

Abstract
Primary blast injury (caused by the initial rapid increase in pressure following an explosive blast) to the retina and optic nerve (ON) causes progressive visual loss and neurodegeneration. Military personnel are exposed to multiple low-overpressure blast waves, which may be in quick succession, such as during breacher training or in combat. We investigated the necroptotic cell death pathway in the retina in a mouse repeated primary ocular blast injury (rPBI) model using immunohistochemistry. We further evaluated whether intravitreal injections of a potent necroptosis inhibitor, Necrostatin-1s (Nec-1s), protects the retina and ON axons by retinal ganglion cells (RGC) counts, ON axonal counting and optical coherence tomography (OCT) analysis of vitreous haze. Receptor interacting protein kinase (RIPK) 3, increased in the inner plexiform layer 2 days post injury (dpi) and persisted until 14 dpi, whilst RIPK1 protein expression did not change after injury. The number of degenerating ON axons was increased at 28 dpi but there was no evidence of a reduction in the number of intact ON axons or RNA-binding protein with multiple splicing (RBPMS)+ RGC in the retina by 28 dpi in animals not receiving any intravitreal injections. But, when intravitreal injections (vehicle or Nec-1s) were given there was a significant reduction in RBPMS+ RGC numbers, suggesting that rPBI with intraocular injections is damaging to RGC. There were fewer RGC lost after Nec-1s than vehicle injection, but there was no effect of Nec-1s or vehicle treatment on the number of degenerating axons. OCT analysis demonstrated no effect of rPBI on vitreous haze, but intravitreal injection combined with rPBI increased vitreous haze (P = 0.004). Whilst necroptosis may be an active cell death signalling pathway after rPBI, its inhibition did not prevent cell death, and intravitreal injections in combination with rPBI increased vitreous inflammation and reduced RBPMS+ RGC numbers, implying intravitreal injection is not an ideal method for drug delivery after rPBI.
Keywords: Retina, Retinal ganglion cells, Optic nerve, Primary blast injury, Traumatic optic neuropathy, Necroptosis, Cell death
1. Introduction
Primary blast injuries (PBI) are caused by the initial rapid overpressure wave during an explosive blast (Scott, 2011; Scott et al., 2015) and are common in the military, with ocular PBI occuring in ~9% of US war related eye injuries (Blanch et al., 2011; Cockerham et al., 2011; Phillips et al., 2013; Rex, 2014; Ritenour and Baskin, 2008; Vlasov et al., 2015; Warden, 2006), but, this may be an underestimation, as initial physical signs may be lacking and visual loss may be delayed and progressive. Repeated primary ocular blast injury (rPBI), due to repeated blast wave exposure can occur in breacher training, where soldiers can be exposed to 12 sub-threshold blasts per day and during combat operations with an average of 13 blasts during deployment (Peskind et al., 2011). During terrorist attacks, civilians may be exposed to blast injuries which are more common in war zones (Champion et al., 2009). The prevalence of retinal injury after ocular trauma is greater in military compared to civilian injuries (60% vs. 2%) (Blanch et al., 2011; Jones et al., 1986; Weichel et al., 2008).
In vivo models of ocular blast injury (DeMar et al., 2016; DeWalt and Eldred, 2017) vary in intensity and mechanism including the method of blast-wave induction, blast-wave localisation (head, eye, body), the quantity and sequence of blast waves (single or repeated) and the pressure of the blast-wave. Previous studies using our PBI model, administered a single 26 pounds per square inch (psi) blast-wave directly to the mouse eye and induced optic nerve (ON) axonal degeneration and collapsed myelin at 28 days post injury (dpi) (Bricker-Anthony et al., 2016). We were then able to refine this model, while still replicating aspects of traumatic brain inury to show synergistic ON axonal damage with 15% axonal degeneration after repeated blasts of 15 psi at 0.5 s inter-blast intervals, and that there was additive cumulative vision loss with 3% axonal degeneration after 15 psi blasts once per day for 6 consecutive days, but no photoreceptor damage (Bernardo-Colon et al., 2018; Vest et al., 2019). These mice showed increased vision loss and ON damage compared to exposure to a single 26 psi blast exposure (Vest et al., 2019). When the blast overpressure wave is repeated there is a reduction in DAPI+ cells in the ganglion cell layer (GCL) at 2 dpi which persists until 28 dpi (Bernardo-Colon et al., 2019). Others demonstrate progressive and delayed retinal degeneration between 4 and 10 months after a single 20 psi blast wave exposure (Dutca et al., 2014; Mohan et al., 2013).
Necroptosis is a form of regulated necrosis resulting in cell leakage, organelle swelling, cytoplasmic granulation and initiation of an inflammatory response (Gizycka and Chorostowska-Wynimko, 2015; Vandenabeele et al., 2010). In tumour necrosis factor (TNF)-induced necroptosis, Complex I is formed at the TNF receptor dependent on polyubiquitinated receptor interacting protein kinase (RIPK) 1 and NF-κβ pathway activation (Vanden Berghe et al., 2014). A reduction in pro-survival signals causes Complex I dissociation and Complex IIa formation, and downstream apoptosis. However, when caspase-8 is inhibited through endogenous or pharmacological inhibitors, then RIPK1 and RIPK3 associate with mixed lineage kinase domain-like (MLKL), forming complex IIb. RIPK3 phosphorylates MLKL at Ser345 (Rodriguez et al., 2016) inducing MLKL translocation to the plasma membrane, triggering membrane permeabilisation and necroptotic death (Weinlich et al., 2017).
Necroptosis can be blocked by Necrostatin-1 (Nec-1), an allosteric inhibitor of RIPK1, which also inhibits immunomodulator indoleamine-2,3-dioxygenase (IDO), which is involved with neuroinflammation not directly related to RIPK1 activation (Takahashi et al., 2012). An alternative to Nec-1 is a stable modified form, Nec-1 stable (Nec-1s), which has a higher affinity and specificity for RIPK1 (> 1000 fold more selective for RIPK1 than other human kinases), and does not inhibit IDO (Linkermann and Green, 2014; Takahashi et al., 2012).
Necroptosis drives neuronal cell death in models of trauma, neurodegeneration and ocular disease (Cougnoux et al., 2016; Ito et al., 2016; Politi and Przedborski, 2016; Shen et al., 2017; Wang et al., 2014; You et al., 2008; Zhang et al., 2017), including in our rat model of secondary blast injury (Thomas et al., 2019). Necroptosis has also been implicated in primary blast injury, with RIPK1 and RIPK3 protein levels elevated throughout the retina at 3 dpi after a single 26-psi blast wave (Bricker-Anthony et al., 2014a). Other necroptotic markers such as MLKL and the effect of a necroptotic inhibitor have not yet been investigated. In this study, we aimed to define the contribution of necroptotic pathways to retinal ganglion cell (RGC) death in a blast injury mouse model. There are currently no effective treatments to prevent RGC loss after ocular PBI, highlighting the pressing need for an effective therapy.
2. Methods
2.1. Experimental design
This study investigates whether necroptosis promotes RGC death and ON axonal pathology in a mouse model of rPBI (Bernardo-Colon et al., 2018, 2019; Vest et al., 2019). To assess the time course of necroptosis induction, retinal tissue was collected at 2, 14 and 28 dpi and immunohistochemical staining performed for necroptotic markers RIPK1 and RIPK3 (Supplementary Figs. 1A–B). Necroptosis was inhibited by bilateral intravitreal injections of Nec-1s (2 μl of 3.6 mM reconstituted in 10% dimethyl sulphoxide (DMSO), 0.9% methyl-β-cyclodextrin in 0.1M phosphate buffered saline (PBS) as previously reported (Thomas et al., 2019), at 5 h after the first blast and followed by two further blast waves and repeated every 7 dpi until experimental endpoint at 28 dpi (Supplementary Fig. 1 C–E). OCT retinal scans were performed at baseline (1 day before rPBI) and endpoint (27 dpi). Mice were culled at 28 dpi and the eyes were processed for immunohistochemistry (IHC) and the number of RNA binding protein with multiple splicing (RBPMS)+ RGC quantified in the central, middle and peripheral portion of the retina. The far proximal (closest to the eye) section of the ON was processed for resin semi-thin cross-sections and intact and degenerating ON axons quantified, whilst the remaining ON was processed as longitudinal cryosections for IHC analysis.
2.2. Animal care and procedures
Male 12-week old C57/Bl6 mice purchased from Jackson Laboratory (Bar Harbor, Maine, USA) were used in this study. Animal procedures were performed under regulation of the Institutional Animal Care and Use Committee of Vanderbilt University according to the Association for Assessment and Accreditation of Laboratory Animal Care guidelines and conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Animals were randomly assigned to experimental groups with the experimenter masked to the treatment and procedural conditions. All procedures and investigations were performed between 07:00 and 12:00.
2.3. Repeated primary blast injury mouse model
Anaesthetised (3% isoflurane in O2) male 12-week old C57/Bl6 mice were exposed to a blast overpressure wave produced by a device modified to produce an air blast wave, as previously described (Bernardo-Colon et al., 2018, 2019; Hines-Beard et al., 2012; Vest et al., 2019). Repeated blast injury was chosen to more closely approximate clinical blast injury, using a less severe, and so more refined, insult than single blast exposure requires, whilst increasing RGC axonal degeneration compared to single blast exposure. The left eye of mice was exposed to a repeated blast injury consisting of 2 blasts of 16 psi (peak positive pressure ± SD, pre-blast 16.99 ± 0.75 psi/117.14 ± 5.17 kPa and post-blast 16.55 ± 0.69 psi/114.11 ± 35.65 kPa, Supplementary Fig. 2Ai) in quick succession (1 s interblast intervals) repeated 3 times with 24 h between each blast. The time to peak was 4 ms and remained at peak pressure for 2 ms (Supplementary Fig. 2Aii). The mouse eye was positioned 162 mm from the end of the device. Separate mice were exposed to all procedures excluding the blast wave which was blocked and recorded with a pressure of < 2 psi (peak positive pressure ± SD, pre-blast 1.92 ± 0.15 psi/ 13.24 ± 1.03 kPa and post-blast 1.79 ± 0.10 psi/12.34 ± 0.69 kPa, Supplementary Fig. 2A). This sham condition is referred to as blast blocked. A pressure transducer recorded the blast overpressure wave and was viewed using LabVIEW software (National Instruments ™. Austin, TX, USA). GenTeal® Tears (Alcon, Novartis, Fortworth, Texas, USA) eye drops were applied after the blast to prevent corneal dehydration from anaesthetic exposure and the mice were allowed to recover.
2.4. Intravitreal injection of Nec-1s
Intravitreal injections were performed using a 31-gauge needle with bevelled tip attached to a 10 μl Gastight Syringe (Hamilton, Reno, NV, USA) under inhalational anaesthetic of 2–3% isoflurane at a 45° angle 1 mm peripheral to the limbus and lens avoided. Unilateral rPBI was performed and 2 μl of Nec-1s administered by bilateral intravitreal injection or 2 μl of vehicle (10% DMSO, 0.09% methyl-β-cyclodextrin in PBS) bilaterally injected into the vehicle control group. Injections were performed 5 h after injury and every 7 days until euthanasia and tissue collection at 28 dpi (n = 10 animals per group). Animals were perfused under terminal anaesthesia and tissues processed for immunohistochemistry (IHC).
2.5. OCT imaging, retinal thickness and vitreal haze measurements
Optical coherence tomography (OCT) imaging was performed at 28 dpi to construct a high resolution cross-sectional retinal image using a Bioptigen ultra-high-resolution SD-OCT system with a mouse retinal bore (Bioptigen, North Carolina, USA). Pupils were dilated using 1% tropicamide and GenTeal ™ lubricant gel was used to maintain corneal clarity. All images were acquired with the same level of A-scan averaging (100 averages per A scan) and with the retinal position central to the image. A total of 2 B-scans were analysed per eye either side of the ON head. Whole retinal thickness and RGC complex thickness (ganglion cell layer; GCL and inner plexiform layer; IPL) were measured in OCT images in line with the optic nerve head (ONH). Image J was used to manually segment the layers and measure the area, which was divided by the length of the retinal segment measured to calculate the layer thickness. The number of eyes in which hyper reflective dots were observed in the vitreous is displayed as a percentage of total number of eyes that were imaged in each experimental group. Further analysis to quantify vitreous inflammation was performed using Image J (http://rsbweb.nih.gov/ij), based on the method previously described (Keane et al., 2014) (Supplementary Fig. 2B): two images either side of the ONH were analysed per eye and the pixel intensity in five regions of interest in the vitreous were measured and then displayed as a percentage of the average of retinal pigment epithelium (RPE) intensity.
2.6. Tissue preparation for IHC
At 2, 14 and 28 dpi (n = 4 per group), mice were euthanised by overdose of anaesthetic and intracardially perfused with 4% paraformaldehyde (PFA) in PBS. The anterior segment was removed from eyes and retinal cups cryoprotected in ascending concentrations of sucrose (10%, 20%, 30%) in PBS at 4 °C before embedding in optimal cutting temperature compound (OCTc) and storing at −80 °C. Sections were cut at a thickness of 15 μm using a cryostat (Brights Instruments, Huntingdon, UK) and adhered onto SuperFrost ™ (Fisher Scientific, Loughborough, UK) coated glass microscope slides and stored at −20 °C until required.
2.7. IHC protocol
Frozen sections were left to thaw for 20 min and washed 3 × 5 min in PBS, followed by 20 min permeabilisation and non-specific binding site blocking in 1% Triton-X-100 (Sigma, Poole, UK) and 3% bovine serum albumin (BSA; Sigma). Tissue sections were incubated overnight at 4 °C with primary antibody (Table 1) in 0.5% Tween-20 and 3% BSA before washing 3 × 5 min in PBS and incubating with secondary antibodies (Table 1) at room temperature (RT). Tissue sections were washed 3 × 5 min in PBS then mounted in Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain (Vector Laboratories, Peterborough, UK). Controls with omitted primary antibody were included in each run and these were used to set the background threshold levels for image capture. Sections were viewed under an epi-fluorescent microscope equipped with an AxioCam HRc, controlled using Axiovision Software (All from Zeiss, Hertfordshire, UK).
Table 1.
Immunohistochemical antibodies.
| Antibody | Detects | Host | Dilution | Source | Catalogue number |
|---|---|---|---|---|---|
| ED1 | Macrophages | Rabbit | 1:200 | Abcam | AB125212 |
| GFAP | Activated Müller glia, type-2 astrocytes | Mouse | 1:200 | Sigma | G3893 |
| MLKL (clone 3H1) | Necroptotic protein | Rat | 1:500 | Millipore | MABC604 |
| MLKL-P | Phosphorylated MLKL at Ser 345 | Rabbit | 1:500 | Cell Signaling | 62233S |
| RIPK1 | Necroptotic protein | Mouse | 1:200 | BD Pharmigen | 551041 |
| RIPK3 | Necroptotic protein | Rabbit | 1:500 | Abcam | AB56164 |
| RBPMS | RGC marker | Rabbit | 1:400 | Millipore | ABN1362 |
| Alexa Fluor 488 | Mouse IgG | Donkey | 1:400 | Invitrogen, Molecular Probes | A21202 |
| Alexa Fluor 594 | Rabbit IgG | Donkey | 1:400 | Invitrogen, Molecular Probes | A21207 |
| Alexa Fluor 488 | Rat IgG | Goat | 1:1000 | Invitrogen, Molecular Probes | A11006 |
2.8. Assessment of RGC survival
The number of RBPMS+ (a specific RGC marker) (Kwong et al., 2011; Rodriguez et al., 2014) RGC were counted in the GCL across a retinal section taken in line with the ONH and quantified as RBPMS+ RGC per mm of retina. RBPMS+ RGC were counted on × 20 magnification images, the distance from the ONH to the edge of the retina was measured and was split into 3 regions; central (closest to the ON), middle or peripheral areas (Supplementary Fig. 2C). A total of 4 images were analysed from 2 cryosections per animal.
2.9. Resin-embedded optic nerves and PPD staining
ON tissue (n = 10 per group) was collected and fixed in 4% EM-grade PFA (Electron Microscopy Sciences, Hatfield, PA, USA) in PBS then 2% glutaraldehyde in 0.1M cacaodylate buffer for 2 h on ice, then post fixed in 2% osmium tetroxide in cacodylate buffer and dehydrated in ascending concentrations of 200-proof ethanol (50%, 70%, 95%, 100%) and further dehydrated in propylene oxide. ON tissue was embedded in Epon epoxy resin (Electron Microscopy Sciences) and 1 μm thick semi-thin sections cut using Leica Microtome and stained with 1% p-phenylenediamine (PPD) (Sigma-Aldrich Corp., St. Louis, MO, USA) and 1% toluidine blue (Fisher, Watham, Massachusetts, USA). Slides were imaged using 100x oil emersion objective on AxioPlan 2 microscope (Carl Zeiss) and manually compiled using the photomerge function in Adobe Photoshop (Adobe Systems Inc., San Jose, CA, USA).
2.10. Counting intact and degenerative ON axonal profiles
The total ON area (μm2) was measured using Image J and representative counting grids, representing 1/5th of the total ON area, were superimposed onto the image using the Counting Grid Image J plugin. The number of ON axons with intact and degenerative profiles were quantified and displayed as the total number of axons in the whole ON or as axon density per mm2.
2.11. Statistics
Statistical analysis was carried out using SPSS 21 (IBM Corp, Armonk, NY, USA) and GraphPad Prism version 7.00. (GraphPad Software, La Jolla, CA, USA). The data were tested for normality using the Shapiro-Wilk test. Normally distributed data without linkage were analysed using one-way ANOVA and post-hoc Tukey tests with P values corrected for multiple comparisons (RBPMS+ RGC quantification, ON axon counts, ED1+ cells in ON, retinal thickness in OCT). Data including measurements from two eyes of each animal (vitreal intensity in OCT) were modelled using generalised estimating equations (GEE; normal distribution with identity link function and independent correlation matrix). To construct a model to fit the data, all available factors were included in the initial model with 2-way interaction terms and then terms with P > 0.05 were serially removed from the model, starting with the least significant and interaction terms. In experiments where multiple comparisons were made, the P values were corrected with Bonferroni correction. Data is reported as mean ± standard error of the mean (SEM). Sample size was based on previous studies demonstrating that 10 animals per group detected treatment effects on axon counts (Bernardo-Colon et al., 2018). No animals were excluded / euthanised due to reaching humane end points before study completion.
3. Results
3.1. Increased RIPK3 immunoreactivity after rPBI but there were no changes in RIPK1
To investigate evidence for necroptosis after rPBI, we first explored immunohistological localisation of RIPK1 and RIPK3 in retinal sections at different time points after injury. RIPK1 staining did not show specific changes in immunostaining after rPBI (Fig. 1A, Supplementary Fig. 3). RIPK3+ immunoreactivity increased in the GCL, OPL and IPL at 2, 14 and 28 dpi (Fig. 1A). Meanwhile, no detectable changes were observed in MLKL-P (Ser 345) immunofluorescence (not shown).
Figure 1.
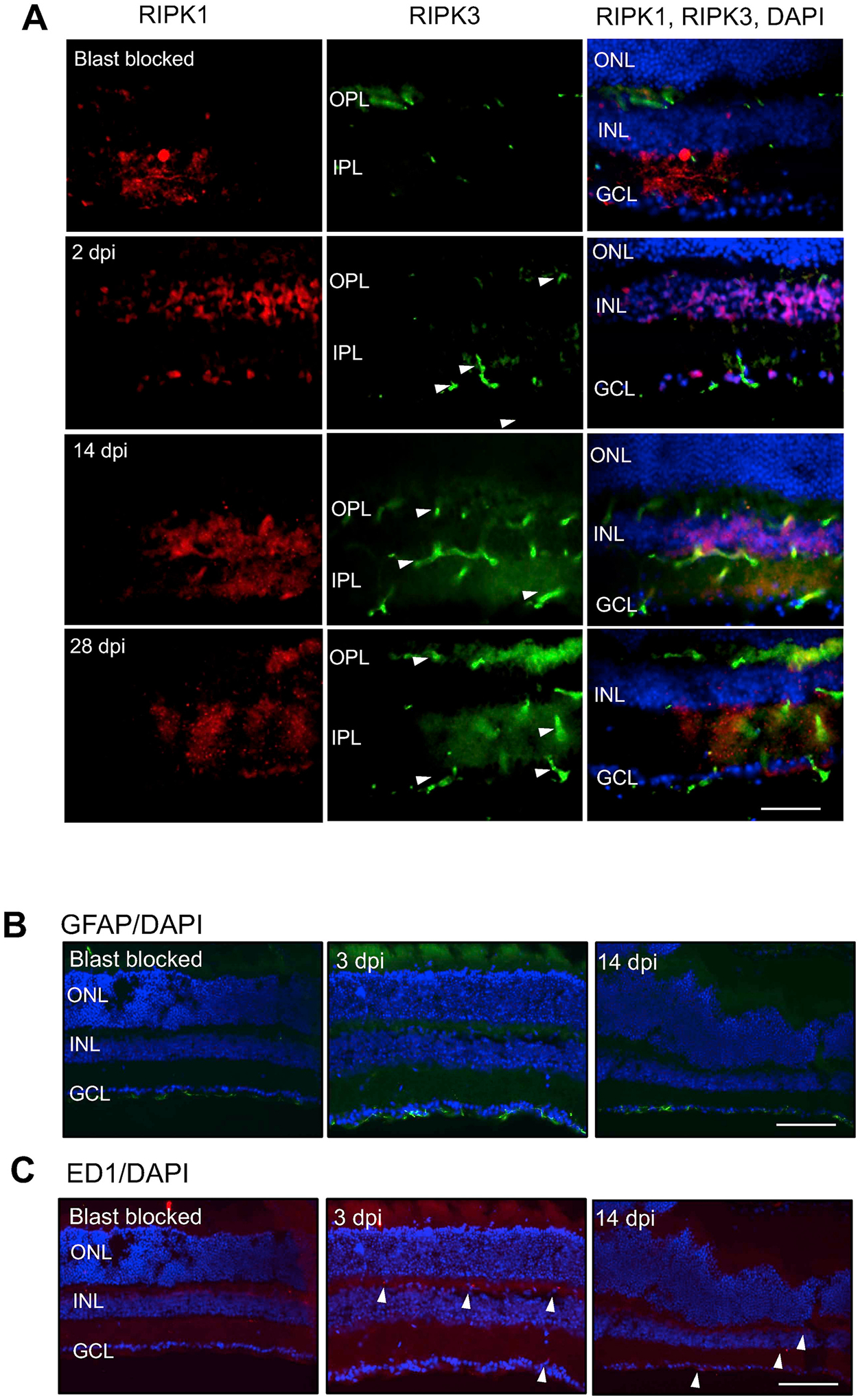
A) RIPK1 is localised to cells of the GCL and INL and its expression remains constant after blast-wave exposure. RIPK3 is present in the OPL in blast blocked eyes, but at 2, 14 and 28 dpi it is localised to the GCL, OPL and IPL. B) Müller glial GFAP+ processes at 3 and 14 dpi show no changes between blast blocked or experimental eyes (n = 4). C) Retinal ED1 immunostaining at 3 and 14 dpi demonstrating a few ED1+ cells in the OPL of rPBI injured eyes (n = 4). Scale bar for Fig. 2A represents 50 μm and for Fig. 2B and C represents 50 μm.
We next determined glial activation at 3 dpi after blast injury and observed glial fibrillary acidic protein (GFAP)+ immunoreactivity confined to astrocytes in the retinal nerve fiber layer (RNFL) in both blast blocked controls and rPBI treated eyes at 3 and 14 dpi with no apparent changes in the pattern of immunoreactivity in control and rPBI-treated eyes (Fig. 1B). In addition, a few ED1+ cells (arrowheads), likely to be macrophages, were observed at 3 and 14 dpi after rPBI with no ED1+ cells detected in blast blocked control eyes (Fig. 1C).
These results demonstrate that some RIPK3 immunoreactivity is present in eyes after rPBI with no apparent change in glial activation but influx of a few ED1+ macrophages into the eye were observed.
3.2. Intravitreal injections reduce the number of RBPMS+ RGC compared to vehicle treated eyes
To determine if inhibition of necroptosis promotes RGC survival, we used RBPMS immunohistochemistry in retinal sections taken at 28 dpi and RBPMS+ RGC counted in the central (closest to ONH), middle and peripheral retina in blast blocked and rPBI treated groups (no intravitreal injections and rPBI with vehicle or Nec-1s injections) (Fig. 2A–C). There were no reductions in RGC number after rPBI 28 dpi without intravitreal injection (Fig. 2A–C). However, rPBI in combination with intravitreal injection caused a reduction in the number of RBPMS+ RGC both in vehicle and Nec-1s injected eyes. There was statistical evidence of differences across all groups in the central, middle and peripheral portion of the retina (Fig. 2 A–C, Supplementary Table 1A, central P = 0.0412; middle P = 0.0003; peripheral P = 0.0330). In the middle and peripheral retina, there was strong evidence of a decrease in the number of RBPMS+ RGC after rPBI with vehicle injection compared to rPBI without injections (P = 0.0008 and P = 0.0306, respectively). In the middle retinal section, there was an increased number of RBPMS+ RGC after rPBI with Nec-1s treatment compared to rPBI with vehicle injection (Fig. 2B, P = 0.0155). There were no differences between rPBI groups with vehicle or Nec-1s injections in the central or peripheral retinal portions (P = 0.9691 and P = 0.7040, respectively). Mean values are displayed in Supplementary Table 1A. Representative RBPMS+ RGC immunostaining in the GCL is displayed in Fig. 2 D–F.
Figure 2. The number of RBPMS+ RGC were reduced after intraocular injections in combination with rPBI and increased in the middle portion of the retina after Nec-1s treatment compared to vehicle.
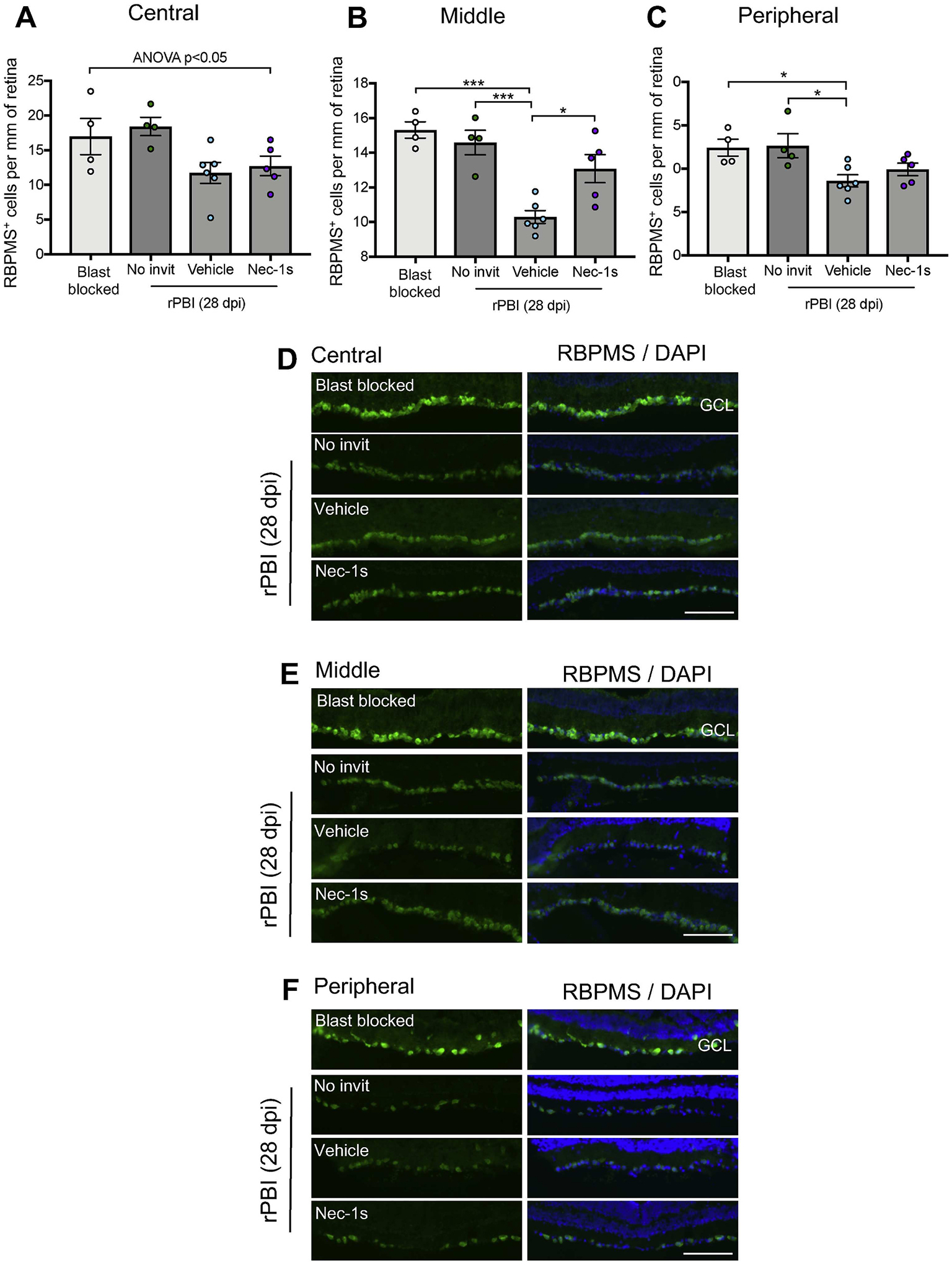
The number of RBPMS+ RGC were counted in three regions of left directly blasted eyes, in the central (close to the ONH), middle and peripheral retina. A) In the central retina, there was a significant difference in RBPMS+ cells across all groups (P = 0.0412). B) In the middle portion of the retina there were differences in RBPMS+ RGC numbers (P = 0.0003), with a reduction in the number of RBPMS+ RGC after rPBI with vehicle injection compared to rPBI without injections (P = 0.0008). There was an increased number of RBPMS+ RGC after rPBI with Nec-1s treatment compared to rPBI with vehicle injection (P = 0.0155). C) In the peripheral retina there was weak evidence of changes in the number of RBPMS+ RGC after rPBI (P = 0.0330). Representative RBPMS+ cells in the ganglion cell layer (GCL) from the D) central, E) middle and F) peripheral retina. Scale bar represents 100 μm n = 4 for blast blocked and rPBI, n = 6 for vehicle and n = 5 for Nec-1s. Error bars represent mean ± SEM. ***P < 0.001, *P < 0.05.
These results suggest that Nec-1s prevents loss of some RBPMS+ RGC, however, the intravitreal injection route combined with rPBI damages more RGC and causes their death.
3.3. The number of degenerating axons increased after rPBI but there was no effect of Nec-1s on ON axonal survival
To determine the number of degenerating axons in the ON, semi-thin cross-sections of the proximal ON were stained with PPD and toluidine blue to quantify intact and degenerating ON axons, gliosis and infiltrating cells (Fig. 3A–E). There were no significant differences at 28 dpi in total intact axons in the ON between blast blocked and rPBI groups with and without intravitreal injections (P = 0.1729; Fig. 3B). There were differences in degenerating ON axons across groups (P = 0.0014), with more degenerating ON axons in the rPBI eyes without intravitreal injection compared to the blast blocked group, there were 86.7 ± 26.9 degenerating axons in blast blocked compared to 467.0 ± 75.3 in rPBI (P = 0.0008; Fig. 3C). However, there was no evidence of a difference between rPBI treated eyes with vehicle or Nec-1s injection, with 275.0 ± 42.3 degenerating axons in vehicle treated eyes vs. 307.5 ± 91.5 degenerating axons in Nec-1s treated eyes (P = 0.9862). There were no differences in axonal density between any groups (P = 0.4539; Fig. 3D). There were also no changes in the total area of the ON (P = 0.3120; Fig. 3E), suggesting minimal ON edema or atrophy at 28 dpi. Mean values are displayed in Supplementary Table 1B.
Figure 3. The number of degenerating ON axons and the number of ED1+ cells increased after rPBI.
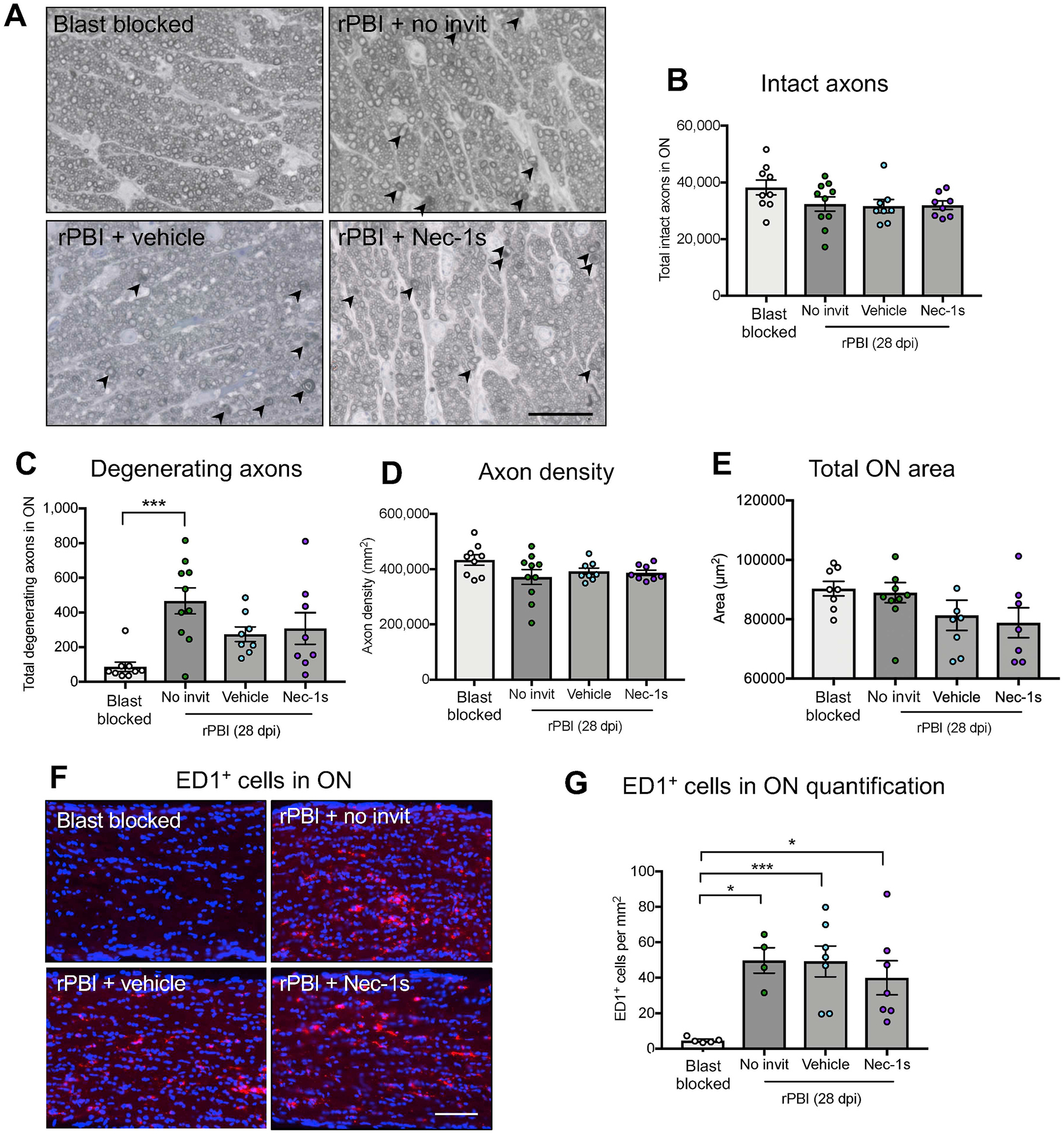
Total number of intact and degenerating axons were counted in the ON in blast blocked and rPBI treated eyes after intravitreal injections of vehicle and Nec-1s. A) Representative PPD and toluidine blue stained ON semi-thin cross sections showing degenerating axons (arrowheads). B) The number of intact axons was not significantly changed at 28 dpi (P = 0.1729). C) The number of degenerating axons in the ON increased after rPBI compared to blast blocked (P = 0.0014; P = 0.0008 post hoc). There were no differences between Nec-1s or vehicle treated eyes (P = 0.9862, post hoc). D) The axon density was not statistically different 28 dpi (P = 0.4539). E) The total ON area was not changed after injury or Nec-1s treatment (P = 0.3120). Error bars represent mean ± SEM. n = 10 per group. F) Immunostaining of ED1+ cells in the ON. G) The number of ED1+ cells in the ON increased after rPBI with no intravitreal injections compared to blast blocked (P = 0.0057; P = 0.0166, post-hoc) and was also more frequent compared to blast blocked after rPBI with vehicle treatment (P = 0.0064) and Nec-1s treatment (P = 0.0344). Fig. 4A scale bar represents 20 μm and Fig. 4F scale bar represents 100 μm. Error bars represent mean ± SEM. ***P < 0.001, *P < 0.05.
3.4. There were ED1+ cells infiltrating the ON at 28 dpi
ED1+ immunostaining was performed in longitudinal ON sections and quantified (Fig. 3F–G). We showed more frequent ED1+ cells in the ON after rPBI (P = 0.0057), with 4.8 ± 0.68 ED1+ cells/mm2 in blast blocked eyes vs. 49.79 ± 7.18 ED1+ cells/mm2 in rPBI without intravitreal injection The number of ED1+ cells were more frequent after rPBI with intravitreal injections of vehicle (49.26 ± 8.70 ED1+ cells/ mm2) and Nec-1s treatment (40.03 ± 9.59 ED1+ cells per mm2) compared to blast blocked (P = 0.0064 and P = 0.0344, respectively). This data suggests that ED1+ macrophages infiltrate into the ON after rPBI regardless of presence or absence of intravitreal injection with vehicle or Nec-1s.
3.5. There were no changes in retinal thickness after rPBI
OCT retinal scans were performed at baseline and 28 dpi in blast blocked and rPBI with and without intravitreal injections (Fig. 4A–B). There was no evidence of a change in total retinal thickness (Fig. 4C; P = 0.6999) or GCC thickness (Fig. 4D; P = 0.9601) following rPBI or with Nec-1s treatment, suggesting there was minimal retinal edema or gliosis and no retinal thickening or thinning at 28 dpi.
Fig. 4. There was vitreous and glial inflammation observed after rPBI with intraocular injections of Nec-1s and vehicle treatment.
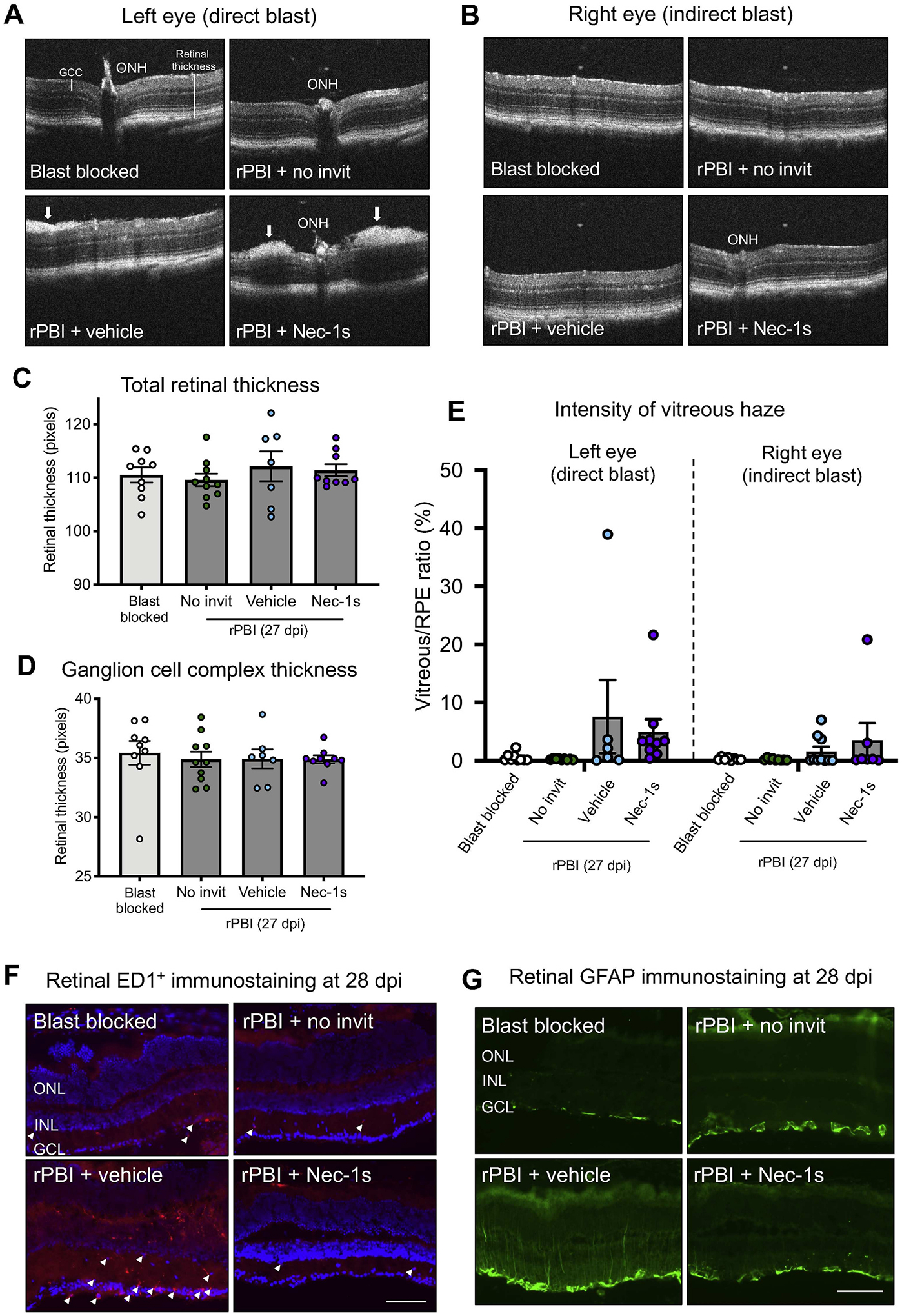
A) Representative OCT images at the ONH showing no gross structural changes comparing blast blocked and rPBI treated eyes but some hyper-reflective density at the vitreal retinal surface (arrows) in rPBI and vehicle or Nec-1s intravitreal injections. B) Representative OCT images at the ONH in the right eyes which received indirect blast injury. C) There was weak evidence of changes in total retinal thickness (P = 0.085) D) and no changes in GCC thickness (P = 0.929). E) There was strong evidence that intravitreal injection increased vitreous intensity (P = 0.004), with vitreous intensity higher in both Nec-1s and vehicle injected eyes after rPBI than after rPBI alone. F) The number of ED1+ cells (arrow heads) were increased after rPBI with intravitreal injections of vehicle but not after rPBI with no intravitreal injections or with Nec-1s injections at 28 dpi. G) GFAP immunostaining increased after rPBI with intravitreal injection of vehicle, but not after rPBI with no intravitreal injections or with Nec-1s injections at 28 dpi. For Fig. 4F and G the scale bar represents 100 μm. Images of representative of n = 4 animals per group. Error bars represent mean ± SEM.
3.6. Vitreal inflammation was observed in blast-injured eyes treated with intravitreal injections
There was no evidence that rPBI affected vitreous intensity without intravitreal injection (Fig. 4E). There was also no evidence of a difference between rPBI and blast blocked eyes and these factors were not therefore included in the final model. There was strong evidence that intravitreal injection increased vitreous intensity (1.93% ± 0.38; P < 0.001), with vitreous intensity higher in both Nec-1s and vehicle injected left (directly blast-injured) and right (indirectly injured) eyes after rPBI than after rPBI alone (Fig. 4E). There was weak evidence that directly blast-injured left eyes had greater vitreous intensity than indirectly-injured right eyes (1.29% ± 0.62; P = 0.038) (Fig. 4E).
3.7. There were more ED1+ cells and glial reactivity at 28 dpi in the retina after rPBI and vehicle injection, which was prevented in Nec-1s treated eyes
ED1 IHC was performed for the presence of infiltrating macrophages in the retina at 28 dpi. There were few retinal ED1+ cells in blast blocked and rPBI with no intravitreal injections and rPBI with Nec-1s injection at 28 dpi (Fig. 4F). However, there were more ED1+ cells after rPBI and vehicle injections. In contrast, few ED1+ cells were detected in rPBI treated eyes with Nec-1s injection at 28 dpi. Furthermore, retinal GFAP+ immunoreactivity was increased after rPBI with vehicle injection, but not in any other groups, with GFAP+ staining spanning the INL and reaching the IPL (Fig. 4G). Together, this data suggests that intravitreal injection causes greater retinal pathology after rPBI, and that Nec-1s may provide some neuroprotective or anti-inflammatory properties.
4. Discussion
We have demonstrated increases in necroptotic protein RIPK3 localisation in the GCL, OPL and IPL compared to blast blocked controls. RIPK1 immunolocalised to cells of the INL and GCL in blast blocked and remains at a constant expression up to 28 dpi. In our experiments, rPBI did not cause detectable RGC or axonal loss but did increase the number of degenerating ON axons observed. However, when rPBI was preceded and followed by intravitreal injection, vitreous inflammation and RGC degeneration increased with retinal infiltration with ED1+ macrophages and increased GFAP immunoreactivity. Nec-1s inhibition reduced this RGC loss in the central retina compared to vehicle injection but this difference was not reflected in axonal counts or the number of degenerating axons. The RGC-neuroprotective effect of Nec-1s treatment was greater in mid-peripheral than central or peripheral retina. As this was a post-hoc analysis in a small subgroup, differences may reflect random variation, differing injury susceptibility in different RGC subtypes, such as the intrinsically photosensitive RGC of the M2 subtype, which is most numerous in the mid-dorsal mouse retina (Hughes et al., 2013), or structural retinal differences affecting drug penetration (Vaney, 1994). The incomplete protective effect of Nec-1s on RGC degeneration after rPBI plus intravitreal injection could also be associated with the activity of alternative cell death pathways involving inflammatory mediators, such as the extrinsic pathway to apoptosis. There was no evidence of an effect of Nec-1s inhibition on vitreous inflammation. Nec-1s inhibition did reduce the number of ED1+ macrophages in the retina and also reduced retinal GFAP reactivity.
OCT imaging was performed to assess retinal layer thickness and vitreous inflammation. We have not previously reported assessment of retinal thickness in our model and did not observe a change in these data, in contrast to Allen et al., who observed increased retinal thickness, associated with increased GFAP staining and retinal hyper-reactivity on electroretinographic measured two months after blast injury (Allen et al., 2018). It is likely that higher energy injury (80 psi vs 16 psi) and later assessment (2 months vs 28 days) explain these differing results.
We observed increased vitreous haze, suggesting inflammation, in eyes exposed to rPBI and injected with either Nec-1s or vehicle. rPBI alone did not affect vitreous haze. The qualitative appearances of the OCT scans after rPBI were similar to those described in other studies in which focal hyperreflective dots correlated with the number of mononuclear cells detected in histological sections, suggestive of macrophage infiltration in response to the combination of rPBI and intravitreal injection and consistent with our finding of ED1+ cells in retinal sections after rPBI and vehicle injection (Kokona et al., 2017). There was no effect of Nec-1s vs vehicle on the increase in vitreous haze, consistent with this being a mechanical effect of injection, but there were fewer ED1+ cells after Nec-1s than vehicle injection indicating that the increased vitreous haze observed in our model after rPBI with intravitreal injection was not completely explained by retinal macrophage infiltration. There was weak evidence of a difference between right and left eyes, which were subject to direct and indirect blast respectively, suggesting that the pro-inflammatory effect of blast and intravitreal injection was related to blast-dose.
In addition, ED1 infiltration of the ON was observed in the rPBI alone group and did not show evidence of an effect of intravitreal injection indicating that the ON ED1 cell infiltration was not completely explained by our assessment of vitreous haze. An inflammatory effect of the blast injury on the ON would be consistent with previous findings of CD68+ cell brain infiltration after blast injury (Zanier et al., 2014) and macrophage ON infiltration after ultrasonic injury (Tao et al., 2017). Macrophages also have a role in clearing myelin after Wallerian degeneration of the ON (Stoll et al., 1989).
RBPMS+ RGC were quantified on retinal cryosections, which has been validated in models of ONC and ocular hypertension (Hill et al., 2015; Mead et al., 2014; Mead and Tomarev, 2016). At 28 dpi there was no reduction in the number of RBPMS+ RGC after rPBI compared to the blast blocked condition. Possible explanations are that: (1) repeated exposure to a low overpressure blast wave might not be sufficient without other associated injuries to cause RGC death within 28 dpi; (2) RGC degenerate at a later time point; (3) the blast blocked condition may also cause some RGC death; (4) multiple blast exposures may also be relatively protective compared to a single blast exposure (Harper et al., 2019). While repeated low level blast exposure has been raised as a clinical concern (Sajja et al., 2019), cell death associated with ≤3 psi blasts have not been reported (Engel et al., 2019) and 5 psi blast preconditioning may have neuroprotective effects (Harper et al., 2019). We previously demonstrated that a single blast at different magnitudes up to 26 psi did not cause as significant ON damage as detected with repeated exposure to 15 psi blasts (Hines-Beard et al., 2012; Vest et al., 2019). Protection from repeated blast injury might be caused by the variations in interval between blast exposures. In our model we show a smaller inter-blast interval is more deleterious, with intervals of < 1 min causing synergistic axon damage, and 1-day interval causing accumulative damage (Vest et al., 2019). We did not test one-week intervals in our model, although it would seem that the tissue would have rebounded entirely from the insult and compensatory mechanisms to protect from future injuries could have been enacted. Delayed and progressive RGC degeneration has been demonstrated between 4 and 10 months after a single 26 psi blast-wave exposure (Dutca et al., 2014; Mohan et al., 2013), shown through a reduction in GCL thickness. We previously showed a decrease in the number of DAPI+ cells in the GCL and ON axons up to 28 days (Bernardo-Colon et al., 2019). We might not have detected sufficient RGC degeneration if the effects were subtle with focal pockets of retinal injury, as demonstrated by focal areas of Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL)+ retinal cells after a single blast exposure (Bricker-Anthony et al., 2014b).
Consistent with the fact that we did not detect loss of RGC soma or axons by 28 days after rPBI alone, we did not detect MLKL phosphorylation, which would imply that detectable levels of necroptosis did not occur by 28 dpi in this group. Given that degenerating axonal profiles were detected by this time point it is possible either that cell death would be detected in the retina after 28 dpi, or that axonal degeneration processes were dissociated from RGC soma death processes (Bernardo-Colon et al., 2019), which may be the case in the fluid percussion model of TBI (Wang et al., 2013) for instance.
PPD-stained resin embedded ON semi-thin sections were used to assess the number of ON axons with a degenerative profile (unravelling or collapsed myelin sheath) and intact axons following rPBI and necroptosis inhibition. At 28 dpi, there increased number of degenerating axons, but no reduction in RGC at this time point, indicating that rPBI primarily injures the ON and that RGC degeneration was still evolving by the end of the study. Consistent with this hypothesis, the number of intact axons were slightly reduced across all rPBI groups but did not reach statistical significance. Thus, axonal degeneration after rPBI may be delayed and progressive and a reduction may be detected at later time points (Dutca et al., 2014; Mohan et al., 2013).
We demonstrate decreased numbers of RBPMS+ RGC after injury in combination with intravitreal injections, irrespective of the injected compound. We suggest the decrease in RGC numbers with the combination of injury and injection could replicate a penetrating ocular injury in close proximity with the primary blast wave exposure, possibly replicating a combination of primary and secondary blast injury (blast-wave exposure and blunt, penetrating, or perforating impact injury due to projectiles) (Blanch et al., 2011; Scott, 2011).
Nec-1s was reconstituted in DMSO and DMSO concentrations > 1% DMSO have caused caspase-3 independent RGC death in vivo (Galvao et al., 2014) and concentrations > 10% DMSO caused apoptosis through plasma membrane permeabilisation in vitro (Notman et al., 2006). However, other studies have used similar concentrations DMSO in vivo without additive degeneration (Huang et al., 2018; Rosenbaum et al., 2010), suggesting that this was not the likely cause of additive RGC loss.
In the military, blast wave exposure is associated with delayed visual deficits (Capo-Aponte et al., 2015); this could be due, in part, to injury of the visual cortex and brain. In rats exposed to a single acoustic blast overpressure wave of ~80 psi generated using a shock-tube device, retinal thickness increased in eyes directly exposed to the blast compared to blast blocked eyes and there was an increase in retinal GFAP immunostaining, suggesting that reactive gliosis may have caused the increase in retinal thickness up to 8-months post blast (Allen et al., 2018). In our study, we investigated retinal changes at 28 dpi and did not detect any change in retinal thickness, but did detect increased retinal GFAP staining after rPBI and vehicle injection only, suggesting that, similar to the increase in vitreous inflammation, this effect was increased by the combination of rPBI and intravitreal injection, but was modulated by Nec-1s.
5. Conclusion
Collectively, our study indicates that the combination of intravitreal injections and rPBI can cause additive injury, with increased cell death and inflammation. However, treatment with Nec-1s may provide some RGC neuroprotection to the additive death that occurred when rPBI and intravitreal injection were combined, and this may be related to clinical situations where blast and penetrating injuries coexist.
Supplementary Material
Acknowledgments
All animal work was undertaken at Vanderbilt University. The authors would like to thank the university’s animal facilities for assistance with animal care. The authors thank Purnima Ghose for histological processing.
Funding
Supported by Fight for Sight PhD Studentship, grant number 1560/1561, DoD W81XWH-15-1-0096, DoD W81XWH-17-2-0055, NEI R01 EY022349, NEI P30 EY008126, Vanderbilt University Medical Center Cell Imaging Shared Resource core facility (Clinical and Translational Science Award Grant UL1 RR024975 from National Center for Research Resources), Research to Prevent Blindness Unrestricted Funds (VEI), Ret. Maj. General Stephen L. Jones, MD Fund, Potoscnak Family-CSC Research Fund, Ayers Research Fund in Regenerative Visual Neuroscience.
Abbreviations
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- DAPI
4′,6-diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- Dpi
days post injury
- GCL
ganglion cell layer
- GFAP
glial fibrillary acidic protein
- GEE
generalised estimating equations
- IDO
indoleamine-2,3-dioxygenase
- IHC
immunohistochemistry
- INL
inner nuclear layer
- IPL
inner plexiform layer
- MLKL
mixed lineage kinase domain like pseudokinase
- Nec-1
Necrostatin-1
- Nec-1s
Necrostatin-1s
- OCT
optical coherence tomography
- OCTc
optimal cutting temperature compound
- ON
optic nerve
- ONH
optic nerve head
- ONL
outer nuclear layer
- PBI
primary blast injuries
- PBS
phosphate buffered saline
- PFA
paraformaldehyde
- Psi
pounds per square inch
- PPD
p-phenylenediamine
- RBPMS
RNA binding protein with multiple splicing
- RGC
retinal ganglion cells
- RNFL
retinal nerve fiber layer
- rPBI
repeated primary ocular blast injury
- RPE
retinal pigment epithelium
- RT
room temperature
- RIPK1
receptor interacting kinase 1
- RIPK3
receptor interacting kinase 3
- SEM
standard error of the mean
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- TNF
tumor necrosis factor
Footnotes
Declaration of competing interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.exer.2020.108102.
References
- Allen R, Motz CT, Feola A, Chesler K, Haider R, Ramachandra Rao S, Skelton L, Fliesler S, Pardue MT, 2018. Long-term functional and structural consequences of primary blast overpressure to the eye. J. Neurotrauma 35, 2104–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo-Colon A, Vest V, Clark A, Cooper ML, Calkins DJ, Harrison FE, Rex TS, 2018. Antioxidants prevent inflammation and preserve the optic projection and visual function in experimental neurotrauma. Cell Death Dis. 9, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo-Colon A, Vest V, Cooper ML, Naguib SA, Calkins DJ, Rex TS, 2019. Progression and pathology of traumatic optic neuropathy from repeated primary blast exposure. Front. Neurosci 13, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanch RJ, Bindra MS, Jacks AS, Scott RAH, 2011. Ophthalmic injuries in British armed forces in Iraq and Afghanistan. Eye (London, England) 25, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker-Anthony C, Hines-Beard J, D’Surney L, Rex TS, 2014a. Exacerbation of blast-induced ocular trauma by an immune response. J. Neuroinflammation 11, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker-Anthony C, Hines-Beard J, Rex TS, 2014b. Molecular changes and vision loss in a mouse model of closed-globe blast trauma. Investig. Ophthalmol. Vis. Sci 55, 4853–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker-Anthony C, Hines-Beard J, Rex TS, 2016. Eye-directed overpressure airwave-induced trauma causes lasting damage to the anterior and posterior globe: a model for testing cell-based therapies. J. Ocul. Pharmacol. Therapeut 32, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capo-Aponte JE, Jurek GM, Walsh DV, Temme LA, Ahroon WA, Riggs DW, 2015. Effects of repetitive low-level blast exposure on visual system and ocular structures. J. Rehabil. Res. Dev 52, 273–290. [DOI] [PubMed] [Google Scholar]
- Champion HR, Holcomb JB, Young LA, 2009. Injuries from explosions: physics, biophysics, pathology, and required research focus. J. Trauma 66, 1468–1477 discussion 1477. [DOI] [PubMed] [Google Scholar]
- Cockerham GC, Rice TA, Hewes EH, Cockerham KP, Lemke S, Wang G, Lin RC, Glynn-Milley C, Zumhagen L, 2011. Closed-eye ocular injuries in the Iraq and Afghanistan wars. N. Engl. J. Med 364, 2172–2173. [DOI] [PubMed] [Google Scholar]
- Cougnoux A, Cluzeau C, Mitra S, Li R, Williams I, Burkert K, Xu X, Wassif CA, Zheng W, Porter FD, 2016. Necroptosis in Niemann-Pick disease, type C1: a potential therapeutic target. Cell Death Dis. 7, e2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMar J, Sharrow K, Hill M, Berman J, Oliver T, Long J, 2016. Effects of primary blast overpressure on retina and optic tract in rats. Front. Neurol 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWalt GJ, Eldred WD, 2017. Visual system pathology in humans and animal models of blast injury. J. Comp. Neurol 525, 2955–2967. [DOI] [PubMed] [Google Scholar]
- Dutca LM, Stasheff SF, Hedberg-Buenz A, Rudd DS, Batra N, Blodi FR, Yorek MS, Yin T, Shankar M, Herlein JA, Naidoo J, Morlock L, Williams N, Kardon RH, Anderson MG, Pieper AA, Harper MM, 2014. Early detection of subclinical visual damage after blast-mediated TBI enables prevention of chronic visual deficit by treatment with P7C3-S243. Investig. Ophthalmol. Vis. Sci 55, 8330–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel CC, Hoch E, Simmons M, 2019. The Neurological Effects of Repeated Exposure to Military Occupational Blast: Implications for Prevention and Health: Proceedings, Findings, and Expert Recommendations from the Seventh Department of Defense State-Of-The-Science Meeting. RAND Corporation. [Google Scholar]
- Galvao J, Davis B, Tilley M, Normando E, Duchen MR, Cordeiro MF, 2014. Unexpected low-dose toxicity of the universal solvent DMSO. Faseb. J 28, 1317–1330. [DOI] [PubMed] [Google Scholar]
- Gizycka A, Chorostowska-Wynimko J, 2015. Programmed necrosis and necroptosis - molecular mechanisms. Postepy Hig. Med. Dosw. (Online) 69, 1353–1363. [DOI] [PubMed] [Google Scholar]
- Harper MM, Woll AW, Evans LP, Delcau M, Akurathi A, Hedberg-Buenz A, Soukup DA, Boehme N, Hefti MM, Dutca LM, Anderson MG, Bassuk AG, 2019. Blast preconditioning protects retinal ganglion cells and reveals targets for prevention of neurodegeneration following blast-mediated traumatic brian injury. Invest. Ophthalmol. Vis. Sci 60, 4159–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill LJ, Mead B, Blanch RJ, Ahmed Z, De Cogan F, Morgan-Warren PJ, Mohamed S, Leadbeater W, Scott RA, Berry M, Logan A, 2015. Decorin reduces intraocular pressure and retinal ganglion cell loss in rodents through fibrolysis of the scarred trabecular meshwork. Invest. Ophthalmol. Vis. Sci 56, 3743–3757. [DOI] [PubMed] [Google Scholar]
- Hines-Beard J, Marchetta J, Gordon S, Chaum E, Geisert EE, Rex TS, 2012. A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp. Eye Res 99, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Zhou T, Sun X, Zheng Y, Cheng B, Li M, Liu X, He C, 2018. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 25, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S, Watson TS, Foster RG, Peirson SN, Hankins MW, 2013. Nonuniform distribution and spectral tuning of photosensitive retinal ganglion cells of the mouse retina. Curr. Biol 23, 1696–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan JB, Pasparakis M, Kelliher MA, Ravits J, Yuan J, 2016. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NP, Hayward JM, Khaw PT, Claoue CM, Elkington AR, 1986. Function of an ophthalmic “accident and emergency” department: results of a six month survey. Br. Med. J. (Clin Res Ed) 292, 188–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane PA, Karampelas M, Sim DA, Sadda SR, Tufail A, Sen HN, Nussenblatt RB, Dick AD, Lee RW, Murray PI, Pavesio CE, Denniston AK, 2014. Objective measurement of vitreous inflammation using optical coherence tomography. Ophthalmology 121, 1706–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokona D, Haner NU, Ebneter A, Zinkernagel MS, 2017. Imaging of macrophage dynamics with optical coherence tomography in anterior ischemic optic neuropathy. Exp. Eye Res 154, 159–167. [DOI] [PubMed] [Google Scholar]
- Kwong JMK, Quan A, Kyung H, Piri N, Caprioli J, 2011. Quantitative analysis of retinal ganglion cell survival with rbpms immunolabeling in animal models of optic neuropathies. Investig. Ophthalmol. Vis. Sci 52, 9694–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Green DR, 2014. Necroptosis. N. Engl. J. Med 370, 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead B, Thompson A, Scheven BA, Logan A, Berry M, Leadbeater W, 2014. Comparative evaluation of methods for estimating retinal ganglion cell loss in retinal sections and wholemounts. PloS One 9, e110612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead B, Tomarev S, 2016. Evaluating retinal ganglion cell loss and dysfunction. Exp. Eye Res 151, 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan K, Kecova H, Hernandez-Merino E, Kardon RH, Harper MM, 2013. Retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury. Invest. Ophthalmol. Vis. Sci 54, 3440–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notman R, Noro M, O’Malley B, Anwar J, 2006. Molecular basis for dimethylsulfoxide (DMSO) action on lipid membranes. J. Am. Chem. Soc 128, 13982–13983. [DOI] [PubMed] [Google Scholar]
- Peskind ER, Petrie EC, Cross DJ, Pagulayan K, McCraw K, Hoff D, Hart K, Yu CE, Raskind MA, Cook DG, Minoshima S, 2011. Cerebrocerebellar hypometabolism associated with repetitive blast exposure mild traumatic brain injury in 12 Iraq war Veterans with persistent post-concussive symptoms. Neuroimage 54 (Suppl. 1), S76–S82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BN, Chun DW, Colyer M, 2013. Closed globe macular injuries after blasts in combat. Retina 33, 371–379. [DOI] [PubMed] [Google Scholar]
- Politi K, Przedborski S, 2016. Axonal degeneration: RIPK1 multitasking in ALS. Curr. Biol 26, R932–R934. [DOI] [PubMed] [Google Scholar]
- Rex TS, 2014. Delayed vision loss and therapeutic intervention after blast injury. Invest. Ophthalmol. Vis. Sci 55, 8342. [DOI] [PubMed] [Google Scholar]
- Ritenour AE, Baskin TW, 2008. Primary blast injury: update on diagnosis and treatment. Crit. Care Med 36, S311–S317. [DOI] [PubMed] [Google Scholar]
- Rodriguez AR, Muller LPD, Brecha NC, 2014. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J. Comp. Neurol 522, 1411–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR, 2016. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI, 2010. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J. Neurosci. Res 88, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajja V, LaValle C, Salib JE, Misistia AC, Ghebremedhin MY, Ramos AN, Egnoto MJ, Long JB, Kamimori GH, 2019. The role of very low level blast overpressure in symptomatology. Front. Neurol 10, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R, 2011. The injured eye. Phil. Trans. Biol. Sci 366, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RAH, Blanch RJ, Morgan-Warren PJ, 2015. Aspects of ocular war injuries. Trauma-England 17, 83–92. [Google Scholar]
- Shen H, Liu C, Zhang D, Yao X, Zhang K, Li H, Chen G, 2017. Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro. Cell Death Dis. 8, e2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Trapp BD, Griffin JW, 1989. Macrophage function during Wallerian degeneration of rat optic nerve: clearance of degenerating myelin and Ia expression. J. Neurosci 9, 2327–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P, 2012. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 3, e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Dvoriantchikova G, Tse BC, Pappas S, Chou TH, Tapia M, Porciatti V, Ivanov D, Tse DT, Pelaez D, 2017. A novel mouse model of traumatic optic neuropathy using external ultrasound energy to achieve focal, indirect optic nerve injury. Sci. Rep 7, 11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CN, Thompson AM, Ahmed Z, Blanch RJ, 2019. Retinal ganglion cells die by necroptotic mechanisms in a site-specific manner in a rat blunt ocular injury model. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P, 2014. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol 15, 135–147. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G, 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol 11, 700–714. [DOI] [PubMed] [Google Scholar]
- Vaney DI, 1994. Territorial organization of direction-selective ganglion cells in rabbit retina. J. Neurosci 14, 6301–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest V, Bernardo-Colon A, Watkins D, Kim B, Rex TS, 2019. Rapid repeat exposure to subthreshold trauma causes synergistic axonal damage and functional deficits in the visual pathway in a mouse model. J. Neurotrauma 36, 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasov A, Ryan DS, Ludlow S, Weichel ED, Colyer MH, 2015. Causes of combat ocular trauma-related blindness from operation Iraqi freedom and enduring freedom. J. Trauma Acute Care Surg 79, S210–S215. [DOI] [PubMed] [Google Scholar]
- Wang J, Fox MA, Povlishock JT, 2013. Diffuse traumatic axonal injury in the optic nerve does not elicit retinal ganglion cell loss. J. Neuropathol. Exp. Neurol 72, 768–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang H, Tao Y, Zhang S, Wang J, Feng X, 2014. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 266, 91–101. [DOI] [PubMed] [Google Scholar]
- Warden D, 2006. Military TBI during the Iraq and Afghanistan wars. J. Head Trauma Rehabil 21, 398–402. [DOI] [PubMed] [Google Scholar]
- Weichel ED, Colyer MH, Ludlow SE, Bower KS, Eiseman AS, 2008. Combat ocular trauma visual outcomes during operations Iraqi and enduring freedom. Ophthalmology 115, 2235–2245. [DOI] [PubMed] [Google Scholar]
- Weinlich R, Oberst A, Beere HM, Green DR, 2017. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol 18, 127–136. [DOI] [PubMed] [Google Scholar]
- You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ, 2008. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cerebr. Blood Flow Metabol 28, 1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanier ER, Pischiutta F, Riganti L, Marchesi F, Turola E, Fumagalli S, Perego C, Parotto E, Vinci P, Veglianese P, D’Amico G, Verderio C, De Simoni MG, 2014. Bone marrow mesenchymal stromal cells drive protective M2 microglia polarization after brain trauma. Neurotherapeutics 11, 679–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Tang MB, Luo HY, Shi CH, Xu YM, 2017. Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis. 8, e2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.