

Abstract
Background
Basic helix-loop-helix (bHLH) is a superfamily of transcription factors that is widely found in plants and animals, and is the second largest transcription factor family in eukaryotes after MYB. They have been shown to be important regulatory components in tissue development and many different biological processes. However, no systemic analysis of the bHLH transcription factor family has yet been reported in Sorghum bicolor.
Results
We conducted the first genome-wide analysis of the bHLH transcription factor family of Sorghum bicolor and identified 174 SbbHLH genes. Phylogenetic analysis of SbbHLH proteins and 158 Arabidopsis thaliana bHLH proteins was performed to determine their homology. In addition, conserved motifs, gene structure, chromosomal spread, and gene duplication of SbbHLH genes were studied in depth. To further infer the phylogenetic mechanisms in the SbbHLH family, we constructed six comparative syntenic maps of S. bicolor associated with six representative species. Finally, we analyzed the gene-expression response and tissue-development characteristics of 12 typical SbbHLH genes in plants subjected to six different abiotic stresses. Gene expression during flower and fruit development was also examined.
Conclusions
This study is of great significance for functional identification and confirmation of the S. bicolor bHLH superfamily and for our understanding of the bHLH superfamily in higher plants.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-021-07652-9.
Keywords: Sorghum bicolor, bHLH gene family, Genome-wide analysis, Abiotic stress
Background
Transcription factors (TFs) play an important role in controlling plant growth and environmental adaptation [1, 2]. They regulate gene expression by combining with specific cis-promoter elements that specifically regulate certain genes or transcription rates, thereby playing a unique regulatory role in plant morphogenesis, cell-cycle processes, and the like [3, 4]. Structurally, the typical TF includes a DNA-binding site, a transcription-activation or repression domain, an oligomerization site, and a nuclear-localization site. TF genes, such as members of the bHLH, WRKY, MYB, bZIP and other TF families, constitute a high proportion of all plant genomes, and their target genes are widely involved in physiological processes, such as plant development and stress responses [5, 6].
Basic helix-loop-helix (bHLH) is a superfamily of TFs that is widely found in plants and animals; it is the second largest TF family among eukaryotic proteins after MYB [7, 8]. The first discovered bHLH family member was the c-myc proto-oncogene of avian myeloid cell carcinoma virus [9]. The bHLH TFs are so named because of their structural feature of a bHLH domain in all family members. The amino acid sequence of this domain is highly conserved. There are about 50 to 60 amino acid residues that can be divided into two regions based on their functions: a basic region and the HLH [9, 10]. The basic domain is located at the N terminus of the conserved domain of bHLH and contains about 15 amino acids. It can bind to the cis-acting element E-box (5′-canntg-3′). Therefore, the number of basic and key amino acid residues in the basic region determine whether the bHLH TF has DNA-binding activity. The HLH domain is distributed at the C terminus of the gene sequence, where two α-helices are connected by a low-conserved loop, which is essential for the formation of homodimers or heterodimers of bHLH TFs [11, 12, 13]. Based on their ability to bind DNA, bHLH TFs can be divided into two categories: DNA binding and non-DNA binding. These can be further divided into E-box binding and non-E-box binding. The most common method of E-box binding is G-box binding (5′-cacgtg-3′) [10, 14, 15]. According to Atchley et al. [10, 16], Glu and Arg at positions 9 and 13 of the basic region, namely E9 and R13, are essential amino acid residues that bind to E-box and H/K5-E9-R13 patterns, and bind to G-box. The study of bHLH gene family in different species will help to understand the evolutionary process and biological function. Previous phylogenetic results showed that bHLH proteins in plants were divided into 26 subfamilies, 20 of which were found in the common ancestor of vascular and bryophytes plants [17]. Toledo Ortiz et al. [15] divided 147 AtbHLH proteins into 21 subfamilies; and Li et al. [18] divided 167 OsbHLH proteins into 22 subfamilies.
The bHLH TF family is involved in plants’ perception of the external environment, cell-cycle regulation, and tissue differentiation [18, 19]. Different subfamilies regulate different biological processes, such as transduction of light signals [20, 21] and hormone signals [22, 23], and organ development [24–26, 27]. Under stress conditions, certain bHLH TFs are activated; they combine with the promoters of key genes involved in various signaling pathways, and regulate the transcription level of these target genes, thereby regulating the plants’ stress tolerance. For example, some researchers have found that the homologous bHLH genes bhlh068 of Oryza sativa and bHLH112 of Arabidopsis thaliana play an active role in the response to salt stress, but have opposite effects on regulation of plant flowering [28]. Appropriate TFs, together with AtbHLH38 and AtbHLH39, can regulate iron metabolism in Arabidopsis [29]. Atbhlh112 is a transcriptional activator of drought and other stress signal-transduction pathways, but it has an inhibitory effect on root development [30]. In Nicotiana tabacum, plants overexpressing Ntbhlh123 have enhanced resistance under low-temperature stress [31]. bHLH TFs are involved in regulating the accumulation of secondary metabolites in plants [32]. These examples all show the roles of bHLH TFs in the plant response to stress.
The expansion of this family is closely related to plant evolution and diversity [33, 34], not only in higher plants, but also in lower plants or non-plants, such as algae, mycobacteria, lichens and mosses [34]. With regards to abiotic stresses, bHLH is mainly involved in the defense responses to drought, high temperature, low temperature, and high salinity, which are unique to the terrestrial environment. Therefore, the evolution of the bHLH gene family provides clues to understanding the evolution of green algae to flowering plants through their adaptation to environmental changes. In particular, genome-wide analysis of bHLH gene families of different species will help understand the biological function and evolutionary origin of the bHLH genes.
Sorghum bicolor (L.) Moench is an annual row crop in the family Gramineae [35]. It is a common grain crop, which is used to produce food and beverage, widely distributed in the tropical, subtropical and temperate regions of the world and cultivated in the northern and southern provinces of China. S. bicolor seeds serve as a food source in China, North Korea, the former Soviet Union, India and Africa [36]. S. bicolor has rich genetic and phenotypic diversity, especially in plant height, seed color, seed size and branch number. Moreover, S. bicolor is a particularly nutritious crop, high in resistant starch, proteins, vitamins and polyphenols [37, 38], and it is widely used in the brewing industry [39]. In the long-term environmental adaptation, different varieties have been formed on sorghum, and some extreme abiotic stresses still have significant effects on its growth and development. For example, S. bicolor plants show reduced floret fertility and single-grain weight under high temperature, thereby reducing yield [40, 41]; low temperature leads to weakening of this crop’s growth potential, and plants are generally seriously damaged by frost [42]. S. bicolor has a well-developed root system that enables it to survive drought to some extent [43, 44]; nevertheless, long-term extreme drought has a huge impact on growth and yield [43]. In the process of S. bicolor production, pests, diseases, weeds and other biotic stresses will also cause serious yield losses [44]. Because S. bicolor is cultivated throughout the world, it has great economic and research value, and the identification of its functional genes is important.
In 2009, the completion and publication of the whole S. bicolor genome sequence enabled us to further explore, clone and verify the bHLH genes related to its stress resistance [45]. The S. bicolor genome is 750 Mb in length, with about 30,000 genes, ca. 75% more than in rice [46]. The bHLH gene family has been widely studied in many plant species, such as Arabidopsis [15], rice [18], Chinese cabbage [26], tomato [47], common bean [48], apple [49], peanut [50], Brachypodium distachyon [51], potato [52], maize [53], wheat [54], MOSO bamboo [55], Carthamus tinctorius [56], Chinese jujube [57], pepper [58], Jilin ginseng [59], pineapple [60], and tartary buckwheat [61], among others. However, at present, our understanding of gene families in S. bicolor is very limited. The main gene families identified in this plant are MADS-box [62], Dof [63], CBL [64], ERF [65], SBP-box [66], HSP [67], LEA [68], and NAC [69], among others. Because bHLH genes play an important role in various physiological processes, it is of great significance to systematically study the bHLH family in S. bicolor. Here, we identified 174 bHLH genes in S. bicolor and classified them into 24 major groups. Exon–intron structure, motif composition, gene duplication, chromosome distribution, and phylogeny were analyzed. The expression of bHLH family members in S. bicolor under different biological processes and abiotic stresses was also analyzed. This study provides valuable clues to the functional identification and evolutionary relationships of S. bicolor.
Results
Identification of bHLH genes in S. bicolor
To identify all possible bHLH members in the S. bicolor genome, we used two BLAST methods (Additional file 1: Table S1). To better distinguish these genes, we named them SbbHLH001 to SbbHLH174 according to their location on the S. bicolor chromosomes (Additional file 1: Table S1) and provide the genes’ characteristics, including molecular weight, isoelectric point (pI), protein length, domain information, and subcellular localization (http://cello.life. nctu.edu.tw/) (Additional file 1: Table S1).
Of the 174 SbbHLH proteins, SbbHLH031 and SbbHLH168 were the smallest with 87 amino acids, and the largest protein was SbbHLH040 with 1105 amino acids. The molecular mass of the proteins ranged from 9.67 kDa (SbbHLH168) to124.74 kDa (SbbHLH040), and the pI ranged from 4.53 (SbbHLH081) to 12.05 (SbbHLH004), with a mean of 6.70. Of all of the SbbHLH genes, 14 contained the bHLH-MYC-N domain and 172 contained the HLH domain (the exceptions being SbbHLH097 and SbbHLH116). The predicted subcellular localization results showed that 141 SbbHLHs are located in the nucleus, 26 in the cytoplasm, 4 in the mitochondria, 2 (SbbHLH103 and SbbHLH090) in the endoplasmic reticulum, and 1 (SbbHLH095) in the cytoskeleton (Additional file 1: Table S1). The ratio of SbbHLH genes to total genes in the S. bicolor genome was about 0.58%, which is similar to Arabidopsis (0.59%), but more than in rice (0.44%) [18], poplar (0.40%) [27], and tomato (0.46%) [48].
Multiple sequence alignment, phylogenetic analysis, and classification of SbbHLH genes
We constructed a phylogenetic tree using the neighbor-joining (NJ) method with a bootstrap value of 1000 based on the amino acid sequences of 174 SbbHLH and 158 AtbHLH proteins (Fig. 1; Additional file 1: Table S1). According to the topological structure of the tree and classification method proposed by Pires and Gabriela [15, 17], 332 bHLH genes in the phylogenetic tree were divided into 24 clades (groups 1–24) and 1 orphan [1, 6, 7]. The unclassified group (UC) contained 8 SbbHLH and 6 AtbHLH genes, and 149 SbbHLH proteins clustered into 21 subfamilies. This is consistent with the taxonomic group of bHLH proteins in Arabidopsis [18], indicating no loss of those proteins during the long-term evolution in S. bicolor evolution. Seventeen S. bicolor proteins constituted three typical topological structures (groups 22–24), suggesting that these are new characteristics in the evolution of S. bicolor diversity. None of AtbHLHs was assigned into subfamily 23,which contained 7 SbbHLHs (SbbHLH86, SbbHLH87, SbbHLH108, SbbHLH123, SbbHLH124, SbbHLH142, SbbHLH143); this group might indicate a new evolutionary direction for S. bicolor. Among the 24 subfamilies, the subfamily 15 had the largest number of members (17 SbbHLHs), and subfamilies 2 (SbbHLH79), 14 (SbbHLH68), and 20 (SbbHLH34) had the fewest (1 SbbHLH). Eight SbbHLH genes, which are not clearly classified into any subfamily, were classified as “orphans” [15, 16] (Fig. 1, Additional file 1: Table S1). A phylogenetic tree for Arabidopsis showed that some SbbHLHs are tightly grouped with the AtbHLHs (bootstrap support ≥70). These may be orthologous to the AtbHLHs and have similar functions.
Fig. 1.

Unrooted phylogenetic tree showing relationships among bHLH domains of S. bicolor and Arabidopsis. The phylogenetic tree was derived using the NJ method in MEGA7.0. The tree shows the 24 phylogenetic subfamilies and 1 unclassified group (UC) marked with red font on a white background. bHLH proteins from Arabidopsis are marked with the prefix ‘At’
The bHLH domain of Arabidopsis bHLH proteins and those from subgroups 1–21 were randomly selected as representatives of groups and subgroups for further multiple-sequence comparison (Fig. 2, Additional file 1: Table S1). The SbbHLH members from groups 22–24 were selected for the comparison. The bHLH domains of S. bicolor span approximately 50 amino acids. As shown in Fig. 2, although the characteristic bHLH domain is well conserved in Arabidopsis and S. bicolor, the regions outside of this domain in the rest of the protein are usually differentiate and diversify [13, 14, 18]. We considered the basic region to be 17 amino acids long based on Gabriela’s view [15]. In terms of amino acid structure, the loop was the most divergent region of this domain, especially in subfamily 6, 10 and 23, as has been observed for bHLH proteins from other plants, including Arabidopsis [18], potato [26], tomato [48] and buckwheat [61].
Fig. 2.

Multiple sequence alignment of the bHLH domains of the members of 24 phylogenetic subfamilies and 1 unclassified group (UC) of the SbbHLH protein family. The scheme at the top depicts the locations and boundaries of the basic, helix, and loop regions in the bHLH domain
Conserved motifs and gene structure analysis of SbbHLH genes
To understand the structural components of the SbbHLH genes, their exon and intron structures were obtained by comparing the corresponding genomic DNA sequences (Fig. 3, Additional files 1and 2: Tables S1 and S2). A comparison of the number and position of the exons and introns revealed that the 174 SbbHLH genes had different numbers of exons, varying from 1 to 12 (Fig. 3a/b). In addition, 17 (9.77%) genes contained 1 exon, and the remaining genes had 2 or more exons. The 17 intronless genes belonged to four subfamilies (8, 13, 14, 19), but were mainly in subfamilies 8 and 19. The largest proportion of SbbHLH genes (n = 31) had 2 introns. SbbHLH038 and SbbHLH054 had the most introns, with 11. Group 1, 2, 4, 10, 20, 21 and 23 members contained 1 or 2 introns. Further analyses indicated that group 18 showed more diversity in the number of introns. In general, members of the same subfamily had similar gene structures.
Fig. 3.
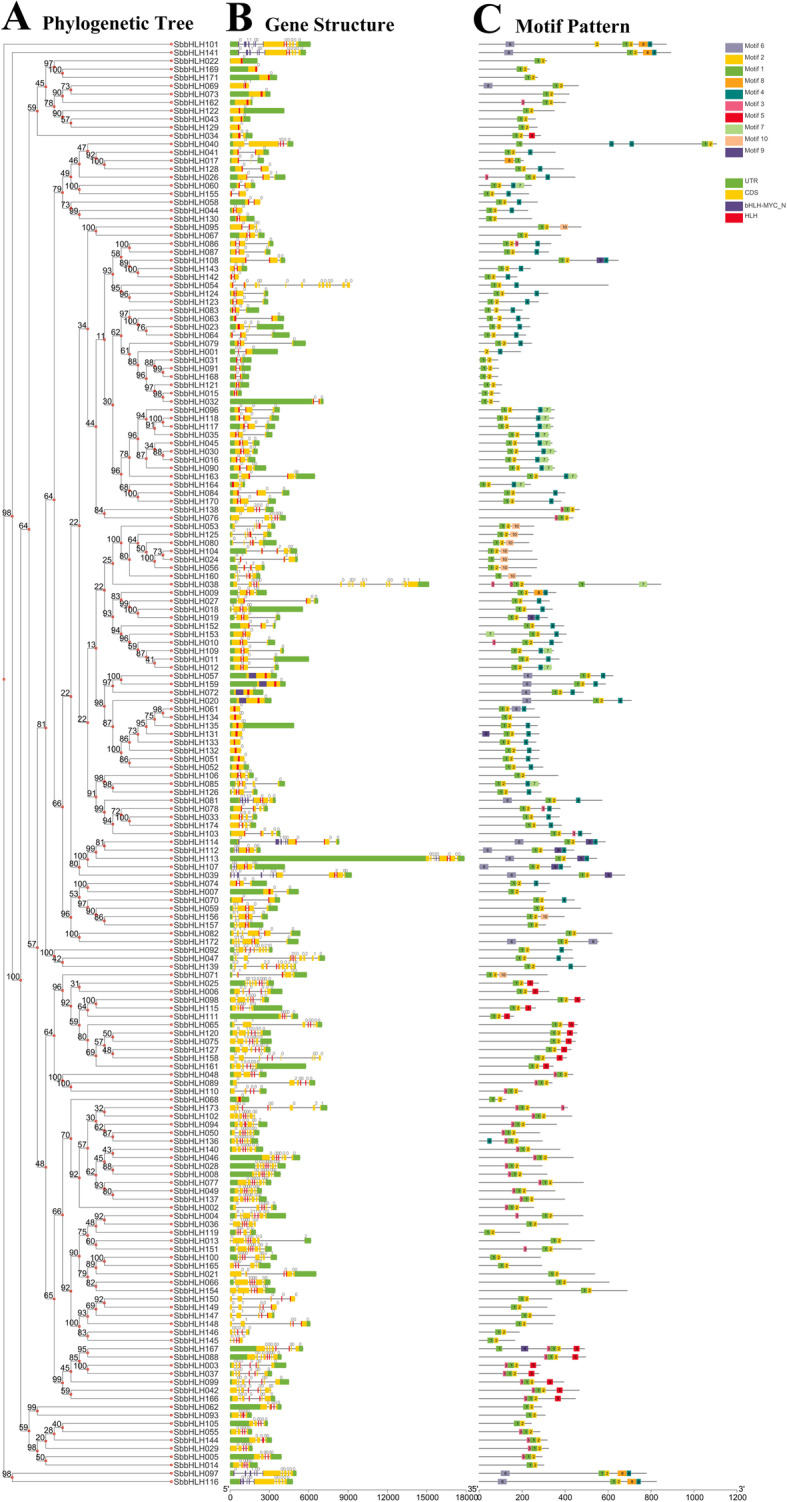
Phylogenetic relationships, gene-structure analysis, and motif distributions of S. bicolor bHLH genes. a Phylogenetic tree was constructed by the NJ method with 1000 replicates on each node. b Exons and introns are indicated by yellow rectangles and gray lines, respectively. c Amino acid motifs in the SbbHLH proteins (1–10) are represented by colored boxes. The black lines indicate relative protein lengths
To further study the characteristic region of the SbbHLH proteins, the motifs of 174 SbbHLH proteins were analyzed using the online tool MEME. A total of 10 distinct conserved motifs (motifs 1–10) were found (Fig. 3c, Additional file 2: Table S2). As exhibited in Fig. 3c, motifs 1 and 2 were widely distributed in the SbbHLHs, except for SbbHLH001 and SbbHLH017, and the two motifs were very close to each other in the bHLH proteins. SbbHLH members within the same groups were usually found to share a similar motif composition. For example, group 1, 2, 3, 5, 7, 9, 11 and 23 members contained motifs 1, 2, and 4; groups 12 and 17 contained motifs 1, 2, and 5; group 16 contained motifs 3, 1, and 2; and group 22 contained motifs 6, 1, 2, 8, and 4. At the same time, we found that some motifs were only present in specific subfamilies. In addition, motif 5 was specific to groups 12, 17 and 20, whereas motif 8 was specific to groups 5, 10 and 22. Further analysis showed that some of the motifs could only be distributed in specific locations of the pattern. For example, motif 1 was always distributed at the start of the pattern in groups 1, 2, 3, 4, 5, 6, 9, 10, 11, 12, 13, 14, 15, 20, 21, 23 and 24; motif 6 was almost always distributed at the start of groups 7 and 22; motif 3 was almost always distributed at the start of groups 16, 17 and 18. Motif 4 was almost always distributed at the end of the pattern in groups 1, 2, 7, 8, 9, 10, 11, 22 and 23; and motif 10 was distributed at the end of the pattern in the group 6. The functions of most of these conserved motifs remain to be elucidated. Overall, members that belonged to the same subfamily had similar gene structure and motif composition, in accordance with the results of the phylogenetic analysis, and supporting the reliability of the population classification.
Chromosomal spread and gene duplication of SbbHLH genes
A map of the physical position of the SbbHLH genes was created based on the latest S. bicolor genome database (Fig. 4, Additional file 3: Table S3). The distribution of the 174 SbbHLH genes on chromosomes (Chr) 1 to 10 was uneven (Fig. 4). Each of the SbbHLHs’ names was given according to its physical position from the top to the bottom on S. bicolor Chr1 to Chr10. Chr1 contained the largest number of SbbHLH genes (35 genes, ~ 20.11%), followed by Chr3 (23, ~ 13.22%), while Chr5 contained the least (5, ~ 2.87%). Chr2 and Chr4 each contained 21 (~ 12.07%) SbbHLH genes. Chr8 and Chr9 each contained 12 (~ 6.90%) SbbHLH genes. Chr6, Chr7, and Chr10 contained 16 (~ 9.20%), 19 (~ 10.92%), and 10 (~ 5.75%) SbbHLH genes, respectively. Interestingly, most SbbHLH genes were distributed at the ends of the 10 chromosomes. In addition, we observed a large number of SbbHLH gene-duplication events. A chromosomal region within 200 kb exhibiting two or more identical genomic regions is defined as a tandem duplication event [35]. On chromosomes 1, 3, 4, 6, 7 and 8, we discovered 13 tandem duplication events involving 20 SbbHLH genes (Fig. 4). SbbHLH132, SbbHLH133, SbbHLH134, SbbHLH147, SbbHLH148 and SbbHLH149 each had two tandem repeat events (SbbHLH132 and SbbHLH131 / SbbHLH133; SbbHLH133 and SbbHLH132 / SbbHLH134; SbbHLH134 and SbbHLH133 / SbbHLH135; SbbHLH147 and SbbHLH146 / SbbHLH148; SbbHLH148 and SbbHLH147 / SbbHLH149; SbbHLH149 and SbbHLH148 / SbbHLH150). All genes that formed tandem repeat events came from the same subfamily. For example, SbbHLH117 and SbbHLH118 were tandem repeat genes and they clustered together in subfamily 3 (Fig. 4, Additional file 3: Table S3).
Fig. 4.

Schematic representation of the chromosomal distribution of the S. bicolor bHLH genes. Vertical bars represent the chromosomes of S. bicolor. The chromosome number is indicated to the left of each chromosome. The scale on the left represents chromosome length
In addition, there were 42 pairs of segmental duplications in the SbbHLH genes (Fig. 5, Additional file 4: Table S4). As shown in Figs. 5, 71 (40.8%) paralogs were identified in the SbbHLH gene family, indicating an evolutionary relationship among these bHLH members. The SbbHLH genes were unevenly distributed in 10 S. bicolor linkage groups (LGs) (Fig. 5). Some LGs had more SbbHLH genes than others (LG2, LG7). LG2 had the most SbbHLH genes (14), and LG5 had the least (1). Further analysis of the subfamilies of these genes showed that most of them are linked within their subfamily, except for SbbHLH024 / UC and SbbHLH056 / 6. For all identified SbbHLH genes, group 18 had the largest number of linked genes (9/71). In addition, the group 15 had 8 genes, while groups 13 and 6 had only 1 (Additional file 4: Table S4). These results suggest that some SbbHLH genes may have been produced by gene-replication events, and that these replication events played a major role in the occurrence of new functions in S. bicolor evolution and the amplification of the SbbHLH gene family.
Fig. 5.

Schematic representation of the chromosomal distribution and interchromosomal relationships of S. bicolor bHLH genes. Colored lines indicate all synteny blocks in the S. bicolor genome and the red lines indicate duplicated bHLH gene pairs. Chromosome number is indicated at the bottom of each chromosome
Synteny analysis of SbbHLH genes
To further infer the phylogenetic mechanisms of the S. bicolor bHLH family, we constructed six comparative synteny maps of S. bicolor’s association with six representative species, including three dicotyledons (A. thaliana, Vitis vinifera and Solanum lycopersicum) and three monocotyledons (B. distachyon, O. sativa and Zea mays) (Fig. 6, Additional file 5: Table S5). A total of 150 SbbHLH genes showed syntenic relationships with those in A. thaliana (16), V. vinifera (46), S. lycopersicum (37), B. distachyon (129), O. sativa (135) and Z. mays (195) (Additional file 5: Table S5). The numbers of orthologous pairs between the other six species (A. thaliana, V. vinifera, S. lycopersicum, B. distachyon, O. sativa and Z. mays) were 20, 66, 59, 194, 208 and 273, respectively. Some SbbHLH genes were associated with at least four syntenic gene pairs (particularly between S. bicolor and Z. mays bHLH), such as SbbHLH043, SbbHLH049, SbbHLH050, SbbHLH101, SbbHLH137, SbbHLH138, SbbHLH141 and SbbHLH166, hinting at these genes’ important role during evolution.
Fig. 6.

Synteny analyses of the bHLH genes between S. bicolor and six representative plant species (Arabidopsis thaliana, Vitis vinifera, Solanum lycopersicum, Brachypodium distachyon, Oryza sativa subsp. indica, Zea mays). Gray lines on the background indicate the collinear blocks in S. bicolor and other plant genomes; red lines highlight the syntenic S. bicolor bHLH gene pairs
As expected, some collinear gene pairs (with 57 SbbHLH genes) identified between S. bicolor and B. distachyon, O. sativa or Z. mays were not found between S. bicolor and A. thaliana, V. vinifera, or S. lycopersicum, such as SbbHLH001 with KQK12528/BGIOSGA013800-TA/Zm00001d034596_T001, and SbbHLH004 with KQK12892/BGIOSGA013672-TA/Zm00001d034298_T001. This suggests that these homologous genes may be gradually formed after the independent differentiation of monocotyledons (Additional file 5: Table S5). Similar patterns were also observed between S. bicolor and O. sativa/ Z. mays, which may be related to the phylogenetic relationships between S. bicolor and the other six plant species. In addition, some SbbHLH genes were found to be associated with at least one syntenic gene pair among the six plants (especially between S. bicolor and Z. mays), such as SbbHLH030, SbbHLH045, SbbHLH050, SbbHLH066, SbbHLH099, SbbHLH136, SbbHLH138, SbbHLH154, SbbHLH166, suggesting that these orthologous pairs already existed before the ancestral divergence, and thus indicating that these genes may have played an important role in the bHLH gene family during evolution. To better understand the evolutionary constraints of the bHLH gene family in S. bicolor, the SbbHLH genes were subjected to Tajima’s D Neutrality Test. Calculations gave D = 7.736378, the large deviation from 0, suggesting that the SbbHLH gene family might have experienced strong purifying selective pressure during evolution.
Evolutionary analysis of the SbbHLH genes and bHLH genes of several different species
To analyze the evolutionary relationship of the trihelix family of bHLH proteins among S. bicolor and six other plants (A. thaliana, V. vinifera, S. lycopersicum, B. distachyon, O. sativa, Z. mays), an unrooted NJ tree with 10 conserved motifs according to the MEME web server was constructed using the NJ method of Geneious R11 according to the protein sequences of 174 SbbHLH genes and the six other plants’ trihelix genes (Fig. 7, Additional file 2: Table S2). The detailed genetic correspondence is presented in Additional files 1 and 2: Tables S1 and S2. The distribution of SbbHLHs in the phylogenetic tree was relatively dispersed. As shown in Fig. 7, the SbbHLH proteins tended to gather with the bHLH proteins of O. sativa and Z. mays. With the exception of a few bHLH proteins, for example ZmbHLH8, ZmbHLH53, SbbHLH001, all other proteins of the six studied plants contained motifs 1 and 2. In addition, several motifs existed only in bHLH proteins of a few specific SbbHLH branches, such as motifs 5, 8 and 10. We also found that the bHLH proteins of O. sativa, Z. mays and S. bicolor on the same branch generally have similar motif compositions, and similar serial motifs tend to cluster in specific bHLH protein families. For example, serial motifs 1, 2, 5 and 10 tended to gather within group 6; and serial motifs 8, 9, 1, 2, 7 and 4 tended to gather within group 8. Thus, SbbHLH proteins may be more closely related to those of O. sativa and Z. mays.
Fig. 7.

Phylogenetic relationship and motif composition of the bHLH proteins from S. bicolor with six different plant species (Arabidopsis thaliana, Vitis vinifera, Solanum lycopersicum, Brachypodium distachyon, Oryza sativa subsp. indica, Zea mays). Outer panel: An unrooted phylogenetic tree constructed using Geneious R11 with the NJ method. Inner panel: Distribution of the conserved motifs in bHLH proteins. The differently colored boxes represent different motifs and their positions in each bHLH protein sequence. The sequence information for each motif is provided in Additional File 2: Table S2.
Expression patterns of SbbHLHs in several plant organs
To investigate the potential roles of the SbbHLH genes, real-time PCR was used to detect the expression of 12 individual members of the gene family which were homologous to, or had close evolutionary relationships with AtbHLH genes with established functions. The accumulation of the transcriptional products of 12 SbbHLH genesfrom different subfamilies in six organs (anthers, styles, roots, leaves, fruit and stems) was evaluated (Fig. 8a). The results showed that some genes exhibited preferential expression in some tissues of S. bicolor. Most of the genes were expressed in all organs, and 4 genes (SbbHLH014, SbbHLH050, SbbHLH079, SbbHLH134) showed their highest expression level in the styles. Two genes (SbbHLH063 and SbbHLH110) showed their highest expression level in the anthers, and the highest expression level of SbbHLH037 and SbbHLH125 was in the leaves. Three genes (SbbHLH045, SbbHLH047 and SbbHLH130) showed highest expression in the S. bicolor stems, and the highest expression of SbbHLH101 was found in fruit. In addition, correlations of SbbHLH expression among the six organs were studied (Fig. 8b). We found that the expression of different genes in the plant organs was significantly correlated, indicating that their roles may be synergistic. Most SbbHLH genes showed significant positive correlations; for example, we observed four genes—SbbHLH050, SbbHLH079, SbbHLH014 and SbbHLH134—that had their highest expression in the styles, and were significantly positively correlated; they also showed significant positive correlations with SbbHLH110, which is most highly expressed in the anthers. However, four pairs of SbbHLH genes (SbbHLH050 and SbbHLH125; SbbHLH110 and SbbHLH045; SbbHLH079 and SbbHLH045; SbbHLH045 and SbbHLH134) were significantly negatively correlated.
Fig. 8.

Tissue-specific gene expression of 12 S. bicolor bHLH genes and the correlation between their expression patterns. a Expression patterns of 12 S. bicolor bHLH genes in the anther, style, leaf, root, stem and fruit organs were examined by qRT-PCR. Error bars were obtained from three measurements. Lowercase letter above the bar indicates significant difference (α = 0.05, LSD) among the treatments. b Positive number: positively correlated; negative number: negatively correlated. Red numbers indicate a significant correlation at the 0.05 level
Expression patterns of SbbHLH genes in response to different treatments
To further determine whether the expression of SbbHLH genes was influenced by different abiotic stresses, the expression of 12 SbbHLH members was examined under six abiotic stresses: strong ultraviolet (UV) radiation, flooding, polyethylene glycol (PEG), NaCl, heat and cold treatments. qRT-PCR analysis was performed to analyze the 12 SbbHLH genes’ expression patterns in roots, leaves and stems in response to the different treatments (Fig. 9a). Some of the SbbHLH genes were significantly induced or repressed by the different treatments. Expression of most of these genes was significantly altered in the early stage of the stress treatment (Fig. 9). Some SbbHLHs showed changes in expression with time or in different organs, depending on the stress. For example, under cold stress, SbbHLH037 and SbbHLH045 were first significantly upregulated, and then downregulated. SbbHLH063 expression was significantly upregulated in the root, while it was significantly downregulated in the stem and leaf. Under flooding stress, SbbHLH045 was significantly upregulated in the root, stem and leaf, but SbbHLH050 was significantly downregulated. Interestingly, several genes showed opposing expression patterns under different treatments. The transcript levels of many SbbHLH genes, such as SbbHLH063, were upregulated in stems and leaves by the heat-stress treatment, but downregulated by the cold-stress treatment. Some other genes showed changes in specific organs. For instance, SbbHLH014 responded significantly to heat treatment in the root. Furthermore, correlations between SbbHLH gene-expression patterns were observed (Fig. 9b). There were negative correlations among most SbbHLH genes. However, a few SbbHLH genes were significantly positively correlated, such as SbbHLH110 and SbbHLH063/SbbHLH134, with P < 0.05 (Fig. 9b).
Fig. 9.

Gene expression of 12 S. bicolor bHLH genes in plants subjected to abiotic stresses (strong UV radiation, flooding, PEG, NaCl, heat and cold treatments) at the seedling stage. a Expression patterns of 12 S. bicolor bHLH genes in leaf, root and stem organs were examined by qRT-PCR. Error bars were obtained from three measurements. Lowercase letter above the bar indicates significant difference (α = 0.05, LSD) among the treatments. b Positive number: positively correlated; negative number: negatively correlated. Red numbers indicate a significant correlation at the 0.05 level
Discussion
,Exploration of the bHLH gene family at the whole-genome level in any species, and the functional identification of some this family’s members, can provide theoretical support for the role of the bHLH gene family in the stress signal-transduction process. In this study, 174 SbbHLH genes were identified, and all of the encoded proteins showed obvious differences in structure, indicating high complexity. According to Atchley et al. [10] and Toledo-Ortiz et al. [15], we analyzed the DNA-binding ability of the basic region of SbbHLHs. The SbbHLH gene sequence can be divided into E-box binding genes and non-E-box binding genes (Additional file 1: Table S1). The E-box binding proteins can be subdivided into G-box binding proteins and non-G-box binding proteins [12, 15]. The basic domain of bHLH contains two essential amino acid residues, Glu-13 and Arg-16. If it contains only one of them, it will be classified as a non-E-box binding protein. The G-box binding protein contains three essential amino acid residues (His/Lys-9, Glu-13 and Arg-17) in the basic domain. If only Glu-13 and Arg-17 are present, it is classified as a non-G-box binding protein. In addition, if the number of basic amino acids is less than 4 in the basic domain, and it contains no or only one of Glu-13 and Arg-16, it will be classified as a non-DNA binding protein. These proteins are thought to have no ability to bind directly to DNA. In this study, 119 (68.4%) SbbHLHs were classified as E-box binding proteins: 99 (56.9%) as G-box binding proteins and 20 (11.5%) as non-G-box binding proteins; 30 (17.2%) members were classified as non-E-box binding genes. The remaining 25 (14.4%) members were not considered to have DNA-binding ability due to the lack of Glu-13 or Arg-16 in the alkaline region (Fig. 2, Additional file 1: Table S1). Similar to the reports of O. sativa (95, 56.9%) and A. thaliana (89, 60.5%), the highest proportion of SbbHLHs were G-box binding proteins [18]. Previous studies have found that some key amino acid residues play important roles in the binding of TFs to DNA and the formation of homodimers or heterodimers between bHLHs or bHLHs and other TFs [15, 70]. For example, His-6, Glu-10, and Arg-14 are related to DNA-binding activity, whereas Leu-25 and Leu-57 in the helical region determine whether bHLH TFs can form homodimers or heterodimers. In SbbHLHs, the conservation rates of Leu-25 and Leu-57 are 94.3 and 96.0%, respectively, which are lower than in S. lycopersicum (99, 97%) [47] and Citrus reticulata (100, 100%) [70]. Previous studies have found that the formation of such heterodimers can change or expand the diversity of molecular interactions, and generate new functions by identifying new DNA-binding sites [15,71]. As already noted, a bHLH protein can form a homodimer with itself or a heterodimer with other TFs, such as R2R3-MYBs, BAR1-BES1 and AP2 [72, 73,74].
,Based on the constructed phylogenetic tree, we identified at least one bHLH protein from S. bicolor in each subgroup of AtbHLHs, indicating that the time of differentiation of the bHLH family may have been earlier than that of S. bicolor and A. thaliana. The bHLH proteins within the reported subfamilies may play a fundamental role in the development, adaptation and evolution in dissimilar plant species, including peanut [51], tomato [48], Chinese cabbage [47], wheat [54], and Carthamus tinctorius [56]. Compared to A. thaliana, SbbHLH genes can be divided into 24 subfamilies and 1 orphan subfamily (UC), 4 more than A. thaliana and 3 more than O. sativa. Among them, group 15 (17, 9.8%) and group 18 (15, 8.6%) have more members, which is similar to the results for A. thaliana [3], and indicates that those bHLH gene groups may have undergone stronger partial differentiation in the long-term evolutionary process. However, there is no research to prove that this kind of differentiation is advantageous in the differentiation process of herbs and woody plants. Seven of the SbbHLHs did not have obvious clusters, so they were all classified into the UC group, and those genes all showed non-DNA binding activity, but still a great deal of variability in the base sequence. The gene-structure analysis revealed that SbbHLH genes in the same subfamily have similar gene structures, which not only supports our classification of the subgroups to a certain extent, but also indicates that all members of a subfamily are close in evolutionary terms (Fig. 3). At the same time, this does not rule out the loss of some independent introns during the long-term evolutionary development of plants, resulting in the loss of some introns in the domains of some bHLH members. For example, SbbHLH153 has fewer introns than other members of the same family. Genes with few or no introns are considered to have lower expression levels in plants [4]. However, the compact gene structure may contribute to the rapid expression of genes in response to endogenous and/or exogenous stimuli [4]. Genome-replication events are considered to have occurred in the process of plant evolution, and the expansion of gene families and genome evolution mechanisms mainly depend on gene-replication events [5, 18,26]. The main replication modes are tandem repeats and fragment replication. These were identified in the SbbHLH genes. We discovered 13 tandem duplication events containing 20 SbbHLH genes (Fig. 48, Additional file 75: Table S3), especially on chromosomes 7 and 8. In addition, there were 42 pairs of segmental duplications of SbbHLH genes (Fig. 76, Additional file 77: Table S4). Therefore, segment duplication may make a higher contribution to the expansion of the bHLH family in S. bicolor. Nevertheless, since there were many duplication events in S. bicolor, it is lower than that of the dicotyledonous plants tomato and potato [78, 79]. Similar situations have been reported in studies of other monocotyledonous species [80]. However, the current conclusions cannot explain the significant differences between monocotyledons and dicotyledons.
Analysis of a gene’s expression profile can provide important clues to understanding its potential biological function. There are many members of the bHLH TF family with diverse functions, but the current research in plants is not particularly thorough, as it focuses mainly on the two model plants A. thaliana and O. sativa. The functions of bHLH TFs in other species still need to be explored. In this study, we used 12 SbbHLH genes with significant differences in clustering on the phylogenetic tree to study their responses to six abiotic stresses in different developmental organs, and found that almost all of the bHLH TF genes have significant differential expression (more than 2-fold difference). For example, under salt stress, 10 SbbHLHs were upregulated in leaves, 7 were upregulated in roots, and 8 were upregulated in stems. The expression pattern results indicate that bHLH TFs participate in a complex cross-regulatory network. SbbHLH079 and SbbHLH045 were responsive, at the same time, to PEG, NaCl and UV treatments, indicating synergistic or antagonistic regulation under a variety of adverse conditions. Further research is needed to explore the relationship between these genes. Interestingly, most of the SbbHLH genes showed significant negative regulation in the expression heat map. If we consider expression patterns and complex protein interactions, then we can suggest that a network of feedback mechanisms coordinates the expression of multiple genes. In addition, flowers and fruit, as plant reproductive organs, are the main structures in all angiosperms [81]. In this study, we explored the expression of 12 bHLH genes in the anthers and styles of S. bicolor flowers, as well as in the main organs of plants at the filling stage. Studies have shown that bHLH TFs play an important role in the development of flowers and fruit. The expression levels of SbbHLH134 and SbbHLH110 in the anther and style were significantly higher than in roots, stems, leaves and fruit, whereas SbbHLH101 showed significantly higher expression in fruit at the filling stage (Fig. 8a). Therefore, we speculate that SbbHLH134, SbbHLH110 and SbbHLH101 may also regulate flower and fruit development in S. bicolor. However, the specific functions still need to be analyzed through in-depth experiments. In summary, these results reveal the functions and regulation of some bHLH TFs.
Conclusion
In summary, we provided the systematic genome-wide analysis of the bHLH gene family in S. bicolor. A total of 174 SbbHLH genes/proteins were characterized and divided into 24 groups. Furthermore, protein motifs and gene structures of the SbbHLHs within the subfamilies were prone to be the similar, which supported the classification predicted. The distribution of the 174 SbbHLH genes on 10 S. bicolor chromosomes was uneven. We found that gene-replication events may have produced some SbbHLH genes, with tandem duplication contributing more to the expansion of the SbbHLH gene family than segmental duplication. The qRT-PCR results showed that the 12 studied SbbHLHs were all affected by abiotic stresses, and their expression during the development of flowers and fruit was studied. It is speculated that SbbHLH134, SbbHLH110 and SbbHLH101 also regulate flower and fruit development in S. bicolor. Taken together, the results and information described in this work provide a good basis for further investigation of the biological functions and evolution of bHLH genes in S. bicolor.
Methods
Gene identification
,We downloaded the complete S. bicolor genome sequence from the Ensembl Genomes website (http://ensemblgenomes.org/). Based on two BLASTp searches [82,83], bHLH family members were identified. First, with BLASTp (score value ≥100 and e-value ≤1e- 10), all possible bHLH proteins were identified from the S. bicolor genome referring to trihelix protein sequences of A. thaliana. Second, the Hidden Markov Model (HMM) profile consistent with the trihelix domain was obtained from the Pfam protein family database (http://pfam.sanger.ac.uk/). We used both HMMER3.0 (default parameters) with a cutoff of 0.01 (http://plants.ensembl. org/hmmer/index.html) [84] and SMART (http://smart.embl-heidelberg.de/) [85,86] to ascertain the presence of the bHLH domain, and to further verify the results. In addition, the basic features of the trihelix proteins of the SbbHLH gene family were identified: coding sequence length, pI, protein molecular mass, and subcellular localization, from the ExPasy website (http://web.expasy.org/protparam/).
bHLH gene structure
,The bHLH domain sequences of the characterized SbbHLH proteins were used to create multiple protein sequence alignments using ClustalW with default parameters [87]. The deduced amino acid sequences in the bHLH domains were then adjusted manually using GeneDoc software. We used Gene Structure Display Server (GSDS: http://GSDS.cbi.pku.edu.cn) [88] to analyze the constituents of the exons/introns of the SbbHLH genes. We used MEME to analyze the motifs of SbbHLH proteins, (http://meme-suite.org/tools/meme) [89,90]. The optimized parameters were as follows: number of repetitions, any; the maximum number of motifs, 10; and the optimum width of each motif, between 6 and 200 residues [83,90,91].
Chromosomal distribution and gene duplication
All SbbHLH genes were mapped to S. bicolor chromosomes based on physical location information from the database of the S. bicolor genome using Circos [92]. The Multiple Collinearity Scan toolkit (MCScanX) was adopted to analyze the gene-duplication events, with the default parameters [93]. We analyzed homology of the bHLH genes between S. bicolor and the other six plants (A. thaliana, V. vinifera, S. lycopersicum, B. distachyon, O. sativa subsp. indica, Z. mays) using Dual Synteny Plotter (https://github.com/CJ-Chen/TBtools). Non-synonymous (ka) and synonymous (ks) substitutions of each duplicated bHLH gene were calculated using Ka/Ks-Calculator 2.0 [94].
Phylogenetic analysis and classification of SbbHLH gene family
According to the classification of AtbHLHs, all of the identified SbbHLH genes were divided into groups. The phylogenetic trees were inferred using the NJ method of MEGA X via Geneious R11 with BLOSUM62 cost matrix, the Jukes–Cantor model, global alignment with free end gaps and bootstrap value of 1000. The full-length amino acid sequences of the bHLH proteins (Additional file 1: Table S1) derived from A. thaliana, V. vinifera, S. lycopersicum, B. distachyon, O. sativa subsp. indica, and Z. mays (UniProthttps://www.uniprot.org/), combined with newly identified SbbHLHs, were used for phylogenetic analysis.
Plant materials, growth conditions, and abiotic stress in S. bicolor
Sorghum bicolor cv. Hongyingzi, a typical cultivated variety, was used throughout the study. Since 2019, ‘Hongyingzi’ has been grown in the greenhouse of Guizhou University. S. bicolor was grown in pots filled with soil and vermiculite (1:1) in a growth room with a 16 h/25 °C day and 8 h/20 °C night regime, and a relative humidity of 75%. We collected the stems, roots, leaves, fruit, anthers and styles separately from five plants with good growth and similar growth conditions, and quickly placed them in liquid nitrogen for storage at -80 °C pending further use. To investigate gene-expression patterns in response to various stresses, several SbbHLH genes were selected for further analysis. S. bicolor plants were subjected to the following abiotic stress treatments at the seedling stage (21 days): salt (5% NaCl), water flooding (whole plant), drought (30% PEG6000), UV exposure (70 μW/cm2, 220 V, 30 W), high temperature (40 °C), and low temperature (4 °C); each stress treatment was performed with five replicates, and qRT-PCR analysis was carried out after sampling at 2 h and 24 h, respectively. The collected samples were stored at -80 °C for further analysis.
Total RNA extraction, cDNA reverse transcription and qRT-PCR analysis
,Total RNA of each sample was extracted with a plant RNA extraction kit (TaKaRa) and used for cDNA library construction. The sequencing was performed in an Illumina GAII sequencer following the manufacturer’s instructions [90,6]. Gene-expression analysis was performed by qRT-PCR, with primers designed by Primer 5.0 (Additional file 91: Table S6). We used the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene, which was stably expressed at each growth stage in almost all tissues, as an internal control [95]. The qRT-PCR with SYBR Premix Ex Taq II (TaKaRa) was repeated at least three times and the data were analyzed using the 2− (ΔΔCt) method [96].
Statistical analysis
Analysis of variance (ANOVA) was performed with JMP6.0 software (SAS Institute), and compared with least significant difference (LSD) at the 0.05 and 0.01 levels. The histogram was drawn with Origin 8.0 software (OriginLab).
Supplementary Information
Additional file 1 Table S1. List of the 174 S. bicolor bHLH genes identified in this study.
Additional file 2 Table S2. Analysis and distribution of the conserved motifs in S. bicolor bHLH proteins.
Additional file 3 Table S3. Tandem duplication events of S. bicolor bHLH genes.
Additional file 4 Table S4. The 42 pairs of segmental duplications in S. bicolor bHLH genes.
Additional file 5 Table S5. One-to-one orthologous genes relationships between S. bicolor and other plants.
Additional file 6 Table S6. Primer sequences for qRT-PCR.
Acknowledgements
We thank all of the colleagues in our laboratory for providing useful discussions and technical assistance. We are very grateful to the editor and reviewers for critically evaluating the manuscript and providing constructive comments for its improvement.
Abbreviations
- bHLH
helix-loop-helix
- SbbHLH
Sorghum bicolor bHLH
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- qRT-PCR
Quantitative real-time polymerase chain reaction
- TF
Transcription factor
- AtbHLH
Arabidopsis thaliana bHLH
- HMM
Hidden Markov Model
- pI
Isoelectric point
- LG
Linkage group
Authors’ contributions
YF planned and designed the research and analyzed the data. YF and DL wrote the manuscript. HY, LF and LC studied gene expression by qRT-PCR. AH identified the S. bicolor bHLH gene family and analyzed gene structure. GX studied chromosome distribution, gene duplication and syntenic analysis of S. bicolor bHLH genes. YF and X-bC analyzed the evolutionary relationship of bHLH genes in several different species. JC supervised the research. JR and JY revised the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the National Science Foundation of China (31560578, Cheng JP, http://www.nsfc.gov.cn), Sichuan International Science and Technology Cooperation and Exchange Research and Development Project (2018HH0116, Yan J, http://kjt.sc.gov.cn), Guizhou Science and Technology Support Project (No. 20201Y125).
Availability of data and materials
The entire Sorghum bicolor genome sequence information was from the Ensembl Genomes website (http://ensemblgenomes.org/). The Sorghum bicolor materials (Hongyingzi) used in the experiment were supplied by Prof. Cheng Jianping of Guizhou University. The datasets supporting the conclusions of this article are included in the article and its Additional files.
Declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by the authors. These methods were carried out in accordance with relevant guidelines and regulations. We confirm that all experimental protocols were approved by Guizhou University.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jun Yan, Email: yanjun62@qq.com.
Jianping Cheng, Email: chengjianping63@qq.com.
References
- 1.Riaño-Pachón DM, Ruzicic S, Dreyer I, Mueller-Roeber B. PlnTFDB: an integrative plant transcription factor database. BMC Bioinformatics. 2007;8(1):42. doi: 10.1186/1471-2105-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang H, Jin J, Tang L, Zhao Y, Gu X, Gao G, et al. PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011;39(suppl_1):D1114–7. 10.1093/nar/gkq1141. [DOI] [PMC free article] [PubMed]
- 3.Riechmann JL, Heard J, Martin G, Reuber L, Jiang C, Keddie J, et al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;290(5499):2105–10. 10.1126/science.290.5499.2105. [DOI] [PubMed]
- 4.Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, et al. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 2003;20(9):1377–419. 10.1093/molbev/msg140. [DOI] [PubMed]
- 5.Schwechheimer C, Zourelidou M, Bevan MW. Plant transcription factor studies. Annu Rev Plant Physiol Plant Mol Biol. 1998;49(1):127–150. doi: 10.1146/annurev.arplant.49.1.127. [DOI] [PubMed] [Google Scholar]
- 6.Jin J, Zhang H, Kong L, Gao G, Luo J. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014;42(D1):D1182–D1187. doi: 10.1093/nar/gkt1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun X, Wang Y, Sui N. Transcriptional regulation of bHLH during plant response to stress. Biochem Biophys Res Commun. 2018;503(2):397–401. doi: 10.1016/j.bbrc.2018.07.123. [DOI] [PubMed] [Google Scholar]
- 8.Ledent V, Vervoort M. The basic helix-loop-helix protein family: comparative genomics and phylogenetic analysis. Genome Res. 2001;11(5):754–770. doi: 10.1101/gr.177001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56(5):777–783. doi: 10.1016/0092-8674(89)90682-X. [DOI] [PubMed] [Google Scholar]
- 10.Atchley WR, Terhalle W, Dress A. Positional dependence, cliques, and predictive motifs in the bHLH protein domain. J Mol Evol. 1999;48(5):501–516. doi: 10.1007/PL00006494. [DOI] [PubMed] [Google Scholar]
- 11.Buck MJ, Atchley WR. Phylogenetic analysis of plant basic helix-loop-helix proteins. J Mol Evol. 2003;56(6):742–750. doi: 10.1007/s00239-002-2449-3. [DOI] [PubMed] [Google Scholar]
- 12.Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20(2):429–440. doi: 10.1128/MCB.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair SK, Burley SK. Recognizing DNA in the library. Nature. 2000;404(6779):717–718. doi: 10.1038/35008182. [DOI] [PubMed] [Google Scholar]
- 14.Shimizu T, Toumoto A, Ihara K, Shimizu M, Kyogoku Y, Ogawa N, et al. Crystal structure of PHO4 bHLH domain-DNA complex: flanking base recognition. EMBO J. 1997;16(15):4689–97. [DOI] [PMC free article] [PubMed]
- 15.Toledo-Ortiz G, Huq E, Quail PH. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell. 2003;15(8):1749–1770. doi: 10.1105/tpc.013839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atchley WR, Fitch WM. A natural classification of the basic helix-loop-helix class of transcription factors. Proc Natl Acad Sci U S A. 1997;94(10):5172–5176. doi: 10.1073/pnas.94.10.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pires N, Dolan L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol Biol Evol. 2010;27(4):862–874. doi: 10.1093/molbev/msp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Duan X, Jiang H, Sun Y, Tang Y, Yuan Z, et al. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 2006;141(4):1167–84. 10.1104/pp.106.080580. [DOI] [PMC free article] [PubMed]
- 19.Amoutzias GD, Robertson DL, Oliver SG, Bornberg-Bauer E. Convergent evolution of gene networks by single-gene duplications in higher eukaryotes. EMBO Rep. 2004;5(3):274–279. doi: 10.1038/sj.embor.7400096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stevens JD, Roalson EH, Skinner MK. Phylogenetic and expression analysis of the basic helix-loop-helix transcription factor gene family: genomic approach to cellular differentiation. Differentiation. 2008;76(9):1006–1022. doi: 10.1111/j.1432-0436.2008.00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roig-Villanova I, Bou-Torrent J, Galstyan A, Carretero-Paulet L, Portolés S, Rodríguez-Concepción M, et al. Interaction of shade avoidance and auxin responses: a role for two novel atypical bHLH proteins. EMBO J. 2007;14(22):4756–67. [DOI] [PMC free article] [PubMed]
- 22.Leivar P, Monte E, Al-Sady B, Carle C, Storer A, Alonso JM, et al. The Arabidopsis phytochrome-interacting factor PIF7, together with PIF3 and PIF4, regulates responses to prolonged red light by modulating phyB levels. Plant Cell. 2008;20(2):337–52. 10.1105/tpc.107.052142. [DOI] [PMC free article] [PubMed]
- 23.Friedrichsen DM, Nemhauser J, Muramitsu T, Maloof JN, Alonso J, Ecker JR, et al. Three redundant brassinosteroid early response genes encode putative bHLH transcription factors required for normal growth. Genetics. 2002;162(3):1445–56. 10.1093/genetics/162.3.1445. [DOI] [PMC free article] [PubMed]
- 24.Lu R, Zhang J, Liu D, Wei YL, Wang Y, Li XB. Characterization of bHLH/HLH genes that are involved in brassinosteroid (BR) signaling in fiber development of cotton (Gossypium hirsutum) BMC Plant Biol. 2018;18(1):304. doi: 10.1186/s12870-018-1523-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Onohata T, Gomi K. Overexpression of jasmonate-responsive OsbHLH034 in rice results in the induction of bacterial blight resistance via an increase in lignin biosynthesis. Plant Cell Rep. 2020;39(9):1175–1184. doi: 10.1007/s00299-020-02555-7. [DOI] [PubMed] [Google Scholar]
- 26.Wang R, Zhao P, Kong N, Lu R, Pei Y, Huang C, et al. Genome-Wide Identification and Characterization of the Potato bHLH Transcription Factor Family. Genes (Basel). 2018;9(1):54. [DOI] [PMC free article] [PubMed]
- 27.Fu Y, Win P, Zhang H, Li C, Shen Y, He F, et al. PtrARF2.1 Is Involved in Regulation of Leaf Development and Lignin Biosynthesis in Poplar Trees. Int J Mol Sci. 2019;20(17):4141. [DOI] [PMC free article] [PubMed]
- 28.Li Z, Liu C, Zhang Y, Wang B, Ran Q, Zhang J. The bHLH family member ZmPTF1 regulates drought tolerance in maize by promoting root development and abscisic acid synthesis. J Exp Bot. 2019;70(19):5471–5486. doi: 10.1093/jxb/erz307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen HC, Hsieh-Feng V, Liao PC, Cheng WH, Liu LY, Yang YW, et al. The function of OsbHLH068 is partially redundant with its homolog, AtbHLH112, in the regulation of the salt stress response but has opposite functions to control flowering in Arabidopsis. Plant Mol Biol. 2017;94(4–5):531–48. 10.1007/s11103-017-0624-6. [DOI] [PMC free article] [PubMed]
- 30.Yuan Y, Wu H, Wang N, Li J, Zhao W, Du J, et al. FIT interacts with AtbHLH38 and AtbHLH39 in regulating iron uptake gene expression for iron homeostasis in Arabidopsis. Cell Res. 2008;18(3):385–97. 10.1038/cr.2008.26. [DOI] [PubMed]
- 31.Liu Y, Ji X, Nie X, Qu M, Zheng L, Tan Z, et al. Arabidopsis AtbHLH112 regulates the expression of genes involved in abiotic stress tolerance by binding to their E-box and GCG-box motifs. New Phytol. 2015;207(3):692–709. 10.1111/nph.13387. [DOI] [PubMed]
- 32.Zhao Q, Xiang X, Liu D, Yang A, Wang Y. Tobacco Transcription Factor NtbHLH123 Confers Tolerance to Cold Stress by Regulating the NtCBF Pathway and Reactive Oxygen Species Homeostasis. Front Plant Sci. 2018;9:381. doi: 10.3389/fpls.2018.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matus JT, Poupin MJ, Cañón P, Bordeu E, Alcalde JA, Arce-Johnson P. Isolation of WDR and bHLH genes related to flavonoid synthesis in grapevine (Vitis vinifera L.) Plant Mol Biol. 2010;72(6):607–620. doi: 10.1007/s11103-010-9597-4. [DOI] [PubMed] [Google Scholar]
- 34.Quattrocchio F, Wing JF, van der Woude K, Mol JN, Koes R. Analysis of bHLH and MYB domain proteins: species-specific regulatory differences are caused by divergent evolution of target anthocyanin genes. Plant J. 1998;13(4):475–488. doi: 10.1046/j.1365-313X.1998.00046.x. [DOI] [PubMed] [Google Scholar]
- 35.Chen F, Hu Y, Vannozzi A, Wu KC, Cai HY, Qin Y, et al. The WRKY transcription factor family in model plants and crops. Crit Rev Plant Sci. 2018;36(5):1–25.
- 36.Ali TM, Hasnain A. Morphological, physicochemical, and pasting properties of modified white Sorghum (Sorghum bicolor) starch. Int J Food Prop. 2014;17(3):523–535. doi: 10.1080/10942912.2012.654558. [DOI] [Google Scholar]
- 37.Pelpolage SW, Han K, Koaze H, et al. Influence of enzyme-resistant fraction of sorghum (Sorghum bicolor L.) flour on gut microflora composition, short-chain fatty acid production and toxic substance metabolism [J] J Food Nutr Res. 2019;58(2):135–145. [Google Scholar]
- 38.Xiong Y, Zhang P, Warner RD, Fang Z. Sorghum Grain: From Genotype, Nutrition, and Phenolic Profile to Its Health Benefits and Food Applications. Compr Rev Food Sci Food Saf. 2019;18(6):2025-46. [DOI] [PubMed]
- 39.Zhao ZY, Che P, Glassman K, Albertsen M. Nutritionally enhanced Sorghum for the arid and semiarid tropical areas of Africa. Methods Mol Biol. 1931;2019:197–207. doi: 10.1007/978-1-4939-9039-9_14. [DOI] [PubMed] [Google Scholar]
- 40.Han Y, Song L, Liu S, Zou N, Li Y, Qin Y, et al. Simultaneous determination of 124 pesticide residues in Chinese liquor and liquor-making raw materials (sorghum and rice hull) by rapid Multi-plug Filtration Cleanup and gas chromatography-tandem mass spectrometry. Food Chem. 2018;241:258–67. [DOI] [PubMed]
- 41.Prasad PV, Djanaguiraman M, Perumal R, Ciampitti IA. Impact of high temperature stress on floret fertility and individual grain weight of grain sorghum: sensitive stages and thresholds for temperature and duration. Front Plant Sci. 2015;6:820. doi: 10.3389/fpls.2015.00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prasad PVV, Pisipati SR, Mutava RN, Tuinstra MR. Sensitivity of grain Sorghum to high temperature stress during reproductive development [J] Crop Sci. 2008;48(5):1911–1917. doi: 10.2135/cropsci2008.01.0036. [DOI] [Google Scholar]
- 43.Tsuji W, Ali MEK, Inanaga S, Sugimoto Y. Growth and gas exchange of three Sorghum cultivars under drought stress [J] Biol Plant. 2003;46(4):583–587. doi: 10.1023/A:1024875814296. [DOI] [Google Scholar]
- 44.Rooney W L . Sorghum improvement—integrating traditional and new technology to produce improved genotypes [J]. Adv Agron, 2004, 83, 37–109, DOI: 10.1016/S0065-2113(04)83002-5.
- 45.Li H, Payne WA, Michels GJ, Rush CM. Reducing plant abiotic and biotic stress: drought and attacks of greenbugs, corn leaf aphids and virus disease in dryland sorghum. Environ Exp Bot. 2008;63(1–3):305–316. doi: 10.1016/j.envexpbot.2007.11.014. [DOI] [Google Scholar]
- 46.Paterson AH, Bowers JE, Bruggmann R, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(7229):551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 47.Song XM, Huang ZN, Duan WK, Ren J, Liu TK, Li Y, et al. Genome-wide analysis of the bHLH transcription factor family in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol Gen Genomics. 2014;289(1):77–91. 10.1007/s00438-013-0791-3. [DOI] [PubMed]
- 48.Sun H, Fan HJ, Ling HQ. Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genomics. 2015;16(1):9. doi: 10.1186/s12864-014-1209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kavas M, Baloğlu MC, Atabay ES, Ziplar UT, Daşgan HY, Ünver T. Genome-wide characterization and expression analysis of common bean bHLH transcription factors in response to excess salt concentration. Mol Gen Genomics. 2016;291(1):129–143. doi: 10.1007/s00438-015-1095-6. [DOI] [PubMed] [Google Scholar]
- 50.Mao K, Dong Q, Li C, Liu C, Ma F. Genome Wide Identification and Characterization of Apple bHLH Transcription Factors and Expression Analysis in Response to Drought and Salt Stress. Front Plant Sci. 2017;8:480. doi: 10.3389/fpls.2017.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao C, Sun J, Wang C, Dong Y, Xiao S, Wang X, et al. Genome-wide analysis of basic/helix-loop-helix gene family in peanut and assessment of its roles in pod development. PLoS One. 2017;12(7):e0181843. [DOI] [PMC free article] [PubMed]
- 52.Niu X, Guan Y, Chen S, Li H. Genome-wide analysis of basic helix-loop-helix (bHLH) transcription factors in Brachypodium distachyon. BMC Genomics. 2017;18(1):619. doi: 10.1186/s12864-017-4044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang T, Lv W, Zhang H, Ma L, Li P, Ge L, et al. Genome-wide analysis of the basic Helix-Loop-Helix (bHLH) transcription factor family in maize. BMC Plant Biol. 2018;18(1):235. [DOI] [PMC free article] [PubMed]
- 54.Wei K, Chen H. Comparative functional genomics analysis of bHLH gene family in rice, maize and wheat. BMC Plant Biol. 2018;18(1):309. doi: 10.1186/s12870-018-1529-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng X, Xiong R, Liu H, Wu M, Chen F. Hanwei Yan, Xiang Y. basic helix-loop-helix gene family: genome wide identification, phylogeny, and expression in Moso bamboo. Plant Physiol Biochem. 2018;132:104–119. doi: 10.1016/j.plaphy.2018.08.036. [DOI] [PubMed] [Google Scholar]
- 56.Yingqi H, Ahmad N, Yuanyuan T, Jianyu L, Liyan W, Gang W, et al. Genome-Wide Identification, Expression Analysis, and Subcellular Localization of Carthamus tinctorius bHLH Transcription Factors. Int J Mol Sci. 2019;20(12):3044. [DOI] [PMC free article] [PubMed]
- 57.Li H, Gao W, Xue C, Zhang Y, Liu Z, Zhang Y, et al. Genome-wide analysis of the bHLH gene family in Chinese jujube (Ziziphus jujuba Mill.) and wild jujube. BMC Genomics. 2019;20(1):568. [DOI] [PMC free article] [PubMed]
- 58.Zhang Z, Chen J, Liang C, Liu F, Hou X, Zou X. Genome-Wide Identification and Characterization of the bHLH Transcription Factor Family in Pepper (Capsicum annuum L.) Front Genet. 2020;11:570156. doi: 10.3389/fgene.2020.570156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu L, Zhao M, Chen M, Li L, Jiang Y, Liu S, et al. The bHLH gene family and its response to saline stress in Jilin ginseng, Panax ginseng C.a. Meyer. Mol Gen Genomics. 2020;295(4):877–90. 10.1007/s00438-020-01658-w. [DOI] [PubMed]
- 60.Aslam M, Jakada BH, Fakher B, Greaves JG, Niu X, Su Z, et al. Genome-wide study of pineapple (Ananas comosus L.) bHLH transcription factors indicates that cryptochrome-interacting bHLH2 (AcCIB2) participates in flowering time regulation and abiotic stress response. BMC Genomics. 2020;21(1):735. [DOI] [PMC free article] [PubMed]
- 61.Sun W, Jin X, Ma Z, Chen H, Liu M. Basic helix-loop-helix (bHLH) gene family in Tartary buckwheat (Fagopyrum tataricum): Genome-wide identification, phylogeny, evolutionary expansion and expression analyses. Int J Biol Macromol. 2020;155:1478–1490. doi: 10.1016/j.ijbiomac.2019.11.126. [DOI] [PubMed] [Google Scholar]
- 62.Zhao Y, Li X, Chen W, Peng X, Cheng X, Zhu S, et al. Whole-genome survey and characterization of MADS-box gene family in maize and sorghum [J]. Plant Cell Tissue Org Cult. 2011;105(2):159–73. 10.1007/s11240-010-9848-8.
- 63.Kushwaha H, Gupta S, Singh VK, Rastogi S, Yadav D. Genome wide identification of Dof transcription factor gene family in sorghum and its comparative phylogenetic analysis with rice and Arabidopsis. Mol Biol Rep. 2011;38(8):5037–5053. doi: 10.1007/s11033-010-0650-9. [DOI] [PubMed] [Google Scholar]
- 64.Yan HW, Hong L, Zhou YQ, Jiang HY, Zhu SW, Fan J, et al. A genome-wide analysis of the ERF gene family in sorghum. Genet Mol Res. 2013;12(2):2038–55. [DOI] [PubMed]
- 65.Chang JZ, Yan FX, Qiao LY, Zheng J, Zhang FY, Liu QS. Genome-wide identification and expression analysis of SBP-box gene family in Sorghum bicolor L. Yi Chuan. 2016;38(6):569–580. doi: 10.16288/j.yczz.16-008. [DOI] [PubMed] [Google Scholar]
- 66.Nagaraju M, Reddy PS, Kumar SA, Kumar A, Rajasheker G, Rao DM, et al. Genome-wide identification and transcriptional profiling of small heat shock protein gene family under diverse abiotic stress conditions in Sorghum bicolor (L.). Int J Biol Macromol. 2020;142:822–34. [DOI] [PubMed]
- 67.Nagaraju M, Kumar SA, Reddy PS, Kumar A, Rao DM, Kavi Kishor PB. Genome-scale identification, classification, and tissue specific expression analysis of late embryogenesis abundant (LEA) genes under abiotic stress conditions in Sorghum bicolor L. PLoS One. 2019;14(1):e0209980. doi: 10.1371/journal.pone.0209980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanjari S, Shirzadian-Khorramabad R, Shobbar ZS, Shahbazi M. Systematic analysis of NAC transcription factors' gene family and identification of post-flowering drought stress responsive members in sorghum. Plant Cell Rep. 2019;38(3):361–376. doi: 10.1007/s00299-019-02371-8. [DOI] [PubMed] [Google Scholar]
- 69.Chunxia Zhang, Mingdi Bian, Hui Yu, Qing Liu, Zhenming Yang, (2011) Identification of alkaline stress-responsive genes of CBL family in sweet sorghum (Sorghum bicolor L.). Plant Physiology and Biochemistry 49 (11):1306-1312 [DOI] [PubMed]
- 70.Geng J, Liu JH. The transcription factor CsbHLH18 of sweet orange functions in modulation of cold tolerance and homeostasis of reactive oxygen species by regulating the antioxidant gene. J Exp Bot. 2018, 27, 69 (10), 2677-2692. [DOI] [PMC free article] [PubMed]
- 71.Dubos C, Le Gourrierec J, Baudry A, Huep G, Lanet E, Debeaujon I, et al. MYBL2 is a new regulator of flavonoid biosynthesis in Arabidopsis thaliana. Plant J. 2008;55(6):940–53. 10.1111/j.1365-313X.2008.03564.x. [DOI] [PubMed]
- 72.Chandler JW, Cole M, Flier A, Werr W. BIM1, a bHLH protein involved in brassinosteroid signalling, controls Arabidopsis embryonic patterning via interaction with DORNROSCHEN and DORNROSCHEN-LIKE. Plant Mol Biol. 2009;69(1–2):57–68. doi: 10.1007/s11103-008-9405-6. [DOI] [PubMed] [Google Scholar]
- 73.Yin Y, Vafeados D, Tao Y, Yoshida S, Asami T, Chory J. A new class of transcription factors mediates brassinosteroid-regulated gene expression in Arabidopsis. Cell. 2005;120(2):249–259. doi: 10.1016/j.cell.2004.11.044. [DOI] [PubMed] [Google Scholar]
- 74.Henriksson M, Lüscher B. Proteins of the Myc network: essential regulators of cell growth and differentiation. Adv Cancer Res. 1996;68:109–182. doi: 10.1016/S0065-230X(08)60353-X. [DOI] [PubMed] [Google Scholar]
- 75.Carretero-Paulet L, Galstyan A, Roig-Villanova I, Martínez-García JF, Bilbao-Castro JR, Robertson DL. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 2010;153(3):1398–1412. doi: 10.1104/pp.110.153593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci U S A. 2003;100(20):11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4:10. doi: 10.1186/1471-2229-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mehan MR, Freimer NB, Ophoff RA. A genome-wide survey of segmental duplications that mediate common human genetic variation of chromosomal architecture. Hum Genomics. 2004;1(5):335–344. doi: 10.1186/1479-7364-1-5-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hudson KA, Hudson ME. A classification of basic helix-loop-helix transcription factors of soybean. Int J Genomics. 2015;2015:603182. doi: 10.1155/2015/603182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gremski K, Ditta G, Yanofsky MF. The HECATE genes regulate female reproductive tract development in Arabidopsis thaliana. Development. 2007;134(20):3593–3601. doi: 10.1242/dev.011510. [DOI] [PubMed] [Google Scholar]
- 81.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed]
- 82.Liu M, Ma Z, Wang A, Zheng T, Huang L, Sun W, et al. Genome-Wide Investigation of the Auxin Response Factor Gene Family in Tartary Buckwheat (Fagopyrum tataricum). Int J Mol Sci. 2018;19(11):3526. [DOI] [PMC free article] [PubMed]
- 83.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2000;28(1):263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46(D1):D493–D496. doi: 10.1093/nar/gkx922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002; Chapter 2: Unit 2. 3. [DOI] [PubMed]
- 87.Guo AY, Zhu QH, Chen X, Luo JC. GSDS: a gene structure display server. Yi Chuan. 2007;29(8):1023–1026. doi: 10.1360/yc-007-1023. [DOI] [PubMed] [Google Scholar]
- 88.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–8. [DOI] [PMC free article] [PubMed]
- 89.Xie T, Chen C, Li C, Liu J, Liu C, He Y. Genome-wide investigation of WRKY gene family in pineapple: evolution and expression profiles during development and stress. BMC Genomics. 2018;19(1):490. doi: 10.1186/s12864-018-4880-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu M, Ma Z, Sun W, Huang L, Wu Q, Tang Z, et al. Genome-wide analysis of the NAC transcription factor family in Tartary buckwheat (Fagopyrum tataricum). BMC Genomics. 2019;20(1):113. [DOI] [PMC free article] [PubMed]
- 91.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed]
- 92.Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49. 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed]
- 93.Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8(1):77–80. doi: 10.1016/S1672-0229(10)60008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sudhakar Reddy P, Srinivas Reddy D, Sivasakthi K, Bhatnagar-Mathur P, Vadez V, Sharma KK. Evaluation of Sorghum [Sorghum bicolor (L.)] Reference Genes in Various Tissues and under Abiotic Stress Conditions for Quantitative Real-Time PCR Data Normalization. Front Plant Sci. 2016;7:529. doi: 10.3389/fpls.2016.00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.海姆MA, 雅各布M, 韦博M, 马丁C, 魏斯哈尔B, 贝利PC. 植物中的基本螺旋-环-螺旋转录因子家族:蛋白质结构和功能多样性的全基因组研究. Mol Biol Evol. 2003;20(5):735–47.
- 96.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (−Delta Delta C (T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Table S1. List of the 174 S. bicolor bHLH genes identified in this study.
Additional file 2 Table S2. Analysis and distribution of the conserved motifs in S. bicolor bHLH proteins.
Additional file 3 Table S3. Tandem duplication events of S. bicolor bHLH genes.
Additional file 4 Table S4. The 42 pairs of segmental duplications in S. bicolor bHLH genes.
Additional file 5 Table S5. One-to-one orthologous genes relationships between S. bicolor and other plants.
Additional file 6 Table S6. Primer sequences for qRT-PCR.
Data Availability Statement
The entire Sorghum bicolor genome sequence information was from the Ensembl Genomes website (http://ensemblgenomes.org/). The Sorghum bicolor materials (Hongyingzi) used in the experiment were supplied by Prof. Cheng Jianping of Guizhou University. The datasets supporting the conclusions of this article are included in the article and its Additional files.