had a history of fertilizer (N) input.
Recovery of metagenome-assembled genomes (MAGs) representative of each site.
In order to test the prevalence of taxa and genes across our sampling sites, the distribution of abundant MAGs recovered from each PR sampling site (assembly and MAG statistics provided in Table S6) was assessed across the sites using read-recruitment plots (55). Taxonomic assignment using the Microbial Genomes Atlas, or MiGA (56), revealed that the most abundant MAG was at site El Verde (lowest elevation), representing 4.39% of the total metagenome, and was affiliated with an unclassified Verrucomicrobia. The second most abundant (1.8% of total) was likely a member of the genus Candidatus Koribacter (Acidobacteria) followed by an unclassified member of Acidobacteria (1.45% of total). The Verrucomicrobium MAG was found at an abundance of 1.03% of the total population at Sabana and at 0.07% and 0.03% in Palm Nido and Pico del Este (highest elevation), respectively. Uneven coverage across the length of the reference sequence and nucleotide sequence identities were observed in the recruitment of short reads from Palm Nido and Pico del Este as well as with all OK data sets, indicating that the related populations in the latter samples were divergent from the reference MAG (Fig. S10). Therefore, at least this abundant low-elevation verrucomicrobial population did not appear to be widespread in the other samples analyzed here (Fig. S10). Similarly, the other abundant MAGs from other sites in the forest soils were unique to the corresponding sites (elevation) from which they were recovered. Almost all MAGs used in the analyses were assignable to a novel family, if not a higher taxonomic rank, according to MiGA analysis (compared to 11,566 classified isolate genomes available in the NCBI prokaryotic genome database), underscoring the large unexplored diversity harbored by the PR tropical rainforest soils. The sequence diversity/complexity as well as sequencing depth limited large-scale recovery of high-quality MAGS.
Functional gene content of the MAGs.
The genome sequences of the most abundant MAGs from each location (n = 6) were analyzed in more detail to assess the functions they encoded, especially with respect to N-cycling pathways (Fig. 4). MAGs from Pico del Este (highest elevation) showed a high abundance of N metabolism related genes compared to MAGs from other sites (Fig. 4). Most notably, genes related to nitrogen fixation were found only in the Pico del Este MAG, which was consistent with the short read analysis data sets showing greater relative abundance of nifH at this site. Nitrification (ammonia oxidation related genes) gene clusters were not detected in any of the recovered MAGs. norB and nosZ genes were found in three out of the six abundant MAGs analyzed. The most abundant El Verde MAG, most closely related to O. terrae (average amino acid identity [AAI] = 40%), possessed a nosZ gene, which was congruent with the nosZ phylogeny described above (i.e., ∼60% of the nosZ-encoded reads from El Verde had a closest match to O. terrae nosZ sequences).
FIG 4.
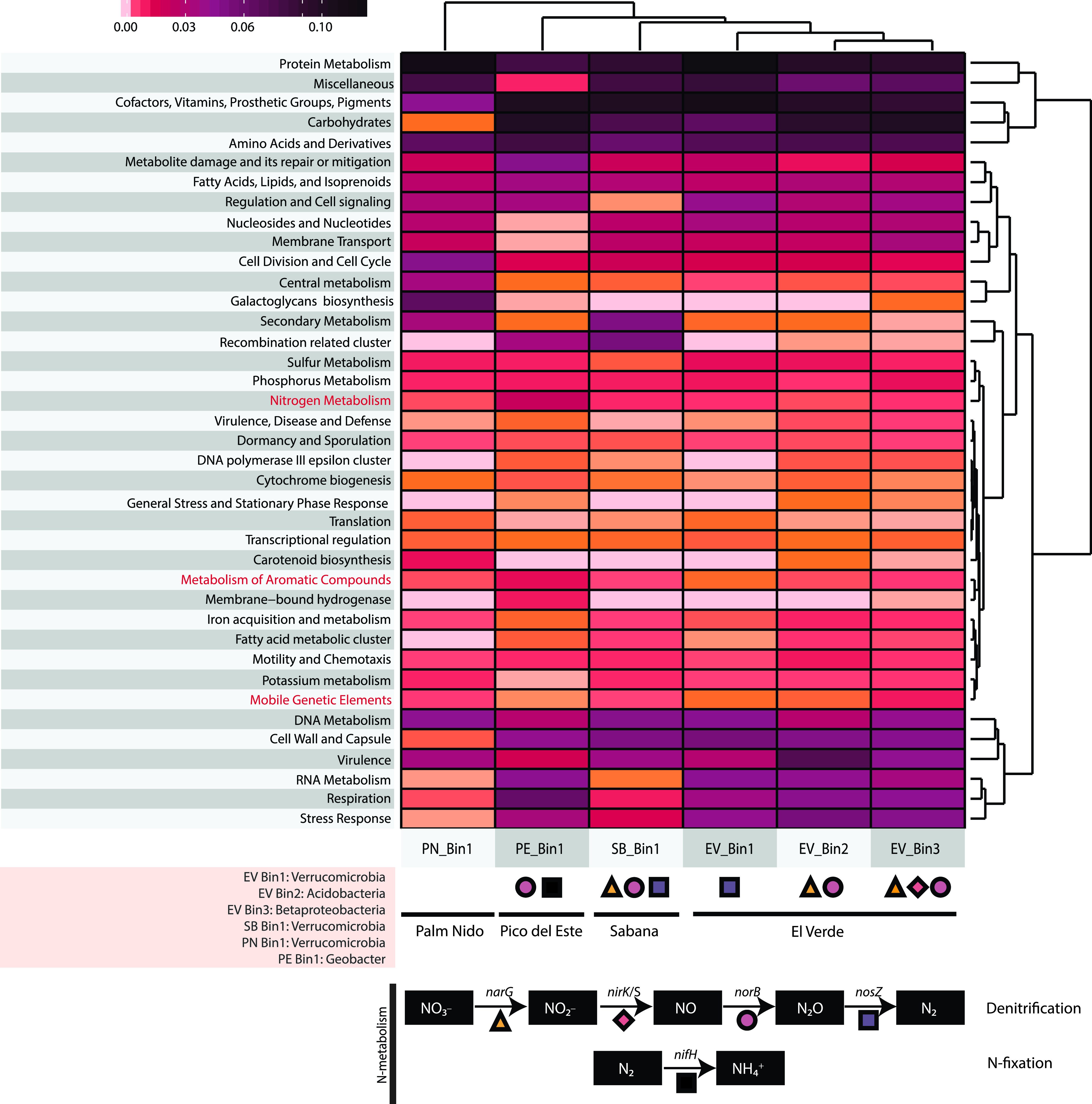
Functions encoded by the recovered population MAGs. Heatmap showing the relative abundance of genes encoding the major metabolic functions (level 1 of the SEED subsystem category) for each MAG recovered from the four sites in Puerto Rico. The taxonomic classification of each MAG based on MiGA is shown on the bottom left. The symbols at the bottom of the heatmap denote the presence (or absence) of specific N-cycling genes, namely, denitrification and nitrogen fixation. No genes involved in nitrification were detected in any of the bins.
DISCUSSION
The present study reported the taxonomic and gene content diversities of poorly characterized tropical rainforest soils by using whole-community, shotgun metagenomic sequencing of samples from the Luquillo Experimental Forest (LEF), Puerto Rico. The recovered nearly complete MAGs represented several abundant and widespread organisms within this ecosystem that could serve as model organisms for future studies. Furthermore, since the LEF is subjected to various natural and experimental (e.g., warming, phosphorus fertilization) perturbations, our study could also provide a baseline for these perturbations and future soil microbial studies at LEF. Our results revealed that the LEF soils harbor distinct microbial communities at sites with elevation distinct from sea level. In contrast, and unlike several other soil ecosystems, sampling depth did not have a substantial impact on structuring community diversity (Fig. 1B and C), revealing no depth stratification in the LEF soils, at least for the depths sampled here (5 to 30 cm). This is most likely due to the lack of distinct soil horizons within the first 30 cm of the sampling sites and indicates that the soil formation and/or physicochemical properties in these ecosystems could differ markedly from those in their temperate counterparts (37).
A recent study examining the dominant bacterial (16S rRNA gene-based) phylotypes across the globe found that the predominant phylotypes were widespread across ecosystems. The only exception to this pattern was the forest tropical soils which harbor distinct phylotypes (7). Consistent with these conclusions, the majority of MAGs recovered from each LEF site appeared to be site-specific (e.g., see Fig. S10 in the supplemental material) and represented at least novel species and genera, further underlining the undertapped microbial diversity harbored by tropical forest soils. Currently, the environmental factors driving these diversity patterns remain poorly understood for tropical forest soils (7), but our study provided several new insights into this issue.
In particular, sites El Verde and Sabana (lowest elevation sites) had community structure and diversity similar to those of the two higher-elevation sampling sites, with certain MAGs being present at both sites but not in any of the other (higher-elevation) sites examined. This is presumably attributable to both sites having similar climate and vegetation patterns (i.e., Tabonuco forest). On the other hand, Pico del Este was the highest-elevation site and experiences almost continuous cloud cover as well as horizontal precipitation. The unique topology of Pico del Este was reflected in distinct and deeply novel MAGs and gene content, which differed markedly from those of the other three sampling sites within the LEF (PCoA plots, Fig. 2B). The high water content of the Pico del Este soils gives rise to a unique ecosystem dominated by epiphytes (e.g., moss) among other plant species (57). The epiphytic community has presumably significant effects on nutrient (e.g., nitrogen) cycling (58) and influences the water input to the soil, thereby shaping a unique habitat/niche for the soil microbes. Free-living microbes have been shown to be one of the highest contributors to biological N fixation in these forests, with high rates of nitrogenase activity associated with the presence of moss/epiphytes (44, 59). Furthermore, lower N-cycling rates and high water content in soils of cloud-immersed forests have been shown to be responsible for the overall low nitrogen levels in these (nitrogen-limited) ecosystems (60).
Consistent with these previous results and interpretations, the Pico del Este showed a high potential for nitrogen fixation, i.e., it was estimated that 1/5 of the total bacterial genomes sampled possessed genes for N fixation, which is at least 10 times greater nitrogenase gene/DNA abundance than that of any other site evaluated herein. Accordingly, we found that site (location) alone explained about half (45%) of the beta diversity differences observed among the four sampling sites, which reached ∼80% when a few physicochemical parameters, namely, pH and moisture, were also included in the analyses (Fig. 2B; Table 1). This is a notably high fraction of beta diversity explained by measured parameters for a soil ecosystem (54) and likely reflected that location and the physical properties that characterized different locations within LEF structured diversity are much stronger than those in other soil ecosystems. Tropical forests have also been shown to have significantly higher rates of nitrogen fixation than other ecosystems, which can exceed the N retention capacity of the soil resulting in large N loss as N2O (61). The findings reported here on denitrification gene abundances were generally consistent with these previous observations as well.
Links between soil community structure and nitrogen cycling can help close the knowledge gaps on how the forest ecosystems affect the release and mitigation of certain highly potent greenhouse gases such as N2O. The gene abundances observed here, e.g., more than 2-fold higher abundance of norB (associated with NO reduction to N2O) and similar nosZ (N2O consumption) abundances in tropical soils relative to those in temperate soils, were consistent with higher N2O emissions observed previously from the tropics. Further, in acidic soils such as the tropical forest soils evaluated in this study, with the possible exception of the cloud-immersed site, lack of N limitation can suppress complete denitrification, thereby leading to higher N2O release compared to that of other soil ecosystems (30). These interpretations were consistent with our observation that the PR soils harbored a relatively high abundance of respiratory (related to denitrification) norB genes as well. Previous studies have also suggested that many denitrifying bacterial genomes possess the genes required to reduce nitrate to nitrous oxide but do not possess the gene responsible for the last step, i.e., N2O reduction to N2, leading to the release of N2O gas (62), consistent with the findings of our study. Alternatively, the higher relative abundance of norB relative to that of nosZ or other denitrification pathway genes may be associated, at least partly, with detoxification of NO and/or dismutation of NO to N2 and O2 and not energy-yielding NO reduction to N2O (reviewed in reference 62). However, this explanation is less likely, because we specifically focused on the respiratory reductase (cnorB), not its detoxifying homolog (qnorB), in our analysis (see Materials and Methods for further details).
It has been established that tropical forest soils are the single highest contributor of natural N2O emissions. While several abiotic and microbial processes can contribute to soil N2O, N2O consumption is an exclusively microbial process, catalyzed by the enzyme product of the nosZ genes (29). Based on the assessment of the nosZ gene phylogeny, it appears that almost all of the nosZ genes from the tropical forest soils studied here belong to a previously overlooked clade II nosZ (29, 63). This clade consists mainly of nondenitrifying N2O reducers (i.e., organisms possessing N2O reductases but lacking all or some of the other reductases in the denitrification pathway). Despite the unique phylogenetic species- and gene-level diversity harbored by tropical soils in general, the nosZ gene sequence diversity appears to be shared between temperate and agricultural soils (Fig. 4). These findings imply strong selection pressure for conservation of nitrous oxide reductase sequences across tropical and temperate soil ecosystems that may not necessarily be applicable to other N-cycling genes and pathways, which warrants further attention in the future.
It would also be interesting to assess how the findings reported here for the LEF apply (or not) to other tropical forests especially because our study is based on a relatively small sample size. Despite the relatively small sample size, however, our results showed statistically significant differences along the elevation gradient sampled at the LEF that are independent of DNA extraction (Fig. S3) or sequencing methods and consistent with our metadata (Fig. 2) and previous process rate measurements. While the diversity in the Puerto Rico soils appears to be lower than that in temperate grassland and agricultural soils and different DNA extraction methods, including phenol-chloroform- and kit-based, provided similar results with our LEF samples (Fig. S3), it is important to note that the DNA of the temperate soil data sets used in our comparisons was extracted using different methods by the original studies (e.g., OK soils were extracted using the PowerSoil kit). Therefore, it would be important to confirm the preliminary findings in terms of α-diversity reported by our study by using the exact same DNA extraction and sequencing procedures in all soils.
Integration of functional (e.g., gene expression) data with in situ rate measurements will provide a more complete picture of the composition and functioning in tropical forest soils. The identification of certain biomarker genes such as nosZ sequences in our study could facilitate future investigations on biogeochemical N-cycling and greenhouse gas emissions. For instance, the assembled MAGs and gene sequences provided here could be useful for the design of specific PCR assays for assessing transcript levels (activity), allowing potential linking of carbon dioxide, methane, nitrogen, and soil organic matter (SOM) turnover to the activity of individual populations and/or their genes. As the gradients at the LEF also provide a natural setting to interpret the potential ramifications of climate change scenarios such as altered precipitation patterns, the DNA sequences provided here could facilitate future manipulation experiments with an emphasis on understanding and predicting the effects of climate change on microbial community dynamics along the elevation gradient.
MATERIALS AND METHODS
Physicochemical analysis of soil samples.
Soil samples were stored in sterile Whirl-Pak bags and kept on ice during transport and until storage at −80°C. Soil pH was determined using an automated LabFit AS-3000 pH analyzer, and soil extractable P, K, Ca, Mg, Mn, and Zn were extracted using the Mehlich-1 method and measured using an inductively coupled plasma spectrograph at the University of Georgia Agricultural and Environmental Services Laboratories (Athens, GA, USA). Soil extractable P using this method is interpreted as the bioavailable fraction of P. NH4-N and NO3-N were measured by first extracting them from soil samples with 0.1 N KCl, followed by the colorimetric phenate method for NH4+ and the cadmium reduction method for NO3. The physicochemical conditions at the sites during the time of sampling are provided in Table S1.
Community DNA extraction and sequencing.
Total DNA from soil was extracted using the FastDNA SPIN kit (MP Biomedicals, Solon, OH) following the manufacturer’s procedure with the following modifications (64). Soils were air dried under aseptic conditions followed by grinding with a mortar and pestle. Cells were lysed by bead beating and DNA was eluted in 50 μl of sterile H2O. DNA sequencing libraries were prepared using the Illumina Nextera XT DNA library prep kit according to the manufacturer’s instructions, except the protocol was terminated after isolation of cleaned double-stranded libraries. Library concentrations were determined by fluorescent quantification using a Qubit HS DNA kit and Qubit 2.0 fluorometer (ThermoFisher Scientific), and samples were run on a high sensitivity DNA chip using the Bioanalyzer 2100 instrument (Agilent) to determine library insert sizes. An equimolar pool of the sequencing libraries was sequenced on an Illumina HiSeq 2500 instrument (located in the School of Biological Sciences, Georgia Institute of Technology) using the HiSeq Rapid PE cluster kit v2 and HiSeq Rapid SBS kit v2 (Illumina) for 300 cycles (2 by 150 bp paired end). Adapter trimming and demultiplexing of sequenced samples was carried out by the HiSeq instrument. In total, 12 metagenomic data sets were generated (3 per site for the three depths), and statistic details on each data set are provided in Table S2.
In order to test for any DNA extraction biases of the kit used above, especially for the high iron/clay content that characterizes tropical forest soils and is known to affect the extraction step, four additional DNA extraction methods were performed in parallel on a small subset of samples collected in 2018 from the same sites (6 samples per extraction method for 5 extraction methods covering the 4 sites). The methods included two manual (as opposed to kit-based) phenol-chloroform-based methods (65, 66) as well as two other kit-based methods, namely, DNeasy PowerSoil and DNeasy PowerSoil Pro (Qiagen Inc.). For this evaluation, the soils were first homogenized and subsequently divided into five subsamples to use with each method (including the FastDNA SPIN kit-based method mentioned above). The libraries were constructed and sequenced the same way as described above for the FastDNA SPIN kit method.
Bioinformatics analysis of metagenomic reads and MAGs.
The paired end reads were trimmed and quality checked using the SolexaQA (67) package with a cutoff at a Phred Q value of >20 (≥99% accuracy per base-position) and a minimum trimmed length of 50 bp.
Assembly and population genome binning. Coassembly of the short reads from the same location was performed using IDBA-UD (68), and only resulting contigs longer than 500 bp in length were used for downstream analysis (e.g., functional annotation and MyTaxa classification). Genes were predicted on the coassembled contigs using MetaGeneMark (69) and the predicted protein-coding regions were searched against the NCBI All Genome database using BLASTP (70). Since the assembly of individual data sets resulted mostly in short contigs (data not shown), the contigs from the coassembly (combining metagenomes from the three sampling depths, for each site) were used for population genome binning. Contigs longer than 1 kbp were binned using MaxBin (71) to recover individual MAGs (default settings). The resulting bins were quality checked for contamination and completeness using CheckM (72) and were further evaluated for their intrapopulation diversity and sequence discreteness using fragment recruitment analysis scripts as part of the Enveomics collection (55) essentially as previously described (73).
Functional annotation of MAGs. Genes were predicted for each MAG using MetaGeneMark and the predicted protein-coding regions were searched against the curated Swiss-Prot (74) protein database using BLASTP (70). Matches with a bitscore higher than 60 or amino acid identity higher than 40% were used in subsequent analysis. The Swiss-Prot database identifiers were mapped to their corresponding metabolic function based on the hierarchical classification subsystems of the SEED subsystem category (level 1) (75). The relative abundance of genes mapping to each function was calculated based on the number of predicted genes from each MAG assigned to the function (for read-based assessment, see below). Relative abundance data were plotted in R using the “superheat” package (https://arxiv.org/abs/1512.01524). Individual biomarker genes for each step of the nitrogen-cycling pathway were manually verified by visually checking the alignment of the identified sequences by the pipeline outlined above against verified reference sequences.
Functional annotation of short reads. Protein-coding sequences present in short reads were predicted using FragGeneScan (76) using the 1% Illumina error model. The predicted genes were then searched against the Swiss-Prot database using BLASTP (best match). Low-quality matches (bitscore < 60) were excluded, and relative abundance of genes mapping to each function was determined as described in the previous section.
Community diversity estimation.
(i) Nonpareil. Nonpareil (53) was used to estimate sequence coverage, i.e., what fraction of the total extracted community DNA was sequenced, and predict the sequencing effort required to achieve “nearly complete coverage” (≥95%). The default parameters in Nonpareil were used for all data sets. Only one of the two paired reads (forward) for each data set was used to avoid dependency of the paired reads, which can bias Nonpareil estimates (53).
(ii) Mash and multidimensional scaling. Mash, a tool employing the MinHash dimensionality reduction technique to compare sample-to-sample sequence composition based on k-mers (77), was used to compute pairwise distances between whole metagenomic data sets and construct the distance matrix to be used in multidimensional scaling. Pairwise Mash distances between the metagenomic data sets were computed from the size-reduced sketches (default parameters). PCoA and NMDS were employed to visualize the distance matrix and evaluate the physicochemical parameters driving community diversity, respectively. Furthermore, dbRDA (distance-based redundancy analysis) was used to obtain a finer resolution on the observed compositional variation. All of the above statistical analyses were performed using the vegan package in R (78), with default settings.
16S rRNA gene fragments recovered from shotgun metagenomes. 16S ribosomal rRNA (16S) gene fragments were extracted from the metagenomic data sets using Parallel-META (79). 16S-carrying reads were classified taxonomically using the GreenGenes database.
Recovered 16S fragments were clustered (“closed-reference OTU picking” strategy using UCLUST [80]) and taxonomically classified based on their best match in the GreenGenes database (81) at an ID of ≥97% in QIIME (82, 83). The relative abundances of the OTUs were calculated based on the number of reads assigned to each OTU. Community composition was assessed based on OTU taxonomic assignments at the genus and the phylum ranks and was compared between the sites based on the relative abundance of OTUs at each site.
Identification of N-cycling genes using ROCker.
ROCker (84) was employed for a precise identification and quantification of nosZ (encoding nitrous oxide reductase), norB (encoding respiratory nitric oxide reductase, cytochrome bc complex associated), nirK (encoding nitrite reductase), narG (encoding nitrate reductase), nrfA (encoding nitrite reductase, DNRA related), amoA (encoding ammonia monooxygenase), and nifH (encoding nitrogenase) encoding metagenomic reads (http://enve-omics.ce.gatech.edu/rocker/models). Briefly, the short-read nucleotide sequences were searched (using BLASTX) against a training set for each above-mentioned protein; training sets were manually curated to encompass experimentally verified reference sequences as suggested previously (84). The resulting matching sequences were then filtered using the ROCker compiled model (model for 150 bp-long reads for PR and OK soils and 100 bp model for IL soils). Protein abundances (based on the number of reads assigned to the protein) were normalized by calculating genome equivalents. For the latter, the ROCker-filtered read counts were normalized by the median length of the sequences of each protein reference, and the corresponding genome equivalents were calculated as the ratio of NosZ (or another protein of interest) read counts to the read counts of RNA polymerase subunit B (rpoB), a universal single copy marker.
NosZ phylogenetic analysis.
The NosZ reference protein sequences were aligned using CLUSTAL Omega (85) and a maximum likelihood reference tree was created using RAxML v8.0.19 (86) with a general time reversible model option, gamma parameter optimization, and “-f a” for the algorithm. The ROCker identified NosZ-encoding reads were extracted from all data sets, translated into protein sequences using FragGeneScan, and then added to the reference alignment using Mafft (87). The reads were placed in the phylogenetic tree using RAxML EPA algorithm and visualized using iTOL (88).
Intrapopulation diversity assessment based on recovered MAGs.
The taxonomic affiliation of individual contig sequences of a MAG was evaluated based on MyTaxa, a homology-based classification tool (89). The MiGA (Microbial Genomes Atlas, www.microbial-genomes.org) webserver was used for the taxonomic classification of the whole MAG using the average nucleotide identity/amino acid identity (ANI/AAI) concept. To assess intrapopulation diversity and sequence discreteness, each target population MAG was searched against all the reads from each location by BLASTN (only contigs longer than 2 kbp were used). Fragment recruitment plots were constructed based on the BLASTN matches (threshold values: nucleotide identity of ≥75% and alignment length of ≥80 bp) using the Enveomics collection of scripts (55). The evenness of coverage and sequence diversity of the reads across the length of the reference genome sequence were used to evaluate the presence and discreteness of the population in the chosen data set.
Data availability.
All metagenomic data sets were deposited in the European Nucleotide Archive (ENA) under project PRJEB26500. Additional data are available at http://enve-omics.ce.gatech.edu/data/prsoils.
ACKNOWLEDGMENTS
This work was supported by the U.S. Department of Energy, Office of Biological and Environmental Research, Genomic Science Program (award DE-SC0006662) and US National Science Foundation (award 1831582). GG was supported by the Luquillo Critical Zone Observatory (National Science Foundation grant EAR-1331841) and the Luquillo Long-Term Ecological Research Site (National Science Foundation grant DEB-1239764).
All research at the USDA Forest Service International Institute of Tropical Forestry (IITF) is done in collaboration with the University of Puerto Rico. We thank María Rivera and Humberto Robles from IITF for their help in soil sampling.
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Konstantinos T. Konstantinidis, Email: kostas@ce.gatech.edu.
Harold L. Drake, University of Bayreuth
REFERENCES
- 1.Luo C, Rodriguez-R LM, Johnston ER, Wu L, Cheng L, Xue K, Tu Q, Deng Y, He Z, Shi JZ, Yuan MM, Sherry RA, Li D, Luo Y, Schuur EAG, Chain P, Tiedje JM, Zhou J, Konstantinidis KT. 2014. Soil microbial community responses to a decade of warming as revealed by comparative metagenomics. Appl Environ Microbiol 80:1777–1786. 10.1128/AEM.03712-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fierer N, Bradford MA, Jackson RB. 2007. Toward an ecological classification of soil bacteria. Ecology 88:1354–1364. 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
- 3.Van Der Heijden MGA, Bardgett RD, Van Straalen NM. 2008. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11:296–310. 10.1111/j.1461-0248.2007.01139.x. [DOI] [PubMed] [Google Scholar]
- 4.DeAngelis KM, Chivian D, Fortney JL, Arkin AP, Simmons B, Hazen TC, Silver WL. 2013. Changes in microbial dynamics during long-term decomposition in tropical forests. Soil Biol Biochem 66:60–68. 10.1016/j.soilbio.2013.06.010. [DOI] [Google Scholar]
- 5.Malhi Y, Phillips OL. 2004. Tropical forests and global atmospheric change: a synthesis. Philos Trans R Soc Lond B Biol Sci 359:549–555. 10.1098/rstb.2003.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemergut DR, Costello EK, Hamady M, Lozupone C, Jiang L, Schmidt SK, Fierer N, Townsend AR, Cleveland CC, Stanish L, Knight R. 2011. Global patterns in the biogeography of bacterial taxa. Environ Microbiol 13:135–144. 10.1111/j.1462-2920.2010.02315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delgado-Baquerizo M, Oliverio AM, Brewer TE, Benavent-González A, Eldridge DJ, Bardgett RD, Maestre FT, Singh BK, Fierer N. 2018. A global atlas of the dominant bacteria found in soil. Science 359:320–325. 10.1126/science.aap9516. [DOI] [PubMed] [Google Scholar]
- 8.Pajares S, Bohannan BJM. 2016. Ecology of nitrogen fixing, nitrifying, and denitrifying microorganisms in tropical forest soils. Front Microbiol 7:1045. 10.3389/fmicb.2016.01045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chambers JQ, Silver WL. 2004. Some aspects of ecophysiological and biogeochemical responses of tropical forests to atmospheric change. Philos Trans R Soc Lond B Biol Sci 359:463–476. 10.1098/rstb.2003.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Templer PH, Silver WL, Pett-Ridge J, M DeAngelis K, Firestone MK. 2008. Plant and microbial controls on nitrogen retention and loss in a humid tropical forest. Ecology 89:3030–3040. 10.1890/07-1631.1. [DOI] [PubMed] [Google Scholar]
- 11.Cusack DF, Silver WL, Torn MS, Burton SD, Firestone MK. 2011. Changes in microbial community characteristics and soil organic matter with nitrogen additions in two tropical forests. Ecology 92:621–632. 10.1890/10-0459.1. [DOI] [PubMed] [Google Scholar]
- 12.Firestone MK, Davidson EA. 1989. Microbiological basis of NO and N2O production and consumption in soil, p 11–27. In Andreae MO, Schimel DOA (ed), Exchange of trace gases between terrestrial ecosystems and the atmosphere. John Wiley & Sons, New York, NY. [Google Scholar]
- 13.Frasier R, Ullah S, Moore TR. 2010. Nitrous oxide consumption potentials of well-drained forest soils in southern Québec, Canada. Geomicrobiol J 27:53–60. 10.1080/01490450903232199. [DOI] [Google Scholar]
- 14.Schmidt J, Seiler W, Conrad R. 1988. Emission of nitrous oxide from temperate forest soils into the atmosphere. J Atmos Chem 6:95–115. 10.1007/BF00048334. [DOI] [Google Scholar]
- 15.Díaz-Pinés E, Werner C, Butterbach-Bahl K. 2018. Effects of climate change on CH4 and N2O fluxes from temperate and boreal forest soils, p 11–27. In Perera AH, Peterson U, Pastur GM, Iverson LR (ed), Ecosystem services from forest landscapes: broadscale considerations. Springer International Publishing, Cham, Switzerland. [Google Scholar]
- 16.Butterbach-Bahl K, Baggs EM, Dannenmann M, Kiese R, Zechmeister-Boltenstern S. 2013. Nitrous oxide emissions from soils: how well do we understand the processes and their controls? Philos Trans R Soc Lond B Biol Sci 368:20130122. 10.1098/rstb.2013.0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houlton BZ, Wang Y-P, Vitousek PM, Field CB. 2008. A unifying framework for dinitrogen fixation in the terrestrial biosphere. Nature 454:327–330. 10.1038/nature07028. [DOI] [PubMed] [Google Scholar]
- 18.Werner C, Butterbach‐Bahl K, Haas E, Hickler T, Kiese R. 2007. A global inventory of N2O emissions from tropical rainforest soils using a detailed biogeochemical model. Global Biogeochem Cycles 21. 10.1029/2006GB002909. [DOI] [Google Scholar]
- 19.Giles M, Morley N, Baggs EM, Daniell TJ. 2012. Soil nitrate reducing processes—drivers, mechanisms for spatial variation, and significance for nitrous oxide production. Front Microbiol 3:407. 10.3389/fmicb.2012.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onley JR, Ahsan S, Sanford RA, Löffler FE. 2018. Denitrification by Anaeromyxobacter dehalogenans, a common soil bacterium lacking the nitrite reductase genes nirS and nirK. Appl Environ Microbiol 84:e01985-17. 10.1128/AEM.01985-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braker G, Tiedje JM. 2003. Nitric oxide reductase (norB) genes from pure cultures and environmental samples. Appl Environ Microbiol 69:3476–3483. 10.1128/aem.69.6.3476-3483.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richardson D, Felgate H, Watmough N, Thomson A, Baggs E. 2009. Mitigating release of the potent greenhouse gas N2O from the nitrogen cycle—could enzymic regulation hold the key? Trends Biotechnol 27:388–397. 10.1016/j.tibtech.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Spiro S. 2012. Nitrous oxide production and consumption: regulation of gene expression by gas-sensitive transcription factors. Philos Trans R Soc Lond B Biol Sci 367:1213–1225. 10.1098/rstb.2011.0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Black A, Hsu PCL, Hamonts KE, Clough TJ, Condron LM. 2016. Influence of copper on expression of nirS, norB and nosZ and the transcription and activity of NIR, NOR and N2OR in the denitrifying soil bacteria Pseudomonas stutzeri. Microb Biotechnol 9:381–388. 10.1111/1751-7915.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garbeva P, Baggs EM, Prosser JI. 2007. Phylogeny of nitrite reductase (nirK) and nitric oxide reductase (norB) genes from Nitrosospira species isolated from soil. FEMS Microbiol Lett 266:83–89. 10.1111/j.1574-6968.2006.00517.x. [DOI] [PubMed] [Google Scholar]
- 26.Zumft WG. 2005. Nitric oxide reductases of prokaryotes with emphasis on the respiratory, heme-copper oxidase type. J Inorg Biochem 99:194–215. 10.1016/j.jinorgbio.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 27.Higgins SA, Schadt CW, Matheny PB, Löffler FE. 2018. Phylogenomics reveal the dynamic evolution of fungal nitric oxide reductases and their relationship to secondary metabolism. Genome Biol Evol 10:2474–2489. 10.1093/gbe/evy187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braker G, Conrad R. 2011. Diversity, structure, and size of N2O-producing microbial communities in soils—what matters for their functioning? Adv Appl Microbiol 75:33–70. 10.1016/B978-0-12-387046-9.00002-5. [DOI] [PubMed] [Google Scholar]
- 29.Hallin S, Philippot L, Löffler FE, Sanford RA, Jones CM. 2018. Genomics and ecology of novel N2O-reducing microorganisms. Trends Microbiol 26:43–55. 10.1016/j.tim.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Cai Z, Cheng Y, Zhu T. 2009. Denitrification and total nitrogen gas production from forest soils of eastern China. Soil Biol Biochem 41:2551–2557. 10.1016/j.soilbio.2009.09.016. [DOI] [Google Scholar]
- 31.Orr CH, James A, Leifert C, Cooper JM, Cummings SP. 2011. Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl Environ Microbiol 77:911–919. 10.1128/AEM.01250-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Townsend AR, Cleveland CC, Houlton BZ, Alden CB, White JWC. 2011. Multi‐element regulation of the tropical forest carbon cycle. Front Ecol Environ 9:9–17. 10.1890/100047. [DOI] [Google Scholar]
- 33.Brown AS, Lugo AE, Silander S, Liegel L. 1983. Research history and opportunities in the Luquillo Experimental Forest. General Technical Report SO-44. US Forest Service, Southern Forest Experiment Station, New Orleans, LA. [Google Scholar]
- 34.Gould WA, Gonzalez G, Rivera GC. 2006. Structure and composition ofvegetation along an elevational gradient in Puerto Rico. J Veg Sci 17:653–664. 10.1111/j.1654-1103.2006.tb02489.x. [DOI] [Google Scholar]
- 35.Aide TM, Zimmerman JK, Herrera L, Rosario M, Serrano M. 1995. Forest recovery in abandoned tropical pastures in Puerto Rico. For Ecol Manage 77:77–86. 10.1016/0378-1127(95)03576-V. [DOI] [Google Scholar]
- 36.Weaver PLG, W A. 2013. Forest vegetation along environmental gradients in northeastern Puerto Rico, p 43–66. In González G, Willig MR, Waide RB (ed), Ecological gradient analyses in a tropical landscape. Ecological Bulletins 54. Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 37.Ping C-LM, Michaelson GJ, Stiles CA, Gonzalez G. 2013. Soil characteristics, carbon stores, and nutrient distribution in eight forest types along an elevational gradient, eastern Puerto Rico, p 67–86. In González G, Willig MR, Waide RB (ed), Ecological gradient analyses in a tropical landscape. Ecological Bulletins 54. Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 38.González G, R Willig M, Waide R. 2013. Ecological gradient analyses in a tropical landscape: multiple perspectives and emerging themes, p 13–20. In González G, Willig MR, Waide RB (ed), Ecological gradient analyses in a tropical landscape. Ecological Bulletins 54. Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 39.González G, Lodge DJ. 2017. Soil biology research across latitude, elevation and disturbance gradients: a review of forest studies from Puerto Rico during the past 25 years. Forests 8:178. 10.3390/f8060178. [DOI] [Google Scholar]
- 40.Van Beusekom AE, González G, Rivera MM. 2015. Short-term precipitation and temperature trends along an elevation gradient in northeastern Puerto Rico. Earth Interact 19:1–33. 10.1175/EI-D-14-0023.1. [DOI] [Google Scholar]
- 41.Liptzin D, Silver WL. 2015. Spatial patterns in oxygen and redox sensitive biogeochemistry in tropical forest soils. Ecosphere 6:1–14. 10.1890/ES14-00309.1. [DOI] [Google Scholar]
- 42.Hall SJ, Liptzin D, Buss HL, DeAngelis K, Silver WL. 2016. Drivers and patterns of iron redox cycling from surface to bedrock in a deep tropical forest soil: a new conceptual model. Biogeochemistry 130:177–190. 10.1007/s10533-016-0251-3. [DOI] [Google Scholar]
- 43.Waldrop MP, Balser TC, Firestone MK. 2000. Linking microbial community composition to function in a tropical soil. Soil Biol Biochem 32:1837–1846. 10.1016/S0038-0717(00)00157-7. [DOI] [Google Scholar]
- 44.Cantrell SA, Lodge DJ, Cruz CA, Garcia LM, Perez-Jimenez JR, Molina M. 2013. Differential abundance of microbial functional groups along the elevation gradient from the coast to the Luquillo Mountains, p 87–100. In González G, Willig MR, Waide RB (ed), Ecological gradient analyses in a tropical landscape. Ecological Bulletins 54. Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 45.Caro-Quintero A, Konstantinidis KT. 2012. Bacterial species may exist, metagenomics reveal. Environ Microbiol 14:347–355. 10.1111/j.1462-2920.2011.02668.x. [DOI] [PubMed] [Google Scholar]
- 46.Orellana LH, Rodriguez-R LM, Higgins S, Chee-Sanford JC, Sanford RA, Ritalahti KM, Löffler FE, Konstantinidis KT. 2014. Detecting nitrous oxide reductase (nosZ) genes in soil metagenomes: method development and implications for the nitrogen cycle. mBio 5:e01193-14. 10.1128/mBio.01193-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shan J, Sanford RA, Chee-Sanford J, Ooi SK, Löffler FE, Konstantinidis KT, Yang WH. 2021. Beyond denitrification: the role of microbial diversity in controlling nitrous oxide reduction and soil nitrous oxide emissions. Glob Chang Biol 10.1111/gcb.15545. [DOI] [PubMed] [Google Scholar]
- 48.Jousset A, Bienhold C, Chatzinotas A, Gallien L, Gobet A, Kurm V, Küsel K, Rillig MC, Rivett DW, Salles JF, van der Heijden MGA, Youssef NH, Zhang X, Wei Z, Hol WHG. 2017. Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J 11:853–862. 10.1038/ismej.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orellana LH, Chee-Sanford JC, Sanford RA, Löffler FE, Konstantinidis KT. 2017. Year-round shotgun metagenomes reveal stable microbial communities in agricultural soils and novel ammonia oxidizers responding to fertilization. Appl Environ Microbiol 84:e01646–17. 10.1128/AEM.01646-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnston ER, Rodriguez-R LM, Luo C, Yuan MM, Wu L, He Z, Schuur EAG, Luo Y, Tiedje JM, Zhou J, Konstantinidis KT. 2016. Metagenomics reveals pervasive bacterial populations and reduced community diversity across the Alaska tundra ecosystem. Front Microbiol 7:579. 10.3389/fmicb.2016.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnston MH. 1992. Soil-vegetation relationships in a Tabonuco forest community in the Luquillo mountains of Puerto Rico. J Trop Ecol 8:253–263. 10.1017/S0266467400006477. [DOI] [Google Scholar]
- 52.Weaver PL. 1995. The Colorado and dwarf forests of Puerto Rico’s Luquillo mountains, p 109–141. In Lugo AE, Lowe C (ed), Tropical forests: management and ecology. Springer New York, New York, NY. [Google Scholar]
- 53.Rodriguez-R LM, Konstantinidis KT. 2014. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 30:629–635. 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- 54.Zhang X, Johnston ER, Li L, Konstantinidis KT, Han X. 2017. Experimental warming reveals positive feedbacks to climate change in the Eurasian Steppe. ISME J 11:885–895. 10.1038/ismej.2016.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez-R LM, Konstantinidis KT. 2016. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr 4:e1900v1. 10.7287/peerj.preprints.1900v1. [DOI] [Google Scholar]
- 56.Rodriguez RL, Gunturu S, Harvey WT, Rossello-Mora R, Tiedje JM, Cole JR, Konstantinidis KT. 2018. The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res 46:W282–W288. 10.1093/nar/gky467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bruijnzeel LA, Proctor J. Hydrology and biogeochemistry of tropical montane cloud forests: what do we really know?, p 38–78. In Hamilton LS, Juvik JO, Scatena FN (ed), Tropical montane cloud forests. Springer, New York, NY [Google Scholar]
- 58.Song L, Lu H-Z, Xu X-L, Li S, Shi X-M, Chen X, Wu Y, Huang J-B, Chen Q, Liu S, Wu C-S, Liu W-Y. 2016. Organic nitrogen uptake is a significant contributor to nitrogen economy of subtropical epiphytic bryophytes. Sci Rep 6:30408. 10.1038/srep30408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cusack DF, Silver W, McDowell WH. 2009. Biological nitrogen fixation in two tropical forests: ecosystem-level patterns and effects of nitrogen fertilization. Ecosystems 12:1299–1315. 10.1007/s10021-009-9290-0. [DOI] [Google Scholar]
- 60.Benner J, Vitousek PM, Ostertag R. 2011. Nutrient cycling and nutrient limitation in tropical montane cloud forests, p 90–100. In Scatena FN, Bruijnzeel LA, Hamilton LS (ed), Tropical montane cloud forests: science for conservation and management. Cambridge University Press, Cambridge. [Google Scholar]
- 61.Hedin LO, Brookshire ENJ, Menge DNL, Barron AR. 2009. The nitrogen paradox in tropical forest ecosystems. Annu Rev Ecol Evol Syst 40:613–635. 10.1146/annurev.ecolsys.37.091305.110246. [DOI] [Google Scholar]
- 62.Graf DRH, Jones CM, Hallin S. 2014. Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O emissions. PLoS One 9:e114118. 10.1371/journal.pone.0114118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanford RA, Wagner DD, Wu Q, Chee-Sanford JC, Thomas SH, Cruz-García C, Rodríguez G, Massol-Deyá A, Krishnani KK, Ritalahti KM, Nissen S, Konstantinidis KT, Löffler FE. 2012. Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc Natl Acad Sci US A 109:19709–19714. 10.1073/pnas.1211238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodríguez-Minguela CM, Apajalahti JHA, Chai B, Cole JR, Tiedje JM. 2009. Worldwide prevalence of class 2 integrases outside the clinical setting is associated with human impact. Appl Environ Microbiol 75:5100–5110. 10.1128/AEM.00133-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Griffiths RI, Whiteley AS, O'Donnell AG, Bailey MJ. 2000. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol 66:5488–5491. 10.1128/aem.66.12.5488-5491.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsai YL, Olson BH. 1991. Rapid method for direct extraction of DNA from soil and sediments. Appl Environ Microbiol 57:1070–1074. 10.1128/AEM.57.4.1070-1074.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng Y, Leung HC, Yiu SM, Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 69.Zhu W, Lomsadze A, Borodovsky M. 2010. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 38:e132. 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421–421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu Y-W, Tang Y-H, Tringe SG, Simmons BA, Singer SW. 2014. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2:26. 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Konstantinidis KT, DeLong EF. 2008. Genomic patterns of recombination, clonal divergence and environment in marine microbial populations. ISME J 2:1052–1065. 10.1038/ismej.2008.62. [DOI] [PubMed] [Google Scholar]
- 74.Bairoch A, Apweiler R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28:45–48. 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res 42:D206–D214. 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rho M, Tang H, Ye Y. 2010. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res 38:e191. 10.1093/nar/gkq747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. 2016. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17:132. 10.1186/s13059-016-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oksanen J. 2011. Multivariate analysis of ecological communities in R: vegan tutorial v.2.02. https://cran.r-project.org/web/packages/vegan/vegan.pdf
- 79.Su X, Pan W, Song B, Xu J, Ning K. 2014. Parallel-META 2.0: enhanced metagenomic data analysis with functional annotation, high performance computing and advanced visualization. PLoS One 9:e89323. 10.1371/journal.pone.0089323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 81.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuczynski J, Stombaugh J, Walters WA, González A, Caporaso JG, Knight R. 2011. Using QIIME to analyze 16S rRNA gene sequences from microbialcommunities. Curr Protoc Bioinformatics 10.1002/0471250953.bi1007s36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Orellana LH, Rodriguez-R LM, Konstantinidis KT. 2017. ROCker: accurate detection and quantification of target genes in short-read metagenomic data sets by modeling sliding-window bitscores. Nucleic Acids Res 45:e14. 10.1093/nar/gkw900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Katoh K, Misawa K, Kuma K-I, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luo C, Rodriguez-R LM, Konstantinidis KT. 2014. MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res 42:e73. 10.1093/nar/gku169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aem.00546-21-s0001.pdf, PDF file, 1.1 MB (1.8MB, pdf)
Data Availability Statement
All metagenomic data sets were deposited in the European Nucleotide Archive (ENA) under project PRJEB26500. Additional data are available at http://enve-omics.ce.gatech.edu/data/prsoils.