

Abstract
Objectives
Innate lymphoid cells (ILCs) secrete cytokines, such as IFN‐γ, IL‐13 and IL‐17, which are linked to chronic obstructive pulmonary disease (COPD). Here, we investigated the role of pulmonary ILCs in COPD pathogenesis.
Methods
Lung ILC subsets in COPD and control subjects were quantified using flow cytometry and associated with clinical parameters. Tissue localisation of ILC and T‐cell subsets was determined by immunohistochemistry. Mice were exposed to air or cigarette smoke (CS) for 1, 4 or 24 weeks to investigate whether pulmonary ILC numbers and activation are altered and whether they contribute to CS‐induced innate inflammatory responses.
Results
Quantification of lung ILC subsets demonstrated that ILC1 frequency in the total ILC population was elevated in COPD and was associated with smoking and severity of respiratory symptoms (COPD Assessment Test [CAT] score). All three ILC subsets localised near lymphoid aggregates in COPD. In the COPD mouse model, CS exposure in C57BL/6J mice increased ILC numbers at all time points, with relative increases in ILC1 in bronchoalveolar lavage (BAL) fluid. Importantly, CS exposure induced increases in neutrophils, monocytes and dendritic cells that remained elevated in Rag2/Il2rg‐deficient mice that lack adaptive immune cells and ILCs. However, CS‐induced CXCL1, IL‐6, TNF‐α and IFN‐γ levels were reduced by ILC deficiency.
Conclusion
The ILC1 subset is increased in COPD patients and correlates with smoking and severity of respiratory symptoms. ILCs also increase upon CS exposure in C57BL/6J mice. In the absence of adaptive immunity, ILCs contribute to CS‐induced pro‐inflammatory mediator release, but are redundant in CS‐induced innate inflammation.
Keywords: innate lymphoid cells, ILC, COPD, innate inflammation
In this article, we investigated the role of pulmonary innate lymphoid cells (ILCs) in the pathogenesis of chronic obstructive pulmonary disease (COPD). We found that the ILC1 subset is increased in COPD patients and correlates with smoking and severity of respiratory symptoms. In C57BL/6 mice, ILCs increase upon cigarette smoke (CS) exposure. In the absence of adaptive immunity, ILCs contribute to CS‐induced pro‐inflammatory mediator release, but they are redundant in CS‐induced innate inflammation.

Introduction
Innate lymphoid cells (ILCs) are relatively rare, tissue‐resident cells that make up the first line of defence at the mucosal barrier surface of the lung. ILCs are activated by epithelial and myeloid cell‐derived cytokines and alarmins, leading to the production of effector cytokines that modulate immune responses. ILCs do not express rearranged antigen‐specific receptors, which make fast expansion and activation possible. 1 Three non‐cytotoxic subsets can be distinguished, which parallel T‐helper cells regarding their transcription factor and cytokine secretion. ILC1 expresses T‐BET and secretes IFN‐γ. ILC2 expresses GATA3 and produces both IL‐5 and IL‐13. ILC3 expresses RORγt and secretes both IL‐17 and IL‐22. 2 , 3 , 4 These cytokines have been associated with the pathogenesis of chronic obstructive pulmonary disease (COPD), which is characterised by abnormal chronic inflammatory responses to inhaled noxious particles and gasses. 5 , 6 IFN‐γ contributes to the activation of alveolar macrophages, and IFN‐γ+CD8+ cells positively correlate with disease severity. 7 , 8 In stable COPD patients with an eosinophilic phenotype, sputum IL‐5 concentrations are increased and associated with the degree of eosinophilia. 9 High levels of IL‐13 are found in serum of patients suffering from an acute exacerbation of COPD. 10 IL‐17A is involved in the recruitment of neutrophils and the formation of lymphoid follicles in COPD. 11 , 12 IL‐22 mRNA and protein levels are increased in COPD patients, and IL‐22‐deficient (−/−) mice have reduced numbers of CS‐induced pulmonary neutrophils and do not show CS‐induced alveolar enlargement and airway remodelling. 13
We previously characterised the different ILC subsets in the human lung. 14 Others have demonstrated increases in ILC1 frequency in the lungs of severe COPD patients compared to healthy subjects or those with mild‐to‐moderate COPD, 15 and increased circulating IFN‐γ‐producing ILC1 in subjects with severe COPD. 16 To what extent specific ILC subsets in the human lung are associated with clinical parameters of COPD has not been investigated. Moreover, localisation of all three ILC subsets by immunohistochemistry within the same human lung has not been previously assessed.
In mice, it has been shown that cigarette smoke (CS) exposure, an important causal factor for COPD, can affect pulmonary ILC numbers, especially T‐BET+ILC1 and RORγt+ILC3 subsets. 16 , 17 Whether CS exposure also affects the numbers of cytokine‐producing ILCs, which shows that ILCs are activated, has not yet been studied. Using an experimental model of COPD, we previously demonstrated that CS‐induced innate inflammation (pulmonary macrophage, neutrophil and dendritic cell influx) is still present in scid mice, which lack B‐ and T‐lymphocytes. 18 We hypothesised that ILCs, as an important source of IFN‐γ, IL‐13 or IL‐17, drive these CS‐induced inflammatory responses in the absence of an adaptive immune system.
We assessed human pulmonary ILC subsets by flow cytometry and immunohistochemistry in lung tissue from well‐characterised COPD patients and control subjects. We also investigated whether pulmonary ILC numbers and activation are altered in our COPD mouse model and whether they contribute to CS‐induced innate inflammatory responses.
Results
Pulmonary ILC1 is elevated in COPD patients
To investigate the involvement of ILCs in COPD, human pulmonary ILC frequencies were analysed using flow cytometry of single‐cell suspensions of freshly isolated lung tissue from COPD patients (n= 15) and control subjects (n = 9) (gating strategy Figure 1a, Supplementary figures 1 and 2 for FMO controls). Subject characteristics are shown in Table 1. The frequency of CD161+CD127+ILCs was similar in COPD and control subjects (Figure 1b). However, discrimination of the different ILC subsets within the CD161+CD127+ILC population based on surface marker expression showed that ILC1 (CRTH2−CD117−) was the dominant subset in COPD. Moreover, ILC1 frequency differed significantly among the groups (Kruskal–Wallis P < 0.05), and the median+IQR showed a numerical increase between never‐smoking controls, smoking controls (ex+current) and COPD patients (Figure 1c, Supplementary table 1). In the COPD group, no apparent differences in the distribution of ILC subsets in ex‐ or current smokers were detected (Supplementary figure 3a). Notably, in this cohort, ILC1 frequencies from COPD GOLD I subjects were relatively high, which coincided with high number of pack‐years. Correlation analysis demonstrated a positive association between the number of pack‐years and the numbers of ILC1 (Supplementary figure 3b). The frequencies of CRTH2+ILC2 and CRTH2−CD117+ (both NKp44+ and NKp44−) ILC3 did not differ significantly within the CD161+CD127+ILC population (Figure 1c).
Figure 1.
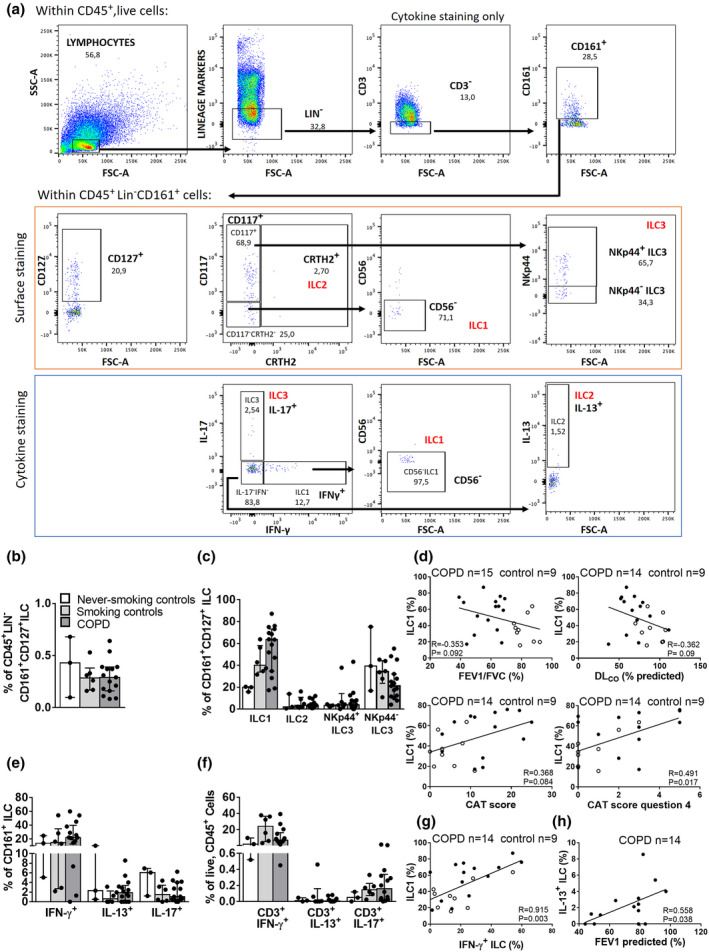
Quantification of pulmonary ILC in COPD patients and correlation with clinical parameters. For flow cytometric analysis, ILCs were surface‐stained and gated as live, CD45+ lymphocytes that are Lin−(CD3, CD19, CD11c, CD11b, CD1a, CD14, CD34, CD123, TCRαβ, TCRγδ, BDCA2, CD235A and FcεR1) CD161+CD127+. ILC subsets were further defined as CD117−CRTH2−CD56− ILC1, CRTH2+ILC2 and CD117+CRTH2−NKp44+/−ILC3 (a, upper panel). For ILC labelling following 4h of stimulation: ILCs were gated as live, CD45+ lymphocytes that were Lin−(CD19, CD11c, CD11b, CD1a, CD14, CD34, CD123, TCRαβ, TCRγδ, BDCA2, CD235A and FcεR1) CD3−CD161+, followed by defining subsets as IFN‐γ+IL‐17−CD56−ILC1, IFN‐γ−IL‐17−IL‐13+ILC2 and IFN‐γ−IL‐17+ILC3 (a, lower panel). Gates were set based on FMO controls (see Supplementary figures 1 and 2). Percentage of CD161+CD127+ ILC of CD45+ live cells (b) and ILC subsets based on surface staining, expressed as % of CD161+CD127+ILC (c). Bar graphs on (b), (c), (e) and (f) represent the median values per group; error bars represent the interquartile range (IQR). Spearman’s correlation between % of ILC1 and FEV1/FVC%, %DLCO, total CAT score and score on CAT question 4 (grade of breathlessness when walking up hill or a flight of stairs) (d). Percentages of ILC subsets based on cytokine staining, expressed as the % of CD161+ ILC (e). Percentage of Lin+CD56−CD3+ lymphocyte subsets based on cytokine staining, expressed as the % of CD45+, live cells: IFN‐γ+IL‐17− T, IFN‐γ−IL‐17−IL‐13+ T and IFN‐γ−IL‐17+ T (f). Spearman’s correlation between % of ILC1 and % of IFN‐γ+ILC (g) and % of IL‐13+ILC and FEV1% predicted (h) in control subjects (clear dots) and COPD patients (black dots). Group sizes: GOLD I = 3 or 4; GOLD II = 8 or 9; GOLD III = 2, never‐smoking controls = 3, ex‐smoking controls = 3, smoking controls = 3.
Table 1.
Subject characteristics
| Characteristics | Controls (n = 9) | COPD (n = 15) |
|---|---|---|
| Median (IQR) | Median (IQR) | |
| Gender (male/female) | 3/6 | 10/5 |
| Age, years | 62 (57–76) | 66 (59–69) |
| Body mass index | 25 (25–28) | 24(23–28) |
| Smoking status (never/current/ex) | 3/3/3 | 0/8/7 |
| COPD GOLD stage | na | I(4)/II(9)/III(2) |
| FEV1% predicted, post‐bronchodilator | 124 (114–126) | 79 (59–80.5) |
| FVC% predicted, post‐bronchodilator | 121.5 (103.5–136.3) | 99 (92–108) |
| FEV1/FVC, post‐bronchodilator | 79 (77–87) | 63 (50–65.5) |
| COPD Assessment Test score | 3 (2–8) | 13 (10.3–18.3) |
| DLCO | 87.9 (84–104) | 67.5 (56.8–75.5) |
na, not applicable; FEV1, forced expiratory volume in 1s; FVC, forced vital capacity; DLCO, diffusing capacity of the lungs for carbon monoxide.
To investigate whether any of all of the ILC subsets were associated with disease severity, a Spearman correlation analysis was performed between ILC subsets and clinical and functional markers of COPD severity. The frequency of ILC1 tended to inversely correlate with FEV1/FVC% (as marker of airflow limitation) and with diffusing lung capacity of carbon monoxide (DLCO, a marker of emphysema and gas exchange) (Figure 1d). The frequency of ILC1 positively correlated with total score in the COPD Assessment Test (CAT); the higher the CAT score, the larger the impact of COPD on a patient’s health status. In addition, regression analysis demonstrated that ILC1 frequency significantly correlated with an indicator of dyspnoea (breathlessness) in the CAT test (Figure 1d), even when adjusted for smoking status (Supplementary table 2).
Using lung single‐cell suspensions from the same cohort, we evaluated intracellular cytokine production within ILCs (gating strategy Figure 1a) and CD3+T cells by stimulating the cells with phorbol myristate acetate (PMA)/ionomycin, followed by flow cytometric analysis. Similar to the data from the surface marker staining, the IFN‐γ+ ILCs represented the largest ILC subset, in both COPD and control subjects (Figure 1e). The CD3+IFN‐γ+ T‐cell frequencies were similar in never‐smoking controls and COPD but tended to increase in smoking controls (Figure 1f). ILC1 frequency identified with surface marker staining strongly correlated with the frequency of IFN‐γ+ILCs (Figure 1g). In COPD patients, IL‐13+ILC frequency positively associated with FEV1% predicted (post‐bronchodilator) (Figure 1h). No significant associations between the frequencies of the other ILC subsets and these clinical parameters were found (data not shown).
Pulmonary ILC localise near lymphoid aggregates in COPD
Next, we determined the presence and localisation of all three ILC subsets, in lung tissue samples of COPD patients and controls using immunohistochemical analysis. ILCs were identified as CD3− cells, positive for the transcription factor T‐BET (ILC1), GATA3 (ILC2) or RORγt (ILC3). B cells, NK cells, macrophages and mast cells were excluded by positivity for CD20, CD56, CD68 and mast cell tryptase, respectively. 15 In assays using isotype controls for the antibody combinations, no staining was observed (Supplementary figure 4). Despite being rare cells, all three ILC subsets could be identified in the same lung tissue. In never‐smoking control lung tissue, ILCs localised in the parenchyma. In lung tissue from smoking controls and COPD patients (GOLD stage II), they localised in and near lymphoid aggregates (Figure 2). Also, the three T‐helper subsets (Th1, Th2 and Th17) could be identified. Using immunofluorescence, which allows visualisation of antibody staining in different channels separately, the presence of the three ILC subsets in the lung was confirmed (Figure 3a–c, Supplementary figures 5, 6 and 7 for isotype staining).
Figure 2.
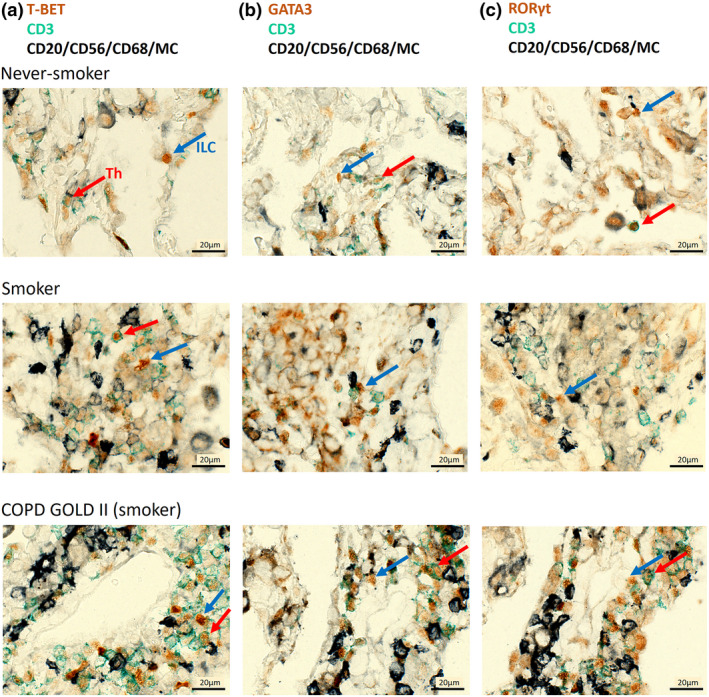
Immunohistochemical detection of pulmonary ILC and Th cells in COPD patients and controls. T‐BET/GATA3/RORγt in brown, CD3 in green and antibodies against CD20, CD56, CD68 and mast cell tryptase (exclusion cocktail) in black. Staining was performed in COPD GOLD II patients and smoking and never‐smoking controls. T‐BET+CD3−ILC1 (in brown, blue arrow) and T‐BET+CD3+Th1 cells (in brown with green border, red arrow) negative for exclusion markers (a). GATA3+CD3−ILC2 (blue arrow) and GATA3+CD3+Th2 cells (red arrow) negative for exclusion markers (b). RORγt+CD3−ILC3 (blue arrow) and RORγt +CD3+Th17 cells (red arrow) negative for exclusion markers (c). Scale bar: 20 µm.
Figure 3.
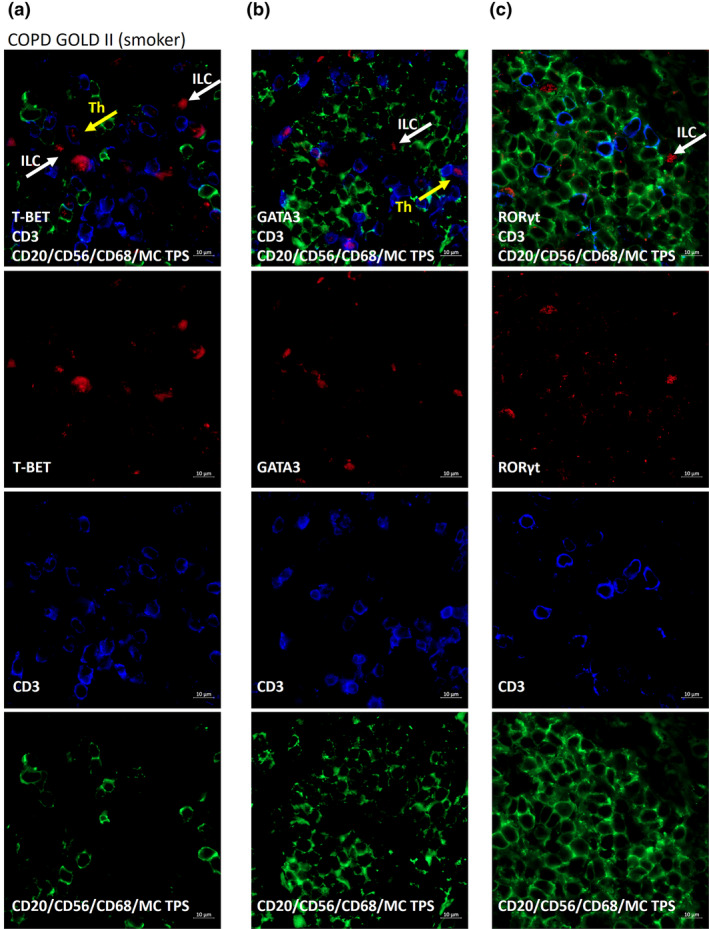
Immunofluorescent detection of pulmonary ILC and Th cells in COPD patients. T‐BET/GATA3/RORγt (red), CD3 (blue), antibodies against CD20, CD56, CD68 and mast cell tryptase (MC TPS) (exclusion cocktail, green). Staining was performed on a COPD GOLD II patient (smoker). T‐BET+CD3−ILC1 (red, white arrow), negative for exclusion markers and T‐BET+CD3+Th1 (red with blue border, yellow arrow) (a). GATA3+CD3−ILC2 (white arrow) and GATA3+CD3+Th2 cell (yellow arrow) negative for exclusion markers (b). RORγt+CD3−ILC3 (white arrow) negative for exclusion markers (c).
CS induces increases in ILCs in a mouse model of COPD
Given that ILCs are an important source of cytokines associated with COPD pathogenesis, 1 and mechanistic studies with human samples have not yet been performed, we investigated whether the total number of ILCs and the different ILC subsets were altered in a COPD mouse model. 19 C57BL/6 mice were exposed to air or CS for 1, 4 or 24 weeks, followed by the evaluation of ILCs in the bronchoalveolar lavage (BAL) fluid and lung tissue. ILC subsets were identified according to the expression of surface markers and specific transcription factors (gating strategy Figure 4a, Supplementary figure 8 for FMO controls): T‐BET+ILC, T‐BET−RORγt−ILC and RORγt+ILC represent ILC1, ILC2 and ILC3, respectively. Notably, CS exposure induced significant increases in the total lineage− CD90+ILCs, as well as all ILC subsets in BAL, at all time points (Figure 4b, c). Also, the % of CD90+ILCs increased within the CD45+ population (Supplementary figure 8b). Within CD90+ILCs, the % ILC2 reduced upon CS exposure, whereas the % of ILC1 increased, after 4 and 24 weeks (Figure 4d). CS exposure also increased the numbers of ST2+ILC2 in BAL, but to a much lower level (Supplementary figure 9). In (lavaged) digested lung tissue, data on ILC frequencies upon CS exposure were inconclusive (not shown).
Figure 4.
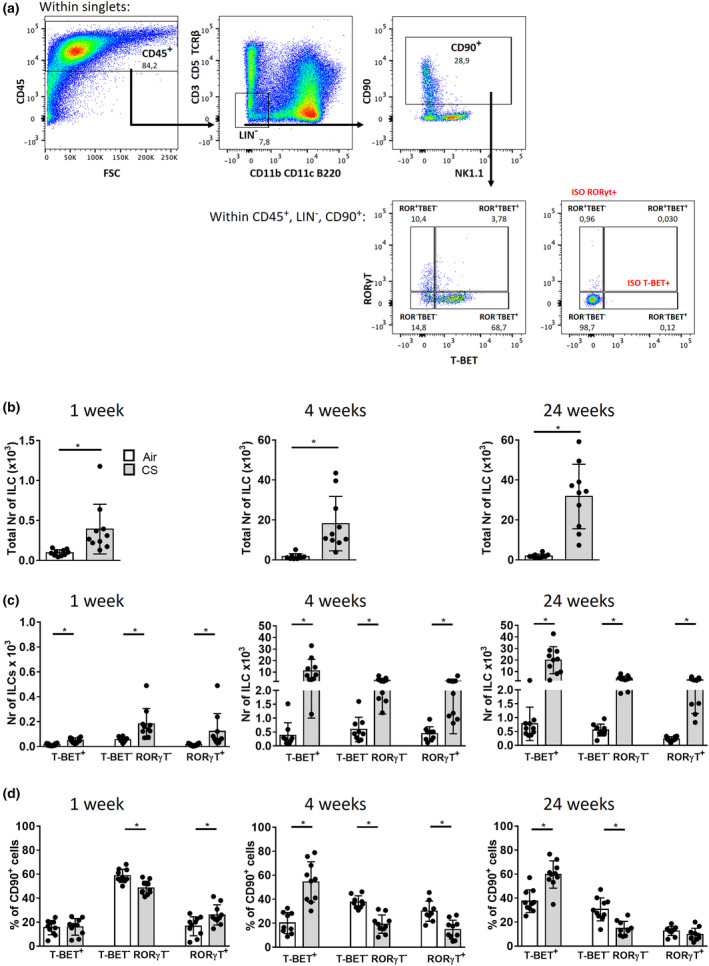
All pulmonary ILC subsets are increased following cigarette smoke exposure in a mouse model. Total ILCs were gated as CD45+Lin− (CD3−CD5−TCR‐β−) Lin−(CD11b−CD11c−B220−) CD90+. ILC subsets were subsequently defined as RORγt−T‐BET+ ILC1, RORγt−T‐BET− ILC2 and RORγt+ T‐BET− ILC3 (a). Total number of ILC (b), ILC subsets (c) and % of ILC within CD45+Lin−Lin− CD90+ cells in mice exposed to CS (grey bars) or air (white bars) for 5 days or 4 or 24 weeks. Results are shown as mean values; error bars represent the standard deviation. N = 10 mice per group. *P < 0.05. Data from the 4‐week experiments are representative of 2 experiments.
Signature cytokine expression upon CS exposure was evaluated in ILC (gating strategy Figure 5a) and in Th cells in BAL fluid. All cytokine‐producing ILC subsets and corresponding Th subsets were increased in BAL following 4 and 24 weeks of CS exposure (Figure 5b–e). Within the CD90+ILC population, there was a predominant increase in the % of IFN‐γ+ILCs both after 4 and 24 weeks (Supplementary figure 10).
Figure 5.
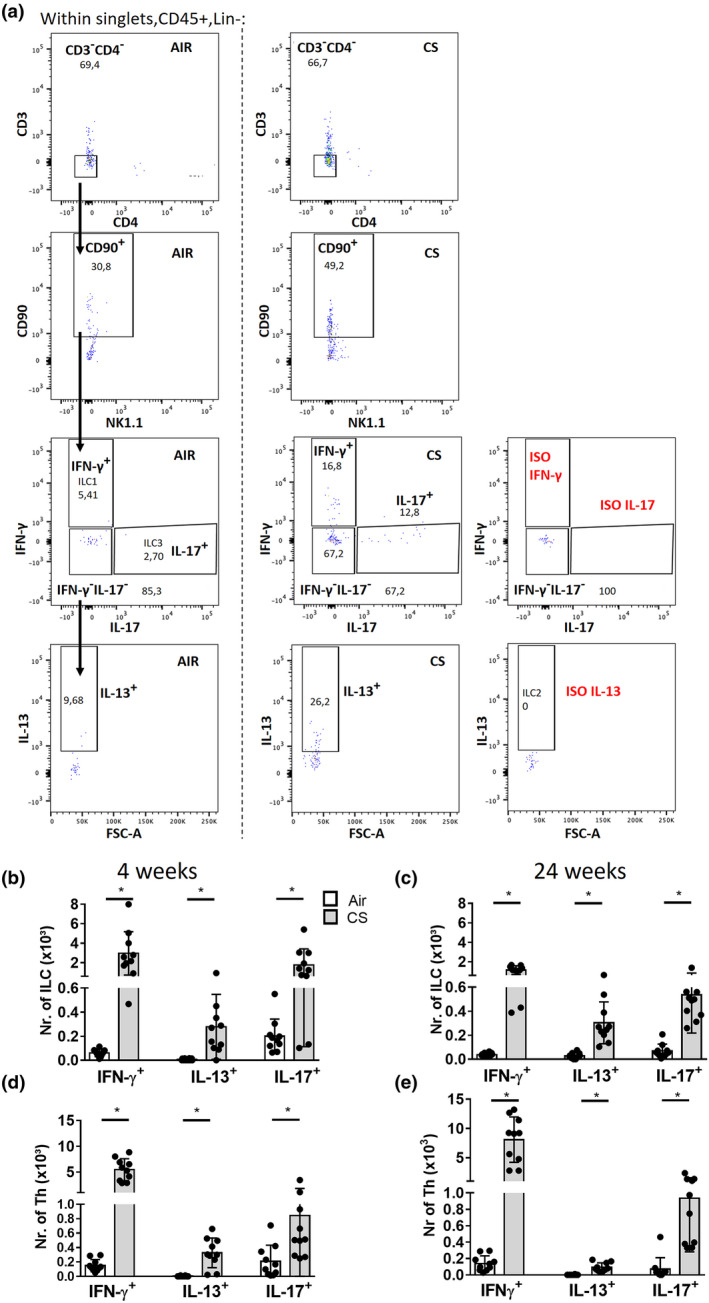
Signature cytokine expression in ILC and Th cells following cigarette smoke exposure in a mouse model. ILC gating following 4 h of stimulation: CD45+Lin−(CD5−TCR‐β−)Lin−(CD11b−CD11c−B220−)CD3−CD4−CD90+ ILCs were gated, followed by defining IFN‐γ+IL‐17−ILC1, IFN‐γ−IL‐17−IL‐13+ILC2 and IFN‐γ−IL‐17+ILC3 (a). Total numbers of cytokine‐producing ILC (b, c) and Th subsets (CD45+, CD11b−CD11c−B220−, CD5+TCR‐β+, CD3+CD4+) (d, e) in mice exposed to CS (grey bars) or air (white bars) for 4 or 24 weeks. Results are shown as mean values; error bars represent the standard deviation. N = 10 mice per group. *P < 0.05. Data of the 4‐week experiments are representative of 2 experiments.
Also, other inflammatory cells were evaluated in the COPD model. As reported previously, a significant increase in the total numbers of inflammatory cells in BAL was observed in CS‐exposed mice after 4 and 24 weeks (Supplementary figure 11a). At all time points, neutrophils, monocytes and dendritic cells were increased in BAL following CS exposure. Lymphoid aggregates were observed after 24 weeks (Supplementary figure 11b–d).
ILC deficiency in Rag2 −/− mice does not prevent CS‐induced innate inflammation
The total numbers of activated ILCs increased in the COPD model (Figure 5), and we previously demonstrated that CS‐induced innate inflammation is still present in scid mice 18 that lack B‐ and T‐lymphocytes but still have ILC. Thus, we hypothesised that ILC, as an important source of IFN‐γ, IL‐13 or IL‐17, could drive CS‐induced inflammatory responses. To evaluate how the additional absence of ILCs would affect CS‐induced inflammation, we exposed WT (C57BL/6NTac background), Rag2 −/− and Rag2/Il2rg −/− mice to air or CS for 4 weeks. Whereas ILCs are present in BAL fluid of Rag2 −/− mice that lack an adaptive immune system, they are absent in Rag2/Il2rg −/− mice (Figure 6a). In air‐exposed Rag2 −/− mice, there were no Th cells, but the numbers of ILCs, monocytes and dendritic cells were higher than in air‐exposed WT mice (Figure 6a, b, d and e). CS exposure of Rag2 −/− mice increased the numbers of total BAL cells, ILCs, dendritic cells and neutrophils (Figure 6a, c, e and f). In Rag2/Il2rg −/− mice, CS‐induced increases in total BAL cells, monocytes, dendritic cells and neutrophils were also present (Figure 6c–f). Hence, ILC deficiency in the absence of a functional adaptive immune system does not prevent CS‐induced innate inflammation.
Figure 6.
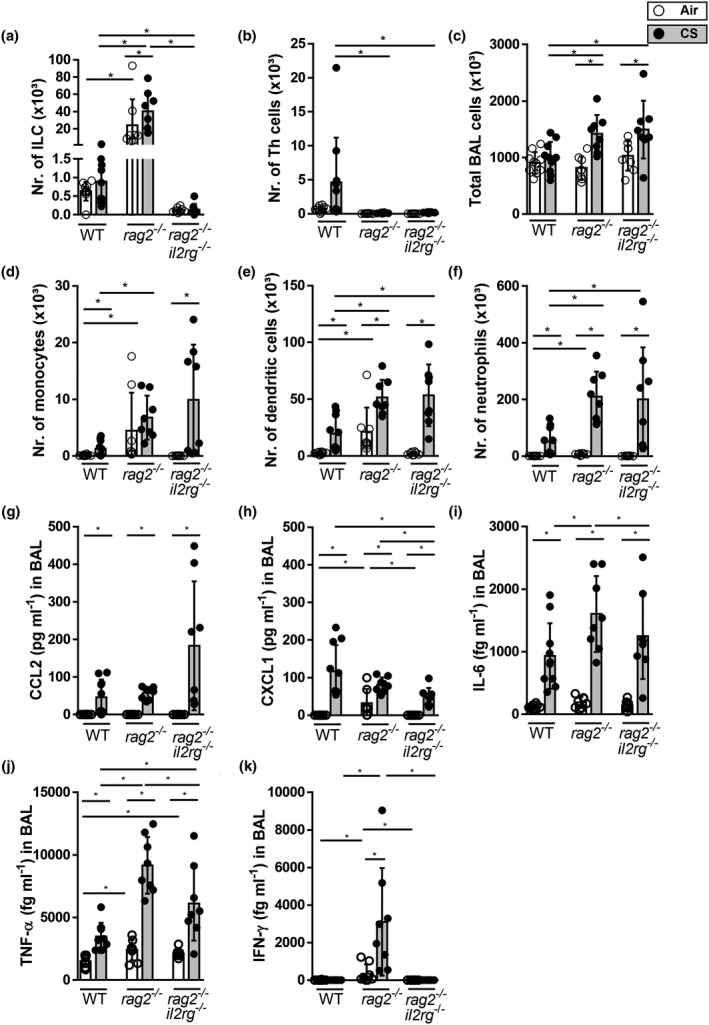
In the absence of adaptive immunity, ILC deficiency does not prevent CS‐induced airway inflammation in a mouse model. C57BL/6NTac, Rag2 −/− and Rag2/Il2rg −/− mice were exposed to CS (grey bars) or air (white bars) for 4 weeks. Total numbers of ILCs (a) and Th cells (b). Total number of BAL cells (c) monocytes, (d) dendritic cells (e) and neutrophils (f). Protein levels of CCL2 (g), CXCL1 (h), IL‐6 (i), TNF‐α (j) and IFN‐γ (k) were determined in BAL fluid. Results are expressed as mean ± SD. N = 8–10 mice per group. *P < 0.05.
The measurement of pro‐inflammatory cytokines and chemokines in BAL fluid demonstrated that CS exposure significantly induced CCL2 (MCP‐1), CXCL1 (KC), IL‐6 and TNF‐α protein levels in WT mice, and Rag2 −/− and Rag2/Il2rg −/− mice compared to air exposure (Figure 6g, h–j). CS‐induced IL‐6, TNF‐α and CXCL1 levels were significantly higher in Rag2−/− mice than in Rag2/Il2rg −/− mice. CS exposure also increased IFN‐γ levels in Rag2 −/− mice, whereas IFN‐γ levels were below the detection limit in both WT‐ and Rag2/Il2rg −/−‐deficient mice (Figure 6k). Thus, although CS‐induced pro‐inflammatory mediators were produced by the three mouse strains, the levels of several cytokines in Rag2 −/− mice were reduced with ILC deficiency.
In lung tissue, the numbers of neutrophils close to the airways were elevated upon CS exposure in all three mouse strains (Figure 7a and b). To evaluate the potential impact of the absence of ILCs on tissue destruction, MMP‐12 mRNA levels in lung tissue were evaluated. CS exposure induced increases in MMP‐12 mRNA levels in the three strains (Figure 7c). MMP‐12 expression was higher in the CS‐exposed Rag2 −/− mice than in the CS‐exposed WT mice. MMP‐12 mRNA levels in the Rag2/Il2rg −/− mice tended to attenuate compared to Rag2 −/− mice.
Figure 7.
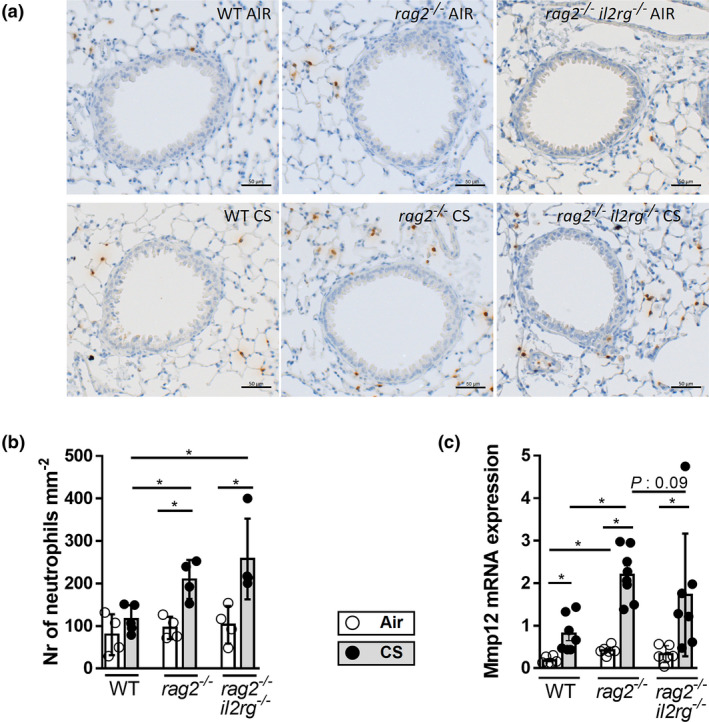
FIn the absence of adaptive immunity, ILC deficiency does not prevent CS‐induced neutrophilic inflammation or MMP‐12 expression in murine lung tissue. C57BL/6NTac, Rag2 −/− and Rag2/Il2rg −/− mice were exposed to CS (grey bars) or air (white bars) for 4 weeks. Myeloperoxidase (MPO)‐positive neutrophils in lung tissue are stained in brown (a) and quantified (numbers mm−2). N = 4 or 5 mice per group (b). MMP‐12 mRNA expression in lung tissue (c). Expression was determined by RT‐PCR and normalised to HPRT and GADPH. N = 6 or 7 mice per group. Results are expressed as mean ± SD. *P < 0.05. Scale bar: 50 µm.
Discussion
Here, we show that ILC1 frequencies in the pulmonary ILC population are elevated in COPD patients and correlate with smoking and severity of respiratory symptoms. We also localised the three ILC subsets in human tissue, particularly close to or in lymphoid aggregates. We also found that CS exposure significantly increased the total numbers of all three ILC subsets, with relative increases in ILC1 frequencies, in an established murine model of COPD. 18 , 19 , 20 However, ILCs appear to be redundant for CS‐induced innate inflammatory responses in the absence of a functional adaptive immune system, given that innate inflammation still occurs, irrespective of the presence of ILC.
Through a combined histological and flow cytometric approach, we assessed ILC subsets in COPD patients. We and others previously reported the presence of ILCs in human lung using flow cytometry. 14 , 15 , 21 The added value of our current findings is that quantification was performed within a more specific cell population (CD127+ and CD161+) on a high number of precious human lung samples and that we correlated ILC numbers with the severity of respiratory symptoms. Moreover, we simultaneously investigated the three signature cytokines in the pulmonary CD161+ ILC population. Pulmonary ILC1 was previously reported to be increased in severe COPD GOLD IV compared to COPD GOLD I‐II subjects and controls. 15 This agrees with our current observations, where we observe an increase in ILC1 in the lungs of COPD GOLD I‐III subjects compared to controls. Notably, ILC1 frequencies seem to be higher in (ex/current) smoking controls than in never‐smoking controls. In these ex‐ and current smokers, who have no features of COPD, several pathogenic processes that could ultimately lead to COPD may already be ongoing. However, the difference between latter two groups was not significant, possibly because of low sample numbers and/ or pronounced smoking effects. A limitation of our study is that the number of pack‐years of (ex and current) smoking controls was lower than in COPD patients (Supplementary table 1) and that sample sizes between groups differed. A larger sample size is needed to confirm statistical significance. The variation in ILC distribution in the COPD group does not seem to be influenced by disease severity (GOLD status, Supplementary figure 3a), but also requires further study. We demonstrate for the first time that pulmonary ILC1 frequency correlates with severity of respiratory symptoms (COPD Assessment Test [CAT] score question 4, an indicator of dyspnoea (breathlessness)). The fact that such a trend already arises in this small‐scale cohort is striking. Future analysis on a larger cohort needs to confirm these findings; however, flow cytometric analyses on fresh human lung tissue is technically difficult and logistically demanding. In the current study, 15 COPD subjects with stable disease were included, to exclude any changes in ILC populations that may occur because of bacterial or viral infections. Possibly, the association between ILC subsets and some clinical parameters would be even more clear during COPD exacerbation. Such lung samples are difficult to obtain, since lung resections for tumors are not performed during an acute exacerbation of COPD. Notably, higher ILC1 frequencies in the peripheral blood of COPD patients correlate with increased susceptibility to exacerbations, poor lung function and disease severity. 16 Furthermore, ILC1 frequencies identified with surface marker staining strongly correlated with the frequency of IFN‐γ‐producing ILCs. Interestingly, CD8 IFN‐γ production has been correlated with COPD severity 7 and circulating IFN‐γ‐producing Th17/Th1 cells have been negatively correlated with FEV1% in COPD patients. 22
In COPD lungs, the IL‐13+ILC frequency positively associated with FEV1% predicted. Circulating ILC2 frequencies (CRTH2+T‐BET−) have previously been correlated with higher lung function (FEV1% predicted and FEV1/FVC%), suggesting that this association with ILC2 is mediated by IL‐13 production. 16 Since the frequency of IL‐13+ILCs was relatively low and ILC2, which are a major source of IL‐5 in the lung, is reported to produce more IL‐5 than IL‐13, it would be of interest to further investigate whether the numbers of pulmonary IL‐5+ILC2 are associated with smoking and respiratory symptoms. In a previous study in a small cohort, we did not find apparent changes in the frequency of IL‐5+ILC2 between controls and COPD. 14 Whether the frequency of IL‐22+ILCs is altered in COPD also requires further investigation.
There is much debate regarding ILC frequencies in the human lung. Monticelli et al. 23 described ILC2 as the predominant ILC population, whereas Bal et al. 15 reported that NKp44+ILC3 was the predominant population (38.2%), followed by ILC2 (29.5%) and ILC1 (10.8%) in healthy and GOLD I‐II subjects. Another study reported that NK cells represent the majority of the ILC population, whereas ILC2 and ILC3 are a minority. 24 The heterogeneity in subjects, processing of the lung samples and the markers used for ILC gating all contribute to data variability. A limitation of our study is that we lack a specific human ILC1 maker, but a similar approach has been used by other groups. 15 Still, we identified CD161+CD127+T‐BET+CD56− cells in our cohort, which increased in COPD. Controversy in reported ILC frequencies may also originate from ILC plasticity. Silver et al. showed that ILC2 displays plasticity and can transform into IFN‐γ‐producing ILC1 upon CS exposure, which is mediated by IL‐12. 16 In addition, TGF‐β can promote the conversion of a subgroup of c‐Kit−ILC2 into IL‐17‐producing ILC3, 25 which could contribute to IL‐17‐mediated pathologies. In our cohort, ILC1 expansion is associated with a reduction in the NKp44−ILC3 population, but is also linked to a reduction in undefined CD161+CD127+ cells, a ‘rest’ population that could contain NK cells, that is pro‐inflammatory CD161+CD56+ NK cells. 26 Also, the relative contribution of ILC2 to the ILC population in the human lung tended to be lower in smokers and in people with COPD than in never smokers, which could support the findings of Silver et al. 16
This is the first study to visualise all three ILC subsets simultaneously in COPD patient lungs. Recently, GATA3+ILC2 was detected within eosinophil foci in the lungs of COPD GOLD IV subjects. 27 In another study, RORγt+NRP1+ILC3 was detected near pulmonary inflammatory aggregates and blood vessels, suggesting a link with lymphoid follicle formation. 21 Interestingly, we detected not only RORγt+ILC3, but also T‐BET+ILC1 and GATA3+ILC2 near lymphoid aggregates.
In our murine COPD model, the increase in ILC1 (in total number and frequency within the CD90+ILC population) was also observed after 4 and 24 weeks of CS exposure in C57BL/6 mice. In addition to increases in T‐BET+ILC1, the numbers of RORγt+ILCs were also increased in BAL upon acute and chronic CS exposure. This agrees with Silver et al. and Donovan et al. who showed elevation of T‐BET+ILCs in the lung and in T‐BET+ILCs and RORγt+ILCs after 8 weeks of CS exposure, respectively. 16 , 17 Also, the numbers of IFN‐γ‐ and IL‐17‐producing ILC and Th subsets were elevated, which corresponds with Th1/Th17 responses associated with COPD. 5 , 28 Within the CD90+ILC population, ILC1 frequencies were particularly elevated. CD90 is often used as an ILC marker 29 , 30 and is the most stable marker on ILC2. 31 Still some ILC populations are CD90+/−, 32 which were not included in our gating and analyses. The numbers of ST2+ILC2 also increased with CS exposure, but were very low. The handling of the samples or analysed compartment (BAL) may have influenced ST2 expression on ILC2. Moreover, different ILC2 subsets exist (natural and inflammatory) and the latter have downregulated ST2 33 and CS exposure can reduce ST2 expression on ILC2. 16 Also, the number of identified IL‐13+ ILC2 was low. Possibly, the experimental approach of ex vivo stimulation and antibody staining – which is rather artificial and dependent on antibody efficiency – has impacted on our data. Indeed, by using IL‐5 and IL‐13 reporter mice, others have shown high numbers of Lin−TCR− ILCs within IL‐5+ or IL‐13+ CD45+ lymphocytes in the lung. 34 Notably, latter study also revealed important differences in murine pulmonary ILC2 numbers depending on age, sex and mouse strain, which could have influenced our findings. ILC2 numbers were shown to be lower in adult mice than in neonatal mice, in C57BL/6 mice than in BALB/C and in male mice than in female mice. 34
Previously our group showed that CS‐induced innate inflammation is still present in scid mice, which lack an adaptive immune system. We hypothesised that ILC, which can produce IFN‐γ, IL‐13 and IL‐17, drives this CS‐induced innate inflammatory response. Thus, we assessed the contribution of ILC to CS‐induced innate inflammation, by comparing inflammatory responses in WT, Rag2 −/− and Rag2/Il2rg −/− mice. Notably, the CS‐induced inflammation in WT mice (C57BL/6NTac background) was not as profound as in WT mice (C57BL/6J background) in our other experiments, which is possibly related to the minor differences in the genetic backgrounds. However, similar to previous reports, 17 , 18 , 35 we demonstrate that Rag2 −/− mice have CS‐induced inflammatory responses, with elevated numbers of total BAL cells and neutrophils. Importantly, in Rag2/Il2rg −/− mice, CS‐induced increases in total BAL cells, neutrophils, monocytes and dendritic cells were still present, implicating that ILC deficiency (in the absence of functional B and T cells) does not prevent CS‐induced innate inflammation. Accordingly, CS exposure induced several pro‐inflammatory mediators such as CCL2 (monocyte chemoattractant), CXCL1 (neutrophil attractant), IL‐6 and TNF‐α not only in WT mice, but also in Rag2 −/− and Rag2/Il2rg −/− mice. Interestingly, CS‐induced IL‐6, TNF‐α and CXCL1 levels were significantly higher in Rag2 −/− mice than in Rag2/Il2rg −/− mice and CS‐induced IFN‐γ was only detectable in the former. These findings suggest that ILCs may be important in CS‐induced IL‐6, TNF‐α, CXCL1 and IFN‐γ release in Rag2 −/− mice and may therefore have roles in pro‐inflammatory responses to CS exposure. Further research is needed to explore this. A limitation of our study is that these experiments were performed in a Rag2 −/− background, where compensatory mechanisms related to the absence of T and B cells likely take place. These mice have significantly elevated ILC numbers and other inflammatory cells at baseline, which may have impacted on our analyses. To further elucidate the role of ILCs in CS‐induced innate inflammation in our COPD model, specific ILC1‐ and ILC3‐deficient mice are needed. To our knowledge, such mice are not yet available.
Our data do not exclude roles for ILCs in emphysema, lymphoid follicle formation or airway remodelling in COPD. We found that both Rag2 −/− and Rag2/Il2rg −/− mice have CS‐induced MMP‐12 expression, suggesting that tissue destruction could still occur in the absence of ILCs. Notably, MMP‐12 mRNA levels tended to be lower in Rag2/Il2rg mice than in Rag2 −/− mice, suggesting that ILC could contribute to tissue destruction. Others showed that ILC2 deficiency does not affect CS‐induced inflammatory responses, but leads to elevated collagen deposition around the small airways and less emphysema. 17 Our group showed that emphysema can still develop in scid mice, that is in the absence of an adaptive immune system. 18 We and others have also shown that B cells and especially B‐cell lymphoid follicles may contribute to the development of CS‐induced emphysema. 20 , 27 , 36 , 37 It remains to be elucidated whether ILCs, particularly ILC1 and ILC3, are implicated in airway remodelling, emphysema or lymphoid follicle neogenesis. In the Rag2/Il2rg −/− mice, the 4‐week CS exposure was too short to study emphysema, nor is this strain suitable to evaluate lymphoid follicle neogenesis because of the absence of B and T cells. Thus, specific ILC‐deficient mice are again needed.
Silencing of ILC function is associated with exaggerated type 1 antiviral responses, 16 and ILC depletion impairs lung tissue repair and function during influenza virus infection. 10 Hence, in these situations, changes in ILC populations and functions do have a notable effect on lung pathology. This suggests that ILCs could be more important in COPD in, for example, exacerbations, which should be investigated further. It is likely that ILC and Th cell populations co‐exist to ensure immune robustness. 38
In conclusion, we showed that ILC1 frequency is increased in COPD patients and is associated with smoking and severity of respiratory symptoms. Moreover, we localised all three ILC subsets in and near lymphoid aggregates in COPD lungs. We also demonstrated that ILC numbers increase with CS exposure in a COPD model in C57BL/6 mice, with relative increases in the ILC1 subset. In the absence of adaptive immunity, ILC contributes to CS‐induced pro‐inflammatory mediator release, but they appear to be redundant in CS‐induced innate inflammation.
Methods
Analysis of human samples
Study population: Subject characteristics are summarised in Table 1. Lung resection tissue was obtained from subjects who underwent lobectomy for solitary pulmonary tumors. Tissue at maximum distance of the pathologic lung lesion was collected and declared free of signs of pneumonia or tumor invasion. Subjects receiving chemo‐ or radiotherapy or suffering from recent COPD exacerbations or infections were excluded. Lung tissue from 9 control subjects (3 never smokers, 3 ex‐smokers and 3 smokers, all without airflow limitation) and 15 COPD patients (4 GOLD I, 9 GOLD II and 2 GOLD III) was used for flow cytometric analysis. Two COPD GOLD II smoking patients, 2 smoking controls and 2 never smokers were selected for IHC analysis. Ex‐smokers were defined as subjects who refrained from smoking for ≤1 year. A COPD Assessment Test was performed prior to surgery. Smoking habits and medication use were evaluated by interview, while COPD diagnosis was made based on preoperative spirometry. Written informed consent was obtained from all donors, according to the protocol approved by the Medical Ethical Committee of Ghent University Hospital (2016/0132).
Sample processing and flow cytometry: Samples of fresh lung resection tissue were fixed in paraformaldehyde 4% (Sigma, Bornem) for 20 h and paraffin‐embedded for later immunohistochemical/immunofluorescent (IHC) analysis. Remaining fresh lung tissue was processed into cell suspensions as described previously. 14 Cell counting was performed with the Coulter Counter (Beckman Coulter, Inc., Fullerton, CA), and cells were stained for flow cytometry. Non‐specific binding was reduced by pre‐incubating the samples with human IgG. The LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Thermo Fisher, Waltham, MA) was used to exclude dead cells. To exclude lineage markers, the following human monoclonal antibodies were used: peridinin chlorophyll protein–cyanine 5.5 (PerCP)‐conjugated anti‐CD1a (HI49), anti‐CD3 (OKT3), anti‐CD14 (63D3), anti‐CD19 (HIB19), anti‐CD11c (3.9), anti‐CD11b (M1/70), anti‐CD123 (6H6), anti‐CD34 (581), anti‐TCRα/β (IP26), anti‐TCRγ/δ (B1), anti‐BDCA2 (201A), anti‐FCεR1 (AER‐37) and anti‐CD235A (HI264).
To distinguish ILC subsets based on surface staining, the following antibodies were used: fluorescein isothiocyanate (FITC)‐conjugated anti‐CD45 (HI30), allophycocyanin (APC)‐conjugated anti‐NKp44 (P44‐8), phycoerythrin (PE)/indotricarbocyanine (Cy7)‐conjugated anti‐CD117 (104D2), Brilliant Violet 421TM‐conjugated anti‐CD127 (A019D5), Brilliant Violet 605TM‐conjugated anti‐CD56 (HCD56) (all from BioLegend, San Diego, CA), PE‐conjugated CD161 (HP3610, eBioscience) and biotinylated anti‐CRTH2 (BM19; Miltenyi Biotec, Bergisch Gladbach, Germany) in combination with streptavidin‐APC‐Cy7 (BD Biosciences, San Jose, CA).
To identify ILC subsets based on cytokine staining, the same PerCP‐conjugated lineage antibodies were used, except for CD3, which was replaced by APC‐Cy7‐conjugated anti‐CD3 (SK7). In addition, FITC‐conjugated anti‐CD45 (HI30), BV605‐conjugated anti‐CD56, APC‐conjugated anti‐CD161 (HP‐3G10, all from BioLegend), PE‐TR‐conjugated anti‐IFN‐γ (EB 45.B3), PE‐conjugated anti‐IL‐13 (EB 85BRD) and PECy7‐conjugated anti‐IL‐17 (EB eBio64DEC17, all from eBioscience, San Diego, CA) were used.
Immunohistochemical/immunofluorescence analysis: Innate lymphoid cell was visualised using specific antibodies against CD3 (Bio‐Rad, Hercules, CA), CD20 (Invitrogen, Rockford, IL), CD68 (Invitrogen), CD56 (Novus Biologicals, Abingdon) and mast cell tryptase (Abcam, Cambridge) in combination with T‐BET (Santa Cruz, Dallas, TX), GATA3 (Biocare Medical, Pacheco, CA) or RORγt (Millipore, County Cork). ILC populations were defined as CD3−T‐BET+(ILC1), CD3−GATA3+(ILC2) and CD3−RORγt+(ILC3), and negative for CD20, CD68, CD56 and mast cell tryptase (deep space black chromogen exclusion).
Data analysis: Statistical analysis was performed with SPSS 24.0 and 26.0 (SPSS Inc. Chicago, IL). Non‐parametric tests (Kruskal–Wallis and Mann–Whitney U) were used to compare independent groups, according to standard statistical criteria. Values were reported as median ± IQR. P‐values < 0.05 (*) were considered statistically significant. Linear regression analyses were performed for the association between CAT scores or lung function and ILC1. GraphPad Prism (GraphPad Software Inc., San Diego, CA) was used to analyse data.
Murine experiments
Mice: Male C57BL/6J mice (8–9 weeks old) were purchased from Charles River (Ecully). To investigate the functional role of ILCs, C57BL/6NTac, Rag2 −/− and Rag2/Il2rg −/−mice were purchased from Taconic (Germantown, NY). Experimental protocols were approved by the Animal Ethical Committee of the Faculty of Medicine and Health Sciences (Ghent University, ECD 14/76 and 16/68) and were carried out in accordance with institutional guidelines for animal care.
Exposure protocol: Mice were exposed to mainstream tobacco smoke from 5 cigarettes (Standard Research Cigarette 3R4F, University of Kentucky, without filters), 4 times daily using a whole‐body exposure system as we have previously performed extensively. 19 , 20 , 36 There were 30‐minute smoke‐free intervals between each exposure. In the smoking chambers, an optimal smoke–air ratio of 1/6 was set. Control groups were exposed to air. This procedure was followed for 5 consecutive days/week for 1, 4 or 24 weeks. Analysis was performed the day after the last smoke exposure, when mice were sacrificed with pentobarbital overdose (Vétoquinol NV/sa, Aartselaar). Results of the 4‐week experiments are representative of ≥ 2 independent experiments.
Lung histology and bronchoalveolar lavage: Haematoxylin/eosin staining and CD3/B220 staining of lung sections, bronchoalveolar lavage, cell counts and RT‐PCR were performed as described previously. 19 , 39 , 40 , 41 Neutrophils on lung tissue were visualised with anti‐mouse myeloperoxidase (R&D Systems, Abingdon, UK) and counterstained with haematoxylin. Protein measurements in BAL were determined by ELISA (CCL2 and CXCL1, R&D Systems, Abingdon, UK) or using the High‐Sensitivity BD Cytometric Bead Array (BD Biosciences, San Jose, CA) according to the manufacturers’ instructions.
Flow cytometry: Cells were pre‐incubated with FcR blocking antibody to reduce non‐specific binding (anti‐CD16/CD32, Clone 2.4G2). All staining procedures were performed in PBS without Ca2+ and Mg2+ but containing 5 mM EDTA and 1% BSA. Cells were stained with a combination of fluorochrome‐conjugated monoclonal antibodies (surface markers) similar to 39 CD3 (145‐2C11), CD5 (53‐7.3), TCR‐β (H57‐597), CD4 (GK1.5), CD45 (30‐F11), CD11b (M1/70), CD11c (N418), CD45R (RA3‐6B2), CD90.2 (30‐H12), Ly6G (1A8), Siglec‐F (E50‐2440), NK1.1 (PK136), Ly6C (BD AL‐21), MHCII (BL M5/114.15.2) and biotinylated anti‐T1/ST2 (MD Bioproducts, St. Paul, MN) in combination with streptavidin–phycoerythrin (SAV‐PE) (BD Biosciences).
For cytokine staining, cells were stimulated for 4 h at 37°C and 5% CO2 with a cocktail of PMA and ionomycin, supplemented with brefeldin A and monensin (eBioscience). Cells were fixed with the Foxp3 transcription factor fixation/permeabilisation set and stained with antibodies against IFN‐γ (XMG1.2), IL‐13 (eBio13A) and IL‐17A (17B7) for cytokine staining and with T‐BET (EBio4B10), GATA3 (EB TWAJ) and RORγt (EB B2D) for nuclear staining (all eBioscience).
Flow cytometric analysis was performed using LSRFortessa, and data were assessed using FlowJo Software.
Conflicts of interest
TM reports grants personal fees from GlaxoSmithKline, outside the submitted work; and is shareholder from Oryzon Genomics and Mendelion Lifesciences SL; and holds a Chiesi chair on Environmental factors in Asthma. GGB reports personal fees from Astra Zeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, Sanofi and Teva, outside the submitted work. EEB, SP, EGDS, KCDG, HPVE, JDV, PMH, MB, MS, SW, FV, KRB and GFJ have no conflict of interest to declare.
Author contribution
Evy E Blomme: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Sharen Provoost: Conceptualization; Funding acquisition; Methodology; Supervision; Writing‐review & editing. Elise G De Smet: Investigation; Writing‐review & editing. Katrien C De Grove: Investigation; Methodology; Writing‐review & editing. Hannelore P Van Eeckhoutte: Investigation; Writing‐review & editing. Joyceline De Volder: Investigation; Visualization; Writing‐review & editing. Philip M Hansbro: Formal analysis; Writing‐review & editing. Matteo Bonato: Methodology; Writing‐review & editing. Marina Saetta: Formal analysis; Methodology; Writing‐review & editing. Sara Wijnant: Data curation; Methodology; Resources; Writing‐review & editing. Fien Verhamme: Data curation; Methodology; Resources; Writing‐review & editing. Guy F Joos: Conceptualization; Formal analysis; Methodology; Writing‐review & editing. Ken R Bracke: Data curation; Formal analysis; Methodology; Resources; Writing‐review & editing. Guy G Brusselle: Conceptualization; Formal analysis; Funding acquisition; Methodology; Resources; Writing‐review & editing. Tania Maes: Conceptualization; Formal analysis; Funding acquisition; Methodology; Project administration; Resources; Supervision; Validation; Writing‐review & editing.
Supporting information
Acknowledgments
The research described here was supported by the Fund for Scientific Research in Flanders (FWO Vlaanderen, G053516N); FWO‐EOS Project G0G2318N; and a Ghent University Grant (BOF19‐GOA‐008). The work of Sara Wijnant was funded by the Funds for Scientific Research Flanders (FWO Vlaanderen, 3G037618). EEB received an FWO Grant for a long stay abroad, which supported her research project in the Priority Research Centre for Healthy Lungs, Hunter Medical Research Institute, Newcastle. MB received an ERS short‐term research training fellowship 2015‐2016, which supported his internship in the Laboratory for Translational Research in Obstructive Pulmonary Diseases, Ghent. PMH is funded by a fellowship from the NHMRC (1175134) and by UTS. TM reports grants from Ghent University, grants from Fund for Scientific Research in Flanders, during the conduct of the study, and holds a Chiesi Chair on Environmental factors in asthma. The authors thank Ann Neesen, Indra De Borle, Katleen De Saedeleer, Anouck Goethals and Greet Barbier from the Laboratory for Translational Research in Obstructive Pulmonary Diseases, Department of Respiratory Medicine (Ghent University Hospital, Ghent, Belgium) for excellent technical assistance.
References
- 1. Starkey MR, McKenzie AN, Belz GT, Hansbro PM. Pulmonary group 2 innate lymphoid cells: surprises and challenges. Mucosal Immunol 2019; 12: 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vivier E, Artis D, Colonna M et al. Innate Lymphoid Cells: 10 Years On. Cell 2018; 174: 1054–1066. [DOI] [PubMed] [Google Scholar]
- 3. Artis D, Spits H. The biology of innate lymphoid cells. Nature 2015; 517: 293–301. [DOI] [PubMed] [Google Scholar]
- 4. Mjosberg J, Spits H. Human innate lymphoid cells. J Allergy Clin Immunol 2016; 138: 1265–1276. [DOI] [PubMed] [Google Scholar]
- 5. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011; 378: 1015–1026. [DOI] [PubMed] [Google Scholar]
- 6. Jones B, Donovan C, Liu G et al. Animal models of COPD: What do they tell us? Respirology 2017; 22: 21–32. [DOI] [PubMed] [Google Scholar]
- 7. Zhu X, Gadgil AS, Givelber R et al. Peripheral T cell functions correlate with the severity of chronic obstructive pulmonary disease. J Immunol 2009; 182: 3270–3277. [DOI] [PubMed] [Google Scholar]
- 8. Schoenborn JR, Wilson CB. Regulation of interferon‐γ during innate and adaptive immune responses. Adv Immunol 2007; 96: 41–101. [DOI] [PubMed] [Google Scholar]
- 9. Bafadhel M, Saha S, Siva R et al. Sputum IL‐5 concentration is associated with a sputum eosinophilia and attenuated by corticosteroid therapy in COPD. Respiration 2009; 78: 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Almansa R, Sanchez‐Garcia M, Herrero A et al. Host response cytokine signatures in viral and nonviral acute exacerbations of chronic obstructive pulmonary disease. J Interferon Cytokine Res 2011; 31: 409–413. [DOI] [PubMed] [Google Scholar]
- 11. Roos AB, Sethi S, Nikota J et al. IL‐17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 192: 428–437. [DOI] [PubMed] [Google Scholar]
- 12. Roos AB, Sandén C, Mori M, Bjermer L, Stampfli MR, Erjefält JS. IL‐17A is elevated in end‐stage chronic obstructive pulmonary disease and contributes to cigarette smoke‐induced lymphoid neogenesis. Am J Respir Crit Care Med 2015; 191: 1232–1241. [DOI] [PubMed] [Google Scholar]
- 13. Starkey MR, Plank MW, Casolari P et al. IL‐22 and its receptors are increased in human and experimental COPD and contribute to pathogenesis. Eur Respir J 2019; 54: 1800174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Grove KC, Provoost S, Verhamme FM et al. Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS One 2016; 11: e0145961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bal SM, Bernink JH, Nagasawa M et al. IL‐1β, IL‐4 and IL‐12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol 2016; 17: 636–645. [DOI] [PubMed] [Google Scholar]
- 16. Silver JS, Kearley J, Copenhaver AM et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol 2016; 17: 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Donovan C, Starkey MR, Kim RY et al. Roles for T/B lymphocytes and ILC2s in experimental chronic obstructive pulmonary disease. J Leukoc Biol 2019; 105: 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. D'Hulst AI, Maes T, Bracke KR et al. Cigarette smoke‐induced pulmonary emphysema in scid‐mice. Is the acquired immune system required? Respir Res 2005; 6: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D'Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke‐induced pulmonary inflammation in mice. Eur Respir J 2005; 26: 204–213. [DOI] [PubMed] [Google Scholar]
- 20. Bracke KR, Verhamme FM, Seys LJ et al. Role of CXCL13 in cigarette smoke‐induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188: 343–355. [DOI] [PubMed] [Google Scholar]
- 21. Shikhagaie MM, Bjorklund AK, Mjosberg J et al. Neuropilin‐1 Is expressed on lymphoid tissue residing LTi‐like group 3 innate lymphoid cells and associated with ectopic lymphoid aggregates. Cell Rep 2017; 18: 1761–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu W, Li R, Sun Y. Increased IFN‐γ‐producing Th17/Th1 cells and their association with lung function and current smoking status in patients with chronic obstructive pulmonary disease. BMC Pulm Med 2019; 19: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Monticelli LA, Sonnenberg GF, Abt MC et al. Innate lymphoid cells promote lung‐tissue homeostasis after infection with influenza virus. Nat Immunol 2011; 12: 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simoni Y, Fehlings M, Kloverpris HN et al. Human innate lymphoid cell subsets possess tissue‐type based heterogeneity in phenotype and frequency. Immunity 2017; 46: 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bernink JH, Ohne Y, Teunissen MBM et al. c‐Kit‐positive ILC2s exhibit an ILC3‐like signature that may contribute to IL‐17‐mediated pathologies. Nat Immunol 2019; 20: 992–1003. [DOI] [PubMed] [Google Scholar]
- 26. Kurioka A, Cosgrove C, Simoni Y et al. CD161 defines a functionally distinct subset of pro‐inflammatory natural killer cells. Front Immunol 2018; 9: 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jogdand P, Siddhuraj P, Mori M et al. Eosinophils, basophils and type 2 immune microenvironments in COPD‐affected lung tissue. Eur Respir J 2020; 55: 1900110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 2016; 138: 16–27. [DOI] [PubMed] [Google Scholar]
- 29. Li S, Heller JJ, Bostick JW et al. Ikaros inhibits group 3 innate lymphoid cell development and function by suppressing the Aryl hydrocarbon receptor pathway. Immunity 2016; 45: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seehus CR, Aliahmad P, de la Torre B et al. The development of innate lymphoid cells requires TOX‐dependent generation of a common innate lymphoid cell progenitor. Nat Immunol 2015; 16: 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Entwistle LJ, Gregory LG, Oliver RA, Branchett WJ, Puttur F, Lloyd CM. Pulmonary group 2 innate lymphoid cell phenotype is context specific: determining the effect of strain, location, and stimuli. Front Immunol 2019; 10: 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller MM, Patel PS, Bao K, Danhorn T, O'Connor BP, Reinhardt RL. BATF acts as an essential regulator of IL‐25‐responsive migratory ILC2 cell fate and function. Sci Immunol 2020; 5: eaay3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang Y, Guo L, Qiu J et al. IL‐25‐responsive, lineage‐negative KLRG1hi cells are multipotential 'inflammatory' type 2 innate lymphoid cells. Nat Immunol 2015; 16: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Loering S, Cameron GJM, Bhatt NP et al. Differences in pulmonary group 2 innate lymphoid cells are dependent on mouse age, sex and strain. Immunol Cell Biol 2021; 99: 542–551. [DOI] [PubMed] [Google Scholar]
- 35. Botelho FM, Gaschler GJ, Kianpour S et al. Innate immune processes are sufficient for driving cigarette smoke‐induced inflammation in mice. Am J Respir Cell Mol Biol 2010; 42: 394–403. [DOI] [PubMed] [Google Scholar]
- 36. Seys LJ, Verhamme FM, Schinwald A et al. Role of B Cell‐activating factor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 192: 706–718. [DOI] [PubMed] [Google Scholar]
- 37. Polverino F, Seys LJ, Bracke KR, Owen CA. B cells in chronic obstructive pulmonary disease: moving to center stage. Am J Physiol Lung Cell Mol Physiol 2016; 311: L687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vivier E, van de Pavert SA, Cooper MD, Belz GT. The evolution of innate lymphoid cells. Nat Immunol 2016; 17: 790–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. De Grove KC, Provoost S, Hendriks RW et al. Dysregulation of type 2 innate lymphoid cells and TH2 cells impairs pollutant‐induced allergic airway responses. J Allergy Clin Immunol 2017; 139: 246–257.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. De Grove KC, Provoost S, Braun H et al. IL‐33 signalling contributes to pollutant‐induced allergic airway inflammation. Clin Exp Allergy 2018; 48: 1665–1675. [DOI] [PubMed] [Google Scholar]
- 41. Van Hove CL, Maes T, Cataldo DD et al. Comparison of acute inflammatory and chronic structural asthma‐like responses between C57BL/6 and BALB/c mice. Int Arch Allergy Immunol 2009; 149: 195–207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials