
Fig. 5. Schematic model for the stepwise folding of the CDR.
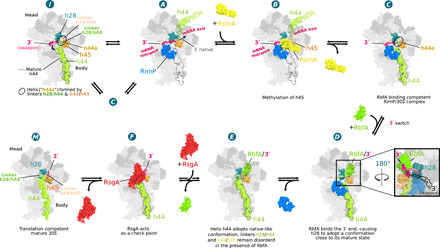
The well-defined cryo-EM structures ordered according to increasing 16S rRNA native structure content illustrates a potential folding pathway for the CDR. State A has the most nonnative CDR fold and shows RimP bound, the 16S 3′-end inside the mRNA entry channel (h28immature), and the decoding h44 positioned in the mRNA exit channel (see fig. S8D discussion). This 30S configuration enables RsmA binding in state B and exposes residues A1518 to A1519 in the h45 loop for methylation by RsmA (28). RsmA release (state C) partially opens the canonical binding site of h44, allowing it to drop into place while its two linkers form a poorly defined helix similar to h44a in state I. Upon RbfA binding (state D) the 3′-end swaps into the exit channel (see inset), stabilized by interactions with the KH domain of RbfA, allowing h28mature to form. After RimP dissociation (state E), h44 adopts a more native-like conformation although both linker regions remain largely disordered. After RsgA binding and release of RbfA (state F), the h44 linkers fold into a nearly native conformation. Last, after guanosine triphosphate hydrolysis, RsgA leaves the ribosome in a mature conformation (state M). In this model, state I represents an inactive state (or kinetic trap) accessed, for instance, when RimP prematurely dissociates from states A or C and h44 drops into its mature position before the 3′-end can swap into the exit channel.