

Abstract
A cyclopenta-fused macrocyclic tetraradicaloid, MC4-S, containing alternating phenanthrene (Phen) and dibenzo[b,d]thiophene (DBTh) units was synthesized and isolated in single-crystal form. Compared with its all-carbon isoelectronic structure, CPTP-M, the incorporation of two sulfur atoms leads to a smaller radical character and a larger singlet–triplet energy gap. X-ray crystallographic analysis reveals that the spin–spin coupling through the DBTh unit is stronger than that through the Phen moiety. In addition, the electron-rich sulfur atoms also raise the energies of both the HOMO and LUMO in MC4-S, but the overall optical and electronic energy gaps are close to that of the CPTP-M. MC4-S displays global anti-aromaticity according to the NMR measurements and theoretical calculations (NICS, ACID and 2D ICSS), with a 36π ring current circuit along the all-carbon periphery excluding the two sulphur atoms. Its dication becomes globally aromatic due to the existence of a dominant 34π-conjugation pathway. This study sheds some light on the effect of heteroatoms on the electronic properties of open-shell polyradicaloids.
The first member of sulfur-heterocycloarene neutral tetraradicaloids, MC4-S, was synthesized in crystalline form, which displays strong global anti-aromaticity and unique properties.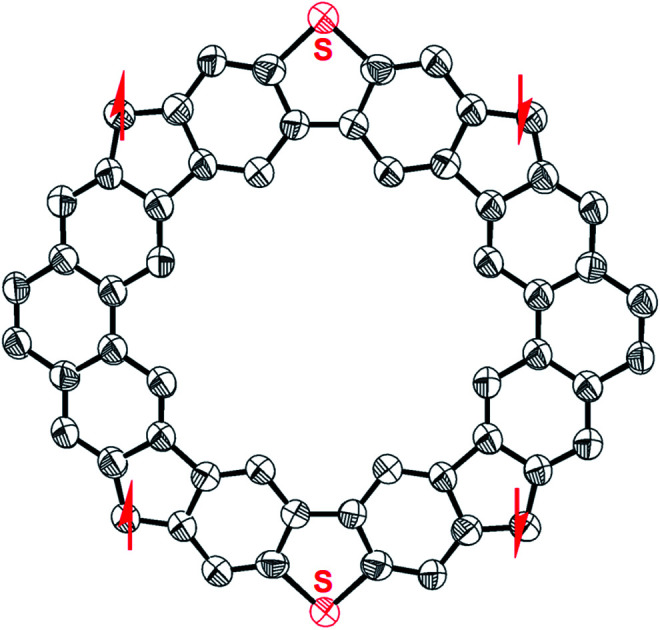
Introduction
Cycloarenes are polycyclic aromatic hydrocarbons consisting of cata-fused benzene rings in a macrocyclic form.1 Kekulene,2 cyclo[d,e,d,e,e,d,e,d,e,e]decakisbenzene,3 septulene,4 and octulene5 are the representative examples of cycloarenes that have been synthesized as model compounds to examine the potential annulene-within-annulene (AWA) electronic structure.6 However, 1H NMR spectra of these synthesized cycloarenes exhibit de-shielded inner protons, suggesting that cycloarenes do not show global aromaticity, but with π-electrons predominantly localized at individual benzene rings.7 On the other hand, our recent studies demonstrated that cyclopenta (CP)-fused cycloarene analogues could show global (anti)aromaticity and even the long-sought AWA superstructure.8 More recently, Stępień's group9 and our group10 independently reported the same cycloarene-like tetraradicaloid, CPTP-M (Fig. 1a), which displayed open-shell radical character and global anti-aromaticity. Replacing one or more benzenoid rings with heterocycles would lead to heterocycloarene-based polyradicaloids, which can be regarded as the isoelectronic structures of the all-carbon analogues but may exhibit different electronic properties. So far, only a handful of cycloarene-based polyradicaloids9,10 and N-heterocycloarene-based polyradicaloids11 have been synthesized. Herein, we report a new CP-fused, sulfur-containing macrocyclic tetraradicaloid, MC4-S, which contains alternating dibenzo[b,d]thiophene (DBTh) and phenanthrene (Phen) units (Fig. 1a). Its conjugated backbone is an isoelectronic structure of that of CPTM-M, and five representative resonance structures can be drawn (Fig. 1b): an open-shell tetraradical form (A), a closed-shell form containing two aromatic Phen units and two quinoidal DBTh units (B), another closed-shell form possessing two quinoidal Phen units and two aromatic DBTh units (C), and two open-shell diradical forms with quinoidal DBTh (D) or Phen (E) units. The contribution of each form to the ground-state electronic structure will be determined by electronic coupling between the neighboring spins. The recovery of two/four Clar's aromatic sextets from the closed-shell to the open-shell diradical/tetraradical form would be the major driving force for the possible open-shell diradical/tetraradical character. In addition, the different spin–spin interactions through the DBTh and Phen unit and the electron-rich character of the sulfur atoms would lift the degeneracy in CPTP-M and result in different electronic structures and physical properties.
Fig. 1. (a) From an all-carbon macrocyclic tetraradicaloid, CMTP-M, to a new S-containing macrocyclic tetraradicaloid, MC4-S. (b) Representative resonance forms of the π-conjugated backbone of MC4-S. Clar's aromatic sextets are shaded in blue color.

Results and discussion
Synthesis
Compound MC4-S was synthesized according to Scheme 1. Suzuki coupling reaction between 1 (ref. 9 and 10) and 2 under optimal conditions (using Pd2(dba)3-[(t-Bu)3PH]BF4 as the catalyst) gave the macrocyclic precursor 3 containing four aldehyde groups in 32% yield after separation by preparative recycling gel permeation chromatography. Then, the solution of the tetraaldehyde 3 in dry tetrahydrofuran (THF) was treated with 2-mesitylmagnesium bromide at room temperature to generate the intermediate tetracarbinol. Without further purification, the crude product was subjected to BF3·Et2O mediated Friedel–Crafts type cyclization in dichloromethane (DCM) to provide the intramolecular-cyclized tetrahydro compound 4. Finally, the oxidative dehydrogenation of 4 with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) in toluene at room temperature afforded the target compound MC4-S in nearly quantitative yield. MC4-S is a stable compound and can be purified by routine silica gel column chromatography.
Scheme 1. Synthetic route of MC4-S: (i) Pd2(dba)3, [(t-Bu)3PH]BF4, NaHCO3, THF/H2O, 80 °C; (ii) (1) MesMgBr, THF; (2) BF3·OEt2, DCM; (iii) DDQ, toluene.

X-ray crystallographic analysis
The single crystals of MC4-S were obtained by slow diffusion of acetonitrile into the DCM solution, and the X-ray diffraction data were collected at 100 K.12 The crystal is aligned in a triclinic unit cell with the space group P1̄, and the asymmetric unit contains half a molecule of MC4-S. As shown in Fig. 2a and b, the molecule possesses a rigid, slightly distorted coplanar π-conjugated skeleton. The dihedral angles between the backbone and the mesityl substituents are found to be 61.3° or 78.7°. The backbone has an overall rectangle-like geometry, with nearly the same interior angles, ∠C28–C13–C28’ = 89.83° and ∠C13–C28–C13’ = 90.17°, but the distance of C13–C28 (9.781 Å) is significantly shorter than that of C13′–C28 (9.941 Å). For comparison, CPTP-M has a nearly square-like backbone, with a side length of about 9.88 Å.10 It is also noted that the bond lengths of the C3–C28 (1.416 Å) and C10–C13 (1.416 Å) in the CP rings linking the DBTh units are significantly shorter than those of the C26–C28 (1.454 Å) and C14–C13 (1.440 Å) in the CP rings linking the Phen units (Fig. 2c). In addition, significant bond length alternation was observed for the two benzenoid rings in the DBTh unit, showing a dominant quinoidal structure as shown in resonance form B in Fig. 1. Moreover, the harmonic oscillator model of aromaticity (HOMA) calculations13 (Fig. 2c) on individual rings revealed smaller HOMA values (0.63/0.63) for the sextet rings in the DBTh unit than that in the Phen unit (0.69/0.82). All these suggest that the anti-ferromagnetic coupling between the neighbouring spin centers through the DBTh spacer is stronger than that through the Phen spacer. This means that MC4-S prefers form B rather than form C. On the other hand, the lengths of the C–C bonds (C3–C28, C26–C28, C10–C13, and C14–C13) in the CP rings linking the Phen and DBTh units all are much longer than that of the typical olefins (1.33–1.34 Å), implying that there are significant contributions from open-shell tetraradical form A and diradical forms D/E to the ground-state electronic structure. DFT calculations are also in consistent with the single crystal structure, indicating that form B is the most preferred (see also Fig. S9 and Table S3† and discussion afterwards).14
Fig. 2. X-ray crystallographic structure of MC4-S: (a) top view with labels; (b) side view; (c) selected bond lengths (in Å) of the backbone. The red numbers in rings A–H are the calculated HOMA values. Hydrogen atoms and aryl/alkoxy substituents are omitted for clarity.

Magnetic properties and multiradical character
The 1H NMR spectrum of MC4-S in CD2Cl2 at room temperature showed broad and weak signals for the protons on the conjugated backbone, while the peaks began to emerge and sharpen upon decreasing the temperature (Fig. 3a). This is a typical phenomenon for many open-shell singlet diradicaloids, in which the NMR broadening is due to the existence of thermally populated triplet species.15 At 193 K, all sharp peaks appeared, and the protons were assigned to the desired structure according to DFT calculations and with the assistance of the 2D ROESY spectrum at low temperature (Table S4 and Fig. S13 in the ESI†). Noticeably, the inner-rim protons a and b are highly de-shielded (δ = 15.48 and 14.92 ppm, respectively), while the outer-rim protons c and d are highly shielded (δ = 4.62 and 4.08 ppm, respectively). In addition, the aromatic protons in the mesityl groups (proton e) and butoxyl groups (proton f) are also shielded (δ = 6.53 and 3.17 ppm, respectively). All these indicate that MC4-S has a strong global anti-aromatic nature (vide infra).
Fig. 3. (a) VT 1H NMR spectra of MC4-S in CD2Cl2 (500 MHz); the peaks labelled * indicate residual solvent. The resonance assignment refers to the structure shown in Scheme 1. (b) I × T–T plot of MC4-S in frozen DCM and the red line is the fitted curve. The inset shows the VT ESR spectra in DCM solution.

Intense and broad ESR signals were observed in DCM at room temperature and even at low temperature (−150 °C), with ge = 2.003 (Fig. 3b), which was typically observed for largely delocalized carbon-centered radicals. The magnetic properties of MC4-S were further investigated by variable-temperature (VT) ESR measurements (Fig. 3a), and the signal intensity (I) in frozen DCM gradually decreased as the temperature (T) was lowered, which is consistent with its open-shell singlet ground state. Fitting of the VT-ESR data using the Bleaney–Bowers equation16 gave a singlet–triplet gap (ΔES–T) of −3.47 kcal mol−1 (Fig. 3b), which is slightly larger than that of the all carbon tetraradicaloid CPTP-M (ΔES–T = −3.04 kcal mol−1) under the same measurement conditions (Fig. S1 in the ESI†). This is also in agreement with the observation that the 1H NMR spectrum of CPTP-M in d8-THF was still significantly broadened even at low temperature (193 K).10 Therefore, replacement of two Phen units by DBTh moieties led to an increased singlet–triplet energy gap.
The radical character and vertical excitation energies of MC4-S were calculated by the restricted active space spin flip (RAS-SF/6-31G*) method,17 which has been demonstrated to be a good method to deal with strongly correlated electrons. Calculations estimate that the excitation energies from the singlet ground state to the lowest triplet and quartet states are 1.61 and 3.04 kcal mol−1, respectively. Natural orbital occupation number analysis of the singlet state reveals large occupancy of the first and second lowest unoccupied natural orbitals (LUNOs), with nLUNO = 0.72 and nLUNO+1 = 0.49 (Fig. 4a), which are significantly smaller than those in CPTP-M (nLUNO = 1.00 and nLUNO+1 = 0.76).10 The number of unpaired electrons (NU) as a quantitative measure of the radical character was calculated to be 2.0, which is smaller than that of CPTP-M (NU = 3.52). Therefore, incorporation of two S atoms led to a smaller radical character. This could be due to the partial charge transfer character from the electron-donating sulfur atom to the electron-deficient quinoidimethane moiety. In addition, the strong antiferromagnetic spin–spin coupling through the DBTh unit also decreases the radical character. The calculated unpaired electron densities of frontier natural orbital profiles (Fig. 4a) as well as spin densities (Fig. 4b) are delocalized throughout the whole backbone, which can explain the good stability of MC4-S well.
Fig. 4. (a) Calculated (RAS-SF/6-31G*) frontier natural orbital profiles of MC4-S at the open-shell singlet ground state. The numbers in the parentheses are the calculated occupation numbers. (b) Calculated (UB3LYP/6-31G**) spin density distribution map of MC4-S at the open-shell singlet state.

Optical properties, electrochemical properties, and global (anti)aromaticity
Compound MC4-S in DCM showed an intense absorption with a maximum (λmax) at 678 nm, together with a long tail extending to 1150 nm (Fig. 5a). The spectrum is similar to that of CPTP-M, but the major absorption peaks are slightly blue shifted (see comparison in Fig. S2 in the ESI†). The broad and weak tail is consistent with its anti-aromaticity and large radical character, and the spectrum is somehow in agreement with the time-dependent DFT calculations (Fig. S4 in the ESI†). The optical energy gap (Eoptg) was estimated to be 1.08 eV from the lowest energy absorption onset, close to that of CPTP-M (Eoptg = 1.09 eV). MC4-S exhibited four quasi-reversible oxidation waves with Eox1/2 at −0.39, −0.26, 0.44 and 0.70 V (vs. Fc+/Fc), respectively, and four quasi-reversible reduction waves with Ered1/2 at −1.44, −1.73, −1.88, and −2.09 V, respectively (Fig. 5b). The HOMO and LUMO energy levels were estimated to be −4.37 eV and −3.53 eV from the onset of the first oxidation and reduction wave, respectively, and the electrochemical energy gap (EECg = LUMO − HOMO) was thus small (0.84 eV). For comparison, CPTP-M has a lower lying HOMO (−4.52 eV) and LUMO (−3.67 eV), with a slightly larger electrochemical energy gap (EECg = 0.85 eV).10 Therefore, incorporation of two sulfur atoms raises the HOMO and LUMO energy levels. The stability of MC4-S in DCM at room temperature under ambient air and light conditions was monitored by UV-vis-NIR measurements (Fig. S3 in the ESI†) and a half-lifetime (t1/2) of about 136 hours was estimated, which is shorter than that of CPTP-M (t1/2 = 424 hours) under the same conditions (Fig. S3 in the ESI†). Such a difference can be explained by the higher lying HOMO of MC4-S containing two electron-rich sulfur atoms. Chemical oxidation of MC4-S with two equivalents of oxidant NO·SbF6 in DCM gave its corresponding dication, which showed two intense absorption bands with λmax = 777 and 1408 nm, respectively (Fig. 5a), and the observed spectrum is in agreement with the TD DFT calculation (Fig. S5 in the ESI†).
Fig. 5. (a) UV-Vis-NIR absorption spectra of MC4-S and its dication (MC4-S2+) in DCM. The inset shows photos of the solutions upon charging. (b) Cyclic voltammogram (CV) and differential pulse voltammogram (DPV) of MC4-S in DCM with 0.1 M n-Bu4N·PF6 as the supporting electrolyte.

The pure dication of MC4-S was isolated, and its 1H NMR spectrum was sharp at room temperature (Fig. 6), indicating a closed-shell singlet ground state. The assignment was conducted with the assistance of the 2D NOESY technique as well as DFT calculation (Fig. S15, S16 and Table S4 in the ESI†), and the inner-rim protons a and b are highly shielded (δ = −4.71 and −4.91 ppm, respectively) while the outer-rim protons c and d are highly de-shielded (δ = 13.73 and 13.17 ppm, respectively) (Fig. 6 and S15 in the ESI†). In addition, the aromatic protons in the mesityl groups (proton e) and butoxyl groups (proton f) are also de-shielded (δ = 8.09 and 5.84 ppm, respectively). This phenomenon is opposite to that of the neutral compound and suggests that MC4-S2+ now has a global aromatic nature.
Fig. 6. 1H NMR spectra of MC4-S (measured at 183 K) and its dication MC4-S2+ (measured at 298 K) in CD2Cl2; the peaks labelled * indicate residual solvent. The dashed lines show the change of the chemical shifts of the same protons.

To better understand the observed global (anti)aromaticity in the neutral compound and dication, nucleus independent chemical shift (NICS),18 anisotropy of the induced current density (ACID),19 and 2D iso-chemical shielding surface (ICSS)20 calculations were performed (Fig. 7). MC4-S shows a large positive NICS(0) value (+15.39 ppm) in the geometric center, while the dication MC4-S2+ has a largely negative NICS(0) value (−25.98 ppm), implying the global anti-aromatic and aromatic character, respectively.
Fig. 7. (a) Calculated ACID plots (contribution from π electrons only) of MC4-S (a) and its dication MC4-S2+ (b) in the singlet ground state. The magnetic field is perpendicular to the XY plane and points out through the paper. The blue and red arrows indicate the counter-clockwise (paramagnetic) and clockwise (diamagnetic) current flow, respectively. Calculated 2D-ICSS maps of MC4-S (c) and its dication MC4-S2+ (d). The top and bottom images are mapped at the XY and XZ planes, respectively.

The ACID plot of the backbone of MC4-S (Fig. 7a) also clearly displays a counter-clockwise (paratropic) ring current circuit along the periphery, and a 36π conjugation pathway can be drawn (Fig. S6 in the ESI†). Therefore, the molecule is globally anti-aromatic following Hückel's rule. On the other hand, the ACID plot of the dication MC4-S2+ exhibits a clockwise (diatropic) ring current flow along the periphery (Fig. 7b), and a 34π conjugation pathway can be drawn along the conjugated pathway (Fig. S6 in the ESI†), indicating its global aromaticity. In both cases, the ring current circuits bypass the sulphur atoms. The 2D-ICSS maps further confirm an inverse of aromaticity from MC4-S to MC4-S2+ (Fig. 7c and d). For the backbone of MC4-S, the inner-rim protons are located in a de-shielding chemical environment (negative ICSS(1)zz value), whereas the outer-rim protons are in a shielding chemical environment (positive ICSS(1)zz value) (Fig. 7c), which is also in agreement with NMR experimental observations. The shielding/de-shielding environments just became inverted in the dication MC4-S2+ (Fig. 7d).
Conclusions
In summary, we have successfully synthesized a stable cyclopenta-fused macrocyclic tetraradicaloid, MC4-S, containing alternating dibenzo[b,d]thiophene and phenanthrene units. The incorporation of two electron-rich sulfur atoms resulted in a small radical character and a larger singlet–triplet energy gap compared with the all-carbon analogue CPTP-M. It also raised the HOMO and LUMO energy levels. However, the optical and electrochemical energy gaps are nearly the same. It was also found that the intramolecular spin–spin coupling through the DBTh spacer is stronger than that via the Phen spacer. The global aromaticity ↔ anti-aromaticity switch was well demonstrated between the neutral compound and its dication. These results gave us a better understanding of the effect of the heteroatom on the electronic properties of open-shell diradicaloids and polyradicaloids.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
X. L. acknowledges financial support from the National Natural Science Foundation of China (51903052, 52073063 and 61890940), the Shanghai Pujiang Project (19PJ1400700) and the Program for Professor of Special Appointment (Eastern Scholar) at the Shanghai Institutions of Higher Learning. J. W. acknowledges financial support from the MOE Tier 2 (MOE2018-T2-2-094) and NRF Investigatorship Award (NRF-NRFI05-2019-0005).
Electronic supplementary information (ESI) available: Synthetic procedures and characterization data of all new compounds; details for all physical characterization and theoretical calculations; and additional spectroscopic data. CCDC 1965057. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06185a
Notes and references
- (a) Buttrick J. C. King B. T. Chem. Soc. Rev. 2017;46:7. doi: 10.1039/C6CS00174B. [DOI] [PubMed] [Google Scholar]; (b) Miyoshi H. Nobusue S. Shimizu A. Tobe Y. Chem. Soc. Rev. 2015;44:6560. doi: 10.1039/C5CS00185D. [DOI] [PubMed] [Google Scholar]
- Staab H. A. Diederich F. Chem. Ber. 1983;116:3487. doi: 10.1002/cber.19831161021. [DOI] [Google Scholar]
- Funhoff D. J. H. Staab H. A. Angew. Chem. Int. Ed. 1986;25:742. doi: 10.1002/anie.198607421. [DOI] [Google Scholar]
- Kumar B. Viboh R. L. Bonifacio M. C. Thompson W. B. Buttrick J. C. Westlake B. C. Kim M.-S. Zoellner R. W. Varganov S. A. Mörschel P. Teteruk J. Schmidt M. U. King B. T. Angew. Chem. Int. Ed. 2012;51:12795. doi: 10.1002/anie.201203266. [DOI] [PubMed] [Google Scholar]
- (a) Majewski M. A. Hong Y. Lis T. Gregoliński J. Chmielewski P. J. Cybińska J. Kim D. Stępień M. Angew. Chem. Int. Ed. 2016;55:14072. doi: 10.1002/anie.201608384. [DOI] [PubMed] [Google Scholar]; (b) Fan W. Han Y. Dong S. Li G. Wu J. CCS Chem. 2020;2:1445. doi: 10.31635/ccschem.020.202000356. [DOI] [Google Scholar]; (c) Yang L. Zhang N. Han Y. Zou Y. Qiao Y. Chang D. Zhao Y. Lu X. Wu J. Liu Y. Chem. Commun. 2020;56:9990. doi: 10.1039/D0CC04289G. [DOI] [PubMed] [Google Scholar]
- Stępień M. Chem. 2018;4:1481. [Google Scholar]
- (a) Clar E., Aromatic Sextet, John Wiley & Sons Ltd., 1972 [Google Scholar]; (b) Randić M. Chem. Rev. 2003;103:3449. doi: 10.1021/cr9903656. [DOI] [PubMed] [Google Scholar]
- Liu C. Sandoval-Salinas M. E. Hong Y. Gopalakrishna T. Y. Phan H. Aratani N. Herng T. S. Ding J. Yamada H. Kim D. Casanova D. Wu J. Chem. 2018;4:1586. [Google Scholar]
- Gregolińska H. Majewski M. Chmielewski P. J. Gregoliński J. Chien A. Zhou J. Wu Y.-L. Bae Y. J. Wasielewski M. R. Zimmerman P. M. Stępień M. J. Am. Chem. Soc. 2018;140:14474. doi: 10.1021/jacs.8b09385. [DOI] [PubMed] [Google Scholar]
- Lu X. Gopalakrishna T. Y. Phan H. Herng T. S. Jiang Q. Liu C. Li G. Ding J. Wu J. Angew. Chem., Int. Ed. 2018;57:13052. doi: 10.1002/anie.201807185. [DOI] [PubMed] [Google Scholar]
- Das S. Herng T. S. Zafra J. L. Burrezo P. M. Kitano M. Ishida M. Gopalakrishna T. Y. Hu P. Osuka A. Casado J. Ding J. Casanova D. Wu J. J. Am. Chem. Soc. 2016;138:7782. doi: 10.1021/jacs.6b04539. [DOI] [PubMed] [Google Scholar]
- Crystallographic data for compound MC4-S (CCDC no.: 1965057†) was deposited in the Cambridge Crystallographic Data Center (CCDC)
- Kruszewski J. Krygowski T. M. Tetrahedron Lett. 1972;13:3839. doi: 10.1016/S0040-4039(01)94175-9. [DOI] [Google Scholar]
- Nobusue S. Miyoshi H. Shimizu A. Hisaki I. Fukuda K. Nakano M. Tobe Y. Angew. Chem. Int. Ed. 2015;54:2090. doi: 10.1002/anie.201410791. [DOI] [PubMed] [Google Scholar]
- (a) Sun Z. Zeng Z. Wu J. Acc. Chem. Res. 2014;47:2582. doi: 10.1021/ar5001692. [DOI] [PubMed] [Google Scholar]; (b) Zeng Z. Shi X. Chi C. Lopez Navarrete J. T. Casado J. Wu J. Chem. Soc. Rev. 2015;44:6578. doi: 10.1039/C5CS00051C. [DOI] [PubMed] [Google Scholar]; (c) Kubo T. Chem. Lett. 2015;44:111. doi: 10.1246/cl.140997. [DOI] [Google Scholar]; (d) Gopalakrishna T. Y. Zeng W. Lu X. Wu J. Chem. Commun. 2018;54:2186. doi: 10.1039/C7CC09949E. [DOI] [PubMed] [Google Scholar]; (e) Liu C. Ni Y. Lu X. Li G. Wu J. Acc. Chem. Res. 2019;52:2309. doi: 10.1021/acs.accounts.9b00257. [DOI] [PubMed] [Google Scholar]; (f) Zeng W. Wu J. Chem. 2021 doi: 10.1016/j.chempr.2020.10.009. [DOI] [Google Scholar]
- Bleaney B. Bowers K. D. Proc. R. Soc. London Ser. A. 1952;214:451. [Google Scholar]
- Casanova D. Head-Gordon M. Phys. Chem. Chem. Phys. 2009;11:9779. doi: 10.1039/B911513G. [DOI] [PubMed] [Google Scholar]
- Chen Z. Wannere C. S. Corminboeuf C. Puchta R. Schleyer P. v. R. Chem. Rev. 2005;105:3842. doi: 10.1021/cr030088+. [DOI] [PubMed] [Google Scholar]
- Geuenich D. Hess K. Köhler F. Herges R. Chem. Rev. 2005;105:3758. doi: 10.1021/cr0300901. [DOI] [PubMed] [Google Scholar]
- (a) Klod S. Kleinpeter E. J. Chem. Soc., Perkin Trans. 2001;2:1893. [Google Scholar]; (b) Lu T. Chen F. J. Comput. Chem. 2012;33:580. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.